

Abstract
PRDM14 is overexpressed in various cancers and can regulate cancer phenotype under certain conditions. Inhibiting PRDM14 expression in breast and pancreatic cancers has been reported to reduce cancer stem‐like phenotypes, which are associated with aggressive tumor properties. Therefore, PRDM14 is considered a promising target for cancer therapy. To develop a pharmaceutical treatment, the mechanism and interacting partners of PRDM14 need to be clarified. Here, we identified the proteins interacting with PRDM14 in triple‐negative breast cancer (TNBC) cells, which do not express the three most common types of receptor (estrogen receptors, progesterone receptors, and HER2). We obtained 13 candidates that were pulled down with PRDM14 in TNBC HCC1937 cells and identified them by mass spectrometry. Two candidates—glucose‐regulated protein 78 (GRP78) and heat shock protein 90‐α (HSP90α)—were confirmed in immunoprecipitation assay in two TNBC cell lines (HCC1937 and MDA‐MB231). Surface plasmon resonance analysis using GST‐PRDM14 showed that these two proteins directly interacted with PRDM14 and that the interactions required the C‐terminal region of PRDM14, which includes zinc finger motifs. We also confirmed the interactions in living cells by NanoLuc luciferase‐based bioluminescence resonance energy transfer (NanoBRET) assay. Moreover, HSP90 inhibitors (17DMAG and HSP990) significantly decreased breast cancer stem‐like CD24− CD44+ and side population (SP) cells in HCC1937 cells, but not in PRDM14 knockdown HCC1937 cells. The combination of the GRP78 inhibitor HA15 and PRDM14 knockdown significantly decreased cell proliferation and SP cell number in HCC1937 cells. These results suggest that HSP90α and GRP78 interact with PRDM14 and participate in cancer regulation.
Keywords: GRP78, heat shock protein, HSP90α, PRDM14, protein‐protein interaction
Abbreviations
- BiP
binding immunoglobulin protein
- C2H2
Cys2His2
- DNMT
DNA methyltransferase
- ER
endoplasmic reticulum
- GRP78
glucose‐regulated protein 78
- HSP
heat shock protein
- HSP90α
heat shock protein 90‐α
- IP
immunoprecipitation
- KLF2
kruppel‐like factor 2
- NanoBRET
NanoLuc luciferase‐based bioluminescence resonance energy transfer
- NLuc
NanoLuc luciferase
- PRC2
polycomb repressive complex 2
- SP
side population
- SPR
surface plasmon resonance
- TET
ten‐eleven translocation
- TNBC
triple‐negative breast cancer
1. INTRODUCTION
PRDM14 is a member of the PR domain‐containing family. It is specifically expressed in ES cells and primordial germ cells and has multiple functions, including a scaffold for chromatin remodeling, a transcription regulator required for maintaining pluripotency, and a required component for epigenetic reprogramming.1, 2, 3, 4, 5 PRDM14 overexpression in several cancers has been shown to be related to cancer properties such as proliferation, drug resistance, and differentiation.6, 7, 8, 9 We have previously reported that knockdown of PRDM14 expression decreased cancer stem‐like phenotypes, which are considered responsible for cancer initiation and progression in breast and pancreatic cancer cells.9, 10 Because siRNA‐based therapy targeting PRDM14 by tail vein injection decreased xenograft tumor and metastasis in mice, PRDM14 is considered a promising target for the treatment of these cancers. However, the working mechanism of PRDM14 in cancers is mostly unknown.
PRDM14 consists of a PR domain and six C2H2‐type zinc finger motifs. The PR domain is related in sequence to a SET methyltransferase domain but histone methyltransferase activity is found in only a subset of the PRDM family.11 Although PRDM14 indirectly associates with a methyltransferase process by recruiting a partner, innate methyltransferase activity has not been detected in PRDM14.4 The zinc finger motif is a DNA‐binding entity12 and, in some cases, also interacts with RNA, protein, and lipid.13, 14, 15, 16, 17 PRDM14 was reported to interact with proteins by IP and/or pull‐down, including components of PRC2 and KLF2 in ES cells, and TET enzymes in epiblast‐like cells. These interactions regulate pluripotency, differentiation, and reprogramming.4, 18, 19 However, binding partners of PRDM14 were not identified in cancer cells.
Breast cancer is one of the most common cancers worldwide.20 Breast cancers that express one or more of the three most common types of receptor (estrogen receptors, progesterone receptors, and HER2) can be treated with hormone and/or trastuzumab therapies to achieve a good prognosis. However, TNBC, which lack all three types of receptor, are often more aggressive with poor prognosis and are more difficult to treat. We have reported that inhibition of PRDM14 decreased s.c. xenografted tumor growth and metastasis of TNBC cell lines, suggesting that PRDM14 is a good target for treating TNBC in patients.10 Elucidation of the binding partners of PRDM14 will help elucidate the mechanism whereby PRDM14 regulates cancer phenotypes, which could contribute to the development of a less costly treatment for patients with these cancers.
Heat shock proteins are highly conserved and are induced in response to several stimuli including environmental stress. HSPs are multifunctional; they possess chaperone activity,21 inhibit apoptosis by associating with factors of the apoptotic machinery,22 and contribute to cell survival by regulating the stability or degradation of selected proteins.23, 24 In cancer cells, HSPs are highly expressed and further increased after stimulation. Moreover, HSP expression correlates with tumor growth, metastasis, and chemotherapy resistance. Thus, HSPs are an emerging target for cancer treatment.25, 26 HSP90α is the most prominent member of the HSP90 family and many client proteins have been identified.27, 28, 29 The client proteins cover various cellular processes and include cancer‐associated proteins, such as tumor suppressor proteins and mediators of invasion, metastasis, and angiogenesis.30, 31 GRP78 is also known as BiP and belongs to the HSP70 family. Its expression is known to increase during ER stress, which takes place when unfolded and/or misfolded proteins accumulate in the ER lumen. GRP78 works as a protein chaperone to mitigate potential damage from the unfolded species.32 In cancer cells, ER stress from microenvironmental factors, such as hypoxia and acidosis, upregulates GRP78 expression.33 In breast cancer, overexpressed GRP78 inhibits cell death by elevating anti‐apoptotic proteins34 and promotes autophagy, which enhances survival and drug resistance.35
To elucidate the molecular mechanism of PRDM14 in cancer cells, we carried out Halo pull‐down from Halo‐PRDM14 transduced TNBC HCC1937 cells, followed by mass spectrometry to identify binding partners. For the binding candidate proteins, we provided SPR analysis supporting that PRDM14 directly bound with HSP90α and GRP78. Furthermore, we carried out a NanoBRET binding assay to assess protein‐protein interactions in living cells,36 which can be made available for screening of binding inhibitors in the future. Moreover, we carried out experiments using HSP90 and GRP78 inhibitors to assess their effects on PRDM14‐mediated cell processes.
2. MATERIALS AND METHODS
2.1. Cell culture
The 293T cell line was purchased from ATCC (Manassas, VA, USA). The cell was identified by the cell banks using short tandem repeat analysis. The cells were cultured in DMEM (Thermo Fisher Scientific, San Jose, CA, USA) containing 10% FBS (GE Healthcare, Uppsala, Sweden). Two TNBC cell lines, HCC1937 and MDA‐MB231 cells, were obtained and cultured as described previously.10 All cell lines were incubated at 37°C in a humidified atmosphere containing 5% CO2.
2.2. Plasmid construction
A DNA fragment encoding PRDM14‐ΔC (amino acids 1‐412) was inserted into pReceiver‐Lv110 vector (GeneCopoeia, Rockville, MD, USA) to produce the plasmid encoding Halo‐PRDM14‐ΔC. For production of proteins tagged with NLuc at either the N‐ or C‐terminus, a DNA fragment containing the ORF sequence was inserted into pFN31K or pFC32K NLuc CMV‐neo Flexi vector (Promega, Madison, WI, USA) at the SgfI and PmeI sites, respectively. The insert sequences of HSP90α and GRP78 are the same as those of FHC25301 and FHC10475, respectively (Kazusa DNA Res. Inst., Chiba, Japan).
2.3. Lentiviral transduction
The plasmid vectors used were HaloTag fusion human PRDM14 (Halo‐PRDM14; EX‐W1089‐Lv110; GeneCopoeia), and HaloTag fusion EGFP (Halo‐con; EX‐EGFP‐Lv110; GeneCopoeia). HCC1937 and MDA‐MB231 cells were transduced using the Lenti‐pack HIV Expression Packaging Kit (GeneCopoeia).10 shRNA‐transduced HCC1937 cells (control shRNA and two anti‐human PRDM14 shRNAs [sh#1 and sh#3]) were prepared as previously described.10
2.4. HaloTag pull‐down and mass spectrometry
The HaloTag pull‐down assay was carried out using HaloTag Mammalian Pull‐Down System (Promega) according to the manufacturer's instructions. Briefly, Halo‐PRDM14 or Halo‐con transduced HCC1937 and MDA‐MB231 cells were collected, washed, and lysed. Equilibrated HaloLink Resin was mixed with the lysate, incubated, washed, and eluted with SDS elution buffer. The eluted samples were analyzed by SDS‐PAGE and detected by silver stain (Invitrogen, Carlsbad, CA, USA). We selected bands that were present in both cell lines expressing Halo‐PRDM14. The selected bands were excised and digested in‐gel with trypsin. Peptides were extracted and analyzed by mass spectrometry. Proteins present in HCC1937 expressing Halo‐con samples were excluded from further consideration.
2.5. Immunoprecipitation
Immunoprecipitation was carried out using HCC1937 and MDA‐MB231 cells expressing Halo fusion proteins. The cell lysates were incubated with each antibody and then TrueBlot anti‐rabbit Ig IP beads (Rockland, Limerick, PA, USA) were added. After incubation, the binding proteins were eluted from the beads with 1× SDS Sample Buffer (62.5 mmol/L Tris pH 6.8, 2% SDS, 10% glycerol, 0.1 mol/L DTT, 0.005% bromophenol blue, 2% 2‐mercaptoethanol). The following antibodies were used for IP: HSP90α (ab2928), and GRP78 (ab21685) from Abcam Inc. (Cambridge, MA, USA), and γ‐catenin (#2309) from Cell Signaling Technology Inc. (Beverly, MA, USA).
2.6. Protein analysis by automated capillary electrophoresis and immunodetection
Protein levels were quantified using an automated capillary electrophoresis and immunoassay system, Wes (ProteinSimple, Santa Clara, CA, USA) or western blotting as described previously.9 We used the following primary antibodies: PRDM14 (ab187881), HSP90α (ab2928), GRP78 (ab21685), and caspase‐14 (ab174847) from Abcam Inc., GAPDH (#5174) and γ‐catenin (#2309) from Cell Signaling Technology Inc., and Halo (G928A) from Promega. Secondary antibody used was TrueBlot HRP‐conjugated anti‐rabbit antibody (Rockland) for immunoprecipitated samples.
2.7. NanoLuc fusion protein detection
NanoLuc fusion proteins were analyzed by luciferase activity. The lysates of NLuc fusion protein‐transfected 293T were separated by non‐reducing SDS‐PAGE and transferred to PVDF membrane. The transferred membrane was treated with the substrate for NLuc and luminescence was detected with an ImageQuant Las‐4010 system (GE Healthcare).
2.8. Proteins
We used the following proteins for SPR analysis: Human PRDM14 (H00063978‐P01, GST‐Tag at N‐terminus; Abnova Inc., Taipei, Taiwan), Human PRDM14 (ΔC; ab196396, GST‐Tag at N‐terminus; Abcam Inc.), Human Hsp90 alpha (ab48801, His‐Tag; Abcam Inc.), Human GRP78 (SPR‐107, His‐Tag; Stress Marq Biosciences Inc., Victoria, BC, Canada), Human caspase‐14 (BML‐SE417; Enzo Life Sciences, Inc., Farmingdale, NY, USA), and Human γ‐catenin (TP304209, MYC/DDK‐Tag; OriGene Technologies, Inc., Rockville, MD, USA).
2.9. Surface plasmon resonance analysis
Interactions between putative partner proteins and PRDM14 were investigated by SPR analysis using a Biacore X100 plus instrument (GE Healthcare). Anti‐GST antibody was immobilized to the surface of a CM5 sensor chip with the GST capture kit (GE Healthcare) according to the manufacturer's instructions. GST or GST‐PRDM14 (ligand) was bound to the immobilized GST antibody on the CM5 sensor chip and the analytes were flowed over the chip. HEPES‐Buffered Saline‐EP + buffer (10 mmol/L HEPES, pH 7.4, 150 mmol/L NaCl, 3 mmol/L EDTA, 0.05% surfactant P20; GE Healthcare) was used as the running buffer. The sensor chip surface was regenerated with regeneration solution (10 mmol/L glycine‐HCl, pH 2.1; GE Healthcare). Specific protein‐protein interactions were identified by binding analysis of the initial screening and the association between recombinant proteins was examined with different concentrations of analyte by single‐cycle kinetic analysis. The data were globally fit to a 1:1 interaction model to determine dissociation constant (K D) for the interactions using BIA evaluation software Version 2.0.1 (GE Healthcare). Measurements were carried out at 25°C.
2.10. NanoLuc luciferase‐based bioluminescence resonance energy transfer
We carried out NanoBRET assays using a NanoBRET Nano‐Glo detection System (Promega) according to the manufacturer's instructions. Briefly, 293T cells were co‐transfected with HaloTag and NLuc fusions using FuGENE reagent (Promega) according to the manufacturer's instructions. The transfected cells were trypsinized, washed with PBS, resuspended in assay medium (Opti‐MEM I Reduced Serum Medium, no phenol red [Thermo Fisher Scientific] + 4% FBS), re‐plated to a 96‐well plate, and incubated with HaloTag NanoBRET 618 Ligand (100 nmol/L final concentration) or DMSO as negative control for greater than 4 hours. NanoBRET Nano‐Glo Substrate was added and donor emission (460 nm) and acceptor emission (618 nm) were measured using the GloMax Discover System (Promega). Raw NanoBRET ratio values with milliBRET units (mBU) were calculated as RawBRET = 618 nm Em/460 nm Em × 1000. Corrected NanoBRET ratio values with milliBRET units were calculated as corrected BRET = RawBRET of experimental sample − RawBRET of negative control sample, which reflected energy transfer from a bioluminescent protein donor to a fluorescent protein acceptor as a result of protein‐protein interactions.
For the initial screening, we carried out the NanoBRET assay with Halo‐PRDM14 and the proteins tagged with NLuc at either N‐ or C‐terminus and chose the combination that yielded a larger BRET ratio. To validate a NanoBRET assay without known inhibitors, we ascertained assay specificity by carrying out a simplified donor saturation assay. 1:1, 1:10, 1:100, and 1:1000 dilutions of NLuc relative to HaloTag DNA were used. If the protein‐protein interactions were real, the donor saturation assay curve would reach an asymptote, whereas a negative control would not.
2.11. Inhibitors
DMAG‐17, HSP990, VER155008, and HA15 were purchased from Selleck Chemicals (Houston, TX, USA). Cells were incubated for 24 hours, and were then treated with inhibitors for 24 hours. DMSO was used as a solvent and the negative control for these experiments. The proliferation assay was carried out using the Cell Counting Kit‐8 (Dojindo, Kumamoto, Japan). Isolation of SP cells and cell surface marker analyses were carried out using a FACSAria or FACSVerse flow cytometer (BD Biosciences, San Jose, CA, USA), as previously described.9
2.12. siRNA transfection
Two DNA‐chimeric siRNAs targeting the coding region of PRDM14 (PRDM14_1082‐1104 and PRDM14_1504‐1526; si#2 and si#3, respectively) as well as a negative control DNA‐chimeric siRNA (siNEGA) were purchased from RNAi Inc. (Tokyo, Japan).9 Cells were transfected with them using the Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific).
3. RESULTS
3.1. Identification of proteins interacting with PRDM14 in TNBC cells
To identify intracellular protein partners of PRDM14 in TNBC cells, we carried out Halo pull‐down assays followed by mass spectrometry. The eluted samples from Halo pull‐down assays of HaloTag protein‐transduced MDA‐MB231 and HCC1937 cells were analyzed by SDS‐PAGE (Figure 1). The 4 selected bands in Halo‐PRDM14‐transduced HCC1937 cells were analyzed by mass spectrometry and 13 proteins were identified as potential binding partners of PRDM14 (Table 1). The candidates included a variety of proteins including four heat shock proteins (HSP90α, GRP78, HSC70, and HSPA1L), 3 proteins of the desmosome (Desmoglein 1, Desmoplakin isoform I, and Dsc1a), a member of Armadillo family γ‐Catenin, which is involved in Wnt signaling, and Hornerin, which is aberrantly expressed and functional in breast cancer.
Figure 1.
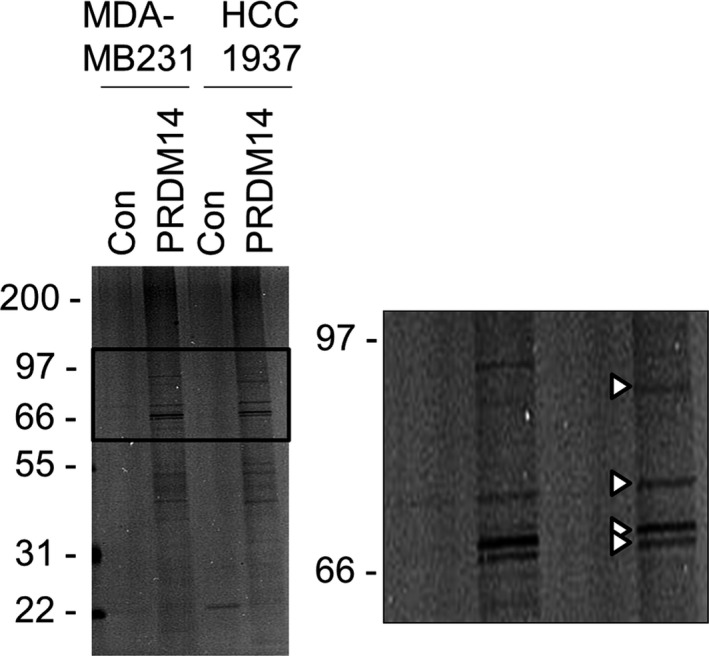
Halo pull‐down assay followed by mass spectrometry. Halo pull‐down assay was carried out using Halo‐PRDM14‐transduced MDA‐MB231 and HCC1937 cells. Pull‐down samples were analyzed by SDS‐PAGE detected by silver stain. The 4 bands in the enlarged view (arrowheads) were analyzed by mass spectrometry
Table 1.
Binding partners of PRDM14 identified by mass spectrometry
| Description | Score | Coverage | # Peptides | # PSM |
|---|---|---|---|---|
| Heat shock protein HSP 90‐alpha, HSP90α | 11.30 | 6.15 | 3 | 4 |
| Glucose‐regulated protein, GRP78 | 118.99 | 30.67 | 19 | 47 |
| Heat shock cognate 71 kDa protein, HSC70 | 141.06 | 27.09 | 17 | 55 |
| Heat shock 70 kDa protein 1‐like, HSPA1L | 85.99 | 22.66 | 14 | 32 |
| Desmoglein‐1 | 34.39 | 7.91 | 7 | 15 |
| Desmoplakin isoform I | 54.96 | 6.13 | 20 | 29 |
| Dsc1a precursor | 7.48 | 2.77 | 2 | 4 |
| γ‐Catenin | 17.41 | 9.81 | 7 | 10 |
| Hornerin | 26.70 | 3.25 | 1 | 9 |
| Caspase‐14 | 11.80 | 13.22 | 3 | 5 |
| Dermcidin | 9.19 | 20.00 | 2 | 3 |
| Serpin Family B Member 12, SERPINB12 | 6.95 | 9.29 | 2 | 3 |
| Profilaggrin | 6.19 | 3.21 | 2 | 3 |
Coverage: The percentage of the protein sequence covered by identified peptides.
# Peptides, total number of distinct peptide sequences identified in the protein group.
# PSM, number of peptide spectrum matches. Number of PSM is the total number of identified peptide spectra matched for the protein. The PSM value may be higher than the number of peptides identified for high‐scoring proteins because peptides may be identified repeatedly.
HSP, heat shock protein.
Halo pull‐down samples from HCC1937 and MDA‐MB231 cells were additionally analyzed by immunodetection. HSP90α, GRP78, and γ‐Catenin were detected, but Caspase‐14 was not (Figure 2A). Subsequently, we carried out an immunoprecipitation assay using antibodies for HSP90α, GRP78, or γ‐Catenin on Halo‐PRDM14 transduced HCC1937 and MDA‐MB231 cell lysates (Figure 2B). Halo‐PRDM14 was detected in all 3 immunoprecipitated samples from both cell lines (Figure 2C). Interactions of Halo‐con and the proteins were not observed in either the Halo pull‐down or the immunoprecipitation assays (Figure S1). These results suggest that HSP90α, GRP78, and γ‐Catenin interacted with PRDM14 in TNBC cells.
Figure 2.
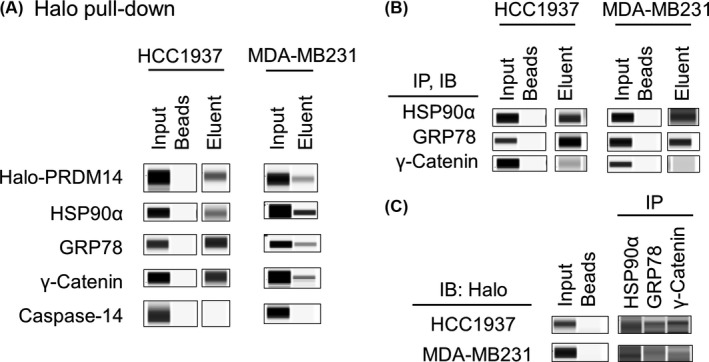
Immunodetection of binding partners of PRDM14. A, Eluents of a Halo pull‐down assay were analyzed by Wes using anti‐PRDM14, anti‐HSP90α, anti‐GRP78, anti‐γ‐Catenin, and anti‐caspase‐14 antibodies. B, C, Co‐immunoprecipitation (Co‐IP) experiments were carried out with lysates prepared from Halo‐PRDM14‐transduced HCC1937 and MDA‐MB231 cells. The lysates were immunoprecipitated with anti‐HSP90α, anti‐GRP78, or anti‐γ‐Catenin antibodies and then immunoblotted (IB) with (B) each antibody and (C) anti‐Halo antibody. GRP78, glucose‐regulated protein 78; HSP, heat shock protein
3.2. Confirmation of direct interactions between PRDM14 and selected proteins by SPR analysis
To determine whether interactions between PRDM14 and the identified proteins were direct or indirect, we carried out SPR experiments using a Biacore instrument. GST‐PRDM14 protein was captured by an anti‐GST antibody‐immobilized sensor chip, which was used as bait for the candidate proteins. Stable binding of GRP78, HSP90α, and γ‐Catenin to GST‐PRDM14 was detected (Figure 3A). Stable binding between PRDM14 and Caspase‐14 was not detected (Figure 3A), which agrees with the immunodetection data from Halo pull‐down samples (Figure 2A). Next, we carried out kinetic analysis using data from different concentrations of the two heat shock proteins (GRP78 and HSP90α) to assess the equilibrium dissociation constant (K D) values (Figure 3B). K D values of GRP78 and HSP90α with PRDM14 were 3.28 × 10−8 and 5.59 × 10−7 mol/L, respectively, suggesting they directly bind with relatively high affinity. To identify the region of PRDM14 that binds to the proteins, we carried out kinetic analysis with GST‐PRDM14‐ΔC protein, which lacks the C‐terminal region including the zinc finger motifs (Figure 3C). Interactions between the chaperone proteins and PRDM14‐ΔC were not detected (Figure 3D). We confirmed that PRDM14‐ΔC protein bound other proteins by SPR analysis (Figure S2). These results indicate that PRDM14 binds directly to GRP78 and HSP90α, and the binding requires the C‐terminus of PRDM14.
Figure 3.
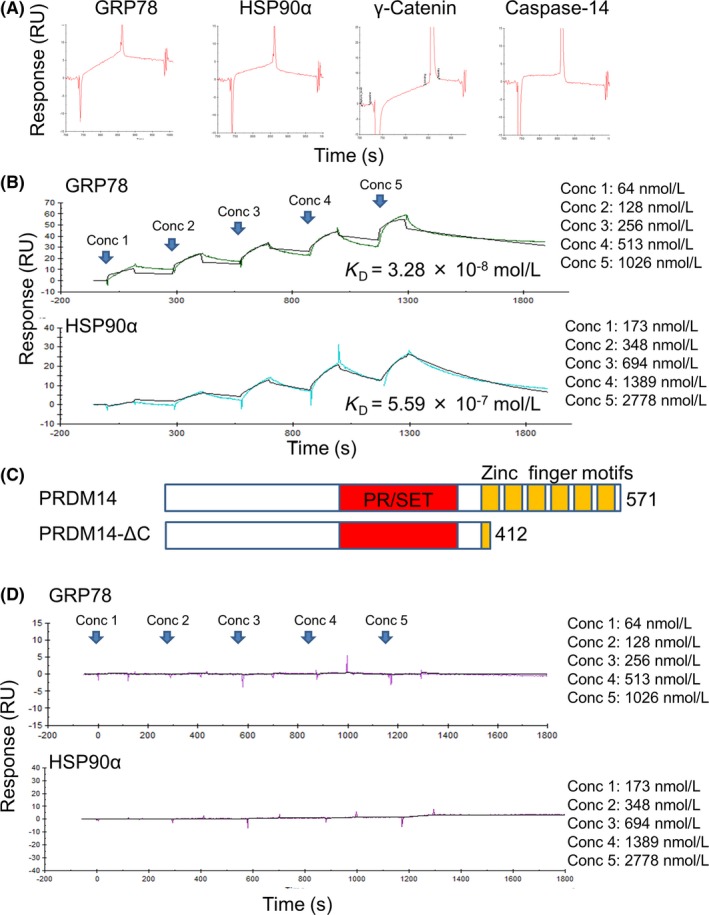
Binding and affinity analysis by surface plasmon resonance. A, Sensorgrams of the proteins (10 μg/mL) binding to surface GST‐PRDM14. B, Different concentrations of HSP90α and glucose‐regulated protein 78 (GRP78) (Conc 1‐5) flowed over GST‐PRDM14 on the anti‐GST‐immobilized sensor chip and analyzed by single‐cycle kinetic analysis. KD, dissociation constant. C, Schematic representation of PRDM14 full length and PRDM14‐ΔC. PRDM14 protein has a PR/SET domain (aa 266‐367) and six zinc finger motifs (aa 402‐426, aa 434‐453, aa 463‐483, aa 491‐511, aa 519‐537, and aa 548‐565). D, Different concentrations of HSP90α and GRP78 flowed over GST‐PRDM14‐ΔC on the anti‐GST‐immobilized sensor chip, analyzed by single‐cycle kinetic analysis. GRP78, glucose‐regulated protein 78; HSP, heat shock protein
3.3. Interactions between PRDM14 and selected proteins assessed by intracellular NanoBRET analysis
To validate the protein‐protein interaction of PRDM14 with HSP90α and GRP78 in vivo, we carried out NanoBRET analysis (Figure 4A). The NanoBRET assay uses an NLuc fusion protein as an energy donor and a fluorescently labeled HaloTag fusion protein as the energy acceptor. The optimized blue‐shifted NLuc donor paired with the red‐shifted HaloTag acceptor minimizes spectral overlap within the assay.36 293T cells were co‐transfected with Halo‐PRDM14 and NLuc fusion HSP90α or GRP78 (tagged with NLuc at either the N‐ or C‐terminus) (Figure 4B). We chose HSP90α‐NLuc and NLuc‐GRP78 because they yielded a larger BRET ratio than the opposite configurations in preliminary experiments with Halo‐PRDM14 (Figure S3A). The donor saturation assay curves of HSP90α‐NLuc and NLuc‐GRP78 with Halo‐PRDM14 reached saturation (Figure 4C). The curves with Halo‐PRDM14‐ΔC and Halo‐con did not reach saturation (Figures 4D and S3B,C). These results also indicate the interactions of PRDM14 with HSP90α and GRP78 require the C‐terminal region of PRDM14 in living cells.
Figure 4.
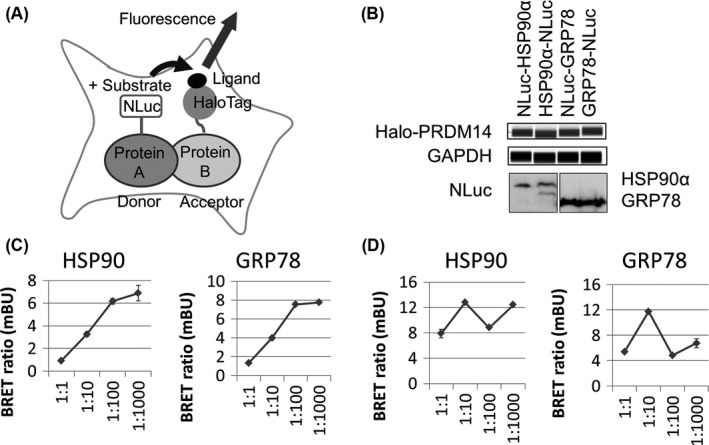
NanoLuc luciferase‐based bioluminescence resonance energy transfer (NanoBRET) analysis in living cells. A, Schematic of NanoBRET assay for detecting intracellular protein‐protein interactions by measuring energy transfer between a NanoLuc (NLuc) donor fusion protein and a HaloTag acceptor fusion protein. NLuc with substrate excites fluorescence‐conjugated HaloTag ligand with HaloTag fusion protein by energy transfer. B, 293T cells were transfected with Halo‐PRDM14 and NLuc fusion proteins. Expression level of Halo‐PRDM14 was analyzed using Wes with anti‐PRDM14. GAPDH was used as a loading control. Expression levels of NLuc fusion proteins expressed in 293T cells were detected by NLuc substrate. C, D, A simplified NanoBRET donor saturation assay was carried out. 1:1, 1:10, 1:100, and 1:1000 dilutions of NLuc relative to HaloTag DNA were used. HSP90α‐NLuc and NLuc‐GRP78 were co‐transfected with (C) Halo‐PRDM14 and (D) Halo‐PRDM14‐ΔC measuring energy transfer. Error bars represent the mean ± SD of triplicate samples. GRP78, glucose‐regulated protein 78; HSP, heat shock protein
To determine whether PRDM14 expression is regulated by the molecular chaperones GRP78 and HSP90α, two TNBC cell lines were transfected with HSP90α‐NLuc or NLuc‐GRP78 alone. PRDM14 expression was not increased by the transfection of HSP90α or GRP78 (Figure S3D). In addition, Halo‐PRDM14 induction had no impact on the expression of HSP90α or GRP78 (Figure S3E). These results suggest that the examined binding partners do not affect each other's expression.
3.4. Effect of inhibitors on HSP90α and GRP78
To determine the physiological relevance of the interactions between PRDM14 and HSP90α or GRP78, we used HSP90 and GRP78 inhibitors. HCC1937 and MDA‐MB231 cells were treated with 2 HSP90 inhibitors, 17DMAG (3 μmol/L) and HSP990 (1 μmol/L), an HSP70 family (HSP70, HSC70, and GRP78) inhibitor, VER155008 (100 μmol/L), and a GRP78 inhibitor, HA15 (100 μmol/L). PRDM14 expression was not changed by the inhibitors (Figure S4A). Proliferation of HCC1937 cells was significantly decreased by the inhibitors (P < .05) (Figure 5A). Combination of HA15 and PRDM14 knockdown reduced proliferation in both shPRDM14‐transduced HCC1937 cells (sh#1 and sh#3) as compared with that in control shRNA‐transduced cells (P < .01) (Figure 5B). Decreased proliferation in HA15‐treated, PRDM14 knockdown cells was also confirmed in DNA‐chimeric siRNA‐transfected HCC1937 and MDA‐MB231 cells (P < .05) (Figure S4B). Next, we assessed the effects of the inhibitors on the numbers of breast cancer stem‐like CD24− CD44+ and SP cells. In HCC1937 cells, two HSP90 inhibitors significantly decreased the number of CD24− CD44+ cells (P < .0001) (Figure 5C), whereas VER155008 and HA15 did not (Figure S4C). Aside from VER155008, all inhibitors decreased the proportion of SP cells in HCC1937 cells (P < .01) (Figure 5D). The inhibitors did not decrease the number of CD24− CD44+ and SP cells in MDA‐MB231 (Figure S4D,E). Subsequently, shRNA‐transduced HCC1937 cells were treated with the inhibitors. The number of CD24− CD44+ cells in shPRDM14 was significantly reduced as compared with that in sh control cells (P < .001) (Figure 5E), as previously reported.10 The inhibitors did not significantly change cell population in shPRDM14 cells (Figure 5E). The number of SP cells in shPRDM14 was significantly reduced as compared with that in sh control cells (P < .001) (Figure 5F) as we have previously reported.10 HSP90 inhibitors did not change the number of SP cells, whereas the GRP78 inhibitor HA15 significantly decreased SP cell number in shPRDM14 as compared with that of the control (P < .05) (Figure 5F).
Figure 5.
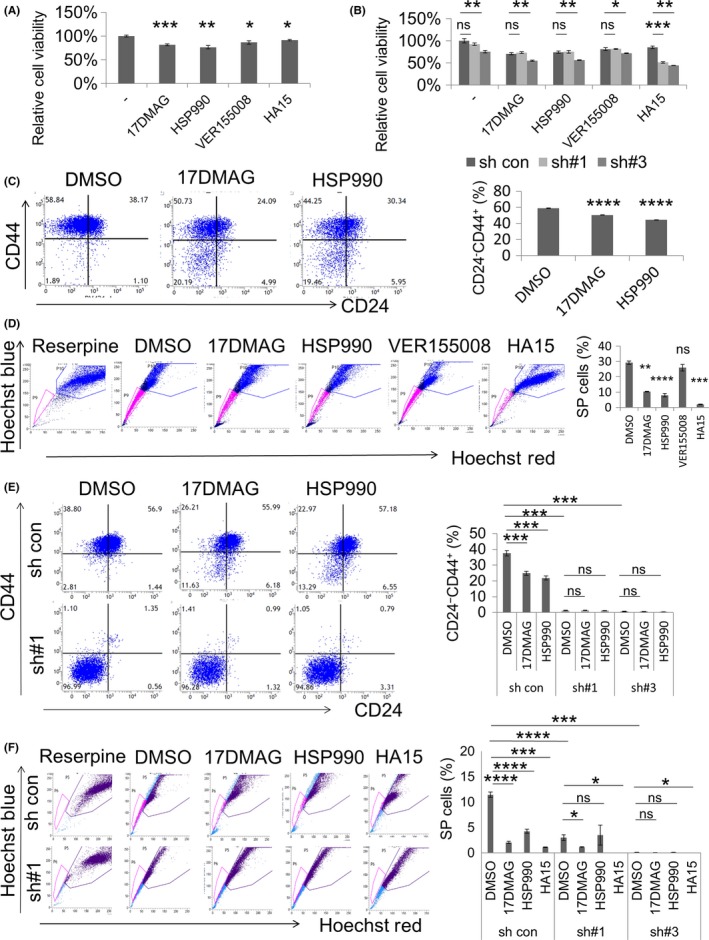
Effects of HSP90 and GRP78 inhibitors. A, Proliferation assay with HSP90 inhibitors (17DMAG and HSP990), an HSP70 family inhibitor (VER155008), and a GRP78 inhibitor (HA15) using HCC1937 cells. B, Proliferation assay with the inhibitors using shRNA‐transduced HCC1937 cells. C, Expression of CD24 and CD44 in HSP90 inhibitor‐treated HCC1937 cells was analyzed by flow cytometry. D, Side population (SP) cells in inhibitor‐treated HCC1937 cells were analyzed by flow cytometry using Hoechst 33342 dye. Reserpine, a multi‐drug transporter inhibitor, was used as a control for SP. E, Expression of CD24 and CD44 in HSP90 inhibitor‐treated, shRNA‐transduced HCC1937 cells was analyzed by flow cytometry. F, SP cells in inhibitor‐treated, shRNA‐transduced HCC1937 cells analyzed by flow cytometry using Hoechst 33342 dye. Data are shown as mean ± SD. ****P < .0001, ***P < .001, **P < .01, *P < .05. GRP78, glucose‐regulated protein 78; HSP, heat shock protein; ns, not significant
4. DISCUSSION
PRDM14 is overexpressed and regulates cancer properties in some cancers including TNBC, which is aggressive and difficult to treat. Because silencing PRDM14 expression in the cancers decreases cancer stem‐like phenotypes including tumor formation and metastasis,9, 10 PRDM14 is considered a promising target for cancer therapy. Identifying the binding partners of PRDM14 in cancers may contribute to the development of inhibitors targeting the PRDM14 working mechanism. We identified HSP90α and GRP78 as direct binding partners of PRDM14 in TNBC cells, whereas the binding patterns of PRDM14 with the other candidates are still under investigation.
Our data show that PRDM14 directly interacts with two heat shock proteins, GRP78 and HSP90α, and the interactions require the C‐terminal region including the zinc finger motifs, which can interact with DNA, RNA, protein, and lipid. Many reports have shown that overexpression of other C2H2 zinc finger‐containing proteins is related to several processes such as cell proliferation, invasion, migration, and drug resistance resulting in tumor progression for various cancers.37, 38, 39, 40 Moreover, a relationship between zinc finger proteins and the heat shock response has been reported;41, 42 a zinc finger protein ZC3H11 binds to mRNAs encoding heat‐shock protein homologues and is required for stabilization of them after heat shock in Trypanosoma brucei.42 Although we observed that PRDM14 induced by transfection did not change the expression of HSP90α or GRP78 (and vice versa) in two TNBC cell lines (Figure S3D,E), PRDM14 may have roles within the heat shock response. The effects of zinc finger motifs on the function of PRDM14 should also be examined.
Heat shock proteins including HSP90α and GRP78 are highly expressed in cancer cells and are further increased after various stimuli. Moreover, they are considered targets in cancer treatment because their expressions are associated with cancer properties such as tumor growth, metastasis, and chemotherapy resistance. HSP inhibitors are emerging as a strategy for cancer treatment, and various inhibitors against HSP90α or GRP78 are used for cancer research and have been used in clinical trials.26, 43, 44 HSP90 inhibitors significantly decreased the number of breast cancer stem‐like CD24− CD44+ (Figure 5C) and SP cells (Figure 5D) in HCC1937. This reduction in cell number by the inhibitors was lower than that by PRDM14 knockdown. The inhibitors did not decrease cell population in shPRDM14 cells (Figure 5E,F), which suggests that HSP90α may be partly associated with regulation of cancer stemness by PRDM14. Interestingly, an HSP90 inhibitor is reported to be specific to cancer cells; it reduced the number of HSP client proteins in cancer cells, but had no effect in normal cells. The difference seems to be caused by the conformation of the HSP90 complex.26, 28, 45 For example, HSP90 complexed with wild‐type p53, which is a short‐lived protein, is transiently activated and maintains protein homeostasis in normal cells. In contrast, most mutant p53 molecules extend their interaction with the complex, which leads to prevention of dynamic regulation in cancer cells.45 Some HSP90 inhibitors seem to prevent an abnormal conformation of the HSP90 complex. PRDM14 is overexpressed in several cancers including TNBC, whereas no or little expression is observed in normal mature tissues. Moreover, our data show that PRDM14 directly interacts with HSP90α and that HSP90 inhibitors do not decrease CD24− CD44+ and SP cells in shPRDM14‐transduced HCC1937 cells. PRDM14 may affect the roles of HSP90 in cancer regulation and the specificity of an HSP90 inhibitor in cancer cells. The GRP78 inhibitor HA15 significantly decreased proliferation in the PRDM14 knockdown TNBC cell lines as compared with that in control cells (Figure 5B), whereas PRDM14 knockdown alone did not decrease proliferation, as previously reported.10 Moreover, HA15 significantly decreased SP cell numbers in both control and PRDM14 knockdown HCC1937 cells (Figure 5D,F). These results suggest that both PRDM14 and GRP78 regulate cancer phenotypes, and that the combination of PRDM14 knockdown and HA15 is useful for treatment. Although GPR78 is reported to be overexpressed in breast cancer stem‐like CD24− CD44+ cells,46 the inhibitors did not have an effect on the number of CD24− CD44+ cells. Because VER155008 also inhibits other members of the HSP70 family, its effects may differ from that of the GRP78 inhibitor HA15, suggesting the importance of direct interaction between PRDM14 and GRP78. However, regulation of cancer stemness by these inhibitors with PRDM14 is a complicated process, as shown by the effects of these inhibitors on CD24− CD44+ and SP cells in MDA‐MB231 cells (Figure S4D,E), and the effect of GRP78 inhibitors on CD24− CD44+ cells in HCC1937 cells (Figure S4C). Nevertheless, our results suggested that HSP90 and GRP78 participate in regulation of cancer stemness by PRDM14. In addition, combination treatment with both inhibitors and PRDM14 knockdown may be especially effective for treatment.
PRDM14 is specifically expressed in primordial germ cells and ES cells. PRDM14 and another member of the PRDM family—PRDM1—work as transcription factors required for primordial germ cell development.47, 48, 49 PRDM14 is related to histone methylation by DNMT in ES cells.4 Previously, HSP90 was reported to interact with many proteins including PRDM1 in a LUMIER (LUminescence‐based Mammalian IntERactome) screening assay.29 Although the binding of HSP90 with PRDM14 was judged insignificant in the report, it may be because of the differences in methods or the strict threshold of binding to show a positive in screening. Moreover, the inhibition of HSP90 was recently reported to modulate DNA methylation by downregulating DNMTs, which are HSP90 clients,50 for both mRNA and protein levels in colorectal and pancreatic cancers.51 The direct interaction of PRDM14 and HSP90 may have an important role in primordial germ cell development and/or epigenetic regulation in ES cells.
In the present study, we identified GRP78 and HSP90α as binding partners of PRDM14 in TNBC cells. The interactions were direct and required the C‐terminal region including the zinc finger motifs of PRDM14 by SPR and intracellular NanoBRET analysis. PRDM14, GRP78, and HSP90α are all overexpressed and are useful targets for cancer treatment in several cancers including TNBC. Moreover, HSP90 and GRP78 inhibitors in HCC1937 cells inhibited some cancer phenotypes, including the number of SP cells; their effects differed depending on PRDM14 expression. Their interactions may have important roles in cancer biology and provide useful targets for cancer treatment.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
Supporting information
ACKNOWLEDGMENTS
This work was supported by Grant‐in‐Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT KAKENHI Grant Number JP16H04710) (H.T., S.N.).
Moriya C, Taniguchi H, Nagatoishi S, Igarashi H, Tsumoto K, Imai K. PRDM14 directly interacts with heat shock proteins HSP90α and glucose‐regulated protein 78. Cancer Sci. 2018;109:373–383. https://doi.org/10.1111/cas.13458
Funding information
Ministry of Education, Culture, Sports, Science and Technology (Grant/Award Number: “KAKENHI Grant Number JP16H04710”).
REFERENCES
- 1. Hohenauer T, Moore AW. The Prdm family: expanding roles in stem cells and development. Development. 2012;139:2267‐2282. [DOI] [PubMed] [Google Scholar]
- 2. Tsuneyoshi N, Sumi T, Onda H, Nojima H, Nakatsuji N, Suemori H. PRDM14 suppresses expression of differentiation marker genes in human embryonic stem cells. Biochem Biophys Res Commun. 2008;367:899‐905. [DOI] [PubMed] [Google Scholar]
- 3. Payer B, Rosenberg M, Yamaji M, et al. Tsix RNA and the germline factor, PRDM14, link X reactivation and stem cell reprogramming. Mol Cell. 2013;52:805‐818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamaji M, Ueda J, Hayashi K, et al. PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell. 2013;12:368‐382. [DOI] [PubMed] [Google Scholar]
- 5. Yamaji M, Seki Y, Kurimoto K, et al. Critical function of Prdm14 for the establishment of the germ cell lineage in mice. Nat Genet. 2008;40:1016‐1022. [DOI] [PubMed] [Google Scholar]
- 6. Nishikawa N, Toyota M, Suzuki H, et al. Gene amplification and overexpression of PRDM14 in breast cancers. Cancer Res. 2007;67:9649‐9657. [DOI] [PubMed] [Google Scholar]
- 7. Bi HX, Shi HB, Zhang T, Cui G. PRDM14 promotes the migration of human non‐small cell lung cancer through extracellular matrix degradation in vitro. Chin Med J (Engl). 2015;128:373‐377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang T, Meng L, Dong W, et al. High expression of PRDM14 correlates with cell differentiation and is a novel prognostic marker in resected non‐small cell lung cancer. Med Oncol. 2013;30:605. [DOI] [PubMed] [Google Scholar]
- 9. Moriya C, Taniguchi H, Miyata K, Nishiyama N, Kataoka K, Imai K. Inhibition of PRDM14 expression in pancreatic cancer suppresses cancer stem‐like properties and liver metastasis in mice. Carcinogenesis. 2017;38:638‐648. [DOI] [PubMed] [Google Scholar]
- 10. Taniguchi H, Hoshino D, Moriya C, et al. Silencing PRDM14 expression by an innovative RNAi therapy inhibits stemness, tumorigenicity, and metastasis of breast cancer. Oncotarget. 2017;8:46856‐46874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mzoughi S, Tan YX, Low D, Guccione E. The role of PRDMs in cancer: one family, two sides. Curr Opin Genet Dev. 2016;36:83‐91. [DOI] [PubMed] [Google Scholar]
- 12. Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183‐212. [DOI] [PubMed] [Google Scholar]
- 13. Font J, Mackay JP. Beyond DNA: zinc finger domains as RNA‐binding modules. Methods Mol Biol. 2010;649:479‐491. [DOI] [PubMed] [Google Scholar]
- 14. Konieczny P, Stepniak‐Konieczna E, Sobczak K. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res. 2014;42:10873‐10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brayer KJ, Kulshreshtha S, Segal DJ. The protein‐binding potential of C2H2 zinc finger domains. Cell Biochem Biophys. 2008;51:9‐19. [DOI] [PubMed] [Google Scholar]
- 16. Gaullier JM, Simonsen A, D'Arrigo A, Bremnes B, Stenmark H, Aasland R. FYVE fingers bind PtdIns(3)P. Nature. 1998;394:432‐433. [DOI] [PubMed] [Google Scholar]
- 17. Matthews JM, Sunde M. Zinc fingers–folds for many occasions. IUBMB Life. 2002;54:351‐355. [DOI] [PubMed] [Google Scholar]
- 18. Okashita N, Suwa Y, Nishimura O, et al. PRDM14 drives OCT3/4 recruitment via active demethylation in the transition from primed to naive pluripotency. Stem Cell Reports. 2016;7:1072‐1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan YS, Göke J, Lu X, et al. A PRC2‐dependent repressive role of PRDM14 in human embryonic stem cells and induced pluripotent stem cell reprogramming. Stem Cells. 2013;31:682‐692. [DOI] [PubMed] [Google Scholar]
- 20. Boyle P. Triple‐negative breast cancer: epidemiological considerations and recommendations. Ann Oncol. 2012;23(Suppl 6):vi7‐vi12. [DOI] [PubMed] [Google Scholar]
- 21. Young JC, Agashe VR, Siegers K, Hartl FU. Pathways of chaperone‐mediated protein folding in the cytosol. Nat Rev Mol Cell Biol. 2004;5:781‐791. [DOI] [PubMed] [Google Scholar]
- 22. Fu Y, Li J, Lee AS. GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation‐induced apoptosis. Cancer Res. 2007;67:3734‐3740. [DOI] [PubMed] [Google Scholar]
- 23. Lanneau D, de Thonel A, Maurel S, Didelot C, Garrido C. Apoptosis versus cell differentiation: role of heat shock proteins HSP90, HSP70 and HSP27. Prion. 2007;1:53‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grkovic S, O'Reilly VC, Han S, Hong M, Baxter RC, Firth SM. IGFBP‐3 binds GRP78, stimulates autophagy and promotes the survival of breast cancer cells exposed to adverse microenvironments. Oncogene. 2013;32:2412‐2420. [DOI] [PubMed] [Google Scholar]
- 25. Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci. 2006;31:164‐172. [DOI] [PubMed] [Google Scholar]
- 26. Jego G, Hazoume A, Seigneuric R, Garrido C. Targeting heat shock proteins in cancer. Cancer Lett. 2013;332:275‐285. [DOI] [PubMed] [Google Scholar]
- 27. McClellan AJ, Xia Y, Deutschbauer AM, Davis RW, Gerstein M, Frydman J. Diverse cellular functions of the Hsp90 molecular chaperone uncovered using systems approaches. Cell. 2007;131:121‐135. [DOI] [PubMed] [Google Scholar]
- 28. Whitesell L, Lindquist SL. HSP90 and the chaperoning of cancer. Nat Rev Cancer. 2005;5:761‐772. [DOI] [PubMed] [Google Scholar]
- 29. Taipale M, Krykbaeva I, Koeva M, et al. Quantitative analysis of HSP90‐client interactions reveals principles of substrate recognition. Cell. 2012;150:987‐1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Eustace BK, Sakurai T, Stewart JK, et al. Functional proteomic screens reveal an essential extracellular role for hsp90 alpha in cancer cell invasiveness. Nat Cell Biol. 2004;6:507‐514. [DOI] [PubMed] [Google Scholar]
- 31. Isaacs JS, Jung YJ, Mimnaugh EG, Martinez A, Cuttitta F, Neckers LM. Hsp90 regulates a von Hippel Lindau‐independent hypoxia‐inducible factor‐1 alpha‐degradative pathway. J Biol Chem. 2002;277:29936‐29944. [DOI] [PubMed] [Google Scholar]
- 32. Garg AD, Kaczmarek A, Krysko O, Vandenabeele P, Krysko DV, Agostinis P. ER stress‐induced inflammation: does it aid or impede disease progression? Trends Mol Med. 2012;18:589‐598. [DOI] [PubMed] [Google Scholar]
- 33. Li Z, Li Z. Glucose regulated protein 78: a critical link between tumor microenvironment and cancer hallmarks. Biochim Biophys Acta. 2012;1826:13‐22. [DOI] [PubMed] [Google Scholar]
- 34. Cook KL, Shajahan AN, Warri A, Jin L, Hilakivi‐Clarke LA, Clarke R. Glucose‐regulated protein 78 controls cross‐talk between apoptosis and autophagy to determine antiestrogen responsiveness. Cancer Res. 2012;72:3337‐3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cook KL, Clarke R. Heat shock 70 kDa protein 5/glucose‐regulated protein 78 “AMP”ing up autophagy. Autophagy. 2012;8:1827‐1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hall MP, Unch J, Binkowski BF, et al. Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012;7:1848‐1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yang L, Hamilton SR, Sood A, et al. The previously undescribed ZKSCAN3 (ZNF306) is a novel “driver” of colorectal cancer progression. Cancer Res. 2008;68:4321‐4330. [DOI] [PubMed] [Google Scholar]
- 38. Jen J, Lin LL, Chen HT, et al. Oncoprotein ZNF322A transcriptionally deregulates alpha‐adducin, cyclin D1 and p53 to promote tumor growth and metastasis in lung cancer. Oncogene. 2016;35:2357‐2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li Y, Tan BB, Zhao Q, Fan LQ, Wang D, Liu Y. ZNF139 promotes tumor metastasis by increasing migration and invasion in human gastric cancer cells. Neoplasma. 2014;61:291‐298. [DOI] [PubMed] [Google Scholar]
- 40. Weng H, Wang X, Li M, et al. Zinc finger X‐chromosomal protein (ZFX) is a significant prognostic indicator and promotes cellular malignant potential in gallbladder cancer. Cancer Biol Ther. 2015;16:1462‐1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rossi A, Riccio A, Coccia M, Trotta E, La Frazia S, Santoro MG. The proteasome inhibitor bortezomib is a potent inducer of zinc finger AN1‐type domain 2a gene expression: role of heat shock factor 1 (HSF1)‐heat shock factor 2 (HSF2) heterocomplexes. J Biol Chem. 2014;289:12705‐12715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Droll D, Minia I, Fadda A, et al. Post‐transcriptional regulation of the trypanosome heat shock response by a zinc finger protein. PLoS Pathog. 2013;9:e1003286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Miao YR, Eckhardt BL, Cao Y, et al. Inhibition of established micrometastases by targeted drug delivery via cell surface‐associated GRP78. Clin Cancer Res. 2013;19:2107‐2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zagouri F, Sergentanis TN, Chrysikos D, Papadimitriou CA, Dimopoulos MA, Psaltopoulou T. Hsp90 inhibitors in breast cancer: a systematic review. Breast. 2013;22:569‐578. [DOI] [PubMed] [Google Scholar]
- 45. Kamal A, Thao L, Sensintaffar J, et al. A high‐affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature. 2003;425:407‐410. [DOI] [PubMed] [Google Scholar]
- 46. Nami B, Ghasemi‐Dizgah A, Vaseghi A. Overexpression of molecular chaperons GRP78 and GRP94 in CD44(hi)/CD24(lo) breast cancer stem cells. Bioimpacts. 2016;6:105‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kurimoto K, Yamaji M, Seki Y, Saitou M. Specification of the germ cell lineage in mice: a process orchestrated by the PR‐domain proteins, Blimp1 and Prdm14. Cell Cycle. 2008;7:3514‐3518. [DOI] [PubMed] [Google Scholar]
- 48. Nakaki F, Hayashi K, Ohta H, Kurimoto K, Yabuta Y, Saitou M. Induction of mouse germ‐cell fate by transcription factors in vitro. Nature. 2013;501:222‐226. [DOI] [PubMed] [Google Scholar]
- 49. Nagamatsu G, Kosaka T, Kawasumi M, et al. A germ cell‐specific gene, Prmt5, works in somatic cell reprogramming. J Biol Chem. 2011;286:10641‐10648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yamaki H, Nakajima M, Shimotohno KW, Tanaka N. Molecular basis for the actions of Hsp90 inhibitors and cancer therapy. J Antibiot (Tokyo). 2011;64:635‐644. [DOI] [PubMed] [Google Scholar]
- 51. Nagaraju GP, Wu C, Merchant N, Chen Z, Lesinski GB, El‐Rayes BF. Epigenetic effects of inhibition of heat shock protein 90 (HSP90) in human pancreatic and colon cancer. Cancer Lett. 2017;402:110‐116. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials