

Abstract
Dysregulation of microRNAs (miRNAs) has been linked to virulence factors of Helicobacter pylori. The role of H. pylori in esophageal disease has not been clearly defined. We previously reported that H. pylori esophageal colonization promotes the incidence of Barrett's esophagus and esophageal adenocarcinoma in vivo. Here, we studied the direct effects of H. pylori on the transformation of esophageal epithelial cells, with particular focus on whether H. pylori exerts its effects by modulating miRNAs and their downstream target genes. The normal human esophageal cell line HET‐1A was chronically exposed to H. pylori extract and/or acidified deoxycholic acid for up to 36 weeks. The miRNA profiles of the esophageal epithelial cells associated with H. pylori infection were determined by microarray analysis. We found that chronic H. pylori exposure promoted acidified deoxycholic acid‐induced morphological changes in HET‐1A cells, along with aberrant overexpression of intestinal metaplasia markers and tumorigenic factors, including caudal‐type homeobox protein 2 (CDX2), mucin 2, and cyclooxygenase 2 (COX2). Helicobacter pylori modified the miRNA profiles of esophageal epithelial cells, particularly aberrant silencing of miR‐212‐3p and miR‐361‐3p. Moreover, in biopsies from Barrett's esophagus patients, esophageal H. pylori colonization was associated with a significant decrease in miR‐212‐3p and miR‐361‐3p expression. Furthermore, we identified COX2 as a target of miR‐212‐3p, and CDX2 as a target of miR‐361‐3p. Helicobacter pylori infection of esophageal epithelial cells was associated with miRNA‐mediated upregulation of oncoprotein CDX2 and COX2. Our observations provide new evidence about the molecular mechanisms underlying the association between H. pylori infection and esophageal carcinogenesis.
Keywords: CDX2, COX2, esophagus, Helicobacter pylori, microRNA
1. INTRODUCTION
Chronic inflammation can promote the initiation and progression of many cancers,1 as exemplified by the gastroesophageal reflux disease (GERD) to Barrett's esophagus to esophageal adenocarcinoma sequence. Chronic gastroesophageal reflux plays a crucial role in the development of Barrett's esophagus, a condition of columnar (intestinal) metaplasia of the esophageal squamous epithelium.2 It has been widely accepted that this esophageal metaplasia is a precursor of esophageal adenocarcinoma.3 The risk of developing esophageal adenocarcinoma in patients with Barrett's esophagus is 30‐125 times higher than in the normal population.4
Helicobacter pylori is a risk factor for the development of gastritis, peptic ulcer disease, and gastric cancer.5 The association between H. pylori infection and Barrett's esophagus or esophageal adenocarcinoma is controversial. A number of studies have concluded that H. pylori infection tends to protect against Barrett's esophagus or esophageal adenocarcinoma.6, 7, 8 In contrast, a meta‐analysis showed no convincing association between H. pylori eradication and the development of GERD,9 and eradication of H. pylori might halt the progress to esophageal adenocarcinoma in patients with GERD and Barrett's esophagus.10 However, these studies did not evaluate the impact of H. pylori colonization sites on the development of esophageal disease. Our previous studies have indicated that, in a rat model of chronic gastroesophageal reflux, H. pylori colonization in the esophagus increased the severity of esophageal inflammation and the incidence of Barrett's esophagus and esophageal adenocarcinoma, whereas H. pylori colonization in the stomach had no influence on the esophageal mucosa.11, 12 Thus, the effect of H. pylori on the esophagus varies with the colonization site. Both chronic gastroesophageal reflux and esophageal H. pylori infection could play important roles in the development of inflammation and Barrett's esophagus‐associated carcinogenesis. Reflux of gastric contents, including acid and bile, induces metaplasia of esophageal mucosa and facilitates colonization of H. pylori in the distal esophagus, and therefore aggravates esophageal injury. Clinical data have shown the high prevalence of H. pylori in the esophagus, and its presence is correlated with signs of inflammation.13 In Barrett's esophagus patients, the presence of metaplasia within the esophagus is a prerequisite for H. pylori colonization, and H. pylori may exacerbate inflammation in Barrett's epithelium.14 However, the molecular mechanisms involved in the effects of H. pylori in the GERD–Barrett's esophagus–esophageal adenocarcinoma sequence remain largely unknown. We have found that H. pylori colonization was associated with overexpression of cyclooxygenase 2 (COX2) in esophageal mucosa. Celecoxib, a selective COX2 inhibitor, significantly inhibited Barrett's esophagus‐associated carcinogenesis.11 The mechanisms for the regulation of H. pylori on COX2 expression in esophageal mucosa may still need to be further investigated.
MicroRNAs (miRNAs) regulate various cellular functions, including proliferation, differentiation, and apoptosis.15 Aberrant miRNA expression has been associated with human diseases such as inflammation and cancer.16 Several miRNAs have been identified as being involved in the development and progression of Barrett's esophagus and esophageal adenocarcinoma.17 Helicobacter pylori infection can modify the expression of more than 50 miRNAs in the gastric mucosa, and these miRNA levels can be restored to normal after H. pylori eradication.18 Recent studies also show that the association of miRNA with esophageal adenocarcinoma prognosis may be influenced by H. pylori infection status, suggesting that miRNA–H. pylori interactions play an important role in the prognosis of esophageal adenocarcinoma.19 However, none of the earlier studies examined the expression signature and the role of miRNA in H. pylori‐infected esophageal epithelial cells.
In this study, we determined the long‐term and direct effects of H. pylori on the phenotype of esophageal epithelia cells by using a well‐established in vitro model.20 We particularly focused on whether H. pylori exerts its effects through modulating miRNAs and their downstream target genes.
2. MATERIALS AND METHODS
2.1. Clinical samples
A total of 16 Barrett's esophagus patients with (n = 10) or without (n = 6) esophageal H. pylori colonization were enrolled in this study. The patients had undergone upper gastrointestinal endoscopy in Peking University First Hospital (Beijing, China). Barrett's esophagus was diagnosed on the basis of endoscopic and histological findings. Histologic changes and esophageal H. pylori colonization were examined by H&E staining. All patients had gastric H. pylori colonization confirmed by a rapid urease test and a 13C‐urea breath test. Clinicopathological characteristics of the study population are presented in Table 1. All subjects gave their informed consent for inclusion before they participated in the study. The study was carried out in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethics Committee of Peking University First Hospital (No. 20161187).
Table 1.
Clinicopathological characteristics of Barrett's esophagus patients with (n = 10) or without (n = 6) esophageal Helicobacter pylori colonization
| Variable | With H. pylori colonization (n = 10) | Without H. pylori colonization (n = 6) |
|---|---|---|
| Age, years | 59.5 ± 6.3 | 62.2 ± 5.1 |
| Male sex (%) | 6 (60) | 4 (66.7) |
| BMI | 25.1 ± 2.1 | 26.3 ± 1.3 |
| BE segment length (cm) | 2.25 ± 0.74 | 1.78 ± 0.48 |
| Presence of low‐grade dysplasia (%) | 6 (60) | 3 (50) |
2.2. Cell culture
The normal human esophageal cell line HET‐1A and the human embryonic kidney cell line HEK‐293T both were obtained from the ATCC (Manassas, VA, USA). Human esophageal adenocarcinoma cell line OE33 was obtained from the European Collection of Cell Cultures (UK). The cells were cultured in RPMI‐1640 medium (Hyclone, Logan, UT, USA) supplemented with 10% FBS at 37°C with 5% CO2. Wild‐type H. pylori 26695 (ATCC) were grown in a microaerobic humidified atmosphere on Columbia agar (Oxoid, Cambridge, UK) plates with 8% defibrinogenated goat blood at 37°C. For co‐culture experiments, H. pylori was harvested and infected HET‐1A or OE33 cells at 100 MOI. For the chronic exposure study, H. pylori was ultrasonically disrupted in serum‐free RPMI‐1640. The breakdown of the bacteria was confirmed by microscopy. Cell debris was removed by centrifugation and the supernatant (H. pylori extract) filtered through a 0.22‐μm filter. Protein concentrations were determined by BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA). Deoxycholic acid (DCA; Sigma‐Aldrich, Saint Louis, MO, USA) was dissolved in RPMI‐1640 and the pH was adjusted to 5.0 with hydrochloric acid. HET‐1A cells were repeatedly exposed to H. pylori extract (600 μg/mL) and/or acidified deoxycholic acid (acidified DCA, 200 μmol/L, pH 5.0) for up to 36 weeks.20 Briefly, cells were treated for 10 minutes each time, twice a week. The treatment duration was increased 10 minutes every week, to a maximum of 2 hours for each treatment. A schematic diagram showing the treatment algorithm is presented in Figure 1A. The control cells were grown in parallel in the RPMI‐1640 medium at pH 7.4. A portion of the cells were harvested every 4 weeks, and morphological changes were examined by H&E staining.
Figure 1.
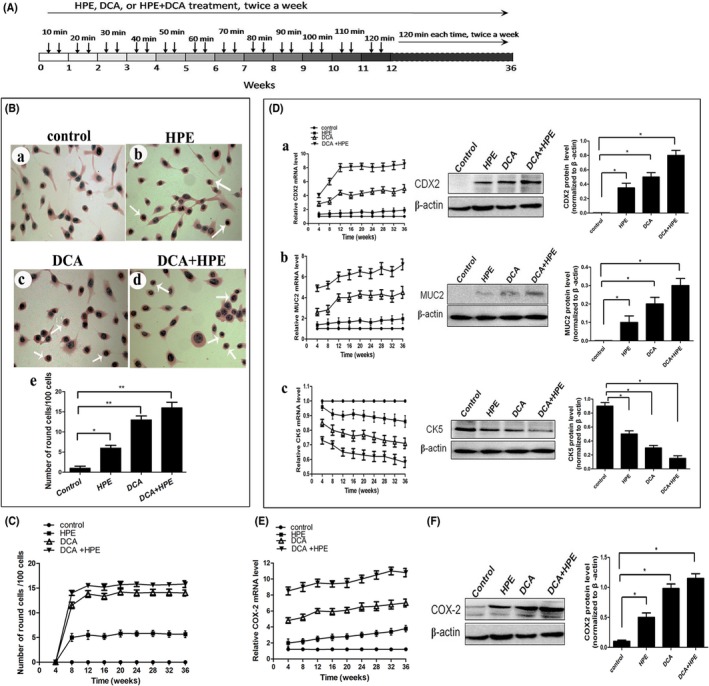
Helicobacter pylori promoted phenotypic changes in esophageal epithelial cells. A, Schematic representation of the chronic exposure experiment. HET‐1A cells were repeatedly treated with H. pylori extract (HPE) and/or acidified deoxycholic acid (DCA) for up to 36 wk. B, Morphological changes in cells were determined by H&E staining. Control (a), HPE‐ (b), acidified DCA‐ (c), and HPE + acidified DCA‐treated (d) cells are shown. The H&E staining data are illustrated by quantitative analysis (e). C, Morphological changes in HET‐1A cells throughout the study. D, Expression of caudal‐type homeobox protein 2 (CDX2) (a), mucin 2 (MUC2) (b), and cytokeratin 5 (CK5) (c) were examined. The mRNA levels during the experiment (left) and the protein levels at 36 wk (middle) are shown, and Western blot data are illustrated by quantitative analysis (right). E, COX2 mRNA expression changes throughout the study. F, Protein levels of COX2 were measured at 36 wk (left), and Western blot data are illustrated by quantitative analysis (right). Data are expressed as mean ± SD. *P < .05, **P < .01
2.3. RNA extraction, miRNA microarray, and target gene prediction
HET‐1A and OE33 cells were co‐cultured with or without H. pylori 26695 for 12 hours. The total RNA of the cells was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA, USA), and the miRNA fraction was further purified using a mirVana miRNA isolation kit (Ambion, Austen, TX, USA). For microarray analysis, the quality of RNA and the RNA integrity number were determined by using RNA 6000 Nano LabChip Kits (Agilent Technologies, Santa Clara, CA, USA). An Agilent Human miRNA microarray chip version, containing 4947 probes corresponding to 1887 unique human miRNAs and 140 human viral miRNAs cataloged in the Sanger database version 18.0 (Agilent Technologies), was used according to the manufacturer's recommendations. The difference in miRNA expression between the cells infected with or without H. pylori was considered significant if the change of expression was more than 1.5‐fold and the t‐test P‐values were <.05. Target gene prediction of the screened miRNA was carried out using the online miRNA target predicting software TargetScan (www.targetscan.org), miRBase (www.mirbase.org), and miRanda (www.microrna.org). Only genes predicted by at least two software programs were considered as potential target genes of the miRNA.
2.4. MicroRNA and transfection
The oligonucleotide of miRNA‐212‐3p mimic (5′‐UAACAGUCUCCAGUCACGGCC‐3′, antisense 5′‐CCGUGACUGGAGACUGUUAUU‐3′), miRNA‐212‐3p inhibitor (5′‐GGCCGUGACUGGAGACUGUUA‐3′), miRNA‐361‐3p mimic (5′‐UCCCCCAGGUGUGAUUCUGAUUU‐3′, antisense 5′‐AUCAGAAUCACACCUGGGGGAUU‐3′), miRNA‐361‐3p inhibitor (5′‐AAAUCAGAAUCACACCUGGGGGA‐3′), mimics negative control (5′‐UUCUCCGAACGUGUCACGUTT‐3′, antisense 5′‐ACGUGACACGUUCGGAGAATT‐3′), and inhibitors negative control (5′‐CAGUACUUUUGUGUAGUACAA‐3′) were synthesized by Ribobio (Guangzhou, China). HET‐1A and OE33 cells were grown to 60% confluence and the oligonucleotide was transfected into cells using Lipofectamine 2000 (Invitrogen). The effects on the mRNA and protein expression were assessed 24 and 48 hours later, respectively.
2.5. Plasmid construction and dual‐luciferase activity assay
The 3′‐UTR of human COX2 containing the putative miR‐212‐3p target site and 3′‐UTR of human CDX2 containing the putative miR‐361‐3p target site were chemically synthesized and cloned into XhoI and NotI sites, respectively, downstream of the luciferase gene in the pmiR‐RB‐Report vector (Ribobio). HEK‐293T cells were transfected with luciferase report vector combined with miRNA mimic or miRNA inhibitor using Lipofectamine 2000 reagent. Luciferase activity was measured 24 hours after transfection using the dual‐luciferase reporter assay system (Promega, Madison, WI, USA) according to the manufacturer's instructions.
2.6. Western blot analysis and real‐time quantitative PCR (RT‐qPCR) assays
Cells were lysed with RIPA buffer. Equal amounts of protein were resolved by 10% SDS‐PAGE and transferred onto PVDF membranes. After blocking, membranes were probed with appropriate primary antibodies at 4°C overnight. Membranes were reacted with species‐specific secondary antibodies conjugated with HRP for 1 hour at room temperature, followed by detection with enhanced chemiluminescence. All primary and secondary antibodies used for Western blot analyses were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The expression of COX2 and caudal‐type homeobox protein 2 (CDX2) was quantified by densitometry analysis using ImageJ software (NIH, Bethesda, MD, USA) and normalized against β‐actin.
Reverse transcription of mRNAs to cDNAs was carried out using the TaqMan MicroRNA Assays kit (Applied Biosystems, Foster City, CA, USA). Amplification assays were undertaken on an Applied Biosystems 7500 Real‐time PCR System. U6 small nuclear RNA or GAPDH expression was assayed for normalization. The relative expression levels were determined with the ΔCT method and reported as 2−ΔΔCT. The sequences of primers used in RT‐qPCR are as follows: CDX2 forward, 5′‐CAGTCGCTACATCACCATCC‐3′ and reverse, 5′‐CTCCTTTGCTCTGCGGTTCT‐3′; COX2 forward, 5′‐CCAACTCCCATGGGTGTGA‐3′ and reverse, 5′‐TCTCTCCTCAGAAGAACCTTTTCC‐3′; CK5 forward, 5′‐CTTGTGGAGTGGGTGGCTAT‐3′ and reverse, 5′‐CCACTTGGTGTCCAGAACCT‐3′; MUC2 forward, 5′‐TGCTCCTACGTGGCTGTTCA‐3′ and reverse, 5′‐GACGTTCTCGGTGATGATGCT‐3′; and GAPDH forward, 5′‐GCCTGGTCACCAGGGCT‐3′ and reverse, 5′‐AATTTGCCATGGGTGGAATC‐3′.
2.7. Statistical analysis
Data are expressed as mean ± SD. Differences were analyzed by Student's t‐test or the non‐parametric Wilcoxon and Mann–Whitney U‐tests. A P‐value of <.05 was considered significant.
3. RESULTS
3.1. Helicobacter pylori promoted acidified DCA‐induced transformation of esophageal epithelial cells
To study the direct effects of H. pylori, human normal esophageal cells (HET‐1A) were exposed to H. pylori extract and/or acidified DCA for up to 36 weeks. As shown in Figure 1B, some HET‐1A cells developed morphological changes, becoming round or oval‐shaped when chronically irritated by H. pylori extract or acidified DCA, whereas untreated cells remained spindle shaped and evenly dispersed on the culture plates. These distinct phenotypic changes were observed at 4‐8 weeks and maintained for up to 36 weeks (Figure 1C). Notably, chronic exposure of HET‐1A cells to H. pylori extract and acidified DCA conferred more prominent morphological changes (Figure 1B,C).
As an intestine‐specific transcription factor, caudal‐type homeobox protein 2 (CDX2) regulates goblet‐specific MUC2 gene expression, resulting in the differentiation of intestinal epithelium.21 Normal esophageal keratinocytes do not express CDX2, which has been reported to be a highly sensitive and specific marker of intestinal metaplasia and adenocarcinoma.22, 23 In agreement with the effect on cell morphology, treatment of HET‐1A cells with H. pylori extract or acidified DCA significantly increased the mRNA expression of CDX2 and mucin 2 (MUC2), and these effects were evident at 8‐12 weeks and maintained for up to 36 weeks (Figure 1Da,b). Moreover, CDX2 and MUC2 protein expression was upregulated in cells treated with these compounds at 36 weeks (Figure 1Da,b). Cytokeratin 5 (CK5) is expressed by normal esophageal keratinocytes but not by intestinal epithelium with metaplasia.24 Coincidentally, with upregulated CDX2 and MUC2, the expression of CK5 also decreased in HET‐1A cells treated with H. pylori extract or acidified DCA (Figure 1Dc). Importantly, the alteration of CDX2, MUC2, and CK5 was enhanced when HET‐1A cells were exposed to both two compounds. As shown in Figure 1D, after 36 weeks, the expression of CDX2 and MUC2 mRNA increased by 8.4‐ and 7.9‐fold, respectively, in H. pylori extract together with acidified DCA‐treated cells, while the expression of CK5 decreased by approximately 50%. These data indicate that H. pylori might synergistically enhance acidified DCA‐induced transformation in normal esophageal epithelial cells.
3.2. Helicobacter pylori enhanced acidified DCA‐induced COX2 expression in esophageal epithelial cells
Esophageal H. pylori colonization is thought to initiate esophageal carcinogenesis through the COX2 pathway.11 As shown in Figure 1E, at the initial stage of exposure (before 12 weeks), H. pylori alone resulted in minor changes of COX2 expression, but it distinctly augmented the acidified DCA‐induced COX2 in HET‐1A cells. Helicobacter pylori extract plus acidified DCA led to a 9.5‐fold increase of COX2 mRNA at 12 weeks, and it was maintained at high levels up to 36 weeks. Similarly, COX2 protein expression significantly increased in cells treated with the two compounds at 36 weeks (Figure 1F). Thus, prolonged exposure of HET‐1A cells to H. pylori seems to aggravate the tumorigenic phenotype conferred by acidified DCA.
3.3. MicroRNA expression profiles associated with Helicobacter pylori infection
To determine the miRNA profiles of esophageal epithelial cells associated with H. pylori infection, we carried out a microarray analysis. Comparison of HET‐1A as well as OE33 cells treated with or without H. pylori revealed that 15 miRNAs were differentially expressed with at least 1.5‐fold changes. A heat map representation of differentially expressed miRNAs is shown in Figure 2. Among the significantly deregulated miRNAs, eight upregulated miRNAs (miR‐1287, miR‐1290, miR‐25‐5p, miR‐205‐3p, miR‐3934, miR‐1202, miR‐3960, and miR‐4516) and seven downregulated miRNAs (miR‐361‐3p, miR‐212‐3p, miR‐4521, miR‐361‐5p, miR‐5100, miR‐455‐5p, and ebv‐miR‐BART13) were identified in both HET‐1A and OE33 cells infected with H. pylori.
Figure 2.
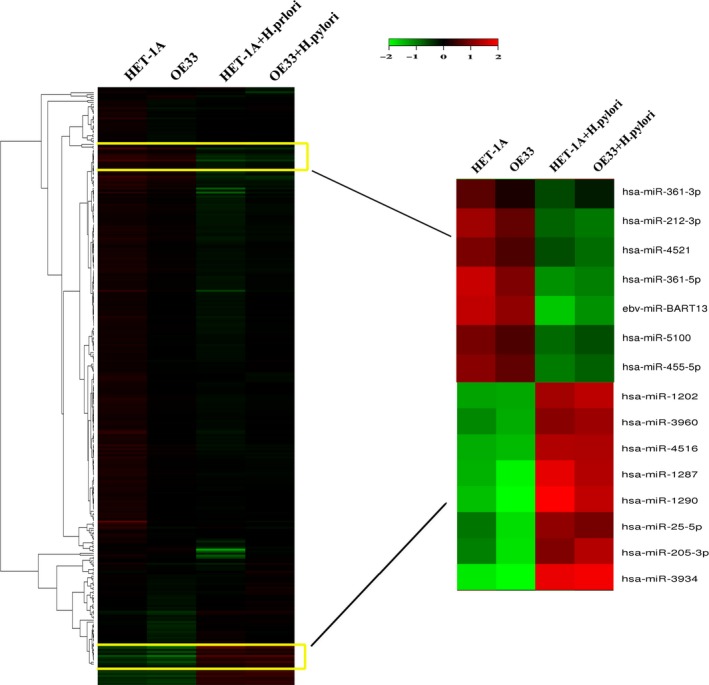
Helicobacter pylori modified the microRNA (miRNA) profiles of esophageal epithelial cells. Hierarchical cluster analysis showed 15 miRNAs differentially expressed by H. pylori treatment in HET‐1A and OE33 cells
3.4. Candidate miRNA selection and RT‐qPCR validation
We further explored whether upregulation of CDX2 and COX2 oncoprotein expression was attributable to aberrant expression of miRNAs induced by H. pylori. Analysis with TargetScan, miRBase, and miRanda identified COX2 as a potential target of miR‐212‐3p and CDX2 as a potential target of miR‐361‐3p. Thus, miR‐212‐3p and miR‐361‐3p were selected for further RT‐qPCR confirmation experiments. We detected the temporal expression of miR‐212‐3p and miR‐361‐3p in esophageal epithelial cells following infection with H. pylori. Consistent with the data obtained from the microarray assay, H. pylori caused pronounced downregulation of miR‐212‐3p and miR‐361‐3p in HET‐1A and OE33 cells (Figure 3A,B). Similarly, exposure to H. pylori extract for 36 weeks significantly decreased miR‐212‐3p and miR‐361‐3p expression in HET‐1A cells (Figure 3C). Furthermore, we measured miRNA expression levels in biopsies obtained from Barrett's esophagus patients with or without H. pylori esophageal colonization. As shown in Figure 3D,E, esophageal H. pylori colonization led to a pronounced decrease of miR‐212‐3p and miR‐361‐3p expression compared with uninfected controls. Moreover, COX2 expression was significantly increased in patients with H. pylori esophageal colonization (Figure 3F). Although these patients showed a trend of increased CDX2 expression, it was not statistically significant (Figure 3G). In Barrett's esophagus, intestinal metaplasia of esophageal epithelium facilitates colonization of H. pylori in the distal esophagus, and therefore exacerbates inflammation. Our data suggested that aberrant silencing of miR‐212‐3p and miR‐361‐3p may be involved in H. pylori‐induced esophageal injury.
Figure 3.
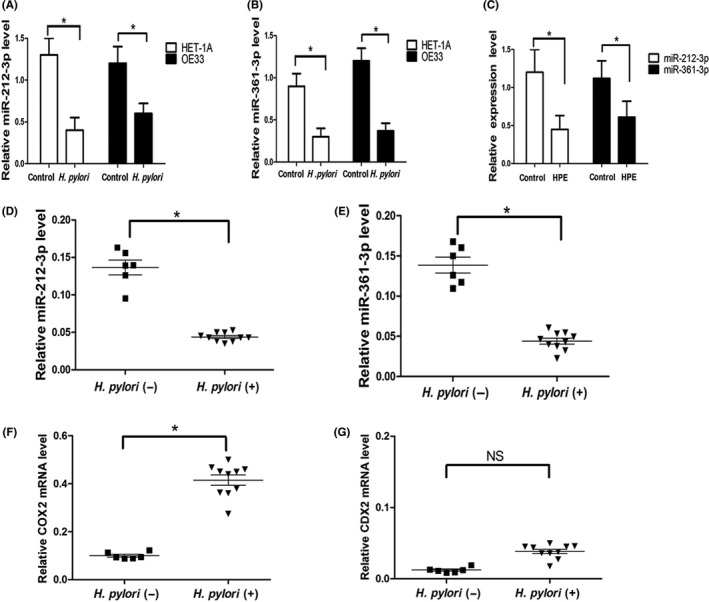
Helicobacter pylori decreased microRNA (miR)‐212‐3p and miR‐361‐3p expression. ET‐1A and OE33 cells were cocultured with H. pylori (100 MOI) for 12 h, relative expression levels of miR‐212‐3p (A) and miR‐361‐3p (B) were analyzed by real‐time quantitative PCR. C, HET‐1A cells were exposed to H. pylori extract (HPE) for 36 wk and relative expression levels of miR‐212‐3p and miR‐361‐3p were determined. In Barrett's esophagus patients with or without H. pylori esophageal colonization, the expression of miR‐212‐3p (D), miR‐361‐3p (E), COX2 (F), and caudal‐type homeobox protein 2 (CDX2) (G) were detected. All miRNA expression was normalized to U6 small nuclear RNA. COX2 and CDX2 mRNA expression was normalized to GAPDH. Data are expressed as mean ± SD. *P < .05. NS, not significant. All results shown are representative of three independent experiments
3.5. MicroRNA‐212‐3p targeted COX2 and miR‐361‐3p targeted CDX2 in esophageal epithelial cells
To explore mechanisms underlying transformation of esophageal epithelial cells induced by H. pylori, we determined whether miR‐212‐3p and miR‐361‐3p could post‐transcriptionally regulate COX2 and CDX2, respectively. Analysis of the human COX2 3′‐UTR sequence predicted that the miR‐212‐3p seed sequence is complementary to a potential binding site of COX2 (Figure 4A). We used a luciferase reporter assay to determine whether miR‐212‐3p could directly target the 3′‐UTR of COX2. As shown in Figure 4B, a significant decreased of luciferase activity was observed when HEK‐293T cells were transfected with the miR‐212‐3p mimic compared to the negative control. In contrast, transfection with miR‐212‐3p inhibitor led to a pronounced increase of luciferase activity, suggesting that COX2 was a direct target of miR‐212‐3p. Furthermore, we transfected HET‐1A cells with the miR‐212‐3p mimic or inhibitor and then examined the COX2 expression. As shown in Figure 4C,D, ectopic expression of miR‐212‐3p did not affect COX2 mRNA level, although it led to a significant decrease of COX2 protein expression. Moreover, miR‐212‐3p inhibitor dose‐dependently increased COX2 protein expression in HET‐1A cells, and COX2 mRNA expression was unaffected. These data suggested that miR‐212‐3p downregulates COX2 in esophageal epithelial cells through inhibition of translation rather than mRNA degradation.
Figure 4.
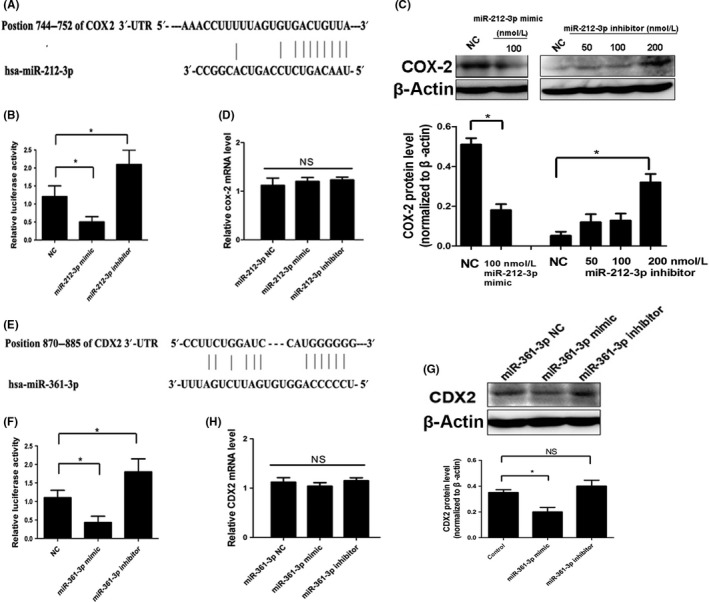
MicroRNA (miR)‐212‐3p targeted COX2 and miR‐361‐3p targeted caudal‐type homeobox protein 2 (CDX2) in esophageal epithelial cells. A, Diagram of the putative binding sequence of miR‐212‐3p in the 3′‐UTR of human COX2. B, Luciferase assay in HEK‐293T cells co‐transfected with report vector and miRNA as indicated. HET‐1A cells were transfected with mimic, inhibitor, or negative control (NC) of miR‐212‐3p. Expression of COX2 was assessed by Western blot (C) and real‐time quantitative PCR (D). E, Diagram of the putative binding sequence of miR‐361‐3p in the 3′‐UTR of human CDX2. F, Luciferase assay in HEK‐293T cells co‐transfected with report vector and miRNA as indicated. OE33 cells were transfected with mimic, inhibitor, or NC of miR‐361‐3p. Protein (G) and mRNA (H) levels of CDX2 were measured. All Western blot bands were quantified and normalized to β‐actin, and illustrated by quantitative analysis of COX2 (C) and CDX2 (G) expression. Data are expressed as mean ± SD. *P < .05. NS, not significant. All results shown are representative of three independent experiments
We found that human CDX2 3′‐UTR contains putative miR‐361‐3p complementary sites (Figure 4E), and further luciferase reporter assays validated the hypothesis that miR‐361‐3p is directly bound to the 3′‐UTR of CDX2 (Figure 4F). Western blot analysis showed that CDX2 expression was decreased after transfection with the miR‐361‐3p mimic in OE33 cells compared to respective controls (Figure 4G). However, because the level of endogenous miR‐361‐3p in OE33 cells is low, miR‐361‐3p inhibitor did not increase CDX2 expression significantly (Figure 4G). The effects of the miR‐361‐3p mimic or inhibitor on CDX2 mRNA level was not obvious in OE33 cells (Figure 4H), indicating that miR‐361‐3p may mediate CDX2 expression by repressing translation.
3.6. Helicobacter pylori upregulated COX2 and CDX2 expression by aberrant silencing of miRNAs
To investigate the effect of miRNAs on COX2 and CDX2 expression during H. pylori infection, we transfected esophageal epithelial cells with mimic or inhibitor, followed by H. pylori infection. As shown in Figure 5, H. pylori infection led to increased COX2 and CDX2 expression in esophageal epithelial cells. Ectopic miR‐212‐3p significantly inhibited H. pylori‐induced COX2, while miR‐212‐3p inhibitor augmented COX2 expression in HET‐1A cells (Figure 5A). Similarly, ectopic miR‐361‐3p dramatically attenuated H. pylori‐induced CDX2 in OE33 cells (Figure 5B). These data indicated that upregulation of COX2 and CDX2 is associated with aberrant miRNA expression induced by H. pylori in esophageal epithelial cells.
Figure 5.
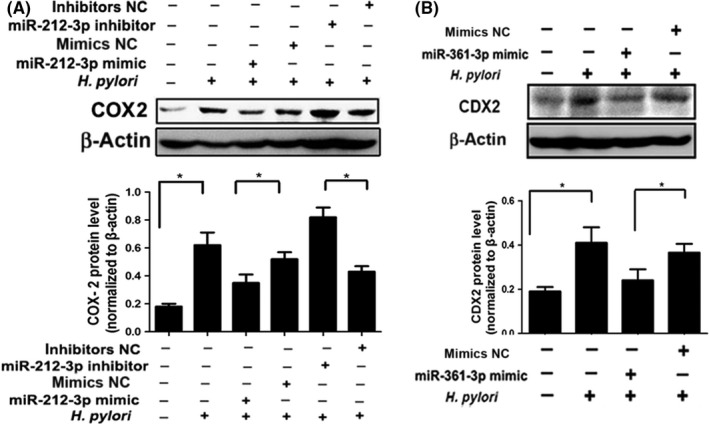
Helicobacter pylori upregulated COX2 and caudal‐type homeobox protein 2 (CDX2) expression by aberrant silencing of microRNAs (miRNAs). A, HET‐1A cells were transfected with mimic, inhibitor, or negative control (NC) of miR‐212‐3p, followed by H. pylori (100 MOI) infection for 48 h. Expression of COX2 was determined by Western blot analysis. B, OE33 cells were transfected with mimic or NC of miR‐361‐3p followed by H. pylori infection, then the expression of CDX2 was detected. All Western blot bands were quantified and normalized to β‐actin, and illustrated by quantitative analysis of COX2 (A) and CDX2 (B) expression. Data are expressed as mean ± SD.*P < .05. All results shown are representative of three independent experiments
4. DISCUSSION
The role of H. pylori in esophageal disease has not been clearly defined. Here, we showed that exposure to H. pylori promoted acidified DCA‐induced transformation of esophageal epithelial cells. Helicobacter pylori infection altered the miRNA profiles of esophageal epithelial cells, particularly aberrant silencing of miR‐212‐3p and miR‐361‐3p expression. Furthermore, we identified COX2 as a target of miR‐212‐3p, and CDX2 as a target of miR‐361‐3p. The miRNA‐mediated upregulation of oncoprotein CDX2 and COX2 was associated with H. pylori infection in esophageal epithelial cells.
In recent years, concerns have arisen that H. pylori is not “protective” against GERD, Barrett's esophagus, or esophageal adenocarcinoma.25, 26 Both gastroesophageal reflux and H. pylori are major etiologic factors in the development of inflammation and intestinal metaplasia of the gastroesophageal junction.27 Long‐standing reflux of gastric contents is thought to induce Barrett's esophagus, and Barrett's esophagus is associated with the increased colonization of H. pylori in the distal esophagus.14, 28 The expected incidence of esophageal adenocarcinoma with persistent H. pylori infection is higher than that of esophageal adenocarcinoma after eradication of infection.29 Thus, H. pylori might be involved in the GERD–Barrett's esophagus–esophageal adenocarcinoma sequence. In the present study, we analyzed the direct effects of H. pylori on the transformation of esophageal epithelial cells. Following repeated exposure to H. pylori extract or acidified DCA, HET‐1A cells showed progressive morphological changes. These changes were coupled with suppressed expression of keratinocyte markers and enhanced expression of intestinal metaplasia markers. It was noteworthy that H. pylori plus acidified DCA led to more prominent morphological and molecular changes in HET‐1A cells, indicating H. pylori and acidified DCA act synergistically, inducing transformation of normal esophageal epithelial cells. These findings are consistent with our earlier study showing that H. pylori colonization can aggravate esophageal injury and promote the incidence of Barrett's esophagus and esophageal adenocarcinoma in a model of chronic gastroesophageal reflux.11, 12
Caudal‐type homeobox protein 2 is a key factor for intestinal development. Ectopic expression of CDX2 occurs during the development and maintenance of Barrett's esophagus.30 Bile acids can directly induce CDX2 expression and activation in Barrett's esophagus.31 Our previous studies showed that H. pylori infection induced CDX2 expression in esophageal epithelium, playing an important role in the inflammation associated with Barrett's esophagus and tumorigenesis.11, 12 In this in vitro model, we found CDX2 was increased in esophageal epithelial cells in response to H. pylori or acidified DCA treatment, implying that the cells underwent transformation. Chronic expression of COX2 plays a crucial role in H. pylori‐associated gastric carcinogenesis.32 Moreover, the expression of COX2 is gradually increased during the neoplastic progression of the esophageal mucosa from Barrett's esophagus to adenocarcinoma.33 In the present study, COX2 expression in esophageal epithelial cells was upregulated by either H. pylori or acidified DCA treatment, and this effect was more pronounced when both factors were present. Thus, alteration of these molecules may contribute to the transformation of the phenotype of the esophageal epithelial cells.
Helicobacter pylori infection could alter the expression and function of miRNAs through epigenetic regulations such as DNA methylation and histone modification.34 The role of miRNA in the progression from normal esophageal mucosa to Barrett's esophagus and finally to esophageal adenocarcinoma has been established.17 However, the contribution of H. pylori‐related miRNA dysregulation to the transformation of esophageal epithelial cells remains unclear. Our microarray data showed that 15 miRNAs are differentially expressed in HET‐1A and OE33 cells after H. pylori infection. To further explore the molecular mechanisms of transformation induced by H. pylori, we searched for miRNAs that target COX2 and CDX2 through bioinformatics analysis and found that miR‐212‐3p could match the sequence of COX2, and miR‐361‐3p could match CDX2. Moreover, we confirmed that both transient and prolonged H. pylori infection significantly downregulated miR‐212‐3p and miR‐361‐3p, suggesting that H. pylori may induce COX2 and CDX2 in esophageal epithelial cells by dysregulating miRNAs.
Helicobacter pylori does not colonize the esophageal squamous epithelium, but does grow on the metaplastic columnar epithelium of Barrett's esophagus that more resembles the gastric mucosa. Our previous animal studies showed that esophageal H. pylori colonization exacerbated inflammation and increased the incidence of esophageal adenocarcinoma. Here, in Barrett's esophagus patients, esophageal H. pylori colonization led to a significant decrease of miR‐212‐3p and miR‐361‐3p expression compared with uninfected controls. Moreover, COX2 and CDX2 were upregulated in patients with H. pylori esophageal colonization. Limited by an inadequate sample, we could not evaluate the relationship between miRNA expression levels and the severity of esophageal injury. Thus, our present observations need to be confirmed and extended by further clinical studies.
The role of miRNA is tissue specific and highly dependent on its targets. Here, we identified COX2 as a new target of miR‐212‐3p, and CDX2 was a target of miR‐361‐3p in esophageal epithelial cells. MicroRNAs control gene expression at the post‐transcriptional level by binding to the 3′‐UTRs of mRNAs, targeting mRNAs for repressing translation or degradation.35 In this study, miRNA mimic or inhibitor caused reciprocal modification of COX2 and CDX2 protein expression, whereas they did not affect COX2 and CDX2 mRNA levels. These data suggested that miR‐212‐3p and miR‐361‐3p downregulated COX2 and CDX2 through translation inhibition rather than mRNA degradation.
To clarify the effect of miRNAs on COX2 and CDX2 expression during H. pylori infection, we transfected esophageal epithelial cells with mimic or inhibitor, followed by H. pylori infection. Ectopic miR‐212‐3p and miR‐361‐3p dramatically reversed H. pylori induction of COX2 and CDX2, respectively. Thus, H. pylori exerted its effects on esophageal epithelial cells, at least in part, by modulating miRNAs and their downstream target genes. It was notable that COX2 and CDX2 mRNA levels were increased in the prolonged H. pylori exposure study, but not in the miRNA mimic and inhibitor experiments. Clearly, the effect of H. pylori is not entirely miRNA‐dependent. Moreover, there are potential differences in the mechanism of acute and chronic H. pylori infection, particularly if epigenetic changes that involve DNA replication are involved. Furthermore, as miRNAs have multiple targets, we cannot assume an effect on a single target (ie, COX2 or CDX2) to be the only consequence. Thus, further studies are needed to validate the effects of altering expression of these miRNAs by both H. pylori and genetic manipulation.
In summary, H. pylori promoted acidified DCA‐induced transformation of the esophageal epithelium through modifying the miRNA profiles. In H. pylori‐infected esophageal epithelial cells, upregulation of oncoprotein COX2 and CDX2 was associated with aberrant silencing of miR‐212‐3p and miR‐361‐3p, respectively. This provides new evidence for better understanding the molecular mechanisms underlying the association between H. pylori infection and esophageal carcinogenesis.
CONFLICT OF INTEREST
The authors have no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by National Natural Science Foundation of China (NSFC #81172271), the Research Fund for the Doctoral Program of Higher Education of China (RFDP #20110001110064) and Beijing Natural Science Foundation (BNSF#7123232) awarded to W. Wang. Y. Dai was supported by NSFC #81472267.
Teng G, Dai Y, Chu Y, et al. Helicobacter pylori induces caudal‐type homeobox protein 2 and COX2 expression by modulating microRNAs in esophageal epithelial cells. Cancer Sci. 2018;109:297–307. https://doi.org/10.1111/cas.13462
Guigen Teng and Yun Dai are contributed equally to this work.
REFERENCES
- 1. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883‐899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Falk GW. Barrett's esophagus. Gastroenterology. 2002;122:1569‐1591. [DOI] [PubMed] [Google Scholar]
- 3. Jankowski JA, Harrison RF, Perry I, Balkwill F, Tselepis C. Barrett's metaplasia. Lancet. 2000;356:2079‐2085. [DOI] [PubMed] [Google Scholar]
- 4. Neumann H, Monkemuller K, Vieth M, Malfertheiner P. Chemoprevention of adenocarcinoma associated with Barrett's esophagus: potential options. Dig Dis. 2009;27:18‐23. [DOI] [PubMed] [Google Scholar]
- 5. Cover TL, Blaser MJ. Helicobacter pylori in health and disease. Gastroenterology. 2009;136:1863‐1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rokkas T, Pistiolas D, Sechopoulos P, Robotis I, Margantinis G. Relationship between Helicobacter pylori infection and esophageal neoplasia: a meta‐analysis. Clin Gastroenterol Hepatol. 2007;5:1413‐1417, 1417 e1‐2. [DOI] [PubMed] [Google Scholar]
- 7. Fischbach LA, Nordenstedt H, Kramer JR, et al. The association between Barrett's esophagus and Helicobacter pylori infection: a meta‐analysis. Helicobacter. 2012;17:163‐175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thrift AP, Pandeya N, Smith KJ, et al. Helicobacter pylori infection and the risks of Barrett's oesophagus: a population‐based case‐control study. Int J Cancer. 2012;130:2407‐2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qian B, Ma S, Shang L, Qian J, Zhang G. Effects of Helicobacter pylori eradication on gastroesophageal reflux disease. Helicobacter. 2011;16:255‐265. [DOI] [PubMed] [Google Scholar]
- 10. Kountouras J, Chatzopoulos D, Zavos C. Eradication of Helicobacter pylori might halt the progress to oesophageal adenocarcinoma in patients with gastro‐oesophageal reflux disease and Barrett's oesophagus. Med Hypotheses. 2007;68:1174‐1175. [DOI] [PubMed] [Google Scholar]
- 11. Liu FX, Wang WH, Wang J, Li J, Gao PP. Effect of Helicobacter pylori infection on Barrett's esophagus and esophageal adenocarcinoma formation in a rat model of chronic gastroesophageal reflux. Helicobacter 2011;16:66‐77. [DOI] [PubMed] [Google Scholar]
- 12. Chu YX, Wang WH, Dai Y, Teng GG, Wang SJ. Esophageal Helicobacter pylori colonization aggravates esophageal injury caused by reflux. World J Gastroenterol. 2014;20:15715‐15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Contreras M, Salazar V, Garcia‐Amado MA, et al. High frequency of Helicobacter pylori in the esophageal mucosa of dyspeptic patients and its possible association with histopathological alterations. Int J Infect Dis. 2012;16:e364‐e370. [DOI] [PubMed] [Google Scholar]
- 14. Henihan RD, Stuart RC, Nolan N, Gorey TF, Hennessy TP, O'Morain CA. Barrett's esophagus and the presence of Helicobacter pylori . Am J Gastroenterol. 1998;93:542‐546. [DOI] [PubMed] [Google Scholar]
- 15. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281‐297. [DOI] [PubMed] [Google Scholar]
- 16. Iorio MV, Croce CM. microRNA involvement in human cancer. Carcinogenesis. 2012;33:1126‐1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yang H, Gu J, Wang KK, et al. MicroRNA expression signatures in Barrett's esophagus and esophageal adenocarcinoma. Clin Cancer Res. 2009;15:5744‐5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matsushima K, Isomoto H, Inoue N, et al. MicroRNA signatures in Helicobacter pylori‐infected gastric mucosa. Int J Cancer. 2011;128:361‐370. [DOI] [PubMed] [Google Scholar]
- 19. Zhai R, Wei Y, Su L, et al. Whole‐miRNome profiling identifies prognostic serum miRNAs in esophageal adenocarcinoma: the influence of Helicobacter pylori infection status. Carcinogenesis. 2015;36:87‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Das KM, Kong Y, Bajpai M, et al. Transformation of benign Barrett's epithelium by repeated acid and bile exposure over 65 weeks: a novel in vitro model. Int J Cancer. 2011;128:274‐282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yamamoto H, Bai YQ, Yuasa Y. Homeodomain protein CDX2 regulates goblet‐specific MUC2 gene expression. Biochem Biophys Res Commun. 2003;300:813‐818. [DOI] [PubMed] [Google Scholar]
- 22. Werling RW, Yaziji H, Bacchi CE, Gown AM. CDX2, a highly sensitive and specific marker of adenocarcinomas of intestinal origin: an immunohistochemical survey of 476 primary and metastatic carcinomas. Am J Surg Pathol. 2003;27:303‐310. [DOI] [PubMed] [Google Scholar]
- 23. Kaimaktchiev V, Terracciano L, Tornillo L, et al. The homeobox intestinal differentiation factor CDX2 is selectively expressed in gastrointestinal adenocarcinomas. Mod Pathol. 2004;17:1392‐1399. [DOI] [PubMed] [Google Scholar]
- 24. DiMaio MA, Kwok S, Montgomery KD, Lowe AW, Pai RK. Immunohistochemical panel for distinguishing esophageal adenocarcinoma from squamous cell carcinoma: a combination of p63, cytokeratin 5/6, MUC5AC, and anterior gradient homolog 2 allows optimal subtyping. Hum Pathol. 2012;43:1799‐1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Graham DY. Helicobacter pylori is not and never was “protective” against anything, including GERD. Dig Dis Sci. 2003;48:629‐630. [DOI] [PubMed] [Google Scholar]
- 26. Lee YY, Mahendra Raj S, Graham DY. Helicobacter pylori infection–a boon or a bane: lessons from studies in a low‐prevalence population. Helicobacter. 2013;18:338‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Correa P, Piazuelo MB, Wilson KT. Pathology of gastric intestinal metaplasia: clinical implications. Am J Gastroenterol. 2010;105:493‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Newton M, Bryan R, Burnham WR, Kamm MA. Evaluation of Helicobacter pylori in reflux oesophagitis and Barrett's oesophagus. Gut. 1997;40:9‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nakajima S, Hattori T. Oesophageal adenocarcinoma or gastric cancer with or without eradication of Helicobacter pylori infection in chronic atrophic gastritis patients: a hypothetical opinion from a systematic review. Aliment Pharmacol Ther. 2004;20(Suppl 1):54‐61. [DOI] [PubMed] [Google Scholar]
- 30. Colleypriest BJ, Palmer RM, Ward SG, Tosh D. Cdx genes, inflammation and the pathogenesis of Barrett's metaplasia. Trends Mol Med. 2009;15:313‐322. [DOI] [PubMed] [Google Scholar]
- 31. Kazumori H, Ishihara S, Rumi MA, Kadowaki Y, Kinoshita Y. Bile acids directly augment caudal related homeobox gene Cdx2 expression in oesophageal keratinocytes in Barrett's epithelium. Gut. 2006;55:16‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sakai T, Fukui H, Franceschi F, et al. Cyclooxygenase expression during Helicobacter pylori infection in Mongolian gerbils. Dig Dis Sci. 2003;48:2139‐2146. [DOI] [PubMed] [Google Scholar]
- 33. Morris CD, Armstrong GR, Bigley G, Green H, Attwood SE. Cyclooxygenase‐2 expression in the Barrett's metaplasia‐dysplasia‐adenocarcinoma sequence. Am J Gastroenterol. 2001;96:990‐996. [DOI] [PubMed] [Google Scholar]
- 34. Hayashi Y, Tsujii M, Wang J, et al. CagA mediates epigenetic regulation to attenuate let‐7 expression in Helicobacter pylori‐related carcinogenesis. Gut. 2013;62:1536‐1546. [DOI] [PubMed] [Google Scholar]
- 35. He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522‐531. [DOI] [PubMed] [Google Scholar]