

Abstract
In recent years, studies in the gastrointestinal (GI) mucosa have taught us a number of important lessons related to tissue oxygenation and metabolism in health and disease. The highly vascularized mucosa lies immediately adjacent to an anaerobic lumen containing trillions of metabolically active microbes (i.e. the microbiome) that results in one of the more austere tissue microenvironments in the body. These studies have also implicated a prominent role for oxygen metabolism and hypoxia in inflammation, so called “inflammatory hypoxia”, that results from the activation of multiple oxygen consuming enzymes. Inflammation-associated shifts in the composition of the microbiome and microbial-derived metabolites have revealed a prominent role for the transcription factor hypoxia-inducible factor (HIF) in the regulation of key target genes that promote inflammatory resolution. Analyses of these pathways have provided a multitude of opportunities for understanding basic mechanisms of both homeostasis and disease and have defined new targets for intervention. Here, we review recent advances in our understanding of metabolic influences on host-microbe interactions in the GI mucosa.
Keywords: Metabolism, Inflammation, Microbiota, Short chain fatty acid, Mucosa, Colitis, Epithelium, Murine model
1. Introduction
Recent investigations of the metabolic demands placed on the mucosa during homeostasis and disease have provided important insights into the biochemical pathways employed during host-microbe interactions. At the center of these metabolic pathways is molecular oxygen utilization. The gastrointestinal (GI) tract, for instance, is characterized by a particularly unique oxygenation profile, experiencing regular intervals of profound blood flow fluctuations [1]. Even at baseline, epithelial cells lining the mucosa exist at a relatively low oxygen tension environment, herein described as ‘physiologic hypoxia’. Countercurrent oxygen exchange mechanisms in the small intestine have revealed that oxygen from arterial blood supply diffuses to adjacent venules, along the crypt villus axis, resulting in graded hypoxia [2]. A steep oxygen gradient has also been documented in distal, colonic regions of the GI tract, spanning from the anaerobic lumen, across the epithelium to the richly vascularized sub-epithelial mucosa [3]. Given the high-energy requirement of the gut and the integral role of the epithelium in maintaining intestinal homeostasis, it is not surprising that these cells have evolved a number of mechanisms to cope with this austere metabolic environment [4]. Here, we will discuss how such metabolic shifts are regulated, particularly as they relate to host-microbe interactions.
2. Oxygen metabolism in healthy and inflamed tissues
The oxygenation profile of the healthy mucosa is an area of significant interest. A comparison of the lung and intestine, for example, provides a number of stark contrasts. Breathable air at sea level contains a partial O2 pressure (pO2) of ~145 mmHg (approximately 21% O2). Measurements of the healthy lung alveolus have revealed a pO2 of 100–110 mmHg [5]. Conversely, the most luminal aspect of the healthy colon exists at a pO2 of less than 10 mmHg [3,6]. This difference is attributed primarily to the source of O2, active local metabolism and the anatomy of blood flow [4].
Tissue oxygenation, particularly at low O2, has been tracked using 2-nitroimidazole dyes, a class of compounds known to undergo intracellular metabolism dependent on the level of tissue oxygenation [7]. These dyes were originally developed to image the low O2 environment of growing tumors [8] and have subsequently been used as tools to monitor levels of tissue oxygenation. Nitroimidazoles form adducts with thiol groups in proteins, peptides and amino acids where all atoms of the ring and side-chain of the 2-nitroimidazole are retained at pO2 < 10 mmHg. Intestinal mucosal localization of these nitroimidazole dyes has revealed two striking observations. First, in the normal GI mucosa, particularly in the colon, “physiologic hypoxia” predominates [6]. Recent studies have shown that these low O2 conditions are critical for the constitutive expression of certain innate immune factors found within the mucosa (e.g. human β defensin-1) [9]. Second, inflammatory lesions within the mucosa are profoundly hypoxic or even anoxic, similar to that seen in some tumors [4]. It is likely that there are multiple contributing factors, i.e. vasculitis, vasoconstriction, edema, increased O2 consumption, predisposing the inflamed intestinal epithelia to decreased oxygen delivery and hypoxia [6]. These nitroimidazole compounds have shown significant clinical utility in tumor imaging and in the identification of stroke regions within the brain of patients [10]. As opposed to other imaging techniques, these molecules have the advantage that they image only viable tissue, are independent of oxygen radical accumulation and are not active in apoptotic/necrotic tissue [11].
Given the substantial shifts in metabolism and oxygen availability during inflammation, a number of studies have shown that stabilization of transcription factor hypoxia-inducible factor (HIF) in low oxygen environments triggers the expression of genes that are essential to epithelial barrier function [12–15]. Additionally, HIF is one of the central regulators of overall tissue metabolism [16] and has profound influences on the inflammatory response [4]. Its activity is dependent on stabilization of an O2-dependent degradation (ODD) domain expressed on the α-subunit and subsequent nuclear translocation to form a functional complex with HIF-1β [17]. In normally oxygenated tissues, iron, alpha-ketoglutarate and O2-dependent hydroxylation of two prolines (Pro564 and Pro402 of HIF-1α) within the ODD of the alpha subunit initiates the association with the von Hippel-Lindau tumor suppressor protein (pVHL) and rapid degradation via ubiquitin-E3 ligase proteasomal targeting [18,19].
Intestinal epithelial cells express both HIF-1α and HIF-2α [20] and genetic studies in mice indicate that these proteins have non-redundant roles [21]. It has been suggested that distinct transcriptional responses mediated by HIF-1α and HIF-2α may have evolved as particular adaptations to hypoxia. For example, the transcriptional responses that coordinate metabolic adaptation through glycolytic pathways are selective for the HIF-1α over the HIF-2α isoform [22]. Conversely, studies addressing selectivity of the two isoforms for increased perfusion and oxygen carrying capacity (e.g. erythropoietin induction) have indicated a more prominent role for HIF-2α [23]. Iron absorption through intestinal epithelial cells also appears to be selective for HIF-2α [24]. While this specificity for individual gene regulation is not well understood, some evidence has shown that binding of HIF-1α or HIF-2α to gene promoters is dependent on interactions with STAT3 and USF2, respectively [25].
The spectrum of basal oxygenation within individual tissues is immense. Given the steep oxygen gradient due to countercurrent blood flow, colonic epithelia exist at very low pO2 [26]. These cells have proven to be remarkably resistant to hypoxia, where even very low levels of oxygenation allow these cells to function normally [27,28]. The importance of HIF to epithelial function was originally shown by microarray analysis of intestinal epithelial cells cultured in low O2 conditions (pO2 ~20 mmHg) [12]. These studies were subsequently validated in murine models of colitis [6,29–33] and in diseased human tissues [34–36]. Notably, the cluster of functional proteins regulated by HIF localizes prominently to the most luminal aspect of polarized epithelia and is composed of proteins important for increased mucin production, [37] molecules that modify mucin function [38], antimicrobial defense [9], barrier function [39], xenobiotic clearance [13] and nucleotide metabolism/signaling (by ecto-5′-nucleotidase and CD73) [14,15] (see Fig. 1). Molecular studies of these hypoxia-regulated pathway(s) have shown a dependence on HIF-mediated transcriptional responses. Original studies by Karhausen, et al. generated mice expressing either mutant Hif-1α (causing constitutive repression of Hif-1α) or mutant von Hippel-Lindau (causing constitutive over-expression of HIF) targeted to the intestinal epithelial cells revealed a more severe colitic phenotype in which increased intestinal permeability was a prominent feature [6]. These findings were somewhat model-dependent, since epithelial HIF-based signaling has also been shown to promote inflammation in another study [33]. Further examination of these pathways revealed that active colonic inflammation, defined by the influx of large numbers of neutrophils, depleted local oxygen levels sufficient enough to stabilize intestinal epithelial cell HIF and imprinted a transcriptional phenotype that strongly reflected HIF stabilization [40]. This phenotype was dependent on neutrophil NADPH oxidase implicated epithelial HIF stabilization in goblet cell differentiation and the production of mucins and mucin-binding elements, which has been suggested by other studies in the colon [12,37]. Overall, these findings confirm that intestinal epithelial cells can adapt to hypoxia and that HIF may play a key role in such an adaptation.
Fig. 1. Preferential oxidation of butyrate by intestinal epithelium stabilizes HIF and contributes to barrier function.
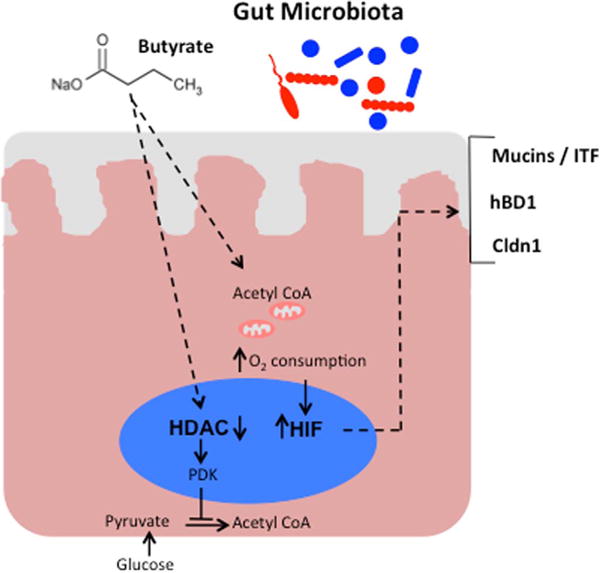
Microbiota-derived butyrate is absorbed apically and acts to inhibit histone deacetylation. One consequence is increased expression of pyruvate dehydrogenase kinase (PDK), which inactivates pyruvate dehydrogenase. This precludes oxidative metabolism of pyruvate, allowing for the majority of acetyl CoA that is used for oxidative metabolism to be derived from β-oxidation of butyrate. Oxidative respiration leads to increased oxygen consumption, stabilization of HIF, and expression of HIF target genes important in barrier regulation (Muc3/ITF, hBD1, Cldn1).
3. Host-microbial metabolism and tissue hypoxia
The gastrointestinal tract of mammals is host to trillions of bacteria. This finely tuned host-microbe relationship exists on the surface of the intestinal mucosa, where microbes are essential for host health, but can also initiate and perpetuate disease [41]. These microbes, in addition to aiding in digestion, produce a number of vitamins and benefit the host through the local synthesis of short-chain fatty acids (SCFAs), including butyrate, propionate, and acetate.
SCFAs are end products of bacterial fermentation, primarily derived from resistant starches, dietary fibers and undigested proteins [42]. Anaerobic bacteria in the colon, particularly members of the Phyla Firmicutes [43], produce butyrate through the conversion of microbial acetyl-CoA to the butyryl-CoA via β-oxidation of fatty acids. The final conversion from butyryl-CoA to butyrate is either catalyzed by butyryl-CoA: acetate CoA transferase or butyrate kinase. Due the presence of highly conserved regions these enzymes can be used for the identification of butyrate-producing bacterial communities in molecular analyses [44–46]. Amino acids can also serve as a substrate for SCFA production. Acetate can be produced through microbial metabolism of several amino acids, including glycine, alanine, threonine, glutamate, lysine and aspartate [42]. Propionate can be synthesized from alanine and threonine and butyrate from lysine and glutamate. Acetate and propionate are utilized primarily by muscle and liver cells, respectively [42], whereas butyrate is the primary metabolic substrate for colonocytes (see below).
The majority of both human and mouse microbiota is composed of members of the Firmicutes and Bacteroidetes phyla [47]. In IBD, there is an overall decrease in microbial diversity, including decreased abundance of these major phyla. The majority of butyrate in the colon is derived from Clostridial clusters IV, XIVa, and XVIII, members of the Firmicutes phylum. Laser capture microdissection experiments demonstrated an enrichment of Firmicutes in the inter-fold regions of the colon when compared to the luminal microbiome population [48].
Potentially related to it biogeographic position in the colon, butyrate is efficiently absorbed and metabolized by the epithelium and very little butyrate is released into portal circulation [49]. Butyrate generates energy by stimulating expression of pyruvate dehydrogenase kinase, which in turn inhibits the pyruvate dehydrogenase complex [50] (see Fig. 1). This inhibition prevents conversion of glucose-derived pyruvate to acetyl-CoA effectively shifting colonocyte metabolism to butyrate from glucose. In addition, it has been suggested that the function of butyrate is dependent on its concentration and location. Butyrate is selectively taken up by the colonocytes (through several transporters including SMCT1 and MCT1) and provides the epithelium with up to 70% of their energy. Other SCFA such as acetate and propionate are primarily transported to muscle and liver, respectively [51]. High concentrations of butyrate are found in the lumen of the colon and may exist at levels exceeding 30 mM [49]. Butyrate concentrations that exceed the metabolic capacity of the cell (~0.5 mM) enter the nucleus to act as an HDAC inhibitor, effectively inhibiting proliferation and inducing apoptosis in colonocytes. In this role, butyrate may promote colonocyte turnover through homeostatic mechanisms [51]. Most cancer cells use glucose as their primary energy source, termed the Warburg effect. Studies comparing the effects of butyrate on normal colonocytes and cancerous colonocytes suggest that butyrate has the capability of exhibiting a differential influence on cells depending on concentration and the metabolic state of the cell. In this report, butyrate stimulated the proliferation of normal colonic epithelia but inhibited cell growth of colorectal cancer cells [52]. This variable function of butyrate is referred to as the “butyrate paradox” and is thought to be, at least in part, related to epigenetic changes exerted by butyrate. A study from 2011 found that butyrate oxidation in UC was significantly lower than controls but, interestingly, saturation of butyrate kinetics was achieved from 1 mM in both UC and control subjects and replacement of butyrate to higher levels does not restore metabolism ([53]). Interestingly, as in cancer cells, actively inflamed colonic epithelia were found to have impaired butyrate oxidation.
Butyrate may exert additional influences on cell growth through the promotion of physiologic hypoxia. Kelly et al., recently demonstrated that butyrate increases epithelial O2 consumption depleting local O2 to levels of hypoxia that stabilize HIF [54,55]. In vivo, these same studies revealed that depletion of the microbiota using antibiotics reduced colonic butyrate and HIF expression, both of which were restored by butyrate supplementation. Moreover, the barrier protection afforded by butyrate was not appreciated in cells lacking HIF, suggesting that butyrate provides barrier protection in a HIF-dependent manner. These results reveal that the butyrate-HIF axis is a novel pathway in butyrate driven host-microbe interactions (Fig. 1).
Analysis of germ-free (GF) mice has revealed nearly no retention of O2-sensitive dyes and significantly decreased HIF stabilization at baseline [57]. Work by Donohoe et al. [58], demonstrated that colonocytes from GF mice are significantly energy deprived, with markedly diminished levels of ATP and NADH/NAD+ in addition to decreased capacity for oxidative phosphorylation. Such metabolic deficits were shown to activate AMP kinase and profoundly increase epithelial autophagy, in which cytoplasmic targets are engulfed by a double-membrane vacuole termed the autophagosome which fuses with lysosomes and undergoes hydrolase-mediated digestion. There is much interest in understanding autophagy in the intestine, as variants in several genes involved in the autophagy pathway have emerged as risk alleles for inflammatory bowel disease (IBD), including autophagy-related 16 like 1 (ATG16L1) [59,60] and immunity-related GTPase family M (IRGM) [61,62].
The metabolic abnormalities associated with GF mice were shown to be proportional to decreased levels of butyrate. The addition of butyrate or butyrate-producing microbes rescued these defects in autophagy and tissue energetics, including the deficits in oxidative phosphorylation [58]. These same studies showed that the protection afforded by butyrate occurred through the provision of energy and not through its function as an HDAC inhibitor.
The hypoxic microenvironment can be shaped as well as perturbed by the microbiome. A recent study by Rivera-Chavez et al. [63] revealed that aerobic respiration enzymes encoded within the bacterial genome might prove beneficial for the expansion of Salmonella, particularly in conditions where anaerobes become depleted (e.g. following antibiotic use). These studies employed Salmonella enterica subsp. Typhimurium (S.Typhimurium) mutants lacking functional cytochrome bd subunits and examined competitive fitness advantages in vitro and in vivo. Results from these studies revealed that mice depleted of major anaerobic microorganisms (especially Clostridia) generate a more aerobic luminal environment that allows for the expansion of aerobic S. Typhimurium. These experiments revealed that the lack of cytochrome bd oxidase resulted in a growth disadvantage for S. Typhimurium that manifested only in antibiotic-treated animals. Further analysis revealed that cytochrome bd oxidase synergizes with nitrate reductases to drive post-antibiotic S. Typhimurium expansion and recovery of targeted S. Typhimurium nitrate respiration mutants were nearly 10-fold less than wild-type S. Typhimurium following antibiotic-mediated depletion of anaerobes. This same group recently demonstrated that the elevation of inducible nitric oxide synthase (iNOS) associated with post-antibiotic treatment results in the selective oxidation of certain monosaccharides by the host, namely galactose and glucose, and provides a fitness advantage for expansion of S. Typhimurium [64]. Finally, it is notable that inflammation-associated luminal expansion of select members of the microbiota can also include facultative anaerobes. It has been shown, for example, that some Enterobacteriacea selectively expand through the local generation of electron acceptors that can be used for anaerobic respiration (esp. nitrate) [65,66]. Conditions associated with ongoing inflammation (i.e. inflammatory hypoxia) provide the ideal environment to promote the localized generation of reactive oxygen and nitrogen-containing electron acceptors.
4. Microbial metabolism in mucosal inflammation
Surprisingly little is known about the influence of tissue metabolism on the expansion and retraction of specific components of the gut microbiome. There is increasing evidence that the pathogenesis of IBD is largely influenced by an intestinal dysbiosis and pertubation of the microbial population, though it remains unclear whether such dysbiosis is a cause or consequence of the inflammation associated with IBD [67]. It is clear, however, that the host immune system exists in close proximity to the microbiome and is capable of detecting and responding to components of its constituents from microbial-associated molecular patterns (MAMPs) to bacterial metabolites, such as SCFAs.
There is increasing evidence supporting a homeostatic role for SCFA in colonic inflammation in both strengthening the intestinal barrier and promoting healing of colitis [49,68]. The protection afforded by high fiber diets in experimental colitis are thought to be dependent on SCFA production, particularly butyrate [69–71]. In support of these data, administration of exogenous butyrate in the form of the triacylglyceride tributyrin has been shown to promote resistance to antibiotic-mediated damage in experimental colitis models [71,72]. Furthermore, pharmacologic inhibition of β-oxidation of fatty acids, thus butyrate production, by sodium 2-bromo-octanoate induces colitis similar to ulcerative colitis (UC) [74].
Ulcerative colitis, a form of IBD, has been described as an energy deficient disease. Two genes associated with the development of UC, SLC22A5 (OCTN2) and uncoupling protein 2 (UCP2) are critical components of mitochondrial energy metabolism and are required for intestinal barrier function [75]. Interestingly, conplastic mice with mitochondrial polymorphisms resulting in increased intracellular ATP production were found to be resistant to chemically-induced colitis likely secondary to a corresponding increased rate of enterocyte proliferation allowing for epithelial repair [75]. Kaiko et al. recently demonstrated that butyrate was a potent inhibitor of intestinal stem cell proliferation through a Foxo3-dependent mechanism and due to intestinal crypt architecture these cells are not exposed to this microbial metabolite except in the setting of damage or colitis, potentially impeding the necessary regeneration required for epithelial repair [76]. Alternatively, these latter studies could implicate the intestinal crypt as a metabolic sensor of energy availability through SCFA.
SCFAs also regulate blood flow in the colon by stimulating perfusion. Mortensen et al. utilized resected colonic segments and determined that SCFA produced dose-dependent vasodilation at concentrations as low as 3 mM [77]. Furthermore, SCFA have been shown to promote post-surgical healing in the human rectum by increasing mucosal blood flow following 10–14 days of SCFA instillation [78]. The concentration of SCFA that colonic vessels are exposed to in vivo is not clear, but SCFA are present in high levels within the healthy intestinal lumen. In the setting of chronic, long-standing inflammation, there is diminished mucosal perfusion and impaired wound healing. In contrast, acute inflammation is characterized by tissue hyperemia resulting from dilation of small vessels [79]. While SCFA stimulates colonic blood flow, the role of low SCFA in diminished blood flow and oxygenation in chronic gut inflammation has not been studied in detail but could represent a possible mechanism to explain this phenomenon.
As in rodent models, there is mounting evidence connecting intestinal dysbiosis to the development of IBD in humans. While monozygotic twin concordance studies suggest a greater environmental component to the development of ulcerative colitis compared to Crohn’s disease, germ-free studies in experimental colitis models strongly suggest that the development of IBD is not purely genetic but requires bacterial colonization. For example, diversion colitis develops after surgery resulting in exclusion of fecal material, the microbiome and its metabolites, from a portion of the colon. This condition is treated most effectively with SCFA enemas. To this point, multiple studies have revealed lower concentrations of luminal butyrate and reduced abundance of butyrate-producing organisms (e.g., certain Roseburia and Faecalibacterium genera) in patients with inflammatory bowel disease [80–82]. This is further supported by data demonstrating that increasing fecal butyrate levels decreases intestinal inflammation and symptoms in ulcerative colitis patients [83]. A recent meta-analysis of 25 studies of patients with UC was notable for a 65% clinical response rate to fecal microbiota transplant (FMT) with a corresponding increase in microbial diversity including an increase in key butyrate producers, Firmicutes and Clostridium clusters IV, XIVa, XVIII [84].
The intestinal microbiota shifts have a profound impact on colonic inflammation, however it remains unclear if inflammation or dysbiosis is the primary driver of colitis [85]. Several lines of evidence support an important role for host-microbe interactions in the regulation of intestinal barrier function. Perturbations in microbial metabolism and epithelial autophagy disrupt multiple aspects of barrier function. Recent studies, for example, defined a central role for autophagy in colonic goblet cell mucus secretion. Conditional epithelial knockout studies of Atg5, Atg7 and Lc3b in mice revealed that autophagy is required for efficient mucin granule accumulation and secretion from goblet cells [86]. Interestingly, this work implicates NADPH oxidase-derived ROS at the autophagosome-endosome interface as critical mediators of mucus secretion. Likewise, butyrate has been shown to enhance epithelial barrier function and selectively induce the tight junction protein claudin-1 through a mechanism involving the transcription factor Sp1 [87]. A so-called “tight claudin,” claudin-1 plays an integral role in determining barrier function in IECs [56]. Given the association between butyrate levels and HIF stabilization [57], our own studies have revealed that HIF is an important regulator of claudin-1 [39]. These studies showed that cells lacking HIF harbored a number of barrier defects. Global HIF-1α chromatin immunoprecipitation (ChIP) analysis identified claudin-1 as a prominent HIF target gene and overexpression of CLDN1 in HIF-deficient cells resulted in resolution of morphological abnormalities and restoration of barrier function. It is noteworthy that claudin-1 was not hypoxia-inducible per se, rather, this gene appears to be regulated basally by mechanisms involving constitutive HIF stabilization (i.e. physiologic hypoxia) [9]. While not completely clear at present, it is intriguing to speculate that such physiologic hypoxia could be proportional to luminal butyrate levels, thereby providing an important host-microbial crosstalk critical for fundamental tissue functions such as barrier.
It has been shown that microbial signals, such as those delivered by a mix of Clostridial species, specifically members of cluster IV, XIVa and XVIII, induce mucosal tolerance and healing by enhancing regulatory T cells expansion and differentiation (Fig. 2). The IL-23/ Th17/IL17 pathway is important in the regulation of IBD; pro-inflammatory Th17 is increased in mucosa and serum of IBD patients. Butyrate is a major product of tolerogenic Clostridial species [88]. Butyrate administration has been shown to increase blood Treg and levels of anti-Th17 cytokines (IL-10 and IL-12) while also suppressing IL-17 levels ([89,90] (see Fig. 2). Furthermore, butyrate is shown to enhance histone H3 acetylation in the promoter and conserved non-coding regions of the Foxp3 locus, which provides a mechanism for butyrate’s regulation of Treg differentiation [89]. Under such conditions, butyrate differentiated T cells attenuated colitis induced by adoptive transfer of CD4+ CD45RBhi T cells in Rag1−/− mice [89]}. In addition to functioning as a direct energy source, SCFAs can signal through a series of G-protein coupled receptors (GPR) to mediate their biological functions [91,92]. In mice, deletion of Gpr41 and Gpr43 mediate protective immunity in inflammatory models [91,92]. Also notable is the observation that treatment of mice with propionate promotes colonic protection during inflammation [92] and that the butyrate receptor (GPR109a) functions to suppress colonic inflammation [93].
Fig. 2.
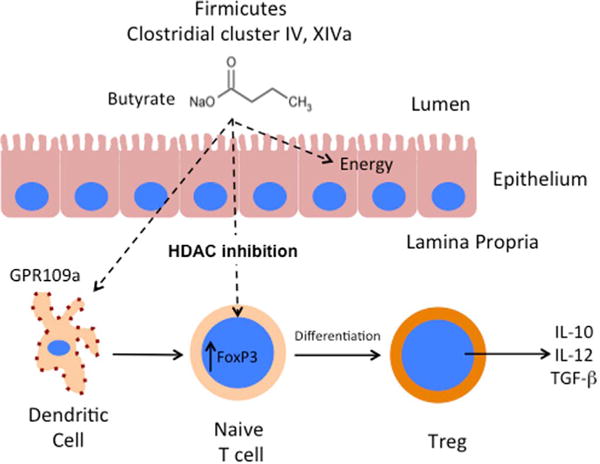
Microbial-derived butyrate enhances regulatory T cell (Treg) expansion and differentiation to promote mucosal tolerance and healing. Butyrate produced from the metabolism of undigested carbohydrates by Firmicutes and Clostridial cluster IV and XIVa are selectively taken up by the colonic epithelium and serves as the primary energy source (~70% of total energy) for healthy epithelial cells. Further, small quantities of butyrate cross the epithelium to bind GPR109a on dendritic cells (DC) and induce Treg generation via HDAC inhibition. Butyrate treatment enhances acetylation at histone H3 lysine 27 (H3K27) at the Foxp3 promoter leading to Foxp3 induction and promotes the conversion of naïve CD4+T cells to Tregs. This action of butyrate in the mucosa results in increased expression of immunosuppressive cytokines within the lamina propia.
5. Conclusions
In the past decade, much work has been accomplished unraveling the host-microbe interactions on the intestinal mucosa however further work is required to deepen this understanding. Of particular interest are the unique locales in which these interactions occur within the length of GI tract: juxtaposed between a microbial-rich anaerobic lumen and the highly vascularized submucosa. The epithelium has evolved and adapted to this “physiologic hypoxia” through the stabilization of HIF. Recently, microbial-derived SCFA butyrate has been discovered to stabilize epithelial HIF and enhance intestinal barrier function through a HIF mechanism. Further, butyrate has also been implicated as a key player in the expansion and differentiation of Tregs by acting as a HDAC inhibitor and suppressing colonic inflammation by binding to surface GPCR’s (e.g. GPR109a). Ongoing studies to better understand the host-microbial crosstalk are becoming increasingly recognized as important to health and disease. Knowledge from these pathways may provide new avenues for the treatment of intestinal disease, such as IBD.
Acknowledgments
This work was supported by NIH grants DK50189/ DK104713/ DK095491 and VA Merit Award 1I01BX002182.
Footnotes
The authors declare no financial interests in any of the work submitted here.
References
- 1.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7:281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shepherd AP. Metabolic control of intestinal oxygenation and blood flow. Fed Proc. 1982;41(6):2084–2089. [PubMed] [Google Scholar]
- 3.Albenberg L, Esipova TV, Judge CP, Bittinger K, Chen J, Laughlin A, Grunberg S, Baldassano RN, Lewis JD, Li H, Thom SR, Bushman FD, Vinogradov SA, Wu GD. Correlation between intraluminal oxygen gradient and radial partitioning of intestinal microbiota. Gastroenterology. 2014;18(14):1055–1063. doi: 10.1053/j.gastro.2014.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colgan SP, Campbell EL, Kominsky DJ. Hypoxia and mucosal inflammation. Ann Rev Pathol. 2016;11:77–100. doi: 10.1146/annurev-pathol-012615-044231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schaible B, Schaffer K, Taylor CT. Hypoxia, innate immunity and infection in the lung. Respir Physiol Neurobiol. 2010;174(3):235–243. doi: 10.1016/j.resp.2010.08.006. http://dx.doi.org/10.1016/j.resp.2010.08.006 (Epub 2010 Aug 13) [DOI] [PubMed] [Google Scholar]
- 6.Karhausen J, Furuta GT, Tomaszewski JE, Johnson RS, Colgan SP, Haase VH. Epithelial hypoxia-inducible factor-1 is protective in murine experimental colitis. J Clin Invest. 2004;114(8):1098–1106. doi: 10.1172/JCI21086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Evans SM, Hahn S, Pook DR, Jenkins WT, Chalian AA, Zhang P, Stevens C, Weber R, Weinstein G, Benjamin I, Mirza N, Morgan M, Rubin S, McKenna WG, Lord EM, Koch CJ. Detection of hypoxia in human squamous cell carcinoma by EF5 binding. Cancer Res. 2000;60:2018–2024. [PubMed] [Google Scholar]
- 8.Lord EM, Harwell L, Koch CJ. Detection of hypoxic cells by monoclonal antibody recognizing 2-nitroimidazole adducts. Cancer Res. 1993;53(23):5721–5726. [PubMed] [Google Scholar]
- 9.Kelly CJ, Glover LE, Campbell EL, Kominsky DJ, Ehrentraut SF, Bowers BE, Bayless AJ, Saeedi BJ, Colgan SP. Fundamental role for HIF-1alpha in constitutive expression of human beta defensin-1. Mucosal Immunol. 2013;6(10):1110–1118. doi: 10.1038/mi.2013.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takasawa M, Moustafa RR, Baron JC. Applications of nitroimidazole in vivo hypoxia imaging in ischemic stroke. Stroke. 2008;39(5):1629–1637. doi: 10.1161/STROKEAHA.107.485938. [DOI] [PubMed] [Google Scholar]
- 11.Kizaka-Kondoh S, Konse-Nagasawa H. Significance of nitroimidazole compounds and hypoxia-inducible factor-1 for imaging tumor hypoxia. Cancer Sci. 2009;100(8):1366–1373. doi: 10.1111/j.1349-7006.2009.01195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Furuta GT, Turner JR, Taylor CT, Hershberg RM, Comerford K, Narravula S, Podolsky DK, Colgan SP. Hypoxia-inducible factor 1-dependent induction of intestinal trefoil factor protects barrier function during hypoxia. J Exp Med. 2001;193(9):1027–1034. doi: 10.1084/jem.193.9.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance MDR1) gene. Cancer Res. 2002;62:3387–3394. [PubMed] [Google Scholar]
- 14.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110(7):993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Ex Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Semenza GL. Regulation of metabolism by hypoxia-inducible factor 1, Cold Spring Harb. Symp. Quant Biol. 2011;2011:22. doi: 10.1101/sqb.2011.76.010678. [DOI] [PubMed] [Google Scholar]
- 17.Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107(1):1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 18.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 19.Tanimoto K, Makino Y, Pereira T, Poellinger L. Mechanism of regulation of the hypoxia-inducible factor-1 alpha by the von Hippel-Lindau tumor suppressor protein. Embo J. 2000;19:4298–4309. doi: 10.1093/emboj/19.16.4298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glover LE, Bowers BE, Saeedi B, Ehrentraut SF, Campbell EL, Bayless AJ, Dobrinskikh E, Kendrick AA, Kelly CJ, Burgess A, Miller L, Kominsky DJ, Jedlicka P, Colgan SP. Control of creatine metabolism by HIF is an endogenous mechanism of barrier regulation in colitis, Proc. Natl Acad Sci USA. 2013;110(49):19820–19825. doi: 10.1073/pnas.1302840110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ratcliffe PJ. HIF-1 and HIF-2: working alone or together in hypoxia? J Clin Invest. 2007;117(4):862–865. doi: 10.1172/JCI31750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu CJ, Sataur A, Wang L, Chen H, Simon MC. The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell. 2007;18(11):4528–4542. doi: 10.1091/mbc.E06-05-0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rankin EB, Biju MP, Liu Q, Unger TL, Rha J, Johnson RS, Simon MC, Keith B, Haase VH. Hypoxia-inducible factor-2 (HIF-2) regulates hepatic erythropoietin in vivo. J Clin Invest. 2007;117(4):1068–1077. doi: 10.1172/JCI30117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mastrogiannaki M, Matak P, Keith B, Simon MC, Vaulont S, Peyssonnaux C. HIF-2alpha, but not HIF-1alpha, promotes iron absorption in mice. J Clin Invest. 2009;119:1159–1166. doi: 10.1172/JCI38499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pawlus MR, Wang L, Murakami A, Dai G, Hu CJ. STAT3 or USF2 contributes to HIF target gene specificity. PLoS One. 2013;8(8):e72358. doi: 10.1371/journal.pone.0072358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karhausen J, Stafford-Smith M. The role of nonocclusive sources of acute gut injury in cardiac surgery. J Cardiothorac Vasc Anesth. 2014;28(2):379–391. doi: 10.1053/j.jvca.2013.04.016. http://dx.doi.org/10.1053/j.jvca.2013.04.016 (Epub 2013 Oct 8) [DOI] [PubMed] [Google Scholar]
- 27.Colgan SP, Curtis VF, Lanis JM, Glover LE. Metabolic regulation of intestinal epithelial barrier during inflammation. Tissue Barriers. 2015;(3):e970936. doi: 10.4161/21688362.2014.970936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pannabecker TL, Layton AT. Targeted delivery of solutes and oxygen in the renal medulla: role of microvessel architecture. Am J Physiol Ren Physiol. 2014;307(6):F649–F655. doi: 10.1152/ajprenal.00276.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cummins EP, Seeballuck F, Keely SJ, Mangan NE, Callanan JJ, Fallon PG, Taylor CT. The hydroxylase inhibitor dimethyloxalylglycine is protective in a murine model of colitis. Gastroenterology. 2008;134(1):156–165. doi: 10.1053/j.gastro.2007.10.012. [DOI] [PubMed] [Google Scholar]
- 30.Han IO, Kim HS, Kim HC, Joe EH, Kim WK. Synergistic expression of inducible nitric oxide synthase by phorbol ester and interferon-gamma is mediated through NF-kappaB and ERK in microglial cells. J Neurosci Res. 2003;73(5):659–669. doi: 10.1002/jnr.10706. [DOI] [PubMed] [Google Scholar]
- 31.Morote-Garcia JC, Rosenberger P, Nivillac NM, Coe IR, Eltzschig HK. Hypoxia-inducible factor-dependent repression of equilibrative nucleoside transporter 2 attenuates mucosal inflammation during intestinal hypoxia. Gastroenterology. 2009;136(2):607–618. doi: 10.1053/j.gastro.2008.10.037. [DOI] [PubMed] [Google Scholar]
- 32.Robinson A, Keely S, Karhausen J, Gerich ME, Furuta GT, Colgan SP. Mucosal protection by hypoxia-inducible factor prolyl hydroxylase inhibition. Gastroenterology. 2008;134(1):145–155. doi: 10.1053/j.gastro.2007.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah YM, Ito S, Morimura K, Chen C, Yim SH, Haase VH, Gonzalez FJ. Hypoxia-inducible factor augments experimental colitis through an MIF-dependent inflammatory signaling cascade. Gastroenterology. 2008;134(7):2036–2048. doi: 10.1053/j.gastro.2008.03.009. 2048 e1–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giatromanolaki A, Sivridis E, Maltezos E, Papazoglou D, Simopoulos C, Gatter KC, Harris AL, Koukourakis MI. Hypoxia inducible factor 1alpha and 2alpha overexpression in inflammatory bowel disease. J Clin Pathol. 2003;56(3):209–213. doi: 10.1136/jcp.56.3.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mariani F, Sena P, Marzona L, Riccio M, Fano R, Manni P, Gregorio CD, Pezzi A, Leon MP, Monni S, Pol AD, Roncucci L. Cyclooxygenase-2 and Hypoxia-Inducible Factor-1alpha protein expression is related to inflammation, and up-regulated since the early steps of colorectal carcinogenesis. Cancer Lett. 2009;279(2):221–229. doi: 10.1016/j.canlet.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Matthijsen RA, Derikx JP, Kuipers D, van Dam RM, Dejong CH, Buurman WA. Enterocyte shedding and epithelial lining repair following ischemia of the human small intestine attenuate inflammation. PLoS One. 2009;4(9):e7045. doi: 10.1371/journal.pone.0007045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Louis NA, Hamilton KE, Canny G, Shekels LL, Ho SB, Colgan SP. Selective induction of mucin-3 by hypoxia in intestinal epithelia. J Cell Biochem. 2006;99(6):1616–1627. doi: 10.1002/jcb.20947. [DOI] [PubMed] [Google Scholar]
- 38.Furuta GT. Clinicopathologic features of esophagitis in children. Gastrointest Endosc Clin N Am. 2001;11(4):683–715. vii. [PubMed] [Google Scholar]
- 39.Saeedi BJ, Kao DJ, Kitzenberg DA, Dobrinskikh E, Schwisow KD, Masterson JC, Kendrick AA, Kelly CJ, Bayless AJ, Kominsky DJ, Campbell EL, Kuhn KA, Furuta GT, Colgan SP, Glover LE. HIF-dependent regulation of claudin-1 is central to intestinal epithelial tight junction integrity. Mol Biol Cell. 2015;26(12):2252–2262. doi: 10.1091/mbc.E14-07-1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, Bayless AJ, Scully M, Saeedi BJ, Golden-Mason L, Ehrentraut SF, Curtis VF, Burgess A, Garvey JF, Sorensen A, Nemenoff R, Jedlicka P, Taylor CT, Kominsky DJ, Colgan SP. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity. 2014;40(1):66–77. doi: 10.1016/j.immuni.2013.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489(7415):220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davila AM, Blachier F, Gotteland M, Andriamihaja M, Benetti PH, Sanz Y, Tome D. Intestinal luminal nitrogen metabolism: role of the gut microbiota and consequences for the host. Pharm Res. 2013;68(1):95–107. doi: 10.1016/j.phrs.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 43.Louis P, Flint HJ. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol Lett. 2009;294(1):1–8. doi: 10.1111/j.1574-6968.2009.01514.x. http://dx.doi.org/10.1111/j.1574-6968.2009.01514.x (Epub 2009 Feb 13) [DOI] [PubMed] [Google Scholar]
- 44.Herrmann G, Jayamani E, Mai G, Buckel W. Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J Bacteriol. 2008;190(3):784–791. doi: 10.1128/JB.01422-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Louis P, Flint HJ. Development of a semiquantitative degenerate real-time pcr-based assay for estimation of numbers of butyryl-coenzyme A (CoA) CoA transferase genes in complex bacterial samples. Appl Environ Microbiol. 2007;73(6):2009–2012. doi: 10.1128/AEM.02561-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vital M, Penton CR, Wang Q, Young VB, Antonopoulos DA, Sogin ML, Morrison HG, Raffals L, Chang EB, Huffnagle GB, Schmidt TM, Cole JR, Tiedje JM. A gene-targeted approach to investigate the intestinal butyrate-producing bacterial community. Microbiome. 2013;1(1):1–8. doi: 10.1186/2049-2618-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Manichanh C, Borruel N, Casellas F, Guarner F. The gut microbiota in IBD. Nat Rev Gastroenterol Hepatol. 2012;9(10):599–608. doi: 10.1038/nrgastro.2012.152. [DOI] [PubMed] [Google Scholar]
- 48.Nava GM, Friedrichsen HJ, Stappenbeck TS. Spatial organization of intestinal microbiota in the mouse ascending colon. ISME J. 2011;5(4):627–638. doi: 10.1038/ismej.2010.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamer HM, Jonkers D, Venema K, Vanhoutvin S, Troost FJ, Brummer RJ. Review article: the role of butyrate on colonic function. Aliment Pharmacol Ther. 2008;27(2):104–119. doi: 10.1111/j.1365-2036.2007.03562.x. [DOI] [PubMed] [Google Scholar]
- 50.Blouin JM, Penot G, Collinet M, Nacfer M, Forest C, Laurent-Puig P, Coumoul X, Barouki R, Benelli C, Bortoli S. Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int J Cancer J Int Cancer. 2011;128(11):2591–2601. doi: 10.1002/ijc.25599. [DOI] [PubMed] [Google Scholar]
- 51.Bultman SJ. Molecular pathways: gene-environment interactions regulating dietary fiber induction of proliferation and apoptosis via butyrate for cancer prevention. Clin Cancer Res. 2014;20(4):799–803. doi: 10.1158/1078-0432.CCR-13-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48(4):612–626. doi: 10.1016/j.molcel.2012.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Preter V, Geboes KP, Bulteel V, Vandermeulen G, Suenaert P, Rutgeerts P, Verbeke K. Kinetics of butyrate metabolism in the normal colon and in ulcerative colitis: the effects of substrate concentration and carnitine on the beta-oxidation pathway. Aliment Pharmacol Ther. 2011;34(5):526–532. doi: 10.1111/j.1365-2036.2011.04757.x. [DOI] [PubMed] [Google Scholar]
- 54.Kelly CJ, Zheng L, Campbell EL, Saeedi BJ, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP. Host-microbe crosstalk between short-chain fatty acids and intestinal epithelial HIF provides a new mechanism to augment tissue barrier function. Cell Host Microbe. 2015;17:662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Inai T, Kobayashi J, Shibata Y. Claudin-1 contributes to the epithelial barrier function in MDCK cells. Eur J Cell Biol. 1999;78(12):849–855. doi: 10.1016/S0171-9335(99)80086-7. [DOI] [PubMed] [Google Scholar]
- 56.Colgan SP, Taylor CT. Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol. 2010;7(5):281–287. doi: 10.1038/nrgastro.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, Weir TL, Ehrentraut SF, Pickel C, Kuhn KA, Lanis JM, Nguyen V, Taylor CT, Colgan SP. Crosstalk between microbiota-derived short-chain fatty acids and intestinal epithelial HIF augments tissue barrier function. Cell Host Microbe. 2015;17(5):662–671. doi: 10.1016/j.chom.2015.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Donohoe DR, Garge N, Zhang X, Sun W, O’Connell TM, Bunger MK, Bultman SJ. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011;13(5):517–526. doi: 10.1016/j.cmet.2011.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hampe J, Franke A, Rosenstiel P, Till A, Teuber M, Huse K, Albrecht M, Mayr G, De La Vega FM, Briggs J, Gunther S, Prescott NJ, Onnie CM, Hasler R, Sipos B, Folsch UR, Lengauer T, Platzer M, Mathew CG, Krawczak M, Schreiber S. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39(2):207–211. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 60.Rioux JD, Xavier RJ, Taylor KD, Silverberg MS, Goyette P, Huett A, Green T, Kuballa P, Barmada MM, Datta LW, Shugart YY, Griffiths AM, Targan SR, Ippoliti AF, Bernard EJ, Mei L, Nicolae DL, Regueiro M, Schumm LP, Steinhart AH, Rotter JI, Duerr RH, Cho JH, Daly MJ, Brant SR. Genome-wide association study identifies new susceptibility loci for Crohn disease and implicates autophagy in disease pathogenesis. Nat Genet. 2007;39(5):596–604. doi: 10.1038/ng2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.McCarroll SA, Huett A, Kuballa P, Chilewski SD, Landry A, Goyette P, Zody MC, Hall JL, Brant SR, Cho JH, Duerr RH, Silverberg MS, Taylor KD, Rioux JD, Altshuler D, Daly MJ, Xavier RJ. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn’s disease. Nat Genet. 2008;40(9):1107–1112. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parkes M, Barrett JC, Prescott NJ, Tremelling M, Anderson CA, Fisher SA, Roberts RG, Nimmo ER, Cummings FR, Soars D, Drummond H, Lees CW, Khawaja SA, Bagnall R, Burke DA, Todhunter CE, Ahmad T, Onnie CM, McArdle W, Strachan D, Bethel G, Bryan C, Lewis CM, Deloukas P, Forbes A, Sanderson J, Jewell DP, Satsangi J, Mansfield JC, C. Wellcome Trust Case Control. Cardon L, Mathew CG. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat Genet. 2007;39(7):830–832. doi: 10.1038/ng2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rivera-Chavez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, Baumler AJ. Depletion of butyrate-producing clostridia from the gut microbiota drives an aerobic luminal expansion of salmonella. Cell Host Microbe. 2016;19(4):443–454. doi: 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Faber F, Tran L, Byndloss MX, Lopez CA, Velazquez EM, Kerrinnes T, Nuccio SP, Wangdi T, Fiehn O, Tsolis RM, Baumler AJ. Host-mediated sugar oxidation promotes post-antibiotic pathogen expansion. Nature. 2016;534(7609):697–699. doi: 10.1038/nature18597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lopez CA, Rivera-Chavez F, Byndloss MX, Baumler AJ. The periplasmic nitrate reductase napABC supports luminal growth of Salmonella enterica serovar typhimurium during colitis. Infect Immun. 2015;83(9):3470–3478. doi: 10.1128/IAI.00351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lopez CA, Winter SE, Rivera-Chavez F, Xavier MN, Poon V, Nuccio SP, Tsolis RM, Baumler AJ. Phage-mediated acquisition of a type III secreted effector protein boosts growth of salmonella by nitrate respiration. MBio. 2012:e00143–2. doi: 10.1128/mBio.00143-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Butto LF, Haller D. Dysbiosis in intestinal inflammation: cause or consequence. Int J Med Microbiol. 2016;306(5):302–309. doi: 10.1016/j.ijmm.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 68.Ploger S, Stumpff F, Penner GB, Schulzke JD, Gabel G, Martens H, Shen Z, Gunzel D, Aschenbach JR. Microbial butyrate and its role for barrier function in the gastrointestinal tract. Ann N Y Acad Sci. 2012;1258:52–59. doi: 10.1111/j.1749-6632.2012.06553.x. [DOI] [PubMed] [Google Scholar]
- 69.Ito H, Tanabe H, Kawagishi H, Tadashi W, Yasuhiko T, Sugiyama K, Kiriyama S, Morita T. Short-chain inulin-like fructans reduce endotoxin and bacterial translocations and attenuate development of TNBS-induced colitis in rats. Dig Dis Sci. 2009;54(10):2100–2108. doi: 10.1007/s10620-008-0599-x. [DOI] [PubMed] [Google Scholar]
- 70.Morita T, Tanabe H, Sugiyama K, Kasaoka S, Kiriyama S. Dietary resistant starch alters the characteristics of colonic mucosa and exerts a protective effect on trinitrobenzene sulfonic acid-induced colitis in rats. Biosci Biotechnol, Biochem. 2004;68(10):2155–2164. doi: 10.1271/bbb.68.2155. [DOI] [PubMed] [Google Scholar]
- 71.Videla S, Vilaseca J, Antolin M, Garcia-Lafuente A, Guarner F, Crespo E, Casalots J, Salas A, Malagelada JR. Dietary inulin improves distal colitis induced by dextran sodium sulfate in the rat. Am J Gastroenterol. 2001;96(5):1486–1493. doi: 10.1111/j.1572-0241.2001.03802.x. [DOI] [PubMed] [Google Scholar]
- 72.Cresci G, Nagy LE, Ganapathy V. Lactobacillus GG and tributyrin supplementation reduce antibiotic-induced intestinal injury. JPEN J Parent Enter Nutr. 2013;37(6):763–774. doi: 10.1177/0148607113486809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leonel AJ, Teixeira LG, Oliveira RP, Santiago AF, Batista NV, Ferreira TR, Santos RC, Cardoso VN, Cara DC, Faria AM, Alvarez-Leite J. Antioxidative and immunomodulatory effects of tributyrin supplementation on experimental colitis. Br J Nutr. 2013;109(8):1396–1407. doi: 10.1017/S000711451200342X. [DOI] [PubMed] [Google Scholar]
- 74.Roediger WE, Nance S. Metabolic induction of experimental ulcerative colitis by inhibition of fatty acid oxidation. Br J Exp Pathol. 1986;67(6):773–782. [PMC free article] [PubMed] [Google Scholar]
- 75.Bar F, Bochmann W, Widok A, von Medem K, Pagel R, Hirose M, Yu X, Kalies K, Konig P, Bohm R, Herdegen T, Reinicke AT, Buning J, Lehnert H, Fellermann K, Ibrahim S, Sina C. Mitochondrial gene polymorphisms that protect mice from colitis. Gastroenterology. 2013;145(5):1055–1063 e3. doi: 10.1053/j.gastro.2013.07.015. [DOI] [PubMed] [Google Scholar]
- 76.Kaiko GE, Ryu SH, Koues OI, Collins PL, Solnica-Krezel L, Pearce EJ, Pearce EL, Oltz EM, Stappenbeck TS. The colonic crypt protects stem cells from microbiota-derived metabolites. Cell. 2016;165(7):1708–1720. doi: 10.1016/j.cell.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mortensen FV, Nielsen H, Mulvany MJ, Hessov I. Short chain fatty acids dilate isolated human colonic resistance arteries. Gut. 1990;31(12):1391–1394. doi: 10.1136/gut.31.12.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mortensen FV, Hessov I, Birke H, Korsgaard N, Nielsen H. Microcirculatory and trophic effects of short chain fatty acids in the human rectum after Hartmann’s procedure. Br J Surg. 1991;78(10):1208–1211. doi: 10.1002/bjs.1800781019. [DOI] [PubMed] [Google Scholar]
- 79.Hatoum OA, Binion DG, Gutterman DD. Paradox of simultaneous intestinal ischaemia and hyperaemia in inflammatory bowel disease. Eur J Clin Invest. 2005;35(10):599–609. doi: 10.1111/j.1365-2362.2005.01567.x. [DOI] [PubMed] [Google Scholar]
- 80.Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, Ferrante M, Verhaegen J, Rutgeerts P, Vermeire S. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2013 doi: 10.1136/gutjnl-2013-304833. [DOI] [PubMed] [Google Scholar]
- 81.Eeckhaut V, Machiels K, Perrier C, Romero C, Maes S, Flahou B, Steppe M, Haesebrouck F, Sas B, Ducatelle R, Vermeire S, Van Immerseel F. Butyricicoccus pullicaecorum in inflammatory bowel disease. Gut. 2013;62(12):1745–1752. doi: 10.1136/gutjnl-2012-303611. [DOI] [PubMed] [Google Scholar]
- 82.Sokol H, Seksik P, Furet JP, Firmesse O, Nion-Larmurier I, Beaugerie L, Cosnes J, Corthier G, Marteau P, Dore J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis. 2009;15(8):1183–1189. doi: 10.1002/ibd.20903. [DOI] [PubMed] [Google Scholar]
- 83.Hallert C, Bjorck I, Nyman M, Pousette A, Granno C, Svensson H. Increasing fecal butyrate in ulcerative colitis patients by diet: controlled pilot study. Inflamm Bowel Dis. 2003;9(2):116–121. doi: 10.1097/00054725-200303000-00005. [DOI] [PubMed] [Google Scholar]
- 84.Shi Y, Dong Y, Huang W, Zhu D, Mao H, Su P. Fecal microbiota transplantation for ulcerative colitis: a systematic review and meta-analysis. PLoS One. 2016;11(6):e0157259. doi: 10.1371/journal.pone.0157259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, Amato MD, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao H, Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford-Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Patel KK, Miyoshi H, Beatty WL, Head RD, Malvin NP, Cadwell K, Guan JL, Saitoh T, Akira S, Seglen PO, Dinauer MC, Virgin HW, Stappenbeck TS. Autophagy proteins control goblet cell function by potentiating reactive oxygen species production. EMBO J. 2013;32(24):3130–3144. doi: 10.1038/emboj.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wang HB, Wang PY, Wang X, Wan YL, Liu YC. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig Dis Sci. 2012;57(12):3126–3135. doi: 10.1007/s10620-012-2259-4. [DOI] [PubMed] [Google Scholar]
- 88.Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly YM, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013;341(6145):569–573. doi: 10.1126/science.1241165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013;500(7461):232–236. doi: 10.1038/nature12331. [DOI] [PubMed] [Google Scholar]
- 90.Zhang M, Zhou Q, Dorfman RG, Huang X, Fan T, Zhang H, Zhang J, Yu C. Butyrate inhibits interleukin-17 and generates Tregs to ameliorate colorectal colitis in rats. BMC Gastroenterol. 2016;16(1):84. doi: 10.1186/s12876-016-0500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kim MH, Kang SG, Park JH, Yanagisawa M, Kim CH. Short-chain fatty acids activate GPR41 and GPR43 on intestinal epithelial cells to promote inflammatory responses in mice. Gastroenterology. 2013;145(2):396–406. doi: 10.1053/j.gastro.2013.04.056. [DOI] [PubMed] [Google Scholar]
- 92.Maslowski KM, Vieira AT, Ng A, Kranich J, Sierro F, Yu D, Schilter HC, Rolph MS, Mackay F, Artis D, Xavier RJ, Teixeira MM, Mackay CR. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature. 2009;461(7268):1282–1286. doi: 10.1038/nature08530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Singh N, Gurav A, Sivaprakasam S, Brady E, Padia R, Shi H, Thangaraju M, Prasad PD, Manicassamy S, Munn DH, Lee JR, Offermanns S, Ganapathy V. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity. 2014;40(1):128–139. doi: 10.1016/j.immuni.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]