

Abstract
DNA replication is one of the fundamental biological processes in which dysregulation can cause genome instability. This instability is one of the hallmarks of cancer and confers genetic diversity during tumorigenesis. Numerous experimental and clinical studies have indicated that most tumors have experienced and overcome the stresses caused by the perturbation of DNA replication, which is also referred to as DNA replication stress (DRS). When we consider therapeutic approaches for tumors, it is important to exploit the differences in DRS between tumor and normal cells. In this review, we introduce the current understanding of DRS in tumors and discuss the underlying mechanism of cancer therapy from the aspect of DRS.
Keywords: chemotherapeutic drugs, DNA damage response, DNA replication stress, genome instability, tumorigenesis
Abbreviations
- 5‐FU
5‐fluorouracil
- ATM
ataxia telangiectasia mutated
- ATR
ATM and Rad3‐related
- ATRIP
ATR‐interacting protein
- Chk1/2
checkpoint kinase 1/2
- CPT
camptothecin
- DDR
DNA damage response
- DRS
DNA replication stress
- dsDNA
double‐stranded DNA
- dTTP
deoxythymidine triphosphate
- FTD
trifluridine
- ssDNA
single‐stranded DNA
- TFTD
trifluridine/tipiracil
- Topo I‐cc
Topo I‐covalent complex
- Topo I
topoisomerase I
- UFB
ultrafine bridge
1. INTRODUCTION
Tumorigenesis is a multistep process by which normal cells progressively evolve to a neoplastic state. During the process, normal cells acquire traits that enable them to become tumorigenic and ultimately malignant. These traits are referred to as “the hallmarks of cancer,” and are proposed to contribute to 8 biological capabilities: (i) sustaining proliferative signaling; (ii) evading growth suppressors; (iii) resistance to apoptosis; (iv) enabling replicative immortality; (v) inducing angiogenesis; (vi) activating invasion and metastasis; (vii) reprogramming of energy metabolism; and (viii) evading immune destruction. Two conditions, genome instability and inflammation, underlie the development of these capabilities.1, 2 Genome instability generates the genetic diversity that accelerates the acquisition of tumorigenic capabilities.
DNA replication and repair are fundamental biological processes that ensure the accurate duplication of the genome and minimal errors within the genetic information. Previous studies have revealed that the stress caused by the perturbations of DNA replication, which is referred to as DRS, is induced in the earliest stages of cancer development. In precancerous lesions, DRS activates DNA damage checkpoints3, 4 and induces cellular senescence,5, 6 which becomes a barrier to malignant progression. In malignant tumors, DRS triggers genome instability and is associated with structural and numerical chromosomal instability that can generate intratumoral heterogeneity.7
Systemic cancer therapy is a prevalent method for the treatment of cancer patients. Numerous chemotherapeutic drugs have been approved for clinical use. In the last 2 decades, a number of molecular targeting drugs, which specifically target a protein and modulate its activity, have also been developed.8 These drugs have expanded our clinical options and improved therapeutic effects, often in combination with classical chemotherapeutic drugs. Although each chemotherapeutic drug has its own, although often unidentified, mechanism of action, there is a general rule that applies to every drug: it must target an essential biological process that is more important to the survival and proliferation of tumor cells than normal cells. DNA replication and repair appear to be one such essential process, because there are many chemotherapeutic drugs that are categorized as DNA damaging agents. In this review, we will focus on DRS in tumors and discuss mechanisms by which we can exploit DRS in tumors for developing therapeutics.
2. DRS IS A SOURCE OF GENOME INSTABILITY
DNA replication is a complex biochemical process that coordinates DNA replication origin licensing and firing (licensing and initiating factors), unwinding (helicases), and relaxation (topoisomerases) with the synthesis through the incorporation of dNTPs (polymerases) (Figure 1A). DNA replication has to be extremely accurate, as inaccurate DNA replication causes the accumulation of genetic mutations. However, the DNA replication process often faces various obstacles of both intracellular and extracellular origin, such as cellular dNTP starvation (Figure 1B), inhibition of DNA polymerase activity (Figure 1C), blockage of DNA unwinding by DNA cross‐links or inhibition of topoisomerases (Figure 1D), or blockage of DNA polymerase progression by DNA adducts or strand breaks (Figure 1E). These obstacles cause the slowing or stalling of replication fork progression and DNA synthesis while also triggering DRS.9
Figure 1.
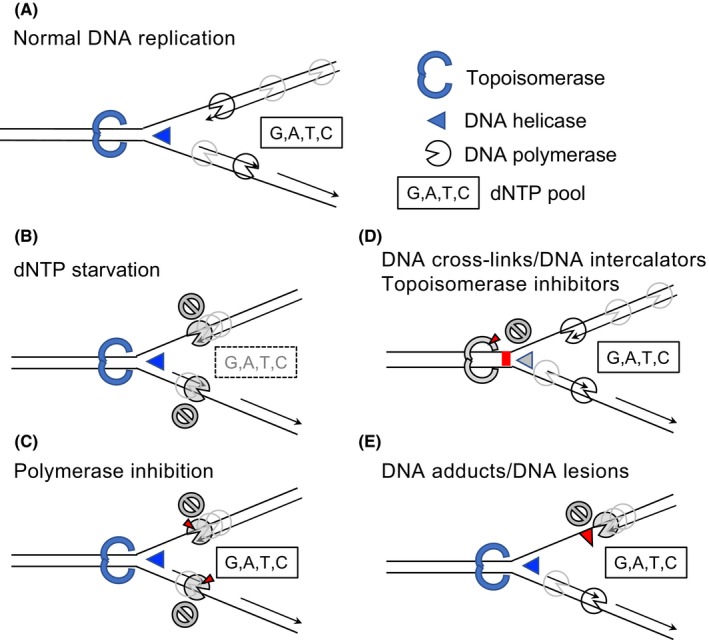
DNA replication stress. A, Replication fork during normal DNA replication process. Topoisomerases, helicases, DNA polymerases, and dNTP are necessary components for the accurate progression of DNA replication. B‐E, Multiple causes of DNA replication stress. B, dNTP starvation: eg, hydroxyurea induces dNTP starvation by inhibition of ribonucleotide reductase activity. C, Polymerase inhibition: eg, aphidicolin inhibits DNA polymerase activity. D, Inhibition of DNA unwinding by DNA cross‐links, DNA intercalators, or topoisomerase inhibitors. DNA lesions caused by the inhibitors in this category are often converted to DNA strand breaks. E, Inhibition of DNA polymerase progression by DNA adducts: eg, cyclobutane pyrimidine dimer produced by UV light exposure
When cells experience DRS, it becomes difficult to complete DNA replication during S phase. Normally, cells with under‐replicated genomic regions activate DNA damage checkpoints and cell cycle arrest before mitosis (Figure 2). If such cells proceed into mitosis, sister chromatids are interlinked as replication intermediates and can be visualized as UFBs during cytokinesis.10 When UFBs are properly processed by endonuclease Mus81, these under‐replicated genomic regions form visible gaps or breaks on condensed chromosomes during mitosis.11, 12 Thus, difficult‐to‐replicate regions in mammalian cells are defined as chromosomal fragile sites because these regions are prone to forming gaps and breaks, particularly following DRS.13 If these UFBs are not properly processed, they can cause unequal segregation of chromosomes, micronuclei formation, and genome instability.14 At an early step of tumorigenesis, oncogene activation by specific mutation, for example, G12V (glycine 12 to valine mutation) in K‐RAS, elicits a growth signal. DNA replication starts in response to the oncogene‐induced growth signal, even when replication materials, such as dNTPs, are not fully available.15 DNA replication under such conditions often causes DRS, which triggers cellular senescence or leads to genome instability and contributes to tumorigenesis.16
Figure 2.
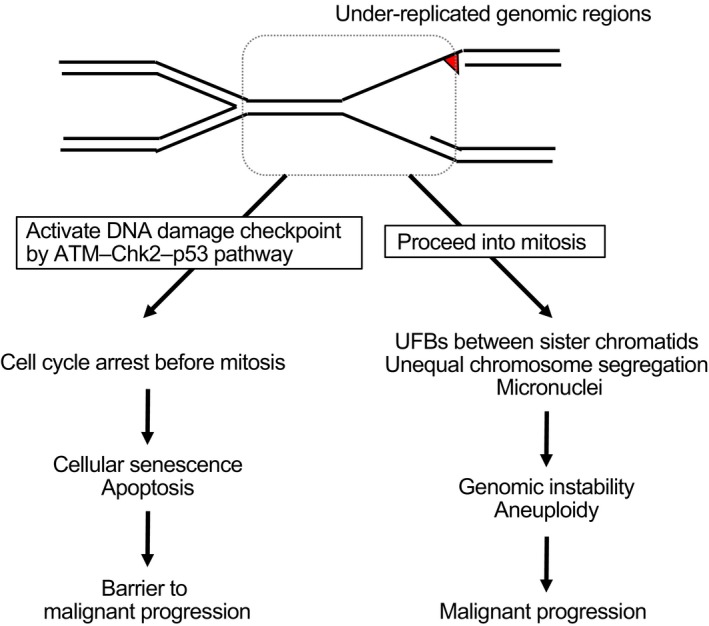
Destiny of cells that have experienced DNA replication stress. Cells with under‐replicated genomic regions, caused by DNA replication stress during S phase, either activate the DNA damage checkpoint before mitosis or proceed into mitosis. When the DNA damage checkpoint is properly activated, cells cease aberrant growth and induce cellular senescence or apoptosis. As the markers for the DNA damage checkpoint or cellular senescence are often detected in the precancerous lesions, this cascade is believed to constitute a barrier to malignant progression. In contrast, when the DNA damage checkpoint is abrogated, cells with under‐replicated genomic regions proceed into mitosis and induce chromosomal abnormalities, possibly caused by ultrafine bridges (UFBs). These cells display genomic instability, which contribute to malignant progression. ATM, ataxia telangiectasia mutated; Chk2, checkpoint kinase 2
3. DDR INDUCED BY DRS
To suppress the genome instability triggered by DRS, cells activate a DDR, including a DNA damage checkpoint, which coordinates cell cycle progression and DNA repair, dictates cellular senescence or apoptosis, and functions as an anticancer barrier in early human tumorigenesis.3, 4 In malignant tumors, DDR is often diminished by additional mutations in the components of DDR. The malignant tumor cells have overridden the anticancer barrier but survive in the presence of severe DRS.16 For these malignant tumors, chemotherapeutic drugs that induce further DRS are thought to be effective.17 In this section, we introduce several key signal transduction pathways of the DDR, a result of DRS, which are regulated by ATM and ATR kinases (Figure 3).
Figure 3.
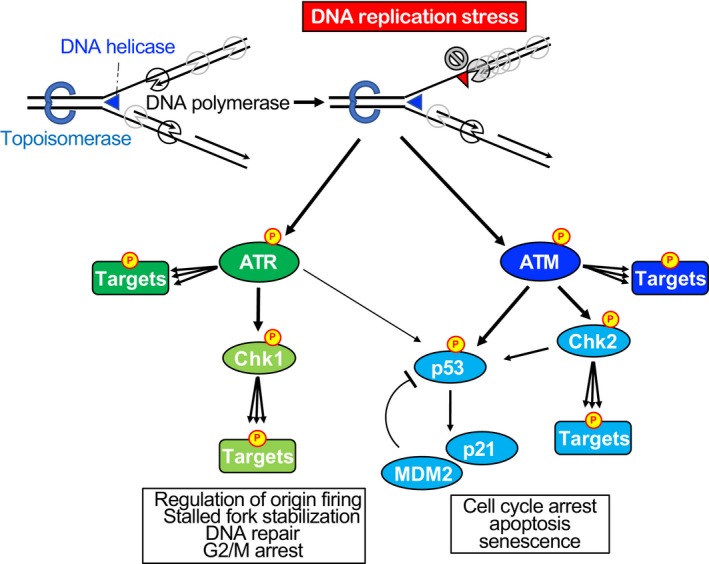
DNA damage response as a result of DNA replication stress. When DNA replication stress is induced, the DNA damage response, governed by the ataxia telangiectasia mutated (ATM) and ATM and Rad3‐related (ATR) kinases, is activated. ATR is activated at the region where single‐stranded DNA is exposed. ATM is also activated in response to DNA replication stress, but in a specific chromatin context. When DNA replication forks collapse and DNA double‐strand breaks are induced, ATM is robustly activated. Both ATM and ATR kinases phosphorylate (represented by P in yellow circle) hundreds of targets that are involved in spreading DNA damage signaling throughout the cell, involving checkpoints for cell cycle progression and activating DNA repair. Checkpoint kinases Chk1 and Chk2 are transducers of DNA damage signaling downstream of ATR and ATM kinase, respectively, and they also phosphorylate multiple targets. p53 is directly phosphorylated by ATM and ATR and functions as a mediator of the DNA damage response
3.1. ATR kinase is activated at stalled replication forks and phosphorylates hundreds of downstream substrates, including Chk1
Both ATM and ATR are phosphoinositide 3‐kinase‐related protein kinases that preferentially phosphorylate serine/threonine residues that are followed by glutamine (Ser/Thr‐Gln) in various downstream target proteins that promote cell cycle arrest or DNA repair.18 In particular, ATR is responsible for the DDR signal transduction pathway when DNA replication is perturbed.19 ATR kinase is mainly activated in replication protein A‐coated ssDNA regions by anchoring ATRIP.19 Optimal ATR activation required the Rad9‐Rad1‐Hus1 sliding clamp, which forms a heterotrimeric ring structure and is loaded at the boundary of the ssDNA and dsDNA regions,20 and DNA topoisomerase 2β‐binding protein 1, which contains 8 BRCA1 C‐terminal domains and an ATR‐activation domain that directly activates the ATR‐ATRIP complex in vitro.21
ATR kinase phosphorylates hundreds of downstream substrates.22, 23, 24, 25 Among these substrates, Chk1 is a key target serine/threonine kinase that spreads the DDR signals to the rest of the nucleus. ATR kinase phosphorylates Chk1 at Ser317 and Ser345, which appear to be a reliable indicator of Chk1 activation.26 Activated Chk1 phosphorylates and inhibits the Cdc25 phosphatases that regulate cell cycle transition at G2/M by removing the inhibitory phosphorylation of cyclin‐dependent kinases.27, 28, 29 The ATR‐Chk1 signaling pathway is also crucial for the checkpoint mechanisms during DNA replication, such as inhibition of origin firing30 and stabilization of stalled replication fork.31, 32
3.2. Activation of ATM and p53 occurs in response to DRS
p53 plays a central role in the response to DNA damage. Following DNA damage, p53 is stabilized and binds to DNA as a transcriptional regulator of genes involved in key cellular processes like DNA repair, cell cycle arrest, senescence, or apoptosis.33 Post‐translational modifications, such as phosphorylation and acetylation, are involved in the activation process of p53.34 Among these, the phosphorylation of p53 at Ser15 inhibits the physical interaction with MDM2, an E3 ligase that polyubiquitinates and degrades p53.35 DNA damage including DNA strand breaks induces the phosphorylation of p53 at Ser15, which is mainly mediated by ATM kinase.36, 37 ATR kinase‐mediated phosphorylation of p53 at Ser15 has also been reported.38
Both ATM and p53 can also be activated by DRS even in the absence of DNA damage. For example, p53 is activated during the process of oncogene‐induced cellular senescence,39 which is triggered by hyperproliferation signals from activated oncogenes and the resulting DRS.5 Consistent with this, chronic exposure to low and clinically relevant levels of DRS by exposure to low concentrations of replication inhibitors, such as hydroxyurea or aphidicolin, also induces p53‐dependent senescence‐like arrest in non‐transformed cells.40 However, the underlying mechanism of p53 activation by DRS in the absence of DNA damage has not yet been fully elucidated. Hypoxia, which occurs in a majority of solid tumors and is associated with tumor development and progression, also induces DRS and activates ATM and p53 in the absence of detectable DNA damage.41 In this case, severe hypoxia induces heterochromatin triggered by histone H3 Lys9 trimethylation as well as DRS. Co‐induction of heterochromatin and DRS activates ATM, and the active ATM kinase induces the phosphorylation of p53 at Ser15 even in the absence of detectable DNA damage.41 As p53 plays a critical role in the suppression of aberrant growth and in the induction of apoptosis, the fact that p53 can be activated under certain circumstances that solid tumors often encounter, even in the absence of detectable DNA damage, may explain why cells harboring p53 mutations are enriched during tumor development and progression.
4. CHEMOTHERAPEUTIC DRUGS THAT INDUCE AND MODULATE DRS
As indicated above, many chemotherapeutic drugs target DNA replication. As DNA replication is a complex process, each drug induces DRS in a different manner. In this section, we describe several examples of the induction and modulation of DRS that are triggered by chemotherapeutic drugs approved for the treatment of colorectal cancer, 5‐FU, oxaliplatin, irinotecan (an analog of CPT) and TFTD (TAS‐102) (Table 1).
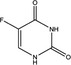
Table 1.
The chemical structures and characteristics of key components or related compounds in chemotherapeutic drugs currently approved for the treatment of metastatic colorectal cancer patients
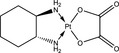
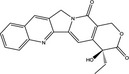
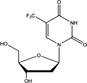
| Nomenclature | 5‐Fluorouracil (5‐FU) | Oxaliplatin | Camptothecin (CPT) | Trifluridine (FTD) |
|---|---|---|---|---|
| Chemical structural formula |
|
|
|
|
| Compositional formula | C4H3FN2O2 = 130.08 | C8H14N2O4Pt = 397.29 | C20H16N2O4 = 348.36 | C10H11F3N2O5 = 296.20 |
| Characteristics | Uracil analogue | DNA crosslinker | Topoisomerase I (Topo I) inhibitor | Nucleoside analogue. A key component of trifluridine/tipiracil (TFTD/TAS‐102) |
| RNA incorporation | Cell cycle arrest at G1 phase via p53‐dependent miR‐34a upregulation | Topo I‐cc (covalent complex) becomes obstacle of replication and triggers 1‐ended dsDNA breaks | DNA incorporation | |
| dTTP starvation via thymidylate synthase inhibition by fluorodeoxyuridine monophosphate | Irinotecan is an analogue of CPT | Cellular senescence‐like growth arrest dependent on p53 |
4.1. 5‐FU compromises DNA and RNA metabolism and exerts its cytotoxicity during DNA replication
Antimetabolites are chemical compounds that compromise normal metabolism. Such compounds are often similar in structure to the metabolites that they interfere with. 5‐FU is the most widely used antimetabolite‐type chemotherapeutic drug. 5‐FU is structurally similar to uracil and invades the uracil metabolic pathway. For the most part, 5‐FU is converted to fluorouridine phosphate and incorporated into RNA.42 5‐FU is also converted to fluorodeoxyuridine phosphate and fluorodeoxyuridine monophosphate, which directly binds and strongly inhibits thymidylate synthase and suppresses de novo thymidylate and dTTP synthesis.42 The effect on the dTTP biosynthesis cascade by 5‐FU triggers DNA damage and subsequent DNA repair, but the cytotoxic effects of 5‐FU are thought to depend mainly on RNA incorporation.43 From our genetic screening using the chicken DT40 cell line, cells deficient in the ATR‐Chk1 signaling pathway and homologous recombinational repair showed severe 5‐FU sensitivity,44 which indicates that 5‐FU exerts cytotoxicity during DNA replication. Thus, 5‐FU affects both DNA and RNA metabolism at the same time and exerts its cytotoxicity in a mixed fashion.
4.2. Oxaliplatin forms unique platinum‐DNA adducts that activate p53 and repress the expression of proteins involved in DNA replication and G2/M progression
The platinum‐type chemotherapeutic drugs preferentially cross‐link 2 closely positioned purine bases. Cisplatin and carboplatin form the same platinum‐DNA adducts with cis‐diammine carrier ligands, whereas oxaliplatin forms a different type of platinum‐DNA adduct with a 1,2‐diaminocyclohexane carrier ligand.45 Possibly reflecting this difference, oxaliplatin suppresses cell growth in a different manner from cisplatin. Oxaliplatin, but not cisplatin, represses the expression of proteins involved in DNA replication and G2/M progression, Cyclin A and Cyclin B, in a p53‐dependent manner.46 Intriguingly, oxaliplatin robustly activates p53 and leads to accumulation of p21 in spite of low levels of activation of ATM and ATR.47 This finding may be consistent with a recent report indicating that oxaliplatin kills cells by inducing ribosome biogenesis stress, but not through the DDR pathway.48 The p53 activation by oxaliplatin causes the transcriptional repression of deoxyuridine triphosphatase, which catalyzes the hydrolysis of dUTP to dUMP and inhibits dUTP‐mediated cytotoxicity,49 as well as the enzymes involved in thymidylate biosynthesis, such as dihydrofolate reductase, thymidine kinase 1, and thymidylate synthase, through miR‐34a‐mediated E2F1 and E2F3 downregulation.47 These effects may elucidate the clinical synergy observed between oxaliplatin and 5‐FU in FOLFOX, a combination regimen of 5‐FU, leucovorin and oxaliplatin, which is superior in the treatment of advanced colorectal cancer patients50 or for adjuvant therapy for colorectal cancer patients who received curable surgery.51
4.3. CPT, a Topo I inhibitor, produces 1‐ended dsDNA breaks during DNA replication and induces ATM‐ and ATR‐dependent cellular response
Topoisomerase is an enzyme that catalyzes the breaking and rejoining of the phosphodiester backbone of DNA strands during the normal cell cycle.52 There are 2 types of topoisomerases: Topo I, which cuts 1 of the 2 strands of dsDNA and relaxes/reanneals the strand, and Topo II, which cuts both strands of dsDNA simultaneously and manages DNA tangles. Topoisomerase inhibitors are chemical compounds that block the action of these topoisomerases and often work as a chemotherapeutic drug.
CPT is a cytotoxic alkaloid that works as a Topo I inhibitor. Irinotecan is a CPT analog that has been approved as a chemotherapeutic drug. CPT binds to Topo I and forms a covalent ternary complex with DNA, called the Topo I‐cc. This complex prevents DNA religation and causes DNA strand breaks. CPT is selectively cytotoxic to the cells that actively replicate their DNA in S phase. This cytotoxicity is a result of the conversion of ssDNA breaks into 1‐ended dsDNA breaks when the replication fork encounters and collides with Topo I‐cc.53 Distinct from the 2‐ended dsDNA breaks formed by ionizing radiation, 1‐ended dsDNA breaks are extremely cytotoxic, possibly because these breaks cannot be repaired by the non‐homologous end‐joining process.54 The Topo I‐cc‐mediated DNA ends induce both ATM and ATR activation and must be properly resected to expose the ssDNA region for repair by homologous recombination. Accordingly, CPT induces hyperphosphorylation of FANCJ, a DNA helicase that participates in DNA replication checkpoint control55, 56 and contributes to suppress microsatellite instability,57 in an end‐resection and ATR‐dependent manner.58 Thus, this CPT‐induced hyperphosphorylation of FANCJ may have certain roles in replication‐coupled DNA repair processes, such as homologous recombination.
4.4. FTD, a chemotherapeutic nucleoside analog, activates DDR and exerts cytotoxicity through incorporation into DNA during DNA replication
FTD, a key cytotoxic component of the novel, orally administrated, nucleoside analog‐type anticancer drug TFTD (TAS‐102), has a unique characteristic. Inside the cell, FTD is activated by phosphorylation and incorporated into DNA during replication process.59, 60, 61 FTD also suppresses dTTP biosynthesis,62 which enhances FTD incorporation into DNA and thus its cytotoxicity. In contrast to other nucleoside analog‐type anticancer drugs like cytarabine or gemcitabine, FTD does not appear to stop elongation of the nascent strand of DNA and DNA synthesis continues. Therefore, FTD is incorporated at a high level into DNA in a time‐dependent manner.59, 61 However, FTD incorporation was not associated with any measurable DNA strand breaks in p53‐proficient human colorectal cancer cell line HCT‐116.61 Intriguingly, FTD activated the DDR associated with DNA replication, which was indicated by Chk1 Ser345 phosphorylation and the robust accumulation of p53 and p21, which were associated with Chk2 Thr68 phosphorylation.61 FTD induced sustained arrest at the G2 phase by downregulating cyclin B1/Cdk1 expression,61 which is also observed when cellular senescence is induced.63 Although the mechanism underlying the induction of DDR by FTD is currently unclear, FTD appears to exert its cytotoxicity in a unique fashion during replication.
5. CONCLUSION
DNA replication is an essential process for cell proliferation; at the same time, it is a fragile system and susceptible to the effects of intrinsic or exogenous environmental factors. Here, we described the underlying mechanism by which DRS is induced at the early stages of cancer development, triggers genome instability, and promotes tumor progression. As induction of DRS appears to be a common event during tumorigenesis, most malignant tumor cells are thought to survive in the presence of severe DRS. Exploiting DRS is a prospective strategy for cancer therapy.17 To develop novel cancer therapeutic regimens that maximize antitumor effects and minimize adverse effects on normal tissues, it will be important to: (i) comprehend the extent of DRS in tumors; (ii) uncover the underlying molecular mechanism of DRS elicited by each drug; and (iii) find optimal ways of modulating or combining chemotherapeutic drugs that enhance cytotoxicity exclusively in tumors.
Molecular targeting drugs that modulate DDR in response to DRS are expected to have antitumor effects. Indeed, several kinase inhibitors against ATR or Chk1 are currently under investigation in clinical trials as anticancer drugs.64 We anticipate that even low doses of these drugs would significantly enhance the anticancer effect of chemotherapeutic drugs that trigger DRS. In addition, genomic instability or hypermutability induced by DRS caused by an intrinsic trait (eg, mismatch‐repair deficiency) or by a chemotherapeutic drug, may enhance tumor susceptibility to therapies like immunocheckpoint blockers.65 Further understanding of DRS in tumors as well as development of novel therapeutic options will pave the way to proceed into the next generation of cancer therapy.
DISCLOSURE STATEMENT
Y.K. and T.W. are employees of Taiho Pharmaceutical Co. Ltd. H.K. and M.I. are staff members of the Joint Research Department funded by Taiho Pharmaceutical Co. Ltd. at Kyushu University. Y.M. reports receiving commercial research grants from Taiho Pharmaceutical Co. Ltd., Yakult Honsha Co. Ltd., Chugai Pharmaceutical Co. Ltd., and Ono Pharmaceutical Co. Ltd. E.O. and Y.M. report receiving honoraria from Taiho Pharmaceutical Co. Ltd. The other authors have no conflict of interest.
ACKNOWLEDGMENT
This study was supported in part by grants‐in‐aid from the Ministry of Education, Culture, Sports, Science, and Technology of Japan (to H. Kitao, Japan Society for the Promotion of Science KAKENHI grant no. 17H03598).
Kitao H, Iimori M, Kataoka Y, et al. DNA replication stress and cancer chemotherapy. Cancer Sci. 2018;109:264–271. https://doi.org/10.1111/cas.13455
Funding information
Ministry of Education, Culture, Sports, Science, and Technology of Japan.
REFERENCES
- 1. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57‐70. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646‐674. [DOI] [PubMed] [Google Scholar]
- 3. Bartkova J, Horejsi Z, Koed K, et al. DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature. 2005;434:864‐870. [DOI] [PubMed] [Google Scholar]
- 4. Gorgoulis VG, Vassiliou LV, Karakaidos P, et al. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 2005;434:907‐913. [DOI] [PubMed] [Google Scholar]
- 5. Di Micco R, Fumagalli M, Cicalese A, et al. Oncogene‐induced senescence is a DNA damage response triggered by DNA hyper‐replication. Nature. 2006;444:638‐642. [DOI] [PubMed] [Google Scholar]
- 6. Bartkova J, Rezaei N, Liontos M, et al. Oncogene‐induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633‐637. [DOI] [PubMed] [Google Scholar]
- 7. Burrell RA, McClelland SE, Endesfelder D, et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492‐496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dobbelstein M, Moll U. Targeting tumour‐supportive cellular machineries in anticancer drug development. Nat Rev Drug Discov. 2014;13:179‐196. [DOI] [PubMed] [Google Scholar]
- 9. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol. 2014;16:2‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chan KL, Palmai‐Pallag T, Ying S, Hickson ID. Replication stress induces sister‐chromatid bridging at fragile site loci in mitosis. Nat Cell Biol. 2009;11:753‐760. [DOI] [PubMed] [Google Scholar]
- 11. Ying S, Minocherhomji S, Chan KL, et al. MUS81 promotes common fragile site expression. Nat Cell Biol. 2013;15:1001‐1007. [DOI] [PubMed] [Google Scholar]
- 12. Di Marco S, Hasanova Z, Kanagaraj R, et al. RECQ5 helicase cooperates with MUS81 endonuclease in processing stalled replication forks at common fragile sites during mitosis. Mol Cell. 2017;66:658‐671. e8. [DOI] [PubMed] [Google Scholar]
- 13. Bhowmick R, Hickson ID. The “enemies within”: regions of the genome that are inherently difficult to replicate. F1000Res. 2017;6:666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mankouri HW, Huttner D, Hickson ID. How unfinished business from S‐phase affects mitosis and beyond. EMBO J. 2013;32:2661‐2671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bester AC, Roniger M, Oren YS, et al. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gaillard H, Garcia‐Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276‐289. [DOI] [PubMed] [Google Scholar]
- 17. Dobbelstein M, Sorensen CS. Exploiting replicative stress to treat cancer. Nat Rev Drug Discov. 2015;14:405‐423. [DOI] [PubMed] [Google Scholar]
- 18. Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177‐2196. [DOI] [PubMed] [Google Scholar]
- 19. Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol. 2008;9:616‐627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parrilla‐Castellar ER, Arlander SJ, Karnitz L. Dial 9‐1‐1 for DNA damage: the Rad9‐Hus1‐Rad1 (9‐1‐1) clamp complex. DNA Repair (Amst). 2004;3:1009‐1014. [DOI] [PubMed] [Google Scholar]
- 21. Kumagai A, Lee J, Yoo HY, Dunphy WG. TopBP1 activates the ATR‐ATRIP complex. Cell. 2006;124:943‐955. [DOI] [PubMed] [Google Scholar]
- 22. Matsuoka S, Ballif BA, Smogorzewska A, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160‐1166. [DOI] [PubMed] [Google Scholar]
- 23. Mu JJ, Wang Y, Luo H, et al. A proteomic analysis of ataxia telangiectasia‐mutated (ATM)/ATM‐Rad3‐related (ATR) substrates identifies the ubiquitin‐proteasome system as a regulator for DNA damage checkpoints. J Biol Chem. 2007;282:17330‐17334. [DOI] [PubMed] [Google Scholar]
- 24. Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome‐wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci U S A. 2007;104:10364‐10369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Stokes MP, Rush J, Macneill J, et al. Profiling of UV‐induced ATM/ATR signaling pathways. Proc Natl Acad Sci U S A. 2007;104:19855‐19860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu Q, Guntuku S, Cui XS, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev. 2000;14:1448‐1459. [PMC free article] [PubMed] [Google Scholar]
- 27. Sanchez Y, Wong C, Thoma RS, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277:1497‐1501. [DOI] [PubMed] [Google Scholar]
- 28. Peng CY, Graves PR, Thoma RS, Wu Z, Shaw AS, Piwnica‐Worms H. Mitotic and G2 checkpoint control: regulation of 14‐3‐3 protein binding by phosphorylation of Cdc25C on serine‐216. Science. 1997;277:1501‐1505. [DOI] [PubMed] [Google Scholar]
- 29. Furnari B, Rhind N, Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495‐1497. [DOI] [PubMed] [Google Scholar]
- 30. Maya‐Mendoza A, Petermann E, Gillespie DA, Caldecott KW, Jackson DA. Chk1 regulates the density of active replication origins during the vertebrate S phase. EMBO J. 2007;26:2719‐2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553‐557. [DOI] [PubMed] [Google Scholar]
- 32. Feijoo C, Hall‐Jackson C, Wu R, et al. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra‐S phase checkpoint monitoring replication origin firing. J Cell Biol. 2001;154:913‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53‐regulated genes. Nat Rev Mol Cell Biol. 2008;9:402‐412. [DOI] [PubMed] [Google Scholar]
- 34. Meek DW, Anderson CW. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb Perspect Biol. 2009;1:a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:296‐299. [DOI] [PubMed] [Google Scholar]
- 36. Banin S, Moyal L, Shieh S, et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998;281:1674‐1677. [DOI] [PubMed] [Google Scholar]
- 37. Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998;281:1677‐1679. [DOI] [PubMed] [Google Scholar]
- 38. Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage‐induced phosphorylation of p53. Genes Dev. 1999;13:152‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593‐602. [DOI] [PubMed] [Google Scholar]
- 40. Marusyk A, Wheeler LJ, Mathews CK, DeGregori J. p53 mediates senescence‐like arrest induced by chronic replicational stress. Mol Cell Biol. 2007;27:5336‐5351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Olcina MM, Foskolou IP, Anbalagan S, et al. Replication stress and chromatin context link ATM activation to a role in DNA replication. Mol Cell. 2013;52:758‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Longley DB, Harkin DP, Johnston PG. 5‐fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer. 2003;3:330‐338. [DOI] [PubMed] [Google Scholar]
- 43. Pettersen HS, Visnes T, Vagbo CB, et al. UNG‐initiated base excision repair is the major repair route for 5‐fluorouracil in DNA, but 5‐fluorouracil cytotoxicity depends mainly on RNA incorporation. Nucleic Acids Res. 2011;39:8430‐8444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fujinaka Y, Matsuoka K, Iimori M, et al. ATR‐Chk1 signaling pathway and homologous recombinational repair protect cells from 5‐fluorouracil cytotoxicity. DNA Repair (Amst). 2012;11:247‐258. [DOI] [PubMed] [Google Scholar]
- 45. Chaney SG, Campbell SL, Bassett E, Wu Y. Recognition and processing of cisplatin‐ and oxaliplatin‐DNA adducts. Crit Rev Oncol Hematol. 2005;53:3‐11. [DOI] [PubMed] [Google Scholar]
- 46. Voland C, Bord A, Peleraux A, et al. Repression of cell cycle‐related proteins by oxaliplatin but not cisplatin in human colon cancer cells. Mol Cancer Ther. 2006;5:2149‐2157. [DOI] [PubMed] [Google Scholar]
- 47. Kiyonari S, Iimori M, Matsuoka K, et al. The 1,2‐diaminocyclohexane carrier ligand in oxaliplatin induces p53‐dependent transcriptional repression of factors involved in thymidylate biosynthesis. Mol Cancer Ther. 2015;14:2332‐2342. [DOI] [PubMed] [Google Scholar]
- 48. Bruno PM, Liu Y, Park GY, et al. A subset of platinum‐containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med. 2017;23:461‐471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wilson PM, Fazzone W, LaBonte MJ, Lenz HJ, Ladner RD. Regulation of human dUTPase gene expression and p53‐mediated transcriptional repression in response to oxaliplatin‐induced DNA damage. Nucleic Acids Res. 2009;37:78‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. de Gramont A, Figer A, Seymour M, et al. Leucovorin and fluorouracil with or without oxaliplatin as first‐line treatment in advanced colorectal cancer. J Clin Oncol. 2000;18:2938‐2947. [DOI] [PubMed] [Google Scholar]
- 51. Andre T, Boni C, Mounedji‐Boudiaf L, et al. Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl J Med. 2004;350:2343‐2351. [DOI] [PubMed] [Google Scholar]
- 52. Wang JC. Cellular roles of DNA topoisomerases: a molecular perspective. Nat Rev Mol Cell Biol. 2002;3:430‐440. [DOI] [PubMed] [Google Scholar]
- 53. Pommier Y, Redon C, Rao VA, et al. Repair of and checkpoint response to topoisomerase I‐mediated DNA damage. Mutat Res. 2003;532:173‐203. [DOI] [PubMed] [Google Scholar]
- 54. Sakasai R, Iwabuchi K. The distinctive cellular responses to DNA strand breaks caused by a DNA topoisomerase I poison in conjunction with DNA replication and RNA transcription. Genes Genet Syst. 2016;90:187‐194. [DOI] [PubMed] [Google Scholar]
- 55. Gong Z, Kim JE, Leung CC, Glover JN, Chen J. BACH1/FANCJ acts with TopBP1 and participates early in DNA replication checkpoint control. Mol Cell. 2010;37:438‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1‐containing complexes. Genes Dev. 2006;20:34‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Matsuzaki K, Borel V, Adelman CA, Schindler D, Boulton SJ. FANCJ suppresses microsatellite instability and lymphomagenesis independent of the Fanconi anemia pathway. Genes Dev. 2015;29:2532‐2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sakasai R, Sakai A, Iimori M, et al. CtIP‐ and ATR‐dependent FANCJ phosphorylation in response to DNA strand breaks mediated by DNA replication. Genes Cells. 2012;17:962‐970. [DOI] [PubMed] [Google Scholar]
- 59. Emura T, Nakagawa F, Fujioka A, et al. An optimal dosing schedule for a novel combination antimetabolite, TAS‐102, based on its intracellular metabolism and its incorporation into DNA. Int J Mol Med. 2004;13:249‐255. [PubMed] [Google Scholar]
- 60. Emura T, Suzuki N, Yamaguchi M, Ohshimo H, Fukushima M. A novel combination antimetabolite, TAS‐102, exhibits antitumor activity in FU‐resistant human cancer cells through a mechanism involving FTD incorporation in DNA. Int J Oncol. 2004;25:571‐578. [PubMed] [Google Scholar]
- 61. Matsuoka K, Iimori M, Niimi S, et al. Trifluridine Induces p53‐Dependent Sustained G2 Phase Arrest with Its Massive Misincorporation into DNA and Few DNA Strand Breaks. Mol Cancer Ther. 2015;14:1004‐1013. [DOI] [PubMed] [Google Scholar]
- 62. Tanaka N, Sakamoto K, Okabe H, et al. Repeated oral dosing of TAS‐102 confers high trifluridine incorporation into DNA and sustained antitumor activity in mouse models. Oncol Rep. 2014;32:2319‐2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Johmura Y, Shimada M, Misaki T, et al. Necessary and sufficient role for a mitosis skip in senescence induction. Mol Cell. 2014;55:73‐84. [DOI] [PubMed] [Google Scholar]
- 64. Zhang J, Dai Q, Park D, Deng X. Targeting DNA replication stress for cancer therapy. Genes. 2016;7:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Le DT, Uram JN, Wang H, et al. PD‐1 blockade in tumors with mismatch‐repair deficiency. N Engl J Med. 2015;372:2509‐2520. [DOI] [PMC free article] [PubMed] [Google Scholar]