

Abstract
TP53 is associated with the resistance of cytotoxic treatment and patient prognosis, and the mutation rate of TP53 in esophageal squamous cell carcinoma (ESCC) is extraordinarily high, at over 90%. PRIMA‐1 (p53 re‐activation and induction of massive apoptosis) has recently been reported to restore the function of mutant TP53; however, its antitumor effect and mechanism in ESCC remain unclear. After evaluating the TP53 mutation status of a panel of 11 ESCC cell lines by Sanger sequencing, we assessed the in vitro effect of PRIMA‐1 administration on cells with different TP53 status by conducting cell viability and apoptosis assays. The expression levels of proteins in p53‐related pathways were examined by Western blotting, while knockdown studies were conducted to investigate the mechanism underlying PRIMA‐1's function. An ESCC xenograft model was further used to evaluate the therapeutic effect of PRIMA‐1 in vivo. PRIMA‐1 markedly inhibited cell growth and induced apoptosis by upregulating Noxa expression in ESCC cell lines with TP53 missense mutations, whereas no apoptosis was induced in ESCC with wild‐type TP53 and TP53 with frameshift and nonsense mutations. Importantly, the knockdown of Noxa canceled the apoptosis induced by PRIMA treatment in ESCC cell lines with TP53 missense mutations. PRIMA‐1 administration, compared with placebo, showed a significant antitumor effect by inducing Noxa in the xenograft model of an ESCC cell line with a TP53 missense mutation. PRIMA‐1 exhibits a significant antitumor effect, inducing massive apoptosis through the upregulation of Noxa in ESCC with TP53 missense mutations.
Keywords: apoptosis, esophageal squamous cell carcinoma, Noxa, PRIMA‐1, TP53
1. INTRODUCTION
Esophageal squamous cell carcinoma (ESCC) is one of the most malignant gastrointestinal cancers and is a leading cause of cancer death.1, 2 Recently, advances in multimodal treatments (ie, preoperative chemotherapy or chemo‐radiotherapy followed by surgery) have improved the clinical outcomes of locally advanced ESCC to some extent; however, the survival benefit is generally confined to responders to these treatments.3, 4, 5, 6 Therefore, the development of a novel promising therapy targeting specific molecules is indispensable to improve the prognosis of ESCC patients.
TP53 is a well‐known tumor suppressor gene that induces apoptosis, cell cycle arrest, senescence, DNA repair or autophagy under stress conditions.7, 8, 9, 10 However, mutant TP53 is known to lose tumor suppressor functions and is associated with tumor malignancy, including invasion, metastasis or resistance to cytotoxic treatments.11, 12, 13 The mutation rate of TP53 is reported to be 30%‐50% in several types of cancer, including multiple myeloma, breast cancer, colorectal cancer and prostate cancer.14, 15, 16, 17 Notably, the TP53 mutation rate of ESCC has been identified to be extraordinarily high, at over 90%.18, 19 In fact, some previous studies have shown that mutation of TP53 is associated with patient prognosis, carcinogenesis, and resistance to chemotherapy or chemo‐radiotherapy in ESCC.20, 21, 22, 23 Therefore, therapeutic strategies targeting mutant TP53 which most ESCC carry may be a promising approach to the improvement of prognosis in ESCC patients.
PRIMA‐1 (p53 re‐activation and induction of massive apoptosis), a low molecular weight compound, has been recently reported to restore the function of wild‐type TP53 to mutant TP53 and to induce p53‐mediated apoptosis.24 The anticancer effects of PRIMA‐1 and its methylated form PRIMA‐1Met (APR‐246) have been demonstrated in some types of cancer, such as osteosarcoma, multiple myeloma, lung cancer, breast cancer and colon cancer.24, 25, 26, 27, 28, 29 In addition, a recent phase I/II clinical trial in hematological malignancies and prostate cancer has demonstrated the feasibility and clinical efficacy of APR‐246 administration in human patients.30 However, to the best of our knowledge, the treatment effect of PRIMA‐1 and its mechanism in ESCC have never been investigated. This study was designed to assess the antitumor effect of PRIMA‐1 treatment in both a panel of ESCC cell lines with different TP53 status and an ESCC xenograft model and to uncover the molecular mechanism of PRIMA‐1.
2. MATERIALS AND METHODS
2.1. Cell lines and culture
The human esophageal squamous cell lines TE1, TE4, TE5, TE6, TE8, TE9, TE10, TE11 and TE14 were obtained from the Riken Bioresource Center Cell Bank (Tsukuba, Japan). KYSE410 and KYSE960 were obtained from the Japanese Cancer Research Resources Bank (Osaka, Japan). All of the cells were cultured in RPMI‐1640 medium (Nakalai Tesque, Kyoto, Japan) containing 10% FBS (Sigma‐Aldrich, St. Louis, MO, USA) and 1% penicillin/streptomycin (Life Technologies, Carlsbad, CA, USA), in a humidified atmosphere under 5% CO2 at 37°C.
2.2. Reagent
PRIMA‐1 was purchased from Calbiochem‐Merck (Milan, Italy), diluted in water and stored at −20°C.
2.3. Sanger sequencing to detect the status of TP53
Genomic DNA was extracted from cultured cells using the DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), following the manufacturer's protocol. The genomic DNA sequence of TP53 was amplified by PCR using primers covering exons 2‐11 of TP53. The primers used for sequencing are detailed in Table S1. PCR products were purified with a MinElute PCR Purification Kit (Qiagen). Cycle sequencing was performed using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Carlsbad, CA, USA).
2.4. Cell growth inhibition assays
Cell growth inhibition was tested using the 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyl tetrazolium bromide (MTT) assay (Sigma‐Aldrich, St Louis, MO, USA). Cells were seeded into 96‐well culture plates at a concentration of 3 × 103 cells/100 μL. After 24 hour, the cells were exposed to PRIMA‐1 (0.5, 1, 5, 10, 25, 50, 100 μmol/L). In the siRNA knockdown experiments, the cells were exposed to various concentrations of PRIMA‐1 at 24 hour after transfection. After an additional 48 hour of incubation, 10 μL (50 μg) of MTT was added to each well, followed by incubation for 3 hour at 37°C. Next, the medium was removed, and 100 μL of acid isopropanol was added to dissolve the resultant formazan crystals. Plate absorbance was measured using an EnSpire Multimode Plate Reader (PerkinElmer, Waltham, MA, USA) at 550 nm, and the absorbance at 665 nm was measured as the reference. The results were expressed as the percentage of absorbance relative to untreated controls.
2.5. Apoptosis analysis
Cells were incubated for 24‐48 hour with PRIMA‐1. The Annexin V Binding 0Buffer (Biovision Research Products, Mountain View, CA, USA) was mixed with 5 μL of Annexin V‐APC (Biolegend, San Diego, CA, USA) and 5 μL of PI (Biovision Research Products) and was incubated for 30 minute at room temperature in the dark. The samples were analyzed by flow cytometry on a FACSCanto II instrument (BD Biosciences, San Jose, CA, USA). Cells stained with Annexin V were considered apoptotic cells.
2.6. Western blot analysis
Total proteins were extracted from cultured cells using RIPA buffer containing protease inhibitor and phosphatase inhibitor (Thermo Fisher Scientific, Waltham, MA, USA). Proteins were subjected to 12% SDS‐PAGE (Bio‐Rad, Hercules, CA, USA). The separated proteins were transferred to Immun‐Blot PVDF membranes (Bio‐Rad) and incubated with anti‐ACTB antibody (A2066; Sigma‐Aldrich; 1:1000 dilution), anti‐cleaved PARP antibody (#5625; Cell Signaling Technology, Beverly, MA, USA; 1:1000 dilution), anti‐p21Waf1/Cip1 antibody (#2947; Cell Signaling Technology; 1:1000 dilution), anti‐Bax antibody (#5023; Cell Signaling Technology, Beverly, MA, USA; 1:1000 dilution), anti‐p53 antibody (Clone DO‐7; DAKO Cytomation, Carpinteria, CA, USA; 1:1000 dilution) and anti‐Noxa antibody (OP180; Calbiochem, Darmstadt, Germany; 1:1000 dilution) at 4°C overnight. Next, the membranes were incubated with HRP‐linked anti‐rabbit IgG (GE Healthcare Biosciences, Piscataway, NJ, USA) and HRP‐linked anti‐mouse IgG (GE Healthcare Biosciences) at a dilution of 1:100 000 for 1 hour at room temperature. The antigen–antibody complex was detected using an ECL Prime Western Blotting Detection Kit (GE Healthcare Biosciences).
2.7. siRNA knockdown
Noxa siRNA and control siRNA‐A were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Approximately 2.5 × 105 cells per well were seeded in 6‐well plates with 2 mL of antibiotic‐free RPMI 1640 medium with 10% FBS. After 24 hour, the cells were transfected with control siRNA‐A or Noxa siRNA using Lipofectamine RNAiMAX Transfection Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's instructions. After transfection for 24 hour, the cells were treated with PRIMA‐1 and incubated. For western blotting analysis and cell viability assays, the cells were collected after 24‐48 hour of incubation.
2.8. Xenograft models
Animal studies were conducted in strict accordance with the principles and procedures approved by the Committee on the Ethics of Animal Experiments of Osaka University. For xenograft models, 6‐week‐old female mice (BALB/c‐nu/nu) were purchased from CLEA Japan (Tokyo, Japan). TE8 (5.0 × 106) cells in a total volume of 100 μL of RPMI 1640/Matrigel (1:1 suspension) were subcutaneously injected into loose skin. When the average size of the subcutaneous tumor reached approximately 200 mm3, all treatments were started. PRIMA‐1 was dissolved in PBS and was administered daily at a dose of 50 mg/kg/d by intraperitoneal injection. Mice in the vehicle control group were treated with PBS daily. These treatments lasted for 10 days. The tumor volume was measured with calipers and was calculated using the formula V = (ab 2)/2, where a is the largest diameter and b is the smallest diameter. Mice were euthanized 24 hour after the last treatment. The tumors were fixed in 10% buffered formalin and processed routinely. Immunohistochemistry staining of the tumor sections with anti‐cleaved caspase‐3 antibody (#9654; Cell Signaling Technology; 1:1000 dilution) and anti‐cleaved PARP antibody (#5625; Cell Signaling Technology; 1:100 dilution) was conducted as described by the manufacturer. For the assessment of PRIMA‐1‐mediated activation of the p53 pathway, tumor lysates were subjected to western blot analysis using TE8 xenograft mice treated for 3 days with PBS or PRIMA‐1 at 100 mg/kg/d daily.
2.9. Statistical analysis
Cell culture experiments were repeated at least 3 times. Statistical analyses were performed using the JMP Pro software program, version 11.0 for Windows (JMP; SAS Institute, Cary, NC, USA). Differences between categories were assessed using Student's t tests or χ2‐tests. P‐values < 0.05 were considered statistically significant.
3. RESULTS
3.1. TP53 status in a panel of esophageal squamous cell carcinoma cell lines
Table 1 shows the summary of the mutation status in the 11 ESCC cell lines. Sanger sequencing of TP53 confirmed the presence of missense mutations in the TE1, TE4, TE5, TE6, TE8, TE10 and TE11 lines, a frameshift mutation in TE9, a nonsense mutation in TE14, and wild‐type status in KYSE410 and 960. All of the detected TP53 mutations in the 11 ESCC cell lines existed in hotspot codons (ie, exons 4‐8). Western blotting detected a p53 protein band in all cell lines with both missense mutation and wild‐type TP53, while protein expression was not identified in TE9 with a frameshift mutation nor in TE14 with a nonsense mutation, as shown in Figure 1A.
Table 1.
TP53 mutation status in esophageal squamous cell carcinoma cell lines
| Cell line | Mutation status | Mutation type | Exon | Codon | Nucleotide | Amino acid |
|---|---|---|---|---|---|---|
| TE1 | Mutant | Missense | 8 | 272 | GTG → ATG | Val → Met |
| TE4 | Mutant | Missense | 7 | 245 | GGC → TGC | Gly → Cys |
| TE5 | Mutant | Missense | 8 | 272 | GTG → TTG | Val → Leu |
| TE6 | Mutant | Missense | 7 | 248 | CGG → CAG | Arg → Gln |
| TE8 | Mutant | Missense | 7 | 237 | ATG → ATT | Met → Ile |
| TE9 | Mutant | Frameshift | 8 | 273 | CGG → delG | Frame shift |
| TE10 | Mutant | Missense | 7 | 242 | TGC → TAC | Cys → Tyr |
| TE11 | Mutant | Missense | 4 | 110 | CGT → CTT | Arg → Leu |
| TE14 | Mutant | Nonsense | 6 | 213 | CGA → TGA | Arg → X |
| KYSE410 | Wild | — | — | — | — | — |
| KYSE960 | Wild | — | — | — | — | — |
Arg, arginine; Cys, cysteine; Gln, glutamine; Gly, glycine; Ile, isoeucine; Leu, leucine; Met, methionine; Tyr, tyrosine; Val, valine.
[Correction added on 19 January 2018, after first online publication: The spelling of “Misssense” was corrected to “Missense” in Table 1.]
Figure 1.
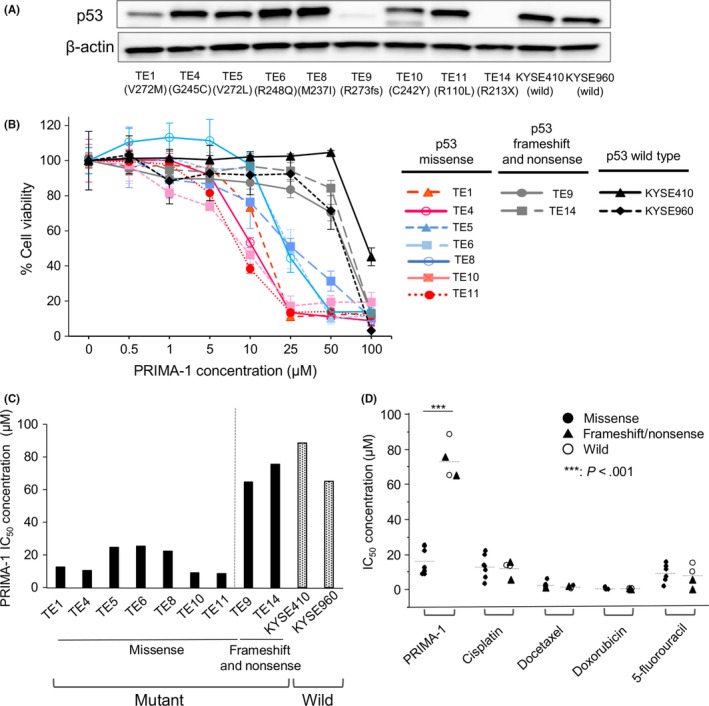
PRIMA‐1 selectively inhibits the proliferation of esophageal squamous cell carcinoma (ESCC) cell lines with TP53 missense mutations. A, Total p53 protein levels were evaluated by western blot analysis. B, PRIMA‐1 dose‐response curves for ESCC cell lines after 48 h of treatment with PRIMA‐1 using the MTT cell viability assay; n = 3‐5. Error bars show the standard deviation. C, IC 50 values of PRIMA‐1 in ECSS cell lines. D, IC 50 values of PRIMA‐1, cisplatin, docetaxel, doxorubicin and 5‐fluorouracil in ESCC with TP53 missense mutation vs frameshift/nonsense mutation and wild‐type ESCC cells. ***P < .001
3.2. PRIMA‐1 selectively inhibits the growth of the esophageal squamous cell carcinoma cell lines with TP53 missense mutation
The effect of PRIMA‐1 treatment was investigated in all 11 ESCC cell lines with different TP53 status using an MTT viability assay. After treatment with PRIMA‐1 for 48 hour, a significant response to PRIMA‐1 was identified in all cell lines with TP53 missense mutations (TE1, TE4, TE5, TE6, TE8, TE10, TE11), while cell lines with a TP53 frameshift mutation (TE9), nonsense mutation (TE14) and wild type (KYSE410, KYSE960) were not sensitive to PRIMA‐1, as shown in Figure 1B,C (missense vs others; mean IC50 16.2 vs 73.6 μmol/L, P < .001). By contrast, the mutation status of TP53 was not significantly influenced by sensitivity to chemotherapy agents, including cisplatin, docetaxel, 5‐fluorouracil and doxorubicin (Figure 1D).
3.3. PRIMA‐1 induces apoptosis in esophageal squamous cell carcinoma cell lines with TP53 missense mutation
Esophageal squamous cell carcinoma cell lines with different TP53 mutation status were treated with PRIMA‐1 for 24‐48 hour to investigate whether PRIMA‐1 induces apoptosis, and the percentage of Annexin V‐positive cells was quantified. Treatment with 25 and 50 μmol/L PRIMA‐1 significantly increased the percentage of Annexin V‐positive cells in the TE1, TE8 and TE10 cell lines with TP53 missense mutation (Figure 2A,B). By contrast, apoptosis was barely induced by PRIMA‐1 treatment in ESCC cells with frameshift (TE9)/nonsense (TE14) mutation or with wild‐type (KYSE410, KYSE960) TP53.
Figure 2.
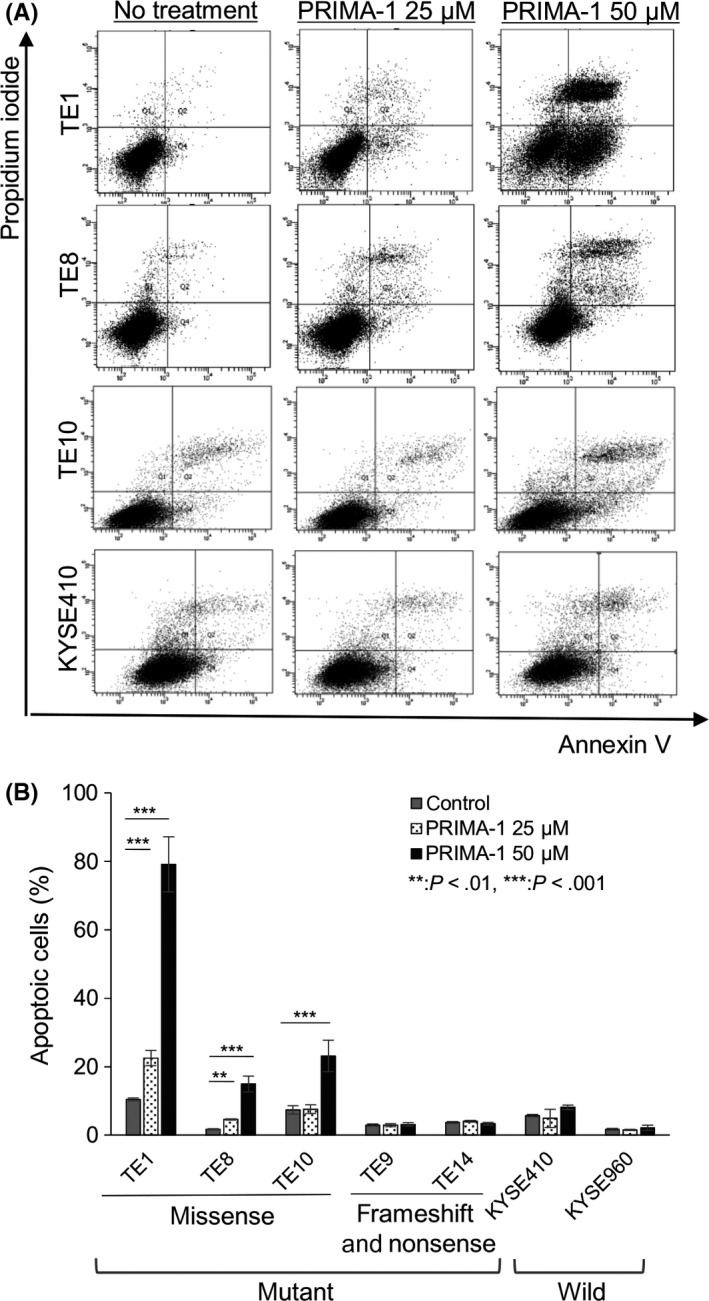
PRIMA‐1 preferentially induces apoptosis in esophageal squamous cell carcinoma (ESCC) cell lines with a TP53 missense mutation. A, Fluorence‐activated cell sorting plots of Annexin V/propidium iodide‐labeled cells treated with 25 and 50 μmol/L PRIMA‐1 for 24‐48 h. B, Percentage of Annexin V‐positive cells after treatment with PRIMA‐1 using flow cytometry; n = 3‐5. Error bars show the standard deviation. P‐values refer to the comparison of PRIMA‐1‐treated samples with untreated samples within each cell line. **P < .01, ***P < .001
3.4. PRIMA‐1 activates the p53‐dependent apoptotic pathway
The expression levels of proteins in p53‐related pathways were examined by western blotting to investigate the mechanism of the antitumor effects of PRIMA‐1 treatment. In ESCC cell lines with TP53 missense mutation, PRIMA‐1 upregulated the pro‐apoptotic protein Noxa in a dose‐dependent manner, leading to an increased level of cleaved PARP, as shown in Figure 3. In addition, the protein expression of Bax was increased in TE8 and TE10, and p21 expression was increased in TE8. However, PRIMA‐1 did not upregulate p53‐dependent apoptotic pathways in ESCC cell lines with a frameshift/nonsense mutation or with the wild‐type TP53 gene, indicating that PRIMA‐1 selectively induces apoptosis in ECSS cell lines with TP53 missense mutation through the upregulation of Noxa. To see if PRIMA‐1 induced Noxa in a p53‐dependent manner, we knocked down p53 in TE10 cells with a TP53 missense mutation, which resulted in the cancellation of the increase in cleaved PARP and Noxa expression caused by PRIMA‐1 treatment (Figure S1A).
Figure 3.
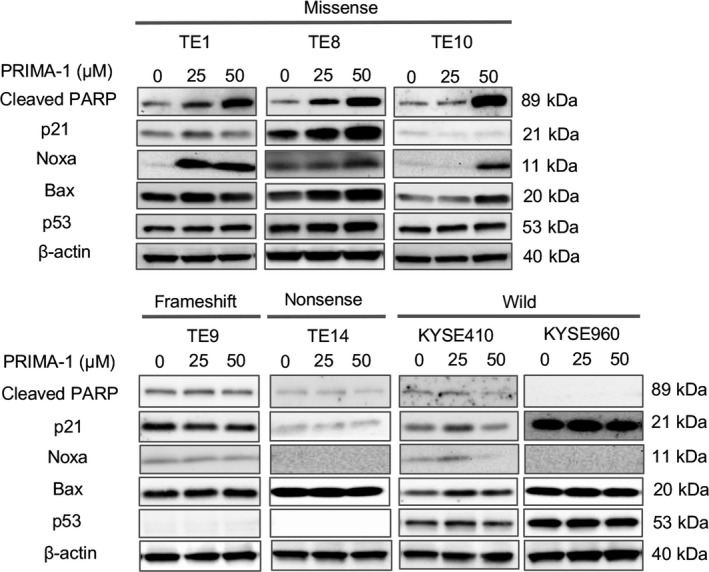
PRIMA‐1 leads to the activation of p53‐dependent apoptotic pathways in esophageal squamous cell carcinoma (ESCC) cell lines with TP53 missense mutations. The total levels of the indicated proteins were evaluated by western blot analysis in ESCC cell lines with different TP53 mutation status after treatment with 25 and 50 μmol/L PRIMA‐1 for 24‐48 h. In TE1, TE8 and TE10 cells with TP53 missense mutations, the expression levels of cleaved PARP and Noxa were increased in a dose‐dependent manner, whereas no increase was observed in TE9 with a frameshift mutation, TE14 with a nonsense mutation, and KYSE410, KYSE960 with wild‐type TP53
3.5. PRIMA‐1 induces apoptosis in esophageal squamous cell carcinoma with TP53 missense mutation and is mediated through Noxa
To further examine whether Noxa plays a pivotal role in apoptosis induced by PRIMA‐1, we knocked down Noxa expression in ESCC cell lines with both a missense mutation and wild‐type TP53. TE1/TE10 (TP53 missense mutation) and KYSE410/KYSE960 (TP53 wild type) cell lines were transfected with siRNA specific to Noxa, followed by treatment with PRIMA‐1 (25 and 50 μmol/L, respectively), and apoptotic induction was assessed. Western blotting demonstrated that the knockdown of Noxa in TE1 and TE10 with TP53 missense mutation inhibited PRIMA‐1‐induced cleaved PARP, while the knockdown of Noxa in KYSE410 and KYSE960 with wild‐type TP53 showed no effect on the level of cleaved PARP (Figure 4A). In addition, p53 knockdown led to a reduction in the apoptosis caused by PRIMA administration in both TE1 and TE10 cells to the same degree as the Noxa knockdown (Figure S1B). Furthermore, flow cytometry analysis identified a decreased percentage of Annexin V‐positive cells following PRIMA‐1 treatment in TE1, TE8 and TE10 cells with TP53 missense mutations but not in the ESCC cells with wild‐type TP53 (Figure 4B). These results agree with the results of the MTT cell viability assay, in which the inhibition of proliferation by PRIMA‐1 treatment in TE1, TE8 and TE10 cells with TP53 missense mutations was abrogated by the Noxa knockdown. This trend was not observed in the ESCC cells with wild‐type TP53 (Figure 4C), indicating that PRIMA‐1‐induced apoptosis is mediated primarily through the upregulation of Noxa.
Figure 4.
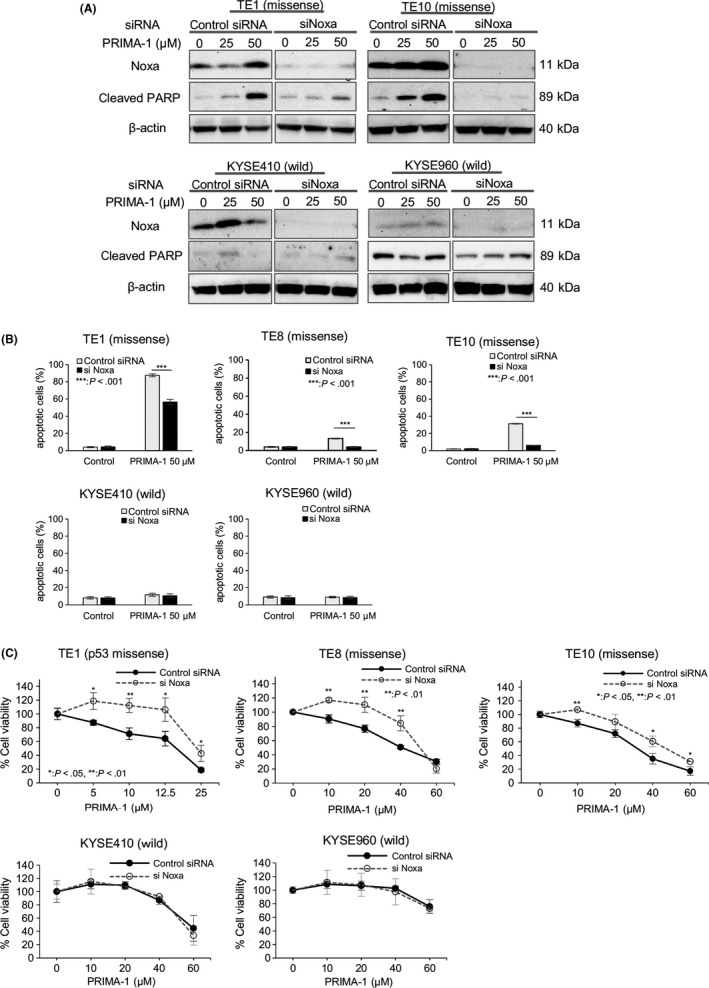
PRIMA‐1 preferentially induces apoptosis in esophageal squamous cell carcinoma (ESCC) cell lines with TP53 missense mutations and is mediated primarily through Noxa. A, Western blot analysis of total Noxa and cleaved PARP levels after treatment with PRIMA‐1 in ESCC cell lines transfected with either control or Noxa siRNA for 24 h. B, Percentage of Annexin V‐positive cells after treatment with PRIMA‐1 in ESCC cell lines transfected with either control or Noxa siRNA for 24 h using flow cytometry. C, The growth suppression effect of PRIMA‐1 was evaluated in ESCC cell lines transfected with either control or Noxa siRNA using the MTT assay; n = 3‐5. Error bars show the standard deviation. P‐values refer to the comparison of Noxa siRNA‐transfected samples with control siRNA‐transfected samples within each cell line. *P < .05, **P < .01, ***P < .001
3.6. PRIMA‐1 suppresses the tumor growth of esophageal squamous cell carcinoma cell lines harboring a TP53 missense mutation in vivo
To examine the anti‐tumorigenic potential of PRIMA‐1 in vivo, we established a subcutaneous xenograft nude mouse model implanted with the TE8 cell line with a TP53 missense mutation, which can form tumors. The mice received ip injections of either PBS (control group, n = 5) or 50 mg/kg PRIMA‐1 (PRIMA‐1 group, n = 5) once daily for 10 days after palpable tumor masses were evident (Figure 5A). The administration of PRIMA‐1 significantly suppressed tumor growth at 10 and 11 days after treatment compared with that in the control group (P < .05). The gross appearance of tumors in the PRIMA‐1 group tended to be smaller than that in the control group (Figure 5A). HE staining showed only scattered tumor cells with fibrotic changes in the PRIMA‐1 group in contrast to the solid tumor area with mostly viable cancer cells in the control group (Figure 5B). Immunohistochemical analysis further identified an increased percentage of both cleaved PARP‐positive and cleaved caspase‐3‐positive cells in xenograft tumors in the PRIMA‐1 group compared with that in the control group (Figure 5B). To assess the re‐activation of p53‐related pathways induced by PRIMA‐1 in vivo, the xenograft mouse model of TE8 with a TP53 missense mutation was used. In fact, treatment with PRIMA‐1 (100 mg/kg/d, for 3 days) induced the protein expression of p21, Bax, Noxa and PARP cleavage in the representative xenograft tumor, as shown in Figure 5C.
Figure 5.
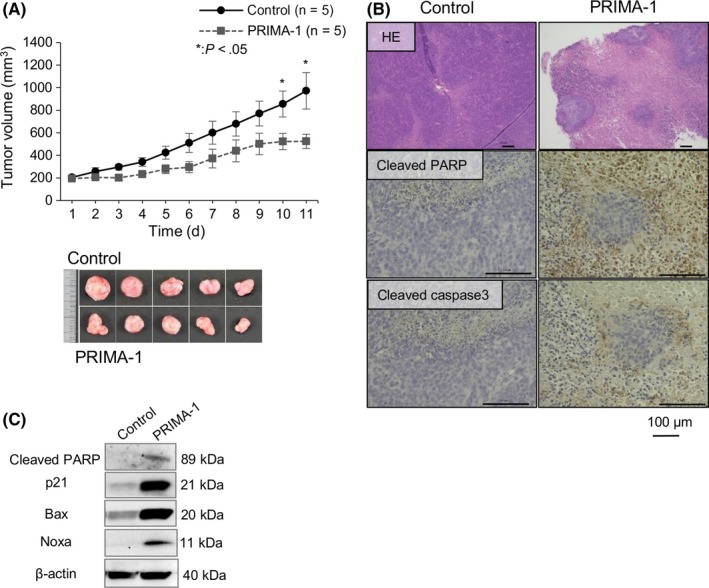
PRIMA‐1 suppresses tumor growth in esophageal squamous cell carcinoma (ESCC) cell line (TE8) xenograft mice with a TP53 missense mutation. A, The growth curves, tumor weight, and images of TE8 tumors from xenograft mice treated with control (PBS) vs PRIMA‐1; n = 5 for each group. Error bars show the SEM. P‐values refer to the comparison of PRIMA‐1‐treated samples with untreated samples. *P < .05. B, HE staining or immunohistochemical staining for apoptotic markers, cleaved PARP and caspase‐3 of tumor sections treated with control and PRIMA‐1. C, Western blot analysis of xenografts treated with control and PRIMA‐1 for 3 days
4. DISCUSSION
In this study, we demonstrated that PRIMA‐1 showed significant antitumor effects on all ESCC cells with TP53 missense mutations, inducing massive apoptosis, while ESCC cells with wild‐type TP53 or other types of TP53 mutations, including frameshift and nonsense mutations, were relatively resistant to PRIMA‐1. This antitumor effect was also confirmed by an in vivo study using the ESCC xenograft model. We further demonstrated that the pro‐apoptotic protein Noxa plays an important role in PRIMA‐1‐mediated apoptosis in ESCC.
The tumor suppressor gene TP53 plays multiple suppressive roles, including apoptosis, cell cycle arrest, senescence, DNA repair, and autophagy, in response to cellular stress. However, this gene is inactivated by mutation in half of all human cancers.31 Most of the TP53 mutations are missense mutations, resulting in amino acid substitutions in the DNA‐binding core domain, and TP53 mutations disrupt p53‐specific DNA binding and transcriptional transactivation, leading to the loss of functions as a tumor suppressor.32 The small molecule PRIMA‐1 has been identified in the low molecular weight compound library to restore mutant p53 to the structure and function of wild‐type p53 by targeting the p53 DNA‐binding core domain, resulting in the induction of p53‐mediated apoptosis.24, 33 However, the mechanisms underlying how PRIMA‐1 induces apoptosis in TP53 mutant cancer cells to change the conformation of p53 mutant protein have not been fully understood.
The present study demonstrated that PRIMA‐1 selectively induced apoptosis in ESCC cell lines with TP53 missense mutations, as previously reported in other types of cancer.24, 25, 33, 34, 35, 36 In addition, PRIMA‐1 showed no considerable effect on TP53 frameshift and nonsense mutations, in which the p53 protein was undetected by western blot analysis. A frameshift mutation, in theory, has a more dramatic effect on the polypeptide than missense mutations, despite the deletion of just 1 nucleotide, while a nonsense mutation produces no p53 protein due to the change in 1 nucleotide. Thereby, our results support that PRIMA‐1 has the capability of inducing the specific restoration of sequence‐specific binding to DNA in p53, similar to that in previous reports.24, 33 As previous studies have reported that methylene quinuclidinone, the active compound converted from PRIMA‐1, binds to cysteine residues in the p53 core domain and stabilizes the wild‐type p53 conformation, the protein expression of p53 might be at least necessary for PRIMA‐1 to function on TP53 mutant ESCC.33 However, recent reports have suggested that PRIMA‐1 can induce cell death through p53‐independent mechanisms. PRIMA‐1MET, the methylated form of PRIMA‐1, was reported to induce ROS, resulting in cell death in a p53‐independent manner.37, 38, 39 Other reports have shown that PRIMA‐1 activates p73, a p53‐related protein, and induces apoptosis irrespective of the TP53 status.25, 40 Therefore, PRIMA‐1 could show different effects on cancer cells according to cancer type or TP53 mutation status, and the mechanisms remain unclear.
For a better understanding of the mechanisms of PRIMA‐1‐induced apoptosis, we examined the expression levels of p53‐mediated apoptotic genes. Noxa, one of the most important pro‐apoptotic target proteins of p53, was found to be the only molecule consistently upregulated by PRIMA‐1 in a dose‐dependent manner in most ESCC cells harboring TP53 missense mutations, while Bax and p21 were upregulated in only some ESCC with TP53 missense mutations after PRIMA‐1 treatment. Silencing Noxa expression by siRNA further showed a significant inhibition of apoptosis in TE1 and TE10 with TP53 missense mutations compared with ESCC with wild‐type TP53. Intriguingly, the knockdown of Noxa caused a significant but partial inhibition of PRIMA‐1‐induced apoptosis in TE1, while PRIMA‐1‐induced apoptosis was completely inhibited to the level of non‐treatment in TE10. This result might imply that p53‐independent pathways also contribute to PRIMA‐1‐induced apoptosis in TE1, which is supported by previous reports showing that PRIMA‐1 induced apoptosis through the upregulation of Noxa in a p53‐independent manner in other types of cancer.25, 28 In this context, we found that the induction of ROS after PRIMA‐1 treatment in TE1, but not in TE10, was inhibited by N‐acetyl cysteine (data not shown). Because Noxa is upregulated in a ROS‐dependent pathway in a p53‐independent manner,41 the PRIMA‐1‐induced upregulation of Noxa in TE1 could be through a ROS‐dependent pathway in addition to a p53‐dependent pathway.
In conclusion, the present study showed the first evidence that PRIMA‐1 inhibits cell viability and tumor growth specifically in ESCC cells with TP53 missense mutations. We further uncovered the molecular mechanism underlying PRIMA‐1‐mediated apoptosis through the upregulation of the pro‐apoptotic protein Noxa, leading to the activation of cleaved PARP. This significant antitumor effect and its mechanism were also confirmed in an in vivo ESCC xenograft model. Although these results should be validated in the future, PRIMA‐1 could be a novel treatment as a potential anticancer agent in clinical trials for ESCC harboring an extraordinarily high frequency of TP53 missense mutation.
DISCLOSURE STATEMENT
The authors have no conflicts of interest to declare.
Supporting information
ACKNOWLEDGMENTS
We thank the members of our laboratories for their helpful discussions.
Furukawa H, Makino T, Yamasaki M, et al. PRIMA‐1 induces p53‐mediated apoptosis by upregulating Noxa in esophageal squamous cell carcinoma with TP53 missense mutation. Cancer Sci. 2018;109:412–421. https://doi.org/10.1111/cas.13454
Funding information
Japan Society for the Promotion of Science (JSPS) Grant‐in‐Aid for Scientific Research grant number 16K19923.
REFERENCES
- 1. Rustgi AK, El‐Serag HB. Esophageal carcinoma. N Engl J Med. 2014;371:2499‐2509. [DOI] [PubMed] [Google Scholar]
- 2. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87‐108. [DOI] [PubMed] [Google Scholar]
- 3. Ando N, Iizuka T, Kakegawa T, et al. A randomized trial of surgery with and without chemotherapy for localized squamous carcinoma of the thoracic esophagus: the Japan Clinical Oncology Group Study. J Thorac Cardiovasc Surg. 1997;114:205‐209. [DOI] [PubMed] [Google Scholar]
- 4. Kelsen DP, Ginsberg R, Pajak TF, et al. Chemotherapy followed by surgery compared with surgery alone for localized esophageal cancer. N Engl J Med. 1998;339:1979‐1984. [DOI] [PubMed] [Google Scholar]
- 5. Medical Research Council Oesophageal Cancer Working Group . Surgical resection with or without preoperative chemotherapy in oesophageal cancer: a randomised controlled trial. Lancet. 2002;359:1727‐1733. [DOI] [PubMed] [Google Scholar]
- 6. Miyata H, Yoshioka A, Yamasaki M, et al. Tumor budding in tumor invasive front predicts prognosis and survival of patients with esophageal squamous cell carcinomas receiving neoadjuvant chemotherapy. Cancer. 2009;115:3324‐3334. [DOI] [PubMed] [Google Scholar]
- 7. Lane DP. p53, guardian of the genome. Nature. 1992;358:15‐16. [DOI] [PubMed] [Google Scholar]
- 8. Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature. 2005;434:864‐870. [DOI] [PubMed] [Google Scholar]
- 9. Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413‐431. [DOI] [PubMed] [Google Scholar]
- 10. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53‐mediated tumour suppression. Nat Rev Cancer. 2014;14:359‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Freed‐Pastor WA, Prives C. Mutant p53: one name, many proteins. Genes Dev. 2012;26:1268‐1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhu J, Sammons MA, Donahue G, et al. Gain‐of‐function p53 mutants co‐opt chromatin pathways to drive cancer growth. Nature. 2015;525:206‐211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Subramanian M, Francis P, Bilke S, et al. A mutant p53/let‐7i‐axis‐regulated gene network drives cell migration, invasion and metastasis. Oncogene. 2015;34:1094‐1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lodé L, Eveillard M, Trichet V, et al. Mutations in <em>TP53 < /em> are exclusively associated with del(17p) in multiple myeloma. Haematologica. 2010;95:1973‐1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lacroix M, Toillon RA, Leclercq G. p53 and breast cancer, an update. Endocr Relat Cancer. 2006;13:293‐325. [DOI] [PubMed] [Google Scholar]
- 16. Li XL, Zhou J, Chen ZR, Chng WJ. P53 mutations in colorectal cancer – molecular pathogenesis and pharmacological reactivation. World J Gastroenterol. 2015;21:84‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ecke TH, Schlechte HH, Hubsch A, et al. TP53 mutation in prostate needle biopsies–comparison with patients follow‐up. Anticancer Res. 2007;27:4143‐4148. [PubMed] [Google Scholar]
- 18. Gao Y‐B, Chen Z‐L, Li J‐G, et al. Genetic landscape of esophageal squamous cell carcinoma. Nat Genet. 2014;46:1097‐1102. [DOI] [PubMed] [Google Scholar]
- 19. Sawada G, Niida A, Uchi R, et al. Genomic landscape of esophageal squamous cell carcinoma in a Japanese population. Gastroenterology. 2016;150:1171‐1182. [DOI] [PubMed] [Google Scholar]
- 20. Kobayashi S, Koide Y, Endo M, Isono K, Ochiai T. The p53 gene mutation is of prognostic value in esophageal squamous cell carcinoma patients in unified stages of curability. Am J Surg. 1999;177:497‐502. [DOI] [PubMed] [Google Scholar]
- 21. Saeki H, Kitao H, Yoshinaga K, et al. Copy‐neutral loss of heterozygosity at the p53 locus in carcinogenesis of esophageal squamous cell carcinomas associated with p53 mutations. Clin Cancer Res. 2011;17:1731‐1740. [DOI] [PubMed] [Google Scholar]
- 22. Yamasaki M, Miyata H, Fujiwara Y, et al. p53 genotype predicts response to chemotherapy in patients with squamous cell carcinoma of the esophagus. Ann Surg Oncol. 2010;17:634‐642. [DOI] [PubMed] [Google Scholar]
- 23. Makino T, Yamasaki M, Miyata H, et al. p53 Mutation status predicts pathological response to chemoradiotherapy in locally advanced esophageal cancer. Ann Surg Oncol. 2010;17:804‐811. [DOI] [PubMed] [Google Scholar]
- 24. Bykov VJ, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low‐molecular‐weight compound. Nat Med. 2002;8:282‐288. [DOI] [PubMed] [Google Scholar]
- 25. Saha MN, Jiang H, Yang Y, Reece D, Chang H. PRIMA‐1Met/APR‐246 displays high antitumor activity in multiple myeloma by induction of p73 and Noxa. Mol Cancer Ther. 2013;12:2331‐2341. [DOI] [PubMed] [Google Scholar]
- 26. Zandi R, Selivanova G, Christensen CL, Gerds TA, Willumsen BM, Poulsen HS. PRIMA‐1Met/APR‐246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res. 2011;17:2830‐2841. [DOI] [PubMed] [Google Scholar]
- 27. Liang Y, Besch‐Williford C, Hyder SM. PRIMA‐1 inhibits growth of breast cancer cells by re‐activating mutant p53 protein. Int J Oncol. 2009;35:1015‐1023. [DOI] [PubMed] [Google Scholar]
- 28. Li XL, Zhou J, Chan ZL, Chooi JY, Chen ZR, Chng WJ. PRIMA‐1met (APR‐246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget. 2015;6:36689‐36699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lu T, Zou Y, Xu G, et al. PRIMA‐1Met suppresses colorectal cancer independent of p53 by targeting MEK. Oncotarget. 2016;7:83017‐83030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lehmann S, Bykov VJ, Ali D, et al. Targeting p53 in vivo: a first‐in‐human study with p53‐targeting compound APR‐246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol. 2012;30:3633‐3639. [DOI] [PubMed] [Google Scholar]
- 31. Soussi T, Ishioka C, Claustres M, Beroud C. Locus‐specific mutation databases: pitfalls and good practice based on the p53 experience. Nat Rev Cancer. 2006;6:83‐90. [DOI] [PubMed] [Google Scholar]
- 32. Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303‐312. [DOI] [PubMed] [Google Scholar]
- 33. Lambert JM, Gorzov P, Veprintsev DB, et al. PRIMA‐1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell. 2009;15:376‐388. [DOI] [PubMed] [Google Scholar]
- 34. Lambert JM, Moshfegh A, Hainaut P, Wiman KG, Bykov VJ. Mutant p53 reactivation by PRIMA‐1MET induces multiple signaling pathways converging on apoptosis. Oncogene. 2010;29:1329‐1338. [DOI] [PubMed] [Google Scholar]
- 35. Messina RL, Sanfilippo M, Vella V, et al. Reactivation of p53 mutants by prima‐1 [corrected] in thyroid cancer cells. Int J Cancer. 2012;130:2259‐2270. [DOI] [PubMed] [Google Scholar]
- 36. Izetti P, Hautefeuille A, Abujamra AL, et al. PRIMA‐1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Invest New Drugs. 2014;32:783‐794. [DOI] [PubMed] [Google Scholar]
- 37. Peng X, Zhang MQ, Conserva F, et al. APR‐246/PRIMA‐1MET inhibits thioredoxin reductase 1 and converts the enzyme to a dedicated NADPH oxidase. Cell Death Dis. 2013;4:e881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tessoulin B, Descamps G, Moreau P, et al. PRIMA‐1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood. 2014;124:1626‐1636. [DOI] [PubMed] [Google Scholar]
- 39. Grellety T, Laroche‐Clary A, Chaire V, et al. PRIMA‐1(MET) induces death in soft‐tissue sarcomas cell independent of p53. BMC Cancer. 2015;15:684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rokaeus N, Shen J, Eckhardt I, Bykov VJ, Wiman KG, Wilhelm MT. PRIMA‐1(MET)/APR‐246 targets mutant forms of p53 family members p63 and p73. Oncogene. 2010;29:6442‐6451. [DOI] [PubMed] [Google Scholar]
- 41. Kim JY, Ahn HJ, Ryu JH, Suk K, Park JH. BH3‐only protein Noxa is a mediator of hypoxic cell death induced by hypoxia‐inducible factor 1alpha. J Exp Med. 2004;199:113‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials