

Abstract
Utility of combined annotation‐dependent depletion (CADD) score was recently reported to rank pathogenicity as C‐scores ranging 1‐99 for both confirmed deleterious mutation. Using C‐scores for BRCA1/2 variants, we tried to constitute the classification system for variant of uncertain significance (VUS), which had been a major problem of genetic testing for hereditary breast and/or ovarian cancer. We analyzed BRCA1/2 genes for 283 patients with breast and/or ovarian cancer. The deleterious mutation and missesne mutations, minor variant, and wild type of BRCA1 and ‐2 were 5, 27, 251 and 15, 85, 183, respectively. Meanwhile, the variants with C‐score ≥10 were involved in 19/283 (6.7%) in BRCA1 and 34/283 (12%) in BRCA2. All deleterious mutations were included in this group. Frequency of personal history and family history of ovarian cancer were significantly high, and frequency of serous adenocarcinoma of ovary and triple negative breast cancer was relatively high in the group with deleterious mutations. Similar findings were seen in patients with variants of C‐score ≥10. According to the C‐score and population frequency, we could define VUS for 11 patients out of 283 patients (3.9 CADD is useful to classify the variant of BRCA1/2 and selecting the patient who needs further segregation studies.
Keywords: BRCA, combined annotation‐dependent depletion, genetic counseling, hereditary breast and/or ovarian cancer, variant of uncertain significance
1. INTRODUCTION
Hereditary breast and/or ovarian cancer (HBOC) syndrome is caused by germline deleterious mutations in BRCA1 and BRCA2.1 Indication for germline genetic testing for BRCA is increasing as a result of directed cancer chemotherapy,2 novel targeted therapies,3, 4 and selection of therapeutic surgery.5 However, a major problem with genetic testing and counseling for the HBOC patient is the finding of variant of uncertain significance (VUS).6 VUS represents a particular challenge as the clinical significance cannot be inferred from sequence information alone. Misinterpretations of VUS can lead to real clinical harm for both patients and families. The number of novel variants without confirmed information in the databases seems to increase in accordance with increased genetic testing.7
The effort of scientific team in Myriad Genetcs Inc. (Salt Lake City, UT, USA) for their accumulated data decreased the ratio of VUS to 2.1%.8 They have a variant classification program that consists of several factors. They reported that most of the variants were classified as benign mutations which had no effect on developing cancer.
In contrast, combined annotation‐dependent depletion (CADD) score was recently reported by Kircher et al9 CADD can rank the pathogenicity as C‐scores ranging 1‐99 for both convinced deleterious mutations (frameshift and nonsense) and missense mutations. This ranking is meaningful for classifying the variants.
Using C‐scores for BRCA1/2 variants, we tried to constitute the classification system for VUS, which is important for genetic counseling to clarify the pathogenesis of variants.
2. MATERIALS AND METHODS
2.1. Patients and methods
We carried out next‐generation analysis for BRCA1/2 genes for 283 patients with breast and/or ovarian cancer from September 2013 to December 2016. Patients were selected according to National Comprehensive Cancer Network (NCCN) criteria for future genetic evaluation.10 This cohort consisted of 177 patients with breast cancer, 12 with breast and ovarian cancer, and 94 with ovarian cancer, in which only 15 patients (5.3%) did not meet the criteria of the NCCN criteria. Our cohort consisted of breast and/or ovarian cancer patients with a high risk for HBOC. The deleterious mutation, minor variant, and wild type of BRCA1 and 2 were 5, 27, 251 and 15, 85, 183, respectively. Next‐generation sequencing for BRCA 1/2 germline gene was done using Ion AmpliSeq™ BRCA1 and BRCA2 Panel (Thermo Fishier Scientific, Waltham, MA, USA) containing 167 primer pairs. Detailed methods were described in our previous reports.11, 12
Classification of deleterious mutation was applied for frameshift, nonsense, and splice site mutations that lead to premature truncation of the protein. Most of the cases were analyzed for their large deletion by the Multiplex Ligation‐dependent Probe Amplification (MLPA) method which was carried out by FALCO Biosystems (Kyoto, Japan). Missense variants with minor allele frequency (MAF) <0.01 were selected as rare variants according to 1000 Genomes Project data,13 in which deleterious mutations were determined by available databases and reports.
Combined annotation‐dependent depletion (CADD) was applied for these rare variants as well as for deleterious mutations. C‐scores were obtained by non‐commercial applications developed by Kircher et al9 CADD annotations were made using a wide range of data types including conservation matrix such as Genomic Evolutionary Rate Profiling (GERP),14 phastCons,15 and phyloP16; functional genomic data such as DNAase hypersensitivity and transcription factor binding; transcript information such as distance to exon‐intron boundaries or expression levels in commonly studied cell lines17; and protein‐level scores such as that of Grantham,18 SIFT,19 and PolyPhen.20 In the CADD system, a support vector machine (SVM) is trained and Phred‐like scores (scaled C‐scores) are defined ranging from 1‐99, based on the rank of each variant relative to all possible 8.6 billion substitutions in the human reference genome.9 The results of this transformation are “scaled” C‐scores. Top 10% in the ranking of CADD scores are assigned C‐score 10, top 1% to C‐score 20 and top 0.1% to C‐score 30 etc.
Patients with variants were divided into 4 groups: deleterious mutations; variants with C‐score ≥10; variants with 10 > C‐score ≥1; and control group including both variants with C‐score <1 and wild type (because we found no difference in clinical features between variants with C‐score <1 and wild type). If the patient had several minor variants, she was divided into a group according to the higher value of the C‐score. Clinical features associated with HBOC were compared between each group and control group. Personal and family history of breast and/or ovarian cancer, histology of ovarian cancer (serous or non‐serous) and breast cancer (triple negative breast cancer [TNBC] or non‐TNBC), age of developing breast cancer (≤45 years or older), and whether bilateral or unilateral breast cancer were selected as the clinical features.
Finally, we showed our classification for the minor variants using C‐score and segregation studies compared with the classification of the other annotation systems (SFIT and PolyPhen2) and representative database of ClinVar.
2.2. Statistical analysis
Frequency of variants were statistically analyzed using Fisher's exact test and χ2‐test as appropriate (StatMate by ATMS, Tokyo, Japan).
3. RESULTS
3.1. Variants with C‐score ≥10 in BRCA1 and BRCA2
Variants with C‐score ≥10 were involved in 19/283 (6.7%) in BRCA1 (Figure 1A) and in 34/283 (12%) in BRCA2 (Figure 1B). All deleterious mutations scored over 10 by CADD, except for one patient with a large deletion of BRCA2 which was excluded from this study.
Figure 1.
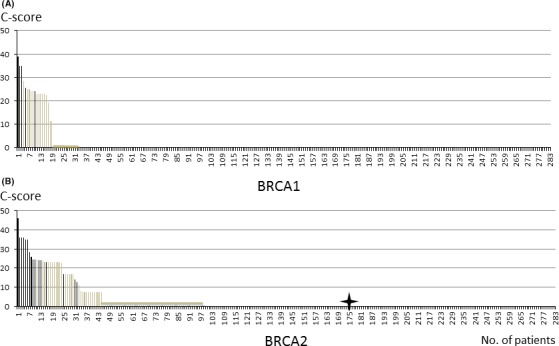
C‐scores of (A) BRCA1 and (B) BRCA2 variants in 283 breast and/or ovarian cancers. Deleterious mutations are indicated by black column, minor variants are indicated by gray. B, A patient with a large deletion detected by Multiplex Ligation‐dependent Probe Amplification method has no C‐score which is indicated by a star
Number of variants including deleterious mutations and rare variants of BRCA1 was 33. Among the 33 variants, 19 cases (57%) were C‐score ≥10, none were 10 > C‐score ≥1, and 14 cases were C‐score <1. Consequently, 5 cases out of the 19 cases with C‐score ≥10 (26%) were defined as deleterious (2 frameshift and 3 nonsense mutations).
Among the 119 variants in BRCA2, C‐score ≥10 was seen in 34/119 (28%) (Figure 1B). Deleterious mutation was defined in 14/34 (41%) variants with C‐score ≥10. However, C‐score was not obtained for 1 patient with a large deletion detected by MLPA that is indicated by a star in Figure 1B.
Frequency of BRCA1 C‐score ≥10 was significantly high (P = .01) in patients with ovarian cancer (including patients with breast and ovarian cancer) with 14.1% (15/106) compared with breast cancer (excluding patients with breast and ovarian cancer) with 2.8% (5/177). However this difference was not observed in BRCA2; 11% (19/177) in breast cancer, and 14% (15/106) in ovarian cancer (Table 1). Furthermore, the frequency of C‐score ≥10 of BRCA1 and BRCA2 is higher in serous adenocarcinoma compared with non‐serous adenocarcinoma of the ovary. Significant difference (P = .05) was seen in BRCA2 (Table 1). In 177 patients with breast cancer, frequency of C‐score ≥10 was not elevated in younger patients (≤45 years) and in bilateral breast cancer. Meanwhile, the frequency of BRCA1 C‐score ≥10 was elevated in TNBC with 12% (2/17) compared with non‐TNBC with 1.9% (3/160) but statistical significance was not seen.
Table 1.
Frequency of BRCA variation with CADD score ≧10 according to the clinical factors
| n | BRCA1 | BRCA2 | |||||
|---|---|---|---|---|---|---|---|
| CADD ≧ 10 | % | P | CADD ≧ 10 | % | P | ||
| Breast ca (without BO) | 177 | 5 | 2.8 | <.01 | 19 | 11 | NS |
| Ovarian ca (with BO) | 106 | 14 | 13.2 | 15 | 14 | ||
| 283 | 19 | 6.7 | 34 | 12 | |||
| Breast ca (177) | |||||||
| Age ≦45 | 91 | 2 | 2.2 | NS | 12 | 13 | NS |
| Age >45 | 86 | 3 | 3.5 | 7 | 8.1 | ||
| Bilateral bc | 7 | 0 | 0 | NS | 0 | 0 | NS |
| Unilateral bc | 170 | 5 | 2.9 | 19 | 11 | ||
| TNBC | 17 | 2 | 12 | NS | 3 | 17.6 | NS |
| NonTNBC | 160 | 3 | 1.9 | (.11) | 16 | 10 | |
| Ovaran cancer (106) | |||||||
| Serous | 77 | 13 | 17 | NS | 14 | 18 | <.05 |
| Non‐serous | 41 | 3 | 7.3 | 1 | 2.4 | ||
| Family History | |||||||
| ≧1 OC family | 19 | 4 | 21 | NS | 3 | 15.8 | NS |
| No OC family | 264 | 15 | 5.7 | (.03) | 31 | 11.7 | |
| ≧2 BC families | 45 | 3 | 6.7 | NS | 4 | 8.9 | NS |
| 1 BC family | 80 | 7 | 8.7 | 14 | 17 | ||
| No BC family | 158 | 10 | 6.3 | 16 | 10 | ||
BO; Breast and Ovarian Cancer; BC; Breast Cancer; OC; Ovarian Cancer; NS, not significant; TNBC; Triple Negative Breast Cancer
P‐value was calculated by Fisher's exact test.
() P‐value by χ2 test.
Regarding the family histories of 283 patients, the number of families with breast cancer did not correlate with the frequency of C‐score ≥10. Also, the frequency of BRCA1 C‐score ≥10 was 21% (4/19) in patients with ovarian cancer family history which was higher than in the patients without ovarian cancer family history at 5.7% (15/264) (P = .03 by χ2‐test, and not significant [NS] by Fisher's exact test).
3.2. Clinical features of variations with deleterious mutations, variants with C‐score ≥10, and 10 > C‐score ≥1
We compared clinical features of HBOC among 4 groups: deleterious mutations; variants with C‐score ≥10; 10 > C‐score ≥1; and control group (including variants with C‐score <1 and wild type). Frequencies of personal history and family history of ovarian cancer were significantly high in the group with deleterious mutations. Also, frequency of serous adenocarcinoma of ovary and TNBC was relatively high (Table 2). Similar findings were seen in the variants with C‐score ≥10 in which personal history of ovarian cancer was significantly high and frequency of serous adenocarcinoma was relatively high. According to clinical features of breast cancer, significant differences were not seen in both the deleterious mutations group and in variants with C‐score ≥10.
Table 2.
Cilinical features of variants with deleterious mutation, variants with C‐score≧10, 10>C‐score≧1
| n | Personal History | Family History | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OC family | BC family | Breast cancer with BO (189) | Ovaran Cancer (with BO) | |||||||||||||||||||||||
| OC with/without BC | BC alone | P | ≧1 OC family | no OC family | P | ≧2 BC families | 1 BC family | No BC families | P | n | Age ≦45 | Age >45 | P | Bilateral | Unilateral | P | TNBC | Non‐TNBC | P | n | Serous | Non‐Serous | P | |||
| Deleterious | BRCA1+2 | 20 | 12 | 8 | .02 | 4 | 16 | .041 | 2 | 9 | 9 | .186 | 14 | 7 | 8 | .944 | 1 | 13 | .819 | 3 | 11 | .224 | 12 | 12 | 0 | .071 |
| BRCA1 | 5 | 5 | 0 | .008 | 2 | 3 | .025 | 0 | 2 | 3 | .537 | 3 | 0 | 3 | .267 | 1 | 2 | .363 | 1 | 2 | .598 | 5 | 5 | 0 | .369 | |
| BRCA2 | 15 | 7 | 8 | .425 | 2 | 13 | .491 | 2 | 7 | 6 | .23 | 11 | 7 | 4 | .546 | 0 | 11 | .966 | 2 | 9 | .531 | 7 | 7 | 0 | .226 | |
| C ≧ 10 | BRCA1+2 | 32 | 17 | 15 | .043 | 3 | 29 | .629 | 3 | 11 | 18 | .401 | 16 | 5 | 11 | .265 | 0 | 16 | .826 | 2 | 15 | .925 | 17 | 14 | 3 | .51 |
| BRCA1 | 13 | 9 | 4 | .018 | 2 | 11 | .385 | 1 | 4 | 8 | .647 | 4 | 2 | 2 | .631 | 0 | 4 | .433 | 1 | 3 | .756 | 9 | 7 | 2 | .403 | |
| BRCA2 | 19 | 8 | 11 | .582 | 1 | 18 | .618 | 2 | 7 | 10 | .526 | 12 | 3 | 9 | .185 | 0 | 12 | .989 | 0 | 12 | .666 | 8 | 7 | 1 | .559 | |
| 10 > C ≧ 1 | BRCA1+2 | 10 | 4 | 6 | .769 | 0 | 10 | .977 | 1 | 2 | 7 | .673 | 7 | 3 | 4 | .959 | 1 | 6 | .794 | 1 | 6 | .923 | 4 | 2 | 2 | .746 |
| BRCA1 | 0 | 0 | 0 | NS | 0 | 0 | NS | 0 | 0 | 0 | NS | 0 | 0 | 0 | 0 | 0 | 0 | NS | 0 | 0 | NS | 0 | 0 | 0 | NS | |
| BRCA2 | 10 | 4 | 6 | .769 | 0 | 10 | .977 | 1 | 2 | 7 | .673 | 7 | 3 | 4 | .959 | 1 | 6 | .794 | 1 | 6 | .923 | 4 | 2 | 2 | .746 | |
| Control | BRCA1+2 | 221 | 73 | 148 | 12 | 209 | 39 | 58 | 124 | 38 | 75 | 77 | 7 | 145 | 12 | 141 | 63 | 51 | 21 | |||||||
| Total | BRCA1+2 | 283 | 106 | 177 | 19 | 263 | 45 | 80 | 158 | 189 | 90 | 99 | 9 | 179 | 18 | 173 | 106 | 79 | 27 | |||||||
C, C‐score; OC, ovarian Cancer; BC, Breast Cancer; BO, Beast and Ovarian Cancer; TNBC, Triple Negtive Breast Cancer.
Significant P value is written by Bold.
3.3. Classifications for minor variants using C‐score and population frequencies
Classifications for deleterious mutations and rare variants of BRCA1 and BRCA2 are listed in Tables 3 and 4, respectively. According to the C‐score and population frequency that was estimated by the number of patients carrying the same variants, we excluded variants as polymorphism and classified VUS for 7 patients with BRCA1 and 4 patients with BRCA2. In total, 11 patients out of 283 patients (3.9%) were classified as VUS.
Table 3.
Classifications for Variations of BRCA1 according to C‐score
| Protein | Position | Coding | MAF | Number of Patients | PolyPhen2 | SIFT | CinVar | C‐score | Our Classification |
|---|---|---|---|---|---|---|---|---|---|
| p.Leu63Ter | chr17:41258497 | c.188T>A | NA | 1 | NA | NA | Deleterious | 39 | Deleterious |
| p.Gln934Ter | chr17:41244748 | c.2800C>T | NA | 2 | NA | NA | Deleterious | 35 | Deleterious |
| p.Glu1257fs | chr17:41243776 | c.3770_3771delAG | NA | 1 | NA | NA | Deleterious | 25.6 | Deleterious |
| p.Lys652fs | chr17:41245594 | c.1952_1953insG | NA | 1 | NA | NA | Deleterious | 24.2 | Deleterious |
| Total Number | 5 | ||||||||
| Protein | Position | Coding | MAF | Number of Patients | PolyPhen2 | SIFT | CinVar | C‐score | Our Classification |
| p.Leu52Phe | chr17:41258531 | c.154C>T | NA | 1 | Probably_damaging | Deleterious | VUS | 28.3 | VUS |
| p.Val1653Leu | chr17:41222974 | c.4957G>T | NA | 2 | Benign | Deleterious | nd | 25 | VUS |
| p.Val271Met | chr17:41246737 | c.811G>A | 0 | 1 | Possibly_damaging | Tolerated | VUS | 24.2 | VUS |
| p.Ala1773Gly | chr17:41209091 | c.5318C>G | NA | 1 | Benign | Deleterious | nd | 24.1 | VUS |
| p.Tyr856His | chr17:41244982 | c.2566T>C | 0.003 | 5 | Possibly_damaging | Tolerated | Benign | 23 | Benign |
| p.Gln94His | chr17:41256904 | c.282G>T | NA | 1 | Benign | Deleterious | nd | 22.3 | VUS |
| p.Ser1125Thr | chr17:41244175 | c.3373T>A | NA | 1 | Probably_damaging | Deleterious | nd | 19.26 | VUS |
| p.Ser1577Pro | chr17:41223202 | c.4729T>C | NA | 3 | Benign | Tolerated | Conflicting | 11.33 | Benign |
| p.Met1628Thr | chr17:41223048 | c.4883T>C | 0.004 | 9 | Benign | Tolerated | Benign | 0.023 | Benign |
| p.Asn1236Ser | chr17:41243841 | c.3707A>G | NA | 1 | Benign | Tolerated | Conflicting | 0.001 | Benign |
| p.Asn1018Ser | chr17:41244495 | c.3053A>G | NA | 1 | Benign | Tolerated | nd | 0.001 | Benign |
| p.Asn1236Ser | chr17:41243841 | c.3707A>G | NA | 1 | Benign | Tolerated | Conflicting | 0.001 | Benign |
| p.Gly401Glu | chr17:41246346 | c.1202G>A | NA | 1 | Benign | Tolerated | Conflicting | 0.001 | Benign |
| Total Number | 38 |
MAF, Minor Allele Frequency; NA, Not Applicable; nd, not documented; VUS, Vaiant of Uncertain Significance.
Table 4.
Classifications for Variants of BRCA2 according to C‐score
| Protein | Position | Coding | MAF | Number of Patients | PolyPhen2 | SIFT | CinVar | C‐score | Our Classification |
|---|---|---|---|---|---|---|---|---|---|
| large_del | chr13:00000000 | NA | NA | 1 | NA | NA | Deleterious | NA | Deleterious |
| p.Arg2318Ter | chr13:32920978 | c.6952C>T | NA | 1 | NA | NA | Deleterious | 46 | Deleterious |
| p.Ser1882Ter | chr13:32914137 | c.5645C>A | NA | 1 | NA | NA | Deleterious | 36 | Deleterious |
| p.Ser1882Ter | chr13:32914137 | c.5645C>A | NA | 2 | NA | NA | Deleterious | 36 | Deleterious |
| p.Gln609Ter | chr13:32907440 | c.1825C>T | NA | 1 | NA | NA | Deleterious | 35 | Deleterious |
| p.Gly2281fs | chr13:32913571 | c.5080_5083delAGAG | 0 | 1 | NA | NA | Deleterious | 35 | Deleterious |
| p.Asn2135fs | chr13:32914893 | c.6402_6406delTAACT | NA | 1 | NA | NA | Deleterious | 28.5 | Deleterious |
| p.Ile2675Val | chr13:32937362 | c.8023A>G | NA | 1 | Probably_damaging | Deleterious | Deleterious | 25.9 | Deleterious |
| p.Ile2149fs | chr13:32914935 | c.6444_6445delTA | NA | 2 | NA | NA | Deleterious | 24.5 | Deleterious |
| p.Gln850fs | chr13:32911039 | c.2547_2548insCC | NA | 1 | NA | NA | Deleterious | 23.7 | Deleterious |
| p.Glu790fs | chr13:32910859 | c.2368_2368delG | NA | 1 | NA | NA | Deleterious | 22.8 | Deleterious |
| p.Gln864fs | chr13:32911080 | c.2589_2589delT | NA | 1 | NA | NA | Deleterious | 14.17 | Deleterious |
| p.Asn1287fs | chr13:32912345 | c.3854_3854delA | NA | 1 | NA | NA | Deleterious | 12.53 | Deleterious |
| Total Number | 15 | ||||||||
| p.Asp1990Ala | chr13:32914461 | c.5969A>C | 0 | 1 | Probably_damaging | Deleterious | nd | 24.6 | VUS |
| p.Arg18His | chr13:32890650 | c.53G>A | 0 | 2 | Possibly_damaging | Tolerated | nd | 24.2 | VUS |
| p.Lys2729Asn | chr13:32937526 | c.8187G>T | 0.003 | 10 | Probably_damaging | Deleterious | nd | 23.1 | Benign |
| p.Lys322Gln | chr13:32906579 | c.964A>C | 0 | 6 | Possibly_damaging | Deleterious | nd | 16.89 | Benign |
| p.His3056Tyr | chr13:32954192 | c.9166C>T | NA | 1 | Benign | Tolerated | nd | 11.31 | VUS |
| p.Thr2766Ala | chr13:32937635 | c.8296A>G | NA | 1 | Benign | Tolerated | nd | 8.269 | Benign |
| p.Gly2044Val | chr13:32914623 | c.6131G>T | 0.001 | 12 | Benign | Deleterious | nd | 7.435 | Benign |
| p.Ile1903Thr | chr13:32914200 | c.5708T>C | 0 | 1 | Benign | Deleterious | nd | 7.344 | Benign |
| p.Val2010Gly | chr13:32914521 | c.6029T>G | NA | 1 | Benign | Tolerated | nd | 0.632 | Benign |
| p.Ile729Thr | chr13:32910678 | c.2186T>C | NA | 1 | Benign | Tolerated | nd | 0.048 | Benign |
| p.Pro1062Ser | chr13:32911676 | c.3184C>T | NA | 1 | Benign | Tolerated | nd | 0.017 | Benign |
| p.Val2109Ile | chr13:32914817 | c.6325G>A | 0 | 7 | Benign | Tolerated | nd | 0.003 | Benign |
| p.Met784Val | chr13:32910842 | c.2350A>G | 0.007 | 12 | Benign | Tolerated | nd | 0.001 | Benign |
| p.Ile1929Val | chr13:32914277 | c.5785A>G | 0 | 1 | Benign | Tolerated | nd | 0.001 | Benign |
| p.Met784Val | chr13:32910842 | c.2350A>G | 0.007 | 1 | Benign | Tolerated | nd | 0.001 | Benign |
| p.Met784Val | chr13:32910842 | c.2350A>G | 0.007 | 38 | Benign | Tolerated | nd | 0.001 | Benign |
| Total Number | 96 |
MAF, Minor Allele Frequency; NA, Not Applicable; nd, not documented; VUS, Variant of Uncertain Significance.
The classifications for the other variants are shown in Table 3. Variants of p.Tyr856His and p.Ser1577Pro were seen in 5 and 3 patients, respectively. The population frequency was estimated as relatively high and classified as benign polymorphism. They were classified as conflicting (likely benign) in the ClinVar database. Meanwhile, variants with C‐score <10 in BRCA1 were observed in 13 cases. The variant of p.Met1628Thr was seen in 9 patients, estimated high population frequency to be classified as benign, and the remaining 4 variants were also classified as benign by other in silico analysis (SIFT and PolyPhen 2). These missense mutations were estimated as benign polymorphism.
The classifications for BRCA2 variants are shown in Table 4. The variant of p.Lys2729Asn with C‐score 23.1 was seen in 10 patients, in whom one patient had another BRCA2 deleterious mutation, so that we classified this variant as benign. Also, p.Lys322Gln with C‐score 16.8 was seen in 6 patients, in whom one patient was offered genetic counseling and her segregation study suggested this mutation was not pathogenic (see Case 2 in the following segregation studies). The remaining 3 variants (3/283 [1%]) were classified as VUS, and they were not documented in the ClinVar database.
We classified 11 variants with C‐score <10 as benign because of the following reasons. The variants of p.Gly2044Val, p.Val2109Ile, p.Met784Val, and p.Met784Val were seen in 12, 7, 12, and 38 patients to be estimated as benign by high population frequency. The other variants were seen individually. All of these variants were not documented in the ClinVar database, but were estimated as benign and/or tolerated in SIFT and PolyPhen2.
3.4. Segregation studies for 3 patients with rare variants
We obtained important information by genetic counseling and segregation studies for patients with rare variants as follows.
3.4.1. Case 1
A 73‐year‐old woman with breast cancer was found to have BRCA1 p.Val271Met with C‐score of 24. This variant was judged benign by Myriad (we had validation data with FALCO Biosystems)11 and uncertain by ClinVar. She had an aunt with breast cancer, a brother with pancreatic cancer and a daughter with ovarian cancer (Figure 2A). Her daughter with ovarian cancer had genetic counseling and testing showed the same variant. She suffers from recurrent serous adenocarcinoma of the ovary. Correlation of the variant and HBOC was suspected in this case. We need further follow up for this family.
Figure 2.
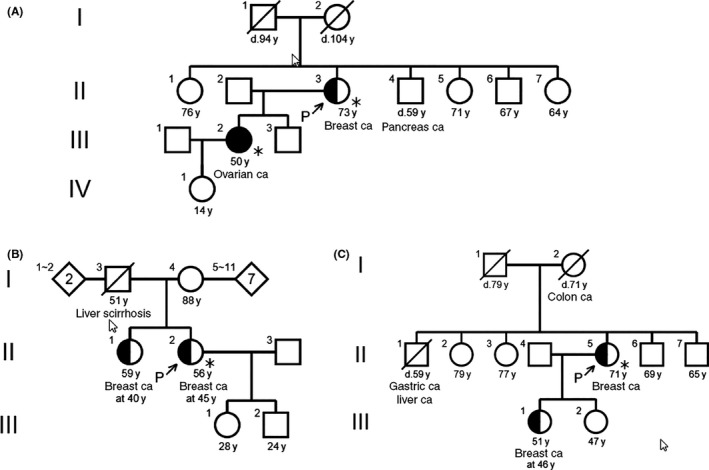
Family trees of 3 patients with minor variants. (A) A 73‐year‐old woman with breast cancer was found to have BRCA1 p.Val271Met with C‐score of 24. Her daughter with ovarian cancer had genetic counseling and testing showed the same variant. She suffered from recurrent serous adenocarcinoma of the ovary. B, A 45‐year‐old woman with breast cancer was found to have BRCA2 p.Lys322Gln with C‐score of 16.89. This variant was identified in 6 patients with breast cancer. Her sister with breast cancer was referred to genetic counseling and testing showed her not to have the same variant. C, A 71‐year‐old woman with breast cancer had a variant of BRCA1 p.Met1628Thr with C‐score of 0.023. This variant was not documented in the ClinVar database. Her daughter was referred to genetic counseling and testing. This variant was not found in the daughter, so the pathogenicity was defined to be low
3.4.2. Case 2
A 45‐year‐old woman with breast cancer was found to have BRCA2 p.Lys322Gln with C‐score of 16.89. This variant was identified in 6 patients with breast cancer. She has a sister suffering from breast cancer since 40 years of age but no other family members with cancer (Figure 2B). Her sister was referred to genetic counseling and testing showed her not to have the same variant. The frequency of this mutation was estimated to be high. According to these findings, we recognized the pathogenicity of this variant as low.
3.4.3. Case 3
A 71‐year‐old woman with breast cancer had a variant of BRCA1 p.Met1628Thr with C‐score of 0.023. This variant was not documented in the ClinVar database. Her mother had colon cancer, a brother had liver and gastric cancer, and a daughter suffered from breast cancer at 46 years of age (Figure 2C). Her daughter was referred to genetic counseling and testing. This variant was not found in the daughter, so the pathogenicity was defined to be low.
4. DISCUSSION
It is important in genetic counseling and testing to provide an appropriate explanation for variants. Most variants may not be associated with a high risk of cancer but a misinterpreted variant has the potential to lead to mismanagement of patients and their relatives. IARC Unclassified Genetic Variants Working Group21 and other researchers22 recommended the following classification for variants. The classification consists of the following 5 categories: (i) ‘deleterious’ (pathogenic); (ii) ‘suspected deleterious’ (likely pathogenic); (iii) ‘VUS’; (iv) ‘genetic variant, favor polymorphism’ (likely not pathogenic); and (v) ‘polymorphism’ (not pathogenic). However, these 5 categories unfortunately confound the clinical direction of the patients.
There are several online database resources that provide some interpretation of BRCA sequence variants, such as The Breast Cancer Information Core (BIC),23 Human Variome Project,24 or ClinVar.25 We referred each variant to ClinVar as a standard reporting database recommended by The American College of Medical Genetics.7 As mentioned earlier, some variants were not documented in the ClinVar database, so that we needed another system to classify the variants.
There are many existing annotations useful for prioritizing causal variants (e.g. PolyPhen,26 SIFT,27 and GERP28), but they have several limitations. First, factors of annotations vary widely, from constitutions to functions. Second, each annotation has its own metric being rarely comparable. Third, each annotation was subject to major ascertainment biases and might not be generalized. Fourth, combined annotations might have only overlapping significance. These limitations have caused many potentially relevant annotations to be ignored.
Combined annotation‐dependent depletion is a framework for estimating the relative pathogenicity of human genetic variants by integrating many diverse annotations into a single, quantitative score. CADD has been implemented as a support vector machine trained to differentiate 14.7 million high‐frequency human‐derived alleles from 14.7 million simulated variants.9 Also, we can compute a “C‐score” for all 8.6 billion possible human single nucleotide variants and short insertion/deletions. C‐score correlates with allelic diversity, annotation of functionality, pathogenicity, disease severity, experimentally measured regulatory effect and complex trait associations, and highly ranks known pathogenic variants within individual genomes.
There are several studies of the power of CADD to classify the variants of familial cancer panels29, 30, 31 in which superiority of the CADD score rather than other in silico analysis is reported. Although limited clinical validity for the identification of pathogenic variants in non‐coding regions has been reported,30 as well as the original report by Kircher,9 the validity of the C‐score ≥10 in clinically relevant genes was also suggested in a dataset of mismatch repair gene variants.31 We still have no reports concerning the validation of CADD in BRCA mutations.
The classification of Myriad (Myriad's New Mutations Committee [NMC]) was made using 8 parameters8: 1, literature review; 2, population frequency; 3, mRNA splice‐site assay; 4, functional assays; 5, evolutionary conservation; 6, segregation analysis; 7, identification of homozygous and compound heterozygous individual (intrans); and 8, mutation co‐occurrence. Re‐classification was made by history‐weighing algorithm and the rate of VUS was reduced at 2.1% for all tested patients.32, 33 Although the classification by Myriad is valuable to determine the pathogenicity of minor variants, not every researcher can access the Myriad database. Furthermore, their algorithm is not designed for low penetrance mutations and newly discovered variants. In addition, it needs a huge clinical database. We need a new tool to classify minor variants despite no definite information being available. It is clear that there is currently no internationally accepted standard for BRCA testing report, and no agreed consistent classification system; some laboratories report variants without interpretation, some use a narrative approach and some use locally developed guidelines or published schemes.34, 35, 36
In our experience, CADD could rank all variants of BRCA by C‐scores in which all deleterious mutations were included except for the large deletion detected by the MLPA method. If we defined C‐Score ≥10 which means top 10% in ranking for pathogenicity, we could reduce the frequency of VUS at 3.9%. This value is satisfactory compared with the VUS rate reported by Myriad at 2.1%.8 Variants with C‐score <10 were estimated benign according to other factors, population frequency, other in silico analysis (SIFT and PolyPhen2), and mutation co‐occurrence. Furthermore, clinical features of patients with C‐score ≥10 supported the utility of CADD. Although significant differences were not observed in family history of ovarian cancer and frequency of serous adenocarcinoma, similar findings with deleterious mutations were seen. Patients with ovarian cancer, TNBC and family history of ovarian cancer were more frequent in variants with C‐score ≥10.
In practice, we could not define pathogenicity as “deleterious” by C‐score individually, as clinical evidence is necessary to determine the pathogenicity. CADD might be useful to select the VUS which is referred for genetic counseling and for segregation studies. Information gained by segregation studies is significant for both patients and clinicians. We can avoid an ambiguous explanation for the patient about variants using C‐score and segregation studies. Although several strategies to classify VUS were reported,37, 38 we established the counseling system using CADD in which targeted VUS were easily selected and we can propose significant segregation studies for the patients. We could reduce the frequency of VUS at 3.9% using CADD and population frequency. We tried further segregation studies for the patients with variants classified as VUS, and determined a benefit of using C‐score for genetic counseling.
ACKNOWLEDGMENTS
We thank Hidetoshi Shigetomo, Yumi Kubota, Rituko Yokouchi, and Nao Sato for their assistance.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Nakagomi H, Mochizuki H, Inoue M, et al. Combined annotation‐dependent depletion score for BRCA1/2 variants in patients with breast and/or ovarian cancer. Cancer Sci. 2018;109:453–461. https://doi.org/10.1111/cas.13464
REFERENCES
- 1. O'Donovan PJ, Livingston DM. BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double‐strand break repair. Carcinogenesis. 2010;31:961‐967. [DOI] [PubMed] [Google Scholar]
- 2. Lips EH, Mulder L, Oonk A, et al. Triple‐negative breast cancer: BRCAness and concordance of clinical features with BRCA1‐mutation carriers. Br J Cancer. 2013;108:2172‐2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in platinum‐sensitive relapsed ovarian cancer. N Engl J Med. 2012;366:1382‐1392. [DOI] [PubMed] [Google Scholar]
- 4. Ledermann J, Harter P, Gourley C, et al. Olaparib maintenance therapy in patients with platinum‐sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15:852‐861. [DOI] [PubMed] [Google Scholar]
- 5. Metcalfe KA, Lubinski J, Ghadirian P, et al. Predictors of contralateral prophylactic mastectomy in women with a BRCA1 or BRCA2 mutation: the Hereditary Breast Cancer Clinical Study Group. J Clin Oncol. 2008;26:1093‐1097. [DOI] [PubMed] [Google Scholar]
- 6. Richter S, Haroun I, Graham TC, Eisen A, Kiss A, Warner E. Variants of unknown significance in BRCA testing: impact on risk perception, worry, prevention and counseling. Ann Oncol. 2013; (24 Suppl 8):viii69‐viii74. [DOI] [PubMed] [Google Scholar]
- 7. Eccles DM, Mitchell G, Monteiro AN, et al. BRCA1 and BRCA2 genetic testing‐pitfalls and recommendations for managing variants of uncertain clinical significance. Ann Oncol. 2015;26:2057‐2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Eggington JM, Bowles KR, Moyes K, et al. A comprehensive laboratory‐based program for classification of variants of uncertain significance in hereditary cancer genes. Clin Genet. 2014;86:229‐237. [DOI] [PubMed] [Google Scholar]
- 9. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Genetic/Familial High‐Risk Assessment: Breast and Ovary. National Comprehensive Cancer Network. Available from https://www.nccn.org/professionals/physician_gls/pdf/genetics_screening.pdf. Accessed October 10, 2017.
- 11. Hirotsu Y, Nakagomi H, Sakamoto I, Amemiya K, Mochizuki H, Omata M. Detection of BRCA1 and BRCA2 germline mutations in Japanese population using next‐generation sequencing. Mol Genet Genomic Med. 2015;3:121‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sakamoto I, Hirotsu Y, Nakagomi H, et al. BRCA1 and BRCA2 mutations in Japanese patients with ovarian, fallopian tube, and primary peritoneal cancer. Cancer. 2016;122:84‐90. [DOI] [PubMed] [Google Scholar]
- 13. McVean GA, Abecasis GR, Auton A, et al. An integrated map of genetic variation from 1,092 human genomes. Nature 2012;491:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Siepel A, Bejerano G, Pedersen JS, et al. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034‐1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010;20:110‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Consortium EP . An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Grantham R. Amino acid difference formula to help explain protein evolution. Science. 1974;185:862‐864. [DOI] [PubMed] [Google Scholar]
- 19. Ng PC, Henikoff S. SIFT: predicting amino acid changes that affect protein function. Nucl Acids Res. 2003;31:3812‐3814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Collaborative Group on Hormonal Factors in Breast Cancer . Familial breast cancer: collaborative reanalysis of individual data from 52 epidemiological studies including 58,209 women with breast cancer and 101,986 women without the disease. Lancet. 2001;358:1389‐1399. [DOI] [PubMed] [Google Scholar]
- 21. Lindor NM, Guidugli L, Wang X, et al. A review of a multifactorial probability‐based model for classification of BRCA1 and BRCA2 variants of uncertain significance (VUS). Hum Mutat. 2012;33:8‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richards CS, Bale S, Bellissimo DB, et al. ACMG recommendations for standards for interpretation and reporting of sequence variations: revisions 2007. Genet Med. 2008;10:294‐300. [DOI] [PubMed] [Google Scholar]
- 23. NIH. Breast Cancer Information Core . Available from https://research.nhgri.nih.gov/bic. Accessed October 10, 2017.
- 24. Global Variome Ltd .The Human Variome Project. Available from http://www.humanvariomeproject.org/activities. Accessed Dec.10, 2017.
- 25. NCBI . ClinVar. Available from https://www.ncbi.nlm.nih.gov/clinvar. Accessed October 10, 2017.
- 26. PolyPhen‐2. Available from http://genetics.bwh.harvard.edu/pph2/. Accessed October 10, 2017.
- 27. SIFT. Available from http://sift.jcvi.org/. Accessed October 10, 2017.
- 28. Genomic Evolutionary Rate Profiling: GERP. Available from http://mendel.stanford.edu/SidowLab/downloads/gerp/. Accessed October 10, 2017.
- 29. Young EL, Feng BJ, Stark AW, et al. Multigene testing of moderate‐risk genes: be mindful of the missense. J Med Genet. 2016;53:366‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mather CA, Mooney SD, Salipante SJ, et al. CADD score has limited clinical validity for the identification of pathogenic variants in noncoding regions in a hereditary cancer panel. Genet Med. 2016;18:1269‐1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van der Velde KJ, Kuiper J, Thompson BA, et al. Evaluation of CADD Scores in Curated Mismatch Repair Gene Variants Yields a Model for Clinical Validation and Prioritization. Hum Mutat. 2015;36:712‐719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rosenthal ET, Bowles KR, Pruss D, et al. Exceptions to the rule: case studies in the prediction of pathogenicity for genetic variants in hereditary cancer genes. Clin Genet. 2015;88:533‐541. [DOI] [PubMed] [Google Scholar]
- 33. Pruss D, Morris B, Hughes E, et al. Development and validation of a new algorithm for the reclassification of genetic variants identified in the BRCA1 and BRCA2 genes. Breast Cancer Res Treat. 2014;147:119‐132. [DOI] [PubMed] [Google Scholar]
- 34. Plon SE, Eccles DM, Easton D, et al. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282‐1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Moghadasi S, Hofland N, Wouts JN, et al. Variants of uncertain significance in BRCA1 and BRCA2 assessment of in silico analysis and a proposal for communication in genetic counselling. J Med Genet. 2013;50:74‐79. [DOI] [PubMed] [Google Scholar]
- 36. Santos C, Peixoto A, Rocha P, et al. Pathogenicity evaluation of BRCA1 and BRCA2 unclassified variants identified in Portuguese breast/ovarian cancer families. J Mol Diagn. 2014;16:324‐334. [DOI] [PubMed] [Google Scholar]
- 37. Easton DF, Deffenbaugh AM, Pruss D, et al. A systematic genetic assessment of 1,433 sequence variants of unknown clinical significance in the BRCA1 and BRCA2 breast cancer‐predisposition genes. Am J Hum Genet. 2007;81:873‐883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kerkhofs CH, Spurdle AB, Lindsey PJ, Goldgar DE, Gomez‐Garcia EB. Assessing biases of information contained in pedigrees for the classification of BRCA‐genetic variants: a study arising from the ENIGMA analytical working group. Hered Cancer Clin Pract. 2016;14:10. [DOI] [PMC free article] [PubMed] [Google Scholar]