

Abstract
Interleukin 15 (IL15) and IL7 are two cytokines essential for T cell development and homeostasis. In order to improve the antitumor activity by Newcastle disease virus (NDV)‐modified tumor vaccine, we generated a recombinant NDV co‐expressing IL15 and IL7 (LX/IL(15+7)) through incorporation of a 2A self‐processing peptide into IL15 and IL7 using reverse genetics. B16 cells infected with LX/IL(15+7) expressed both IL15 and IL7 stably. The cytotoxicity assay showed that murine melanoma cells modified with LX/IL(15+7) could significantly enhance the antitumor immune response in vitro. Then, the antitumor effects of tumor vaccine modified with recombinant virus were tested in the murine tumor models. We observed strong antitumor responses induced by LX/IL(15+7)‐modified tumor cells both in prophylaxis and therapeutic models. Although the tumor‐infiltrating CD4+ T cells and CD8+ T cells were both increased, the antitumor activity of the tumor vaccine modified with LX/IL(15+7) was dependent on CD8+ T cells. Taken together, our data strongly indicated that tumor vaccine modified with NDV strain LX/IL(15+7) is a promising agent for cancer immunotherapy.
Keywords: interleukin‐15, Interleukin‐7, Newcastle disease virus, tumor, vaccine
1. INTRODUCTION
Newcastle disease virus (NDV), a member of the genus Avulavirus, has a non‐segmented, single‐stranded, negative‐sense RNA that contains 6 structural genes in the following order (3′‐5′): nucleoprotein (NP), phosphoprotein (P), matrix protein (M), fusion protein (F), hemagglutinin‐neuraminidase (HN) and large polymerase protein (L).1, 2, 3 NDV has been considered as an effective and safe agent for cancer therapy for more than 50 years.4, 5, 6 NDV can selectively replicate in tumor cells, as most of these cells are defective in interfering pathways.7, 8 Therefore, NDV has been used clinically as an oncolytic agent against many types of cancer.9, 10, 11
Newcastle disease virus has been used to treat cancer in at least the following three ways: (i) lysing tumor cells directly;9 (ii) modifying tumor cells as the vaccine to stimulate antitumor immune responses;12, 13 and as a vector to deliver therapeutic genes.14 Whole‐cell autologous tumor vaccines modified with NDV (ATV‐NDV) are made from nonlytic NDV infected tumor cells, which have been γ‐ray irradiated.15 Clinical trials have demonstrated the antitumor effects of ATV‐NDV in many kinds of cancer. With the development of recombinant DNA technology, many recombinant NDV strains have been successfully generated from cloned cDNA, and most recombinant viruses expressing immune‐modulate genes have exhibited an enhancing antitumor response.
Interleukin 7, one of γ‐chain cytokines, plays a critical role in lymphocyte development and homeostasis of naïve and memory CD8+ T cells.16 IL15, another γ‐chain cytokine, is dedicated to the prolonged maintenance of memory T cell responses, which makes IL15 an attractive candidate for adjuvant immunotherapy.17, 18, 19 IL7 and IL15 support the generation of long‐living memory stem T cells, which could differentiate into potent effectors not only able to efficiently target cancer cells, but also capable of persisting and mediating a dynamic cancer immunosurveillance.20 Therefore, ATV‐NDV co‐expressing IL7 and IL15 may enhance T cell responses and achieve better therapeutic efficacy against tumors.
Foot‐and‐mouth disease virus (FMDV) 2A, a self‐processing peptide from FMDV, has become a useful approach for the expression of multiple proteins under the control of a single promoter.
Previously, we constructed several recombinant viruses using a reverse genetic platform of nonlytic NDV strain LX.13 In this study, we generated recombinant LX strain (LX/IL(15+7)) co‐expressing IL7 and IL15 through incorporation of a 2A self‐processing peptide between the above genes. This recombinant LX/IL(15+7) expressed two heterogenous genes simultaneously. Tumor cells modified with LX/IL(15+7) exhibited increasing preventive and therapeutic effects of cancer both in vitro and in vivo.
2. MATERIALS AND METHODS
2.1. Animals
Pathogen‐free 6‐week‐old female C57BL/6 mice were obtained from SLC (Shanghai, China). Animal care and experimental procedures were performed under specific‐pathogen‐free (SPF) conditions. All animal protocols were approved by the Institutional Laboratory Animal Care and Use Committee at Soochow University.
2.2. Cells and viruses
Newcastle disease virus strain, LX/RFP (recombinant NDV LX strain expressing red fluorescent protein) and LX/IL(15+7) (recombinant LX strain co‐expressing IL7 and IL15) were grown in 10‐day‐old embryonated SPF eggs. The chicken fibroblast cell line (DF‐1) and murine melanoma cell line (B16‐F10) obtained from the American Type Culture Collection (Manassas, VA, USA) were maintained in high‐glucose DMEM supplemented with 10% FBS, penicillin (100 U/mL) and streptomycin (100 mg/mL). Cells were incubated at 37°C in a tissue culture incubator equilibrated with 5% CO2.
2.3. Construction of recombinant Newcastle disease virus co‐expressing interleukin 7 and interleukin 15
To construct pLX/IL(15+7) (based on the NDV LX strain), a 1062 bpcDNA containing IL15‐2A‐1L7 (murine IL15 and IL7 gene separated by 2A‐coding sequence) flanked by NDV gene end (GE), intergenic (IG), gene start (GS) sequences (Figure 1) were constructed by fusion PCR with four pairs of primers (Table 1).
Figure 1.
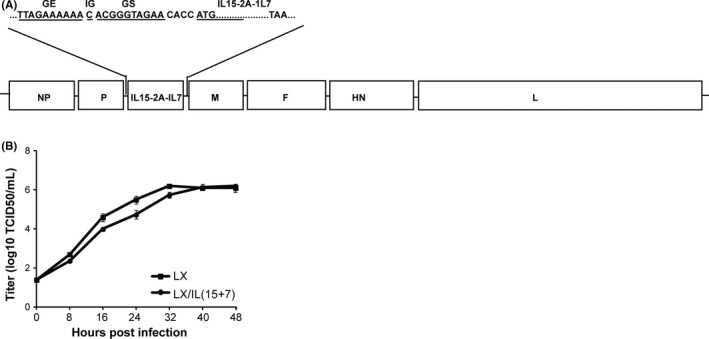
Construction of recombinant virus. A, Schematic representation of recombinant LX construct. The coding sequence interleukin 15 (IL15) and IL7 was inserted into the sequence between P and M genes of LX genome. B, Multistep growth curve for LX and LX/IL(15+7). The virus titers were determined in triplicate by TCID50 in DF‐1 cells at 0, 8, 16, 24, 32 and 40 h post‐infection. The data shown are representative of the 3 experiments
Table 1.
Primers used for the generation of recombinant virus
| Primer | Sequences (5′‐3′) |
|---|---|
| PU | GGGCATGATGAAGATCCTGGACCCTGGTTGTG |
| PD | GGTGGCGTTCTACCCGTGTTTTTTCTAAGAGGAGCTTGGTGCAGATACC‐GTG |
| 15U | TTAGAAAAAACACGGGTAGAACGCCACCATGAAAATTTTGAAACCATA |
| 15R | CCTGCCAACTTGAGCAGGTCAAAGTTCAAAAGCTGTTTCACAGGCGCTC‐TCCTCTTTCTGGACGTGTTGATGAACATTTG |
| 7F | GAACTTTGACCTGCTCAAGTTGGCAGGGGACGTCGAGTCCAACCCTGGGCCTATGTTCCATGTTTCTTTTAGATATATC |
| 7D | TGTTGGACCTTGGGTTTGCAGTTATATA CTGCCCTTCAAAATTTTATTC |
| MU | TGACTGCAAACCCAAGGTCCAAC |
| MD | GCCCTCTACACCGTTGATGTTCAAAGAGC |
The resultant PCR product was digested with AgeI and AvrII and inserted into the AgeI‐to‐AvrII window of a subclone, nucleotides 2311‐8157 in the antigenomic cDNA of NDV strain LX. This subclone encompassed the downstream sequence of the P gene, and the upstream sequence of the M gene. The murine IL7‐coding sequence was inserted into nucleotide position 3143 in the antigenomic cDNA because it allowed flanking by a set of GE, IG and GS. This subclone was digested with Sac II and SpeI, and then assembled into the full‐length antigenomic cDNA to result in the full‐length clone pLX/IL(15+7).
The recombinant virus LX/IL(15+7) was rescued by cotransfecting BSR T7/5 cells with pLX/IL(15+7) and support plasmids (N, P and L) from ZJ1 strain as previously described.21 Then, the supernatant from the transfected BSR T7/5 cells was collected and inoculated into allantoic cavities of 10‐day‐old embryonated SPF eggs. 96 hour later, the allantoic fluid was screened for the virus production by hemagglutination assay (HA) using 0.5% chicken red blood cells. The recombinant viruses were also determined by sequencing for the correct insertion of the heterogenous genes.
2.4. Growth kinetics of the virus
The growth kinetics of the recombinant viruses was determined by a multistep growth assay in DF‐1 cells. Briefly, DF‐1 cells were inoculated with 10‐fold serial diluted viruses at 37°C.
The medium contained 10% uninfected egg allantoic fluid as a source of exogenous protease. The supernatant was collected at 8‐hour intervals and viral titers were determined by limiting dilution on DF‐1 cells.
2.5. Expression of interleukin 7 and interleukin 15 in tumor cells infected with recombinant virus
For the present study, 1 × 106 B16‐F10 cells irradiated with 200 Gy via a 60Go source were infected with 100 hemagglutination units (HAU) of NDV for 1 hour at 37°C. Uninfected viruses were removed by replacing supernatant with equal volume of fresh medium after centrifugation at 350 g for 5 minutes (ms). Then, the supernatants were harvested at 24, 48 and 72 hour post‐infection. The expressions of IL7 and IL15 in the supernatants were measured using ELISA as described by mouse IL7 DuoSet ELISA and IL15 DuoSet ELISA (R&D Systems, Minneapolis, MN USA), respectively.
As for the cleavage efficiency of the two heterogenous genes mediated by 2A, the fusion form protein of IL15‐2A‐IL7 was also measured. Briefly, the fusion proteins in the supernatants were captured using IL15 antibody‐coated plates from IL15 DuoSet ELISA, and the amount of fusion protein was determined using the detecting antibody from IL7 DuoSet ELISA.
2.6. Production of tumor vaccine modified with Newcastle disease virus
B16 cells irradiated with 200 Gy through a 60Go source were infected with recombinant NDV strains at a ratio of 100 HAU per 106 cells as described previously.13
2.7. Cytotoxicity assays
Cytotoxicities of splenocytes were measured using the CytoTox 96 nonradioactive cytotoxicity assay (Promega, Madison, WI, USA). Briefly, B16 (5.5 × 103 cells/well) or EL‐4 (2.5 × 103 cells/well) were plated on 96‐well U‐bottom plates (Corning Costar, Cambridge, MA, USA) as target. The splenocytes (effector) were added to the plates at a total volume of 100 μL in E:T ratios of 100:1, 50:1 and 25:1, respectively. The plates were then incubated for 6 hour at 37°C in a humidified 5% CO2 chamber. After centrifugation, 50 μL supernatants were transferred to fresh 96‐well flat‐bottom plates, and 50 μL substrate mix was added. Next, 50 μL stop solution was added into each well 30 minutes later, and the absorbance was measured at 492 nm. The cytotoxicities of slenocytes at each E:T ratio were calculated using the following formula: (A492 nm[experimental] − A492 nm[effector spontaneous] − A492 nm[target spontaneous]) × 100/(A492 nm[target maximum] − A492 nm[target spontaneous]).
2.8. Prophylaxis experiments
Mice were immunized subcutaneously (s.c.) with a 25G5/8 needle at the left flank with 1 × 106 irradiated B16‐F10 cells, 1 × 106 irradiated B16‐F10 cells modified with LX/RFP or 1 × 106 irradiated B16‐F10 cells modified with LX/IL(15+7) twice with 1‐week (wk) intervals. Two weeks after the last immunization, mice were injected s.c. with 1 × 105 B16‐F10 or EL‐4 cells at the right flank. The tumor sizes were monitored daily from day 5 post‐tumor implant. Tumor volumes were calculated using the formula V = 1/2 (L × W 2), where L is the length (longest dimension) and W is the width (shortest dimension).
2.9. Therapeutic experiments
Mice were inoculated s.c. with 5 × 104 B16‐F10 cells at the right flank with a 25G5/8 needle. The tumor vaccines were injected s.c. into the left flank at days 5 and 9 post‐tumor inoculation. The tumor sizes were measured daily from day 5 post‐challenge.
2.10. In vivo depletion of CD4+ or CD8+ cells
CD4+ or CD8+ cells depletion were performed by i.p. injections of 1 mg GK1.5 (rat anti‐mouse CD4 mAb) or 500 μg 53‐6.7 (rat anti‐mouse CD8 mAb) 2 days before the first immunization in each mouse, and the injections were repeated 7 days later.
2.11. Flow cytometry
Antibodies (Abs) against murine antigens (Ags) were purchased from BD Biosciences (Franklin Lakes, NJ, USA): FITC‐conjugated CD4 (RM4‐5), FITC‐conjugated NK1.1 (PK136), PE‐conjugated anti‐CD3ε (145‐2C11), PerCP Cy5.5–conjugated Gr‐1 (RB6‐8C5), allophycocyanin‐conjugated CD11b (M1/70) and PerCP Cy5.5–conjugated CD8a (53‐6.7). All stainings were performed in FACS buffer (1 ×PBS, 1% BSA and 0.1% NaN3) in the presence of purified anti‐CD16/32 at the saturation to block unspecific staining for 30 minutes at 4°C. The flow cytometric results were analyzed with FACS Calibur (BD Biosciences, San Jose, CA, USA) using CellQuest software.
2.12. Immunohistochemistry and histopathology
After the mice were killed, tumor tissues were removed aseptically and immediately fixed in 4% formalin at room temperature for 2 days. The fixed tissues were processed through graded concentrations of ethanol and xylene and were then embedded in paraffin wax. Tissue sections of 4‐5 mm were mounted on adhesive glass slides and were stained with HE. Tumor sections were then deparaffinized and treated with 0.08% H2O2 for 30 minutes to block endogenous peroxidase. Slides were incubated with rat anti‐CD4 (GK1.5; Abcam, Cambridge, MA, USA) or rat anti‐CD8 (2.43; Abcam, Cambridge, MA, USA) at 4°C overnight, followed by incubation with HRP‐conjugated rabbit anti‐rat Ig. Diaminobenzidine was used to develop the staining reaction, and nuclear counterstaining was performed with hematoxylin. Slides were coded and examined by a pathologist who was blinded for the experimental history of the animals.
2.13. Statistical analysis
All data were analyzed by Student t‐test and expressed as means ± SEM; data were analyzed using GraphPad Prism 5 software for Windows (GraphPad Software, San Diego, CA, USA), and differences were considered statistically significant when P < .05. The significance levels are marked *P < .05 and **P < .01.
3. RESULTS
3.1. Construction of recombinant Newcastle disease virus co‐expressing interleukin 7 and interleukin 15
The heterogeneous gene containing IL15‐2A‐1L7 was inserted into the noncoding region between P and M genes of LX genome according to the rule of six (Figure 1A). Then, recombinant plasmid derived from LX cDNA containing the heterogeneous gene was cotransfected to BSR T7/5 cells with the support plasmids. After the propagation on embryonated chicken eggs, resultant recombinant virus designated as LX/IL(15+7) was readily rescued. Then, the titers of the recombinant virus from allantoic fluids were determined, and the results showed that HA titers of LX/IL(15+7) and LX/RFP are similar. Sequencing of recombinant virus LX/IL(15+7) confirmed that the insertion of the heterologous genes did not induce any mutation during virus rescue.
3.2. Growth kinetics of recombinant virus
The growth kinetics of the parental and recombinant viruses was compared by detecting the virus titers in DF‐1 cells. Growth kinetics of LX/IL(15+7) and LX are similar at the selected time points (Figure 1B). Although, the titers of LX/IL(15+7) were a little lower than that of the parental strains at 16, 18, 24 and 32 hour post‐inoculation, the differences are not significant in each group. The parental and recombinant strains exhibited similar virus titers at 40 and 48 hour post‐inoculation.
3.3. Expression of interleukin 7 and interleukin 15 in tumor cells infected with recombinant virus
In order to define the expression level of IL15 and IL7 by recombinant viruses, B16‐F10 cells were used. IL15 and IL‐7 were simultaneously expressed on the supernatants from B16‐F10 cells infected with LX/IL(15+7), while no product of each protein could be detected in LX/RFP‐infected tumor cells. The highest amount of IL15 and IL7 productions were at 48 h after the infection, which was approximately 9 and 7 ng/mL, respectively (Figure 2A). As shown in Figure 2B, the cleavage efficiency assay showed that <15% heterogenous proteins (approximately 1.5 ng/mL at 48 hour post‐virus infection) are expressed as fusion proteins (IL15‐2A‐IL7).
Figure 2.
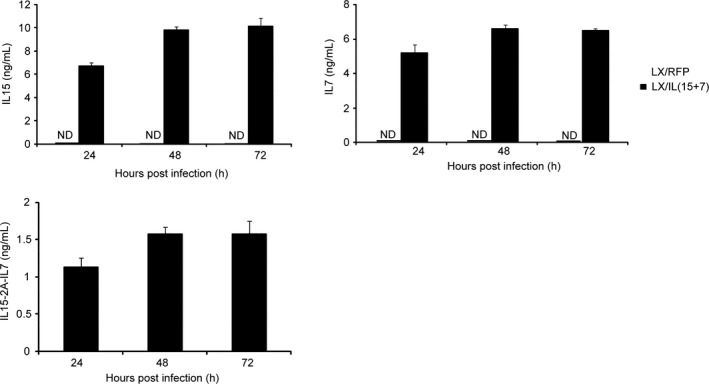
Expression of the interleukin 15 (IL15) and IL7 in tumor cells infected with LX/IL(15+7). A, Irradiated B16 were incubated with LX/IL(15+7) or LX/RFP (100 HAU virus per 106 cells). After incubation for 24, 48, 72 and 96 h, the supernatants were collected and analyzed for the production of IL15 and IL7 (A) or IL15‐2A‐IL7 (B) by ELISA
3.4. Cytotoxicity of splenocytes stimulated by tumor cells modified with LX/IL(15+7)
To determine the antitumor activity induced by LX/IL(15+7)‐modified tumor cells in vitro, splenocytes were stimulated with irradiated B16, B16 cells infected with LX/RFP or B16 cells infected with LX/IL(15+7), and specific killings against either B16‐F10 cells were compared. LX/RFP modified B16‐F10 cells significantly enhanced the cytotoxicity of splenocytes as compared with non‐virus modified cells. Notably, the cytotoxicity was further enhanced as tumor cells were modified with LX/IL(15+7) (Figure 3A).
Figure 3.
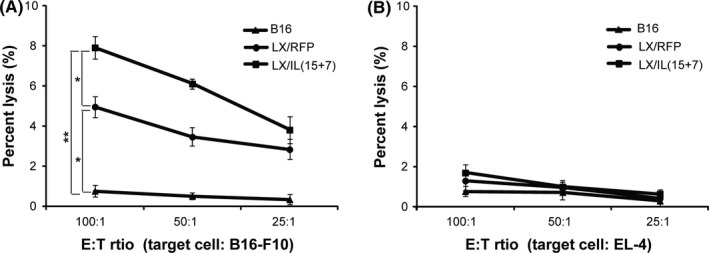
Cytotoxicity assay of tumor vaccine modified with LX/IL(15+7). Splenocytes harvested from C57BL/6 mice were incubated with irradiated B16‐F10 cells or irradiated B16‐F10 cells infected LX/IL(15+7) for 6 d, and used as effector cells. The killing capacity against B16‐F10 (A) or EL‐4 (B) cells was measured using CytoTox 96 nonradioactive cytotoxicity assay kit. The data shown are representative of the 3 experiments. *P < .05 and **P < .01
To determine whether the killing capacities of the stimulated splenocytes are specific, EL‐4 cells were used as the targets. No cytotoxicity was detected in any of the three groups, indicating that the killing capacity stimulated by NDV‐modified tumor cells was tumor‐specific (Figure 3B).
3.5. Prophylaxis with tumor vaccine modified with LX/IL(15+7)
The in vivo protective effects of LX/IL(15+7)‐modified tumor vaccine were assessed in murine melanoma model. C57BL/6 mice were immunized with irradiated B16‐F10 cells (B16), LX/RFP‐modified B16‐F10 cells (B16‐LX/RFP) or LX/IL(15+7)‐modified B16‐F10 cells (B16‐LX/IL(15+7)). Mice immunized with B16‐LX/IL(15+7) or B16‐LX/RFP exhibited a significant reduction in tumor growth, as compared with mice immunized with irradiated B16‐F10 cells (Figure 4A). Moreover, immunization with B16‐LX/IL(15+7) further inhibited tumor growth compared with the B16‐LX/RFP group. To further determine whether the protective immune response is tumor‐specific, immunized mice were challenged with EL‐4 cells. The tumor growth curves of mice immunized with B16‐LX/IL(15+7) and irradiated B16‐F10 cells are similar (Figure 4B), suggesting that the antitumor response induced by B16‐LX/IL(15+7) is tumor‐specific.
Figure 4.
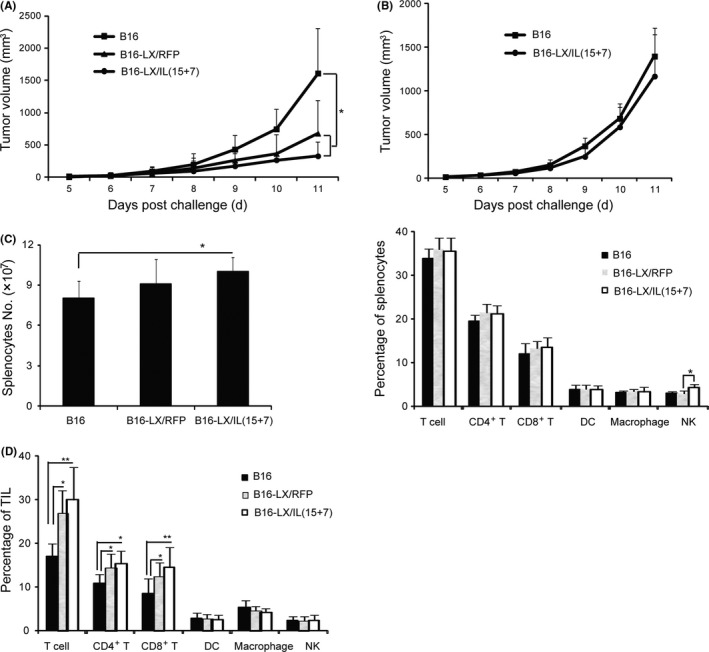
Prophylaxis effect of tumor vaccine modified with LX/IL(15+7). Mice were immunized with irradiated B16, B16‐LX/RFP or B16‐LX/IL(15+7) twice with 1‐week intervals, respectively, then mice were challenged with 1 × 105 B16‐F10 cells (A) or 1 × 105 EL‐4 cells (B) 2 weeks later. The tumor volumes were monitored daily. Mice were killed on day 7 post‐tumor challenge; the total numbers of splenocytes and frequencies of total T, CD4+ T, CD8+ T and NK cells in the spleen (C) and tumor‐infiltrating lymphocytes (TIL) (D) were analyzed. The experiments were performed with 6 mice per group. The data shown are representative of the 3 experiments. *P < .05 and **P < .01
To elucidate the possible immune cells associated with preventive effects induced by B16‐LX/IL(15+7), the phenotype of immune cells from immunized mice were analyzed by flow cytometry 7 days after the tumor challenge. The number of splenocytes were significantly increased in B16‐LX/IL(15+7) immunized mice, as compared with other groups (Figure 4C). The percentages of T cell, CD4+, CD8+, DC and macrophages were similar in each group. The percentage of NK in the B16‐LX/IL(15+7) group was significantly higher than in the B16‐LX/RFP group (Figure 4C).
Tumor‐infiltrating lymphocytes (TIL) were also examined, and a dramatic enhancement of the presences of T, CD4+ T and CD8+ T cells were observed in both B16‐LX/IL(15+7) and B16‐LX/RFP groups, as compared with the B16 group (Figure 4D).
3.6. Therapeutic effects of tumor vaccine modified with LX/IL(15+7)
To determine the therapeutic effects of LX/IL(15+7)‐modified tumor vaccine, murine melanoma model were used. C57BL/6 mice were inoculated s.c. with 5 × 104 B16‐F10 cells. On day 5, each animal was immunized with irradiated B16‐F10 cells, B16‐LX/RFP or B16‐LX/IL(15+7), respectively. The immunization was repeated on day 9. As shown in Figure 5A, mice immunized with B16‐LX/RFP or B16‐LX/IL(15+7) exhibited significant reduction in tumor growth, as compared with mice immunized with irradiated B16‐F10 cells. The tumor growth was further inhibited in the mice immunized with tumor cells modified with NDV co‐expressing IL15 and IL7, as compared with the LX/RFP group. Mice treated with EL‐4‐LX/IL(15+7) were not protected from B16 tumors, suggesting that the therapeutic effect of B16‐LX/IL(15+7)‐modified tumor cells was tumor‐specific.
Figure 5.
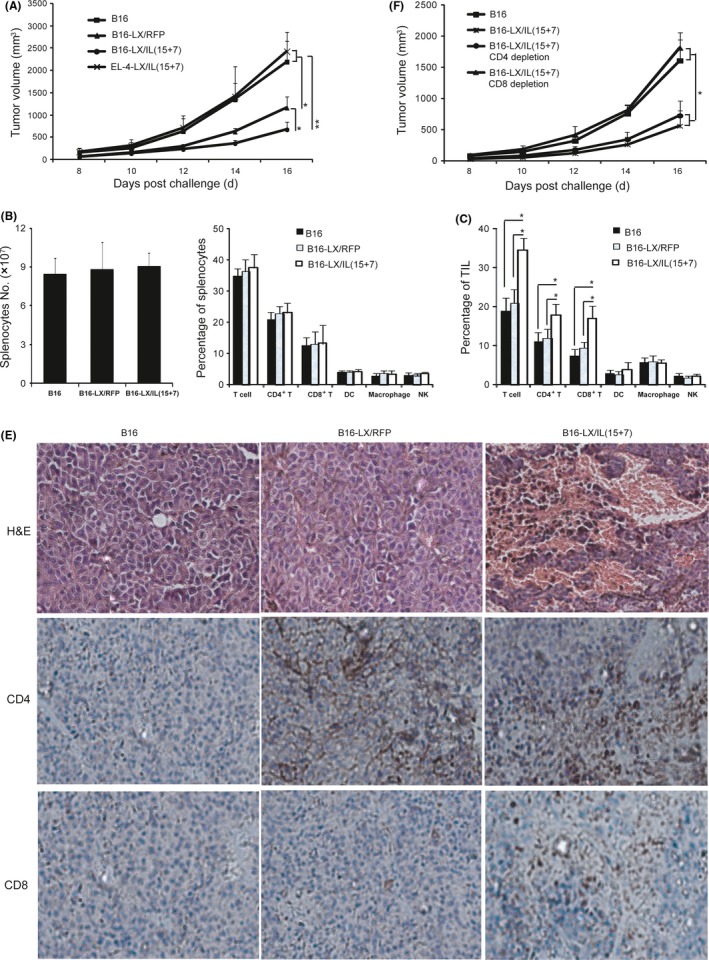
Therapeutic effects of tumor vaccine modified with LX/IL(15+7). Tumor growth of melanoma‐bearing mice. C57BL/6 mice were inoculated s.c. at the right flank with 5 × 104 cultured B16‐F10 cells. On day 5, the left flank of each animal was injected s.c. with irradiated B16 cells, B16‐LX/(RFP), B16‐LX/IL(15+7) or EL‐4‐LX/IL(15+7). The inoculation of vaccines was repeated on day 9. The tumor volumes were monitored daily (A). Mice were sacrificed on day 13 post‐tumor inoculation. Total numbers and frequencies of immune cells in the spleen (B) and frequencies of tumor‐infiltrating lymphocytes (TIL) (C) were analyzed. (D) HE and immunochemistry staining for CD4+ and CD8+ T cells of tumor tissue from melanoma‐bearing mice on day 13 post‐tumor inoculation (×200 magnification). (E) Mice i.p. injections of 1 mg GK1.5 (rat anti‐mouse CD4 mAb) or 500 μg 53‐6.7 (rat anti‐mouse CD8 mAb) 2 d before the first administration of B16‐LX/IL(15+7), and the injections were repeated 7 days later. The tumor volumes were monitored daily. The experiments were performed with 6 mice per group. The data shown are representative of the 3 experiments. *P < .05 and **P < .01
To test whether using IL15 and IL7 as adjuvant is sufficient to induce antitumor responses, the group of mice immunized with irradiated B16 cell plus IL7 (2 μg/mice) and IL15 (1 μg/mice) was also included. Irradiated B16 cells alone or plus IL7 and IL15 exhibited similar tumor growth curve, suggesting that NDV LX strain serves as a better carrier for IL7 and IL15 to generate tumor vaccines than using B16 with exogenous IL7 and 15 proteins (Figure S1).
To gain more insight into the immune responses induced by B16‐LX/IL(15+7), splenocytes and TIL were analyzed by flow cytometry on day 18 after tumor inoculation. The splenocyte numbers and the percentages of CD4+ T, CD8+ T, dendritic cells, macrophages and NK cells were not significantly different among each group (Figure 5B).
Tumor‐infiltrating lymphocytes were also phenotypically analyzed (Figure 5C), and CD3+ T cell frequencies were significantly increased in the B16‐LX/IL(15+7) group, as compared with the B16 group or the B16‐LX/RFP group. CD3+ CD8+ T cells accounted for approximately 13% of TIL in B16‐LX/IL(15+7)‐immunized mice, which was also significantly higher than those in the B16‐F10 or B16‐LX/RFP group. The prevalence of CD3+ CD4+ T cells was also significantly higher in the B16‐LX/IL(15+7) group than that in the B16‐F10 or the B16‐LX/RFP group. The tumor tissues that contained large areas of necrosis in B16‐LX/IL(15+7)‐treated mice were observed by HE staining (Figure 5D). The increased CD4+ and CD8+ T cell infiltration in the tumors of B16‐LX/IL(15+7)‐treated mice was also confirmed by immunohistochemistry (Figure 5D). Taken together, tumor cells modified with LX/IL(15+7) effectively suppressed tumor growth and promoted both CD4+ and CD8+ T cell infiltrations in the tumor.
To determine whether CD4+ or CD8+ T cells are critical for the antitumor activity induced by the B16‐LX/IL(15+7), in vivo depletion of CD4+ or CD8+ T cells were performed using specific Abs (Figure 5E). Depletion of CD4+ T cells did not dampen the therapeutic effects of B16‐LX/IL(15+7), while CD8+ T cell depletion inhibited the therapeutic effects of B16‐LX/IL(15+7). These results indicate that the antitumor response of B16‐LX/IL(15+7) was mainly mediated by CD8+ T cells.
4. DISCUSSION
To enhance the antitumor effects of NDV‐modified tumor vaccines, we generated recombinant virus co‐expressing IL15 and IL7 using a reverse genetics system of nonlytic NDV strain LX. Then, the growth kinetics of the recombinant virus, heterogenous gene expression, and both in vitro and in vivo antitumor effects of virus‐infected tumor cells were determined. Our results showed that the additional expression of these two cytokines significantly enhances the antitumor effects of recombinant NDV‐modified tumor vaccine.
Newcastle disease virus, a bird virus with a genome of non‐segmented negative‐strand RNA, was observed to infect tumor cells of humans and mice. The replication of NDV is confined to cytoplasm of infected cells, and the genome of NDV never integrates into the host, which makes it very safe in cancer therapy.22 NDV can be classified as either lytic or nonlytic. The lytic strains produce infectious progeny virus particles, which infect other tumor cells, whereas nonlytic strains cannot. The lytic NDV strains are often used to lyse tumor cells directly, such as 73‐T.23 The nonlytic strains unable to produce infectious progenies are commonly used to modify tumor cells as ATV‐NDV, such as Ulster and LX.13
Antitumor immunity is mainly mediated by T cells. Therefore, recombinant NDV‐expressing cytokines that can promote T cell responses will exhibit more powerful effects against cancer. IL15 and IL7 play integral roles in T cell development, differentiation and maintenance. IL7 contributes to survival of naïve and memory CD8+ T cells.24 IL15 induces proliferation of naïve and memory CD8+ T cells, and contributes to the maintenance of long‐lasting memory CD8+ T cells.25 Although both IL7 and IL15 have roles in the maintenance and self‐renewal of memory precursors, effector CD8+ T cells actually need both IL7 and IL15 for optimal proliferation and to become long‐lived memory cells.26, 27 Therefore, tumor cells modified with recombinant NDV co‐expressing IL7 and IL15 may have superior effects against tumors than those with parental virus. To co‐express multiple genes by recombinant viruses, an internal ribosome entry site (IRES) is often used. When using IRES to express two genes in one mRNA, the gene upstream or downstream of the IRES is translated by different mechanisms, which often cause a lower expression of the gene downstream of the IRES. These limitations might be solved using self‐processing peptides (2A peptides), which process themselves during the translation, resulting in the “self‐cleavage” of their primary polyproteins. In fact, recombinant NDV LX/IL(15+7) generated by the incorporation of a 2A self‐processing peptide into IL15 and IL7 exhibited a near‐equimolar expression in virus‐infected tumor cells. Although heterogenous genes are expressed stably by the recombinant NDV LX/IL(15+7), the expression level of each gene is much lower compared with recombinant viruses expressing only one of these genes, which were approximately 6‐fold higher for IL15 expression28 and 15‐fold higher for IL7 expression.13 The possible reason is the large burden to the recombinant virus resulting from the insertion of additional genes.
The NDV genome encodes 6 structural proteins, NP‐P‐M‐F‐HN‐L and NP. The insertion of the heterogeneous gene may inhibit the viral replication. Here, we inserted the fusion gene (IL15‐2A‐IL7) between P and M, and found that replication kinetics of the recombinant NDV were similar to those for the parental strain, indicating that the insertion of the foreign gene in the whole genome did not inhibit the viral replication.
To test the antitumor effects of LX/IL(15+7)‐modified tumor cells, both in vitro and in vivo experiments were performed. First, we demonstrated that the tumor‐specific cytotoxicity has been significantly enhanced when using LX/IL(15+7) virus in comparison with LX/RFP virus to modify tumor vaccines in vitro. Then, the preventive and therapeutic effects of recombinant NDV‐modified tumor vaccine were tested on the B16‐F10 melanoma model. CD8+ T cells have a central role in antitumor immunity. The increased percentage of tumor‐infiltrating CD8+ T cells is often correlated with improved clinical outcome in several cancers.13 Although both CD4+ and CD8+ T cells are increased in tumor microenvironment, the antitumor immune response induced by tumor cells modified with LX/IL(15+7) is dependent on CD8+ T cells.
Thus, we demonstrated that tumor cells modified with recombinant NDV co‐expressing IL15 and IL7 genes could greatly promote the prophylaxis and therapeutic efficacy. This warrants future studies on the mechanisms of the antitumor effects of this virus.
DISCLOSURE STATEMENT
The authors have no conflict of interest to declare.
Supporting information
Xu X, Sun Q, Mei Y, Liu Y, Zhao L. Newcastle disease virus co‐expressing interleukin 7 and interleukin 15 modified tumor cells as a vaccine for cancer immunotherapy. Cancer Sci. 2018;109:279–288. https://doi.org/10.1111/cas.13468
Funding information
Training Programs of Innovation and Entrepreneurship for Undergraduates of Soochow University (Grant/Award Number: 201510285105X); Natural Science Fund of Jiangsu Province (Grant/Award Number: BK20170334); Natural Science Foundation of the Jiangsu Higher Education Institutions of China (Grant/Award Number: 15KJB320015); Open Project Program of Jiangsu Key Laboratory of Zoonosis (Grant/Award Number: R1601); Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (Grant/Award Number: PAPD); project funding from Suzhou City (Grant/Award Number: SNG2017047, SYS201677); National Natural Science Foundation of China (Grant/Award Number: 31500746).
Xiaojing Xu and Qing Sun contributed equally to the present study.
REFERENCES
- 1. de Leeuw O, Peeters B. Complete nucleotide sequence of Newcastle disease virus: evidence for the existence of a new genus within the subfamily Paramyxovirinae. J Gen Virol. 1999;80(Pt 1):131‐136. [DOI] [PubMed] [Google Scholar]
- 2. Krishnamurthy S, Samal SK. Nucleotide sequences of the trailer, nucleocapsid protein gene and intergenic regions of Newcastle disease virus strain Beaudette C and completion of the entire genome sequence. J Gen Virol. 1998;79(Pt 10):2419‐2424. [DOI] [PubMed] [Google Scholar]
- 3. Ganarc K, Das M, Sinha S, Kumar S. Newcastle disease virus: current status and our understanding. Virus Res. 2014;184:71‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Csatary LK. Viruses in the treatment of cancer. Lancet. 1971;2:825. [DOI] [PubMed] [Google Scholar]
- 5. Lorence RM, Katubig BB, Reichard KW, et al. Complete regression of human fibrosarcoma xenografts after local Newcastle disease virus therapy. Cancer Res. 1994;54:6017‐6021. [PubMed] [Google Scholar]
- 6. Lorence RM, Reichard KW, Katubig BB, et al. Complete regression of human neuroblastoma xenografts in athymic mice after local Newcastle disease virus therapy. J Natl Cancer Inst. 1994;86:1228‐1233. [DOI] [PubMed] [Google Scholar]
- 7. Reichard KW, Lorence RM, Cascino CJ, et al. Newcastle disease virus selectively kills human tumor cells. J Surg Res. 1992;52:448‐453. [DOI] [PubMed] [Google Scholar]
- 8. Schirrmacher V, Fournier P. Newcastle disease virus: a promising vector for viral therapy, immune therapy, and gene therapy of cancer. Methods Mol Biol. 2009;542:565‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sinkovics JG, Horvath JC. Newcastle disease virus (NDV): brief history of its oncolytic strains. J Clin Virol. 2000;16:1‐15. [DOI] [PubMed] [Google Scholar]
- 10. Ni J, Schirrmacher V, Fournier P. The hemagglutinin‐neuraminidase gene of Newcastle Disease Virus: a powerful molecular adjuvant for DNA anti‐tumor vaccination. Vaccine. 2010;28:6891‐6900. [DOI] [PubMed] [Google Scholar]
- 11. Ramezanpour B, Haan I, Osterhaus A, Claassen E. Vector‐based genetically modified vaccines: exploiting Jenner's legacy. Vaccine. 2016;34:6436‐6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Janke M, Peeters B, Zhao H, et al. Activation of human T cells by a tumorvaccine infected with recombinant Newcastle disease virus producing IL‐2. Int J Oncol. 2008;33:823‐832. [PubMed] [Google Scholar]
- 13. Zhao L, Mei Y, Sun Q, et al. Autologous tumor vaccine modified with recombinant new castle disease virusexpressing IL‐7 promotes antitumor immune response. J Immunol. 2014;193:735‐745. [DOI] [PubMed] [Google Scholar]
- 14. Janke M, Peeters B, de Leeuw O, et al. Recombinant Newcastle disease virus (NDV) withinserted gene coding for GM‐CSF as a new vector for cancer immunogenetherapy. Gene Ther. 2007;14:1639‐1649. [DOI] [PubMed] [Google Scholar]
- 15. Schirrmacher V, Heicappell R. Prevention of metastatic spread by postoperative immunotherapy with virally modified autologous tumor cells. II. Establishment of specific systemic anti‐tumor immunity. Clin Exp Metastasis. 1987;5:147‐156. [DOI] [PubMed] [Google Scholar]
- 16. Lin J, Zhu Z, Xiao H, et al. The role of IL‐7 in immunity and cancer. Anticancer Res. 2017;37:963‐967. [DOI] [PubMed] [Google Scholar]
- 17. Rhode PR, Egan JO, Xu W, et al. Comparison of the superagonist complex, ALT‐803, to IL15 as cancer immunotherapeutics in animal Models. Cancer Immunol Res. 2016;4:49‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Doedens AL, Rubinstein MP, Gross ET, et al. Molecular programming of tumor‐infiltrating CD8+ T cells and IL15 resistance. Cancer Immunol Res. 2016;4:799‐811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Van den Bergh JMJ, Smits ELJM, Berneman ZN, et al. Monocyte‐derived dendritic cells with silenced ligands and transpresenting interleukin‐15 stimulate tumor‐reactive T‐cell expansion. Cancer Immunol Res. 2017;5:710‐715. [DOI] [PubMed] [Google Scholar]
- 20. Cieri N, Camisa B, Cocchiarella F, Forcato M, et al. IL‐7 and IL‐15 instruct the generation of human memory stem T cells from naive precursors. Blood. 2013;121:573‐584. [DOI] [PubMed] [Google Scholar]
- 21. Sun Q, Zhao L, Song Q, et al. Hybrid‐ and complex‐type N‐glycans are not essential for Newcastle disease virus infection and fusion of host cells. Glycobiology. 2012;22:369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Altomonte J, Marozin S, Schmid RM, Ebert O. Engineered newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol Ther. 2010;18:275‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Phuangsab A, Lorence RM, Reichard KW, Peeples ME, Walter RJ. Newcastle disease virus therapy of human tumor xenografts: antitumor effects of local or systemic administration. Cancer Lett. 2001;172:27‐36. [DOI] [PubMed] [Google Scholar]
- 24. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long‐lived memory cells. Nat Immunol. 2003;4:1191‐1198. [DOI] [PubMed] [Google Scholar]
- 25. Schluns KS, Williams K, Ma A, Zheng XX, Lefrancois L. Cutting edge: requirement for IL‐15 in the generation of primary and memory antigen‐specific CD8 T cells. J Immunol. 2002;168:4827‐4831. [DOI] [PubMed] [Google Scholar]
- 26. Bhadra R, Khan IA. IL‐7 and IL‐15 play a synergistic role in the development of CD8+ T cell response against an obligate intracellular parasite. Microbes Infect. 2012;14(13):1160‐1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cha E, Graham L, Manjili MH, Bear HD. IL‐7 + IL‐15 are superior to IL‐2 for the ex vivo expansion of 4T1 mammary carcinoma‐specific T cells with greater efficacy against tumors in vivo. Breast Cancer Res Treat. 2010;122:359‐369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xu X, Sun Q, Yu X, Zhao L. Rescue of nonlytic Newcastle disease virus (NDV) expressing IL‐15 for cancer immunotherapy. Virus Res 2017;233:35‐41. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials