

Abstract
Tumor progression is the main cause of death in patients with breast cancer. Accumulating evidence suggests that dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) functions as a tumor suppressor by regulating cell survival, differentiation, proliferation and apoptosis. However, little is known about the mechanisms of transcriptional regulation by DYRK2 in cancer progression, particularly with respect to cancer proliferation and invasion. Here, using a comprehensive expression profiling approach, we show that cyclin‐dependent kinase 14 (CDK14) is a target of DYRK2. We found that reduced DYRK2 expression increases CDK14 expression, which promotes cancer cell proliferation and invasion in vitro, in addition to tumorigenicity in vivo. CDK14 and DYRK2 expression inversely correlated in human breast cancer tissues. We further identified androgen receptor (AR) as a candidate of DYRK2‐dependent transcription factors regulating CDK14. Taken together, our findings suggest a mechanism by which DYRK2 controls CDK14 expression to regulate tumor cell proliferation and invasion in breast cancer. Targeting of this pathway may be a promising therapeutic strategy for treating breast cancer.
Keywords: breast cancer, cyclin‐dependent kinase 14, dual‐specificity tyrosine‐regulated kinase 2, invasion, proliferation
1. INTRODUCTION
Breast cancer is one of the most common malignancies for women worldwide. From 1980 to 2010, the annual incidence and mortality rates for breast cancer increased 3.1% and 1.8%, respectively.1 Although mammography screening and treatments have advanced in recent decades, cancer progression is the main reason for death in breast cancer patients. Identifying new mechanisms underpinning cancer progression may help to establish a novel strategy for treating breast cancer.
Dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) is a protein kinase that phosphorylates its substrates at serine and threonine residues. Initially, DYRK2 was found to phosphorylate p53 at Ser46 to regulate apoptotic cell death in response to DNA damage.2, 3 We have conducted further studies to understand the functions of DYRK2. DYRK2 shows tumor suppressor functions by modifying c‐Myc, c‐Jun, Snail and mTOR.4, 5, 6, 7 These findings collectively suggest that DYRK2 is involved in post‐transcriptional regulation. With respect to this function, we showed that DYRK2 plays important roles in epithelial–mesenchymal transition (EMT) by degrading Snail in breast cancer and ovarian cancer.5, 6 Recently, we reported for the first time that DYRK2 inhibits breast cancer stem cells through transcriptional downregulation of KLF4.8 However, the mechanism of direct transcriptional regulation by DYRK2 in cancer progression, particularly in cancer proliferation and invasion, remains unclear.
In this study, we comprehensively searched for downstream target genes of DYRK2 using microarray analyses. Silencing of DYRK2 resulted in increased mRNA levels of cyclin‐dependent kinase 14 (CDK14). CDK14, also known as PFTTAIRE1 or PFTK1, is a cell division cycle 2 (Cdc2)‐related serine/threonine protein kinase that promotes cell cycle progression.9 CDK14 shows high expression in the human brain, pancreas, kidneys, heart, testes and ovaries.10 It is also reported that CDK14 promotes cancer progression and chemotherapy resistance in breast cancer, gastric cancer, ovarian cancer, esophageal cancer and hepatocellular carcinoma.11, 12, 13, 14, 15 Therefore, we hypothesize that DYRK2 suppresses the proliferation of breast cancer cells and invasion through CDK14 expression.
2. MATERIALS AND METHODS
2.1. Cell culture and treatment
MCF‐7 cells (human mammary carcinoma cells) were grown in minimal essential medium containing supplemented 10% heat‐inactivated FBS, according to standard protocols. MDA‐MB‐231 cells (human mammary carcinoma cells) were grown in supplemented DMEM containing 10% heat‐inactivated FBS. For androgen receptor (AR) inhibition experiments, cells were treated with the indicated concentration of MDV3100 (Santa Cruz Biotechnology, Dallas, TX, USA) for 24 hour.
2.2. Cell transfection
Plasmids were transfected using X‐tremeGENE HP DNA Transfection Reagent (Roche, Basel, Switzerland), Lipofectamine 2000 (Invitrogen, Waltham, MA, USA) or electroporation. DYRK2‐specific siRNA (Qiagen, Venlo, the Netherlands; Invitrogen), CDK14 siRNA (Sigma‐Aldrich, St. Louis, MO, USA) and control siRNA (Qiagen) were used. The siRNA were transfected using Lipofectamine RNAiMAX (Invitrogen). To isolate stable transfectants, MCF‐7 cells were transfected with pSuper‐puro (Oligoengine, Seattle, WA, USA) or pSuper‐puro DYRK2 shRNA in the presence of puromycin (1 μg/mL). MCF‐7 cells were transfected with pSuper‐neo (Oligoengine) or pSuper‐neo CDK14 shRNA in the presence of G418 (500 μg/mL). For stable overexpression, we used the GFP‐C1 vector. MDA‐MB‐231 cells were transfected with GFP or GFP‐DYRK2 in the presence of G418 (500 μg/mL). Gene knockdown and overexpression were assessed by real‐time RT‐PCR.
2.3. Plasmid construction
pSuper‐puro DYRK2 shRNA was constructed as described previously.5 The sequences of shRNA targeting CDK14 were designed based on those of CDK14 siRNA. CDK14 shRNA were annealed and cloned into pSuper‐neo vector (Oligoengine) according to the manufacturer's instructions. GFP‐tagged DYRK2 was constructed as described previously.2
2.4. Microarray analysis
MCF‐7 cells were transfected with control or DYRK2 siRNA. Total RNA were purified and used for microarray analysis using a SurePrint G3 Human GE Microarray Kit Version 2.0 (50 599 probe sets; Agilent Technologies, Santa Clara, CA, USA). The results are available at the Gene Expression Omnibus database under Accession No. GSE75917.
The pSuper control and shDYRK2 cells were prepared as previously reported.5 Total RNA was purified from the pSuper control cells and shDYRK2 cells. RNA were used for microarray analysis with a GeneChip Human Gene 1.0 ST Array (29 096 probe sets; Affymetrix, Santa Clara, CA, USA). The results from GeneChip analysis are available at the Gene Expression Omnibus Database (http://www.ncbi.nlm.nih.gov/geo/) under Accession No. GSE75918.
2.5. Western blotting
Cells were washed twice in chilled PBS and resuspended in lysis buffer (50 mmol/L Tris‐HCl, pH 7.6; 150 mmol/L NaCl; 10 mmol/L NaF; 1 mmol/L Na3VO4; 1 mmol/L PMSF; 1 mmol/L DTT; 10 μg/mL aprotinin; 1 μg/mL leupeptin; 1 μg/mL pepstatin A; 1% NP‐40). Cell extracts were centrifuged for 5 min at 4°C. Supernatants were separated by SDS‐PAGE and transferred to nitrocellulose membranes. The membranes were incubated with anti‐CDK14 (Santa Cruz Biotechnology), anti‐DYRK2 (Sigma‐Aldrich), anti‐AR (Santa Cruz Biotechnology), anti‐TUBULIN (Sigma‐Aldrich) or anti‐GAPDH (Santa Cruz Biotechnology) antibodies at a 1:1500 dilution. Immune complexes were incubated with secondary antibodies and visualized using ImmunoStar (Wako, Osaka, Japan) or Western Lightning Plus‐ECL (PerkinElmer, Norwalk, CT, USA).
2.6. Real‐time RT‐PCR
Isolation of total RNA from cells was performed using TRIsure (Nippon Genetics, Tokyo, Japan). Total RNA was reverse transcribed into cDNA using the PrimeScript 1st strand cDNA Synthesis Kit (Takara, Shiga, Japan) according to the manufacturer's protocol. PCR was performed using KAPA SYBR FAST qPCR Master Mix (2×) ABI Prism (Kapa Biosystems, Wilmington, MA, USA) according to the instruction manual. The data were normalized to the input control, β‐actin or GAPDH.
2.7. MTS assay
Cells were seeded into 96‐well plates. 3‐(4,5‐dimethylthiazol‐2‐yl)‐5‐(3‐carboxymethoxyphenyl)‐2‐(4‐sulfophenyl)‐2H‐tetrazolium, inner salt (MTS) assays were performed using the CellTiter 96 AQ One Solution Cell Proliferation Assay Kit (Promega) according to the manufacturer's instructions. The absorbance was measured at 490 nm.
2.8. Colony formation assay
MCF‐7 cells or MDA‐MB‐231 cells were seeded into 6‐well culture plates at a density of 4000 cells/well or 500 cells/well, respectively. Cells were then cultured at 37°C in a 5% CO2 incubator. Cells were washed with chilled PBS and fixed with 70% ethanol for 30 min at −20°C. After fixation, cells were stained using Giemsa Solution (Wako) for 10 min at room temperature. The cells were then washed with PBS twice.
2.9. Invasion assay
Invasion assays were performed using the CytoSelect 24‐Well Cell Invasion Assay Kit (Cell Biolabs, San Diego, CA, USA) according to the instruction manual. Invasive cancer cells were stained, extracted and quantified at optical density (OD) 560 nm. Images were obtained using phase contrast microscopy (BZ‐X700; Keyence, Osaka, Japan).
2.10. Xenograft studies
All animal research was conducted in accordance with the guidelines for Animal Experimentation established at the Jikei University (Tokyo, Japan). For the orthotopic model, 5 × 106 MCF‐7‐derived cells were suspended in 100 μL of a 1:1 mixture of PBS and Matrigel (Corning, Bedford, MA, USA) and injected underneath the nipple of the mammary fat pad in the abdomen of 8‐week‐old female BALB/cSlc‐nu/nu mice (Sankyo Labo Service, Tokyo, Japan). Simultaneously, these mice were implanted with pellets containing 17β‐estradiol (Innovative Research of America, Sarasota, FL, USA). Tumor size was monitored three times a week using calipers.
2.11. Upstream transcription factor detection
A promoter analysis was performed using the online tool ExPlain 3.1 (http://explain.biobase-international.com/) to detect overrepresented transcription factor binding sites in MCF‐7 DYRK2 siRNA cells compared to MCF‐7 control siRNA cells (GSE75917). Seventy‐six transcription factors were predicted to have putative binding sites in the promoters of genes suspected to be downstream targets of DYRK2 as described previously.8
2.12. MAPPER database analysis
To identify transcription factors that have binding sites within 3 kb upstream of CDK14, the MAPPER database was used (http://genome.ufl.edu/mapper).16 This identified 1153 putative binding sites and 370 genes in the promoter region of CDK14.
2.13. Androgen receptor reporter assay
Androgen receptor reporter assay was conducted using a Cignal Androgen Receptor Reporter (luc) Kit (Qiagen). A mixture of an inducible AR‐responsive firefly luciferase reporter and constitutively expressing Renilla construct (40:1) was contained in this kit. Luciferase activity was measured 48 h after transfection using the Dual‐Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol. The relative increase in AR transcription activity normalized to Renilla luciferase activity was determined.
2.14. Immunohistochemistry
Sixty samples from surgically treated breast cancer patients were obtained from the Surgery Department at the Jikei University Hospital between 2001 and 2002. The Jikei University School of Medicine Ethics Review Committee approved the study protocol, and informed consent was obtained. All samples were fixed in formalin, embedded in paraffin, and histologically diagnosed as positive for breast cancer by HE staining. We used anti‐DYRK2 (Abgent, San Diego, CA, USA) and anti‐CDK14 antibodies (Santa Cruz Biotechnology) for immunohistochemical studies. DYRK2 expression was detected, as previously described.5 CDK14 antigens were treated at 100°C for 8 min with Tris base buffer (pH 8.5). Next, 3% hydrogen peroxide was used for blocking. Anti‐CDK14 antibody (1:50) was applied to the samples. High versus low expression of DYRK2 was previously quantified.5 For CDK14, cytoplasmic staining intensity was evaluated semi‐quantitatively in 400 high‐powered microscopic fields to obtain three immunohistochemistry (IHC) scores (1 = weak staining, 2 = moderate staining, and 3 = strong staining). We classified the staining intensity into two categories: positive or negative expression. Positive expression was defined as strong staining in more than 20% of the cancer cells, while all other cells were classified as displaying negative expression.
2.15. Statistical analysis
Statistical analyses of continuous variables consisting of two groups or more than two groups were performed by a two‐tailed Student's t‐test or one‐way ANOVA followed by Tukey's multiple comparisons test, respectively. Data are presented as the mean ± SD. χ2 tests were used to examine the associations between DYRK2 expression and CDK14 expression. A P‐value <.05 was considered statistically significant.
3. RESULTS
3.1. Identification of cyclin‐dependent kinase 14 as a direct target of dual‐specificity tyrosine‐regulated kinase 2
To identify the target genes regulated by DYRK2, DYRK2‐expressing MCF‐7 breast cancer cells were transfected with the pSuper‐puro vector (pSuper control cells) or pSuper‐puro DYRK2 shRNA (shDYRK2 cells). We then established a stable cell line in which DYRK2 was depleted. MCF‐7 cells were also transfected with control siRNA or DYRK2 siRNA. To explore the potential target genes regulated by DYRK2, we purified mRNA from these transfected cells for microarray analysis. To isolate DYRK2 target genes, we compared gene expression profiles in stable and transient DYRK2‐depleted cells with those of control cells. We selected 702 and 250 probes that were more than 1.5‐fold increased compared to controls in stable and transiently DYRK2‐depleted cells, respectively. These two data analyses revealed that eight genes were upregulated in DYRK2‐depleted cells (Figure 1A; Table S1). MCF‐7 cells were transfected with two different siRNA or pSuper‐puro shRNA targeting DYRK2 to exclude off‐target effects. Ultimately, one probe was selected that was transcriptionally elevated in a DYRK2‐dependent manner in both stably and transiently knocked‐down cells. The analysis of microarray data indicated that the induction of CDK14 expression in DYRK2‐depleted cells was significantly higher than that of control cells (Figure 1B,D). Moreover, CDK14 was the most upregulated gene in stable DYRK2‐depleted cells (Table S1). We confirmed that stable and transient DYRK2 depletion in MCF‐7 cells increased the levels of CDK14 mRNA and protein using real‐time RT‐PCR and western blotting, respectively (Figure 1C,E). To extend these findings, we evaluated DYRK2 and CDK14 expression in two human breast cancer cell lines: the highly metastatic MDA‐MB‐231 cells and the poorly metastatic MCF‐7 cells. DYRK2 expression was markedly decreased, whereas CDK14 expression was markedly increased in MDA‐MB‐231 cells (Figure 1F). These results indicated that mRNA and protein levels of CDK14 increase following downregulation of DYRK2.
Figure 1.
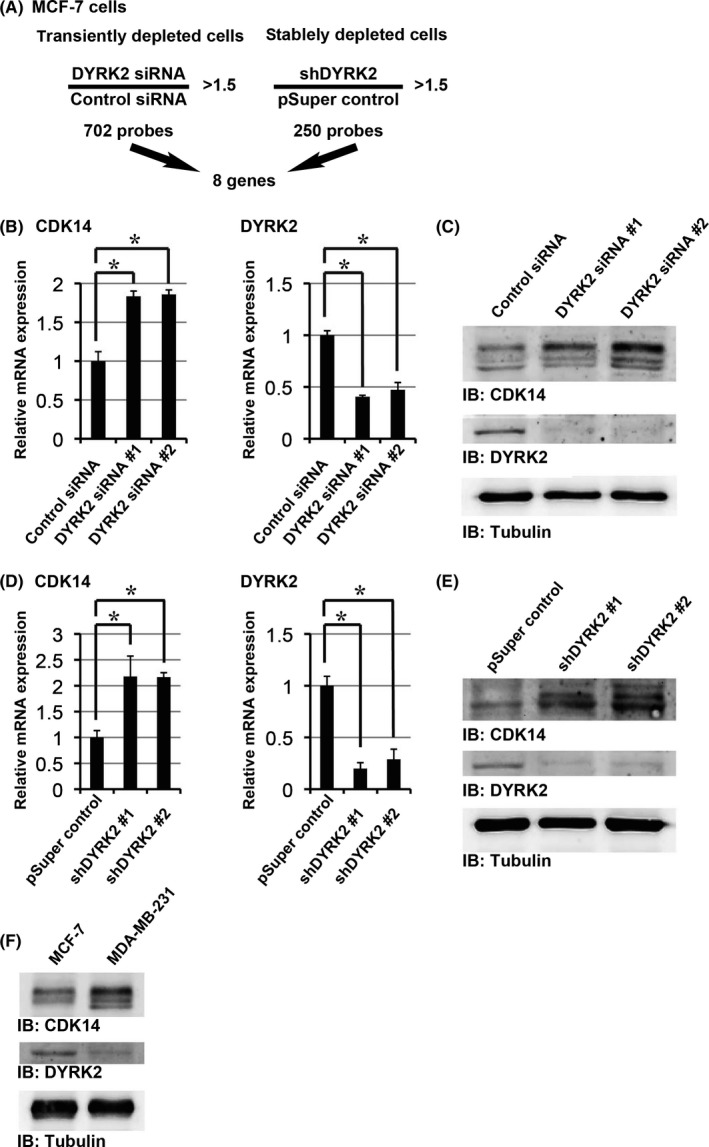
Identification of target genes that are induced in a dual‐specificity tyrosine‐regulated kinase 2 (DYRK2)‐dependent manner. (A) Numbers of genes that are more than 1.5‐fold upregulated by DYRK2 knockdown compared to control cells. Eight genes were identified by microarray analysis after either stable or transient knockdown. (B) MCF‐7 cells were transfected with control siRNA or DYRK2 siRNA #1/#2. The relative mRNA expression was quantitated by real‐time RT‐PCR. Data are presented as the mean ± SD (n = 3; *P < .05). (C) MCF‐7 cells were transfected with control siRNA or DYRK2 siRNA #1/#2. Lysates were analyzed by immunoblotting with the indicated antibodies. (D) The relative mRNA expression in pSuper control, shDYRK2 #1 and shDYRK2 #2 cells were quantitated by real‐time RT‐PCR. Data are presented as the mean ± SD (n = 3; *P < .05). (E) Lysates from the pSuper control, shDYRK2 #1 and shDYRK2 #2 cells were analyzed by immunoblotting with the indicated antibodies. (F) Lysates from the MCF‐7 and MDA‐MB‐231 cells were analyzed by immunoblotting with the indicated antibodies
3.2. Dual‐specificity tyrosine‐regulated kinase 2 expression inversely correlates with tumor cell proliferation
Knocking down DYRK2 accelerated cell growth in MCF‐7 cells (Figure 2A,B). To elucidate the effects of DYRK2 overexpression in a cell line that normally expresses low levels of DYRK2, we generated a stable DYRK2‐overexpressing MDA‐MB‐231 cell line using GFP‐DYRK2 (GFP‐DYRK2 cells). A GFP vector was transfected as a vehicle control (GFP control cells). The mRNA and protein levels of CDK14 in GFP‐DYRK2 cells was significantly lower than that of GFP control cells (Figure 2C,D). The proliferative ability of GFP‐DYRK2 cells was significantly lower than that of GFP control cells (Figure 2E,F). These results suggest that DYRK2 suppresses tumor cell proliferation.
Figure 2.
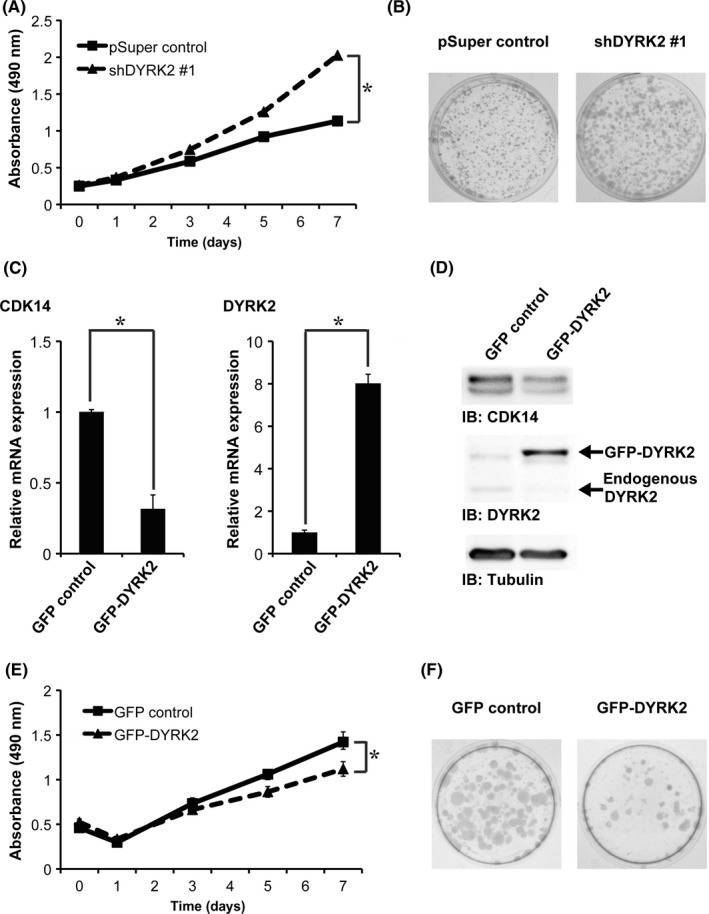
Dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) expression inversely correlates with tumor cell proliferation. (A,B) Proliferation of pSuper control and shDYRK2 #1 cells was analyzed using the MTS assay (A) and the colony formation assay (B). Data are presented as the mean ± SD (n = 3; *P < .05). (C) The relative mRNA expression of GFP control and GFP‐DYRK2 cells was quantitated by real‐time RT‐PCR. Data are presented as the mean ± SD (n = 3; *P < .05). (D) Lysates from the GFP control and GFP‐DYRK2 cells were analyzed by immunoblotting with the indicated antibodies. (E,F) Proliferation of GFP control and GFP‐DYRK2 cells was analyzed using the MTS assay (E) and the colony formation assay (F). Data are presented as the mean ± SD (n = 3; *P < .05)
3.3. Tumor cell proliferation and invasion are regulated by dual‐specificity tyrosine‐regulated kinase 2 via cyclin‐dependent kinase 14 expression
Our previous study showed that silencing of DYRK2 in MCF‐7 cells enhanced cell invasion.5 In xenograft studies, stable depletion of DYRK2 significantly accelerated tumor growth.4 To directly address the role of CDK14 in tumor cell proliferation and invasion, we stably or transiently knocked down endogenous CDK14 in DYRK2‐deficient cells. The shDYRK2 cells were transfected with the pSuper‐neo vector (shDYRK2‐pSuper control cells) or pSuper‐neo CDK14 shRNA (shDYRK2‐shCDK14 cells). The pSuper control cells were transfected with the pSuper‐neo vector (pSuper control‐pSuper control cells) as a vehicle control. The shDYRK2 cells were transiently transfected with control siRNA or CDK14 siRNA. The pSuper control cells were transfected with control siRNA (Figure S1A,B). For the MTS, colony formation and invasion assays, knockdown of CDK14 in shDYRK2 cells significantly decreased cell proliferation and invasive ability (Figure 3A‐C). To determine whether CDK14 plays a crucial role in tumor cell proliferation in vivo, we investigated the effects of DYRK2 in CDK14‐depleted cells in a breast tumor xenograft model. The results revealed that the loss of CDK14 expression significantly inhibited tumor growth in DYRK2‐depleted tumors (Figure 3D). These findings indicate that tumor cell proliferation and invasion are regulated by DYRK2 via CDK14.
Figure 3.

Tumor cell proliferation and invasion are regulated by dual‐specificity tyrosine‐regulated kinase 2 (DYRK2) through modulation of cyclin‐dependent kinase 14 (CDK14) expression. (A,B) Proliferation of pSuper control‐pSuper control, shDYRK2 #1‐pSuper control, shDYRK2 #1‐shCDK14 #1 and shDYRK2 #1‐shCDK14 #2 cells was analyzed using the MTS assay (A) and the colony formation assay (B). Data are presented as the mean ± SD (n = 3; *P < .05). (C) Invasion potential of shDYRK2 #1‐pSuper control and shDYRK2 #1‐shCDK14 #2 cells was analyzed using an invasion assay. Data are presented as the mean ± SD (n = 3; *P < .05). Phase contrast images of invading cells on the membrane. Scale bar represents 100 μm. (D) Tumor growth curves are plotted for shDYRK2 #1‐pSuper control and shDYRK2 #1‐shCDK14 #2 cells. Data are presented as the mean ± SD (n = 4; *P < .05). Representative images of tumor‐bearing nude mice (upper panel) and tumors (lower panel), which were taken 34 days after inoculation. Arrowheads indicate inoculated tumors. Scale bar represents 10 mm
3.4. Androgen receptor is a dual‐specificity tyrosine‐regulated kinase 2‐dependent transcriptional activator of cyclin‐dependent kinase 14
Cyclin‐dependent kinase 14 was found to be regulated at the mRNA level by DYRK2. We therefore explored the transcription factors involved in the regulation of CDK14 expression by DYRK2. We used two approaches to predict the identity of these transcription factors: analysis of upstream transcription factors and binding site analysis. By analyzing the upstream transcription factors, we identified 76 proteins as putative transcription factors that have binding sites in the promoter regions of genes that display expression changes in response to DYRK2. We compared the microarray data from control versus DYRK2‐depleted cells, and predicted upstream transcription factors using the TRANSFAC database (described in the Materials and Methods). We also identified 370 transcription factors that have binding sites less than 3 kb upstream of the CDK14 gene using the MAPPER database (http://genome.ufl.edu/mapper).16 Our previous study showed that AR was a DYRK2‐dependent transcriptional activator.8 In this context, we also identified 5 binding sites of AR within 3 kb upstream of the CDK14 gene using the MAPPER database. AR was also identified by analysis of upstream transcription factors. We thus focused on one of the high‐scoring genes, AR (Figure S2A,B). Real‐time RT‐PCR and western blotting revealed that inhibition of AR activity using the AR inhibitor MDV3100 decreased both mRNA and protein levels of CDK14 in shDYRK2 and GFP control cells (Figure 4A‐D). Of note, silencing or overexpression of DYRK2 had little effect on AR expression in human breast cancer cell lines, such as MCF‐7 and MDA‐MB‐231 (Figure S2C,D). In contast, knocking down DYRK2 induced AR transcription activity in MCF‐7 cells (Figure S2E). We confirmed that DYRK2 depletion in MCF‐7 cells decreased the levels of CDK14 mRNA in a MDV3100 concentration‐dependent manner (Figure 4E). These results imply the possibility that AR acts as a transcriptional activator for CDK14 and is dependent on DYRK2.
Figure 4.
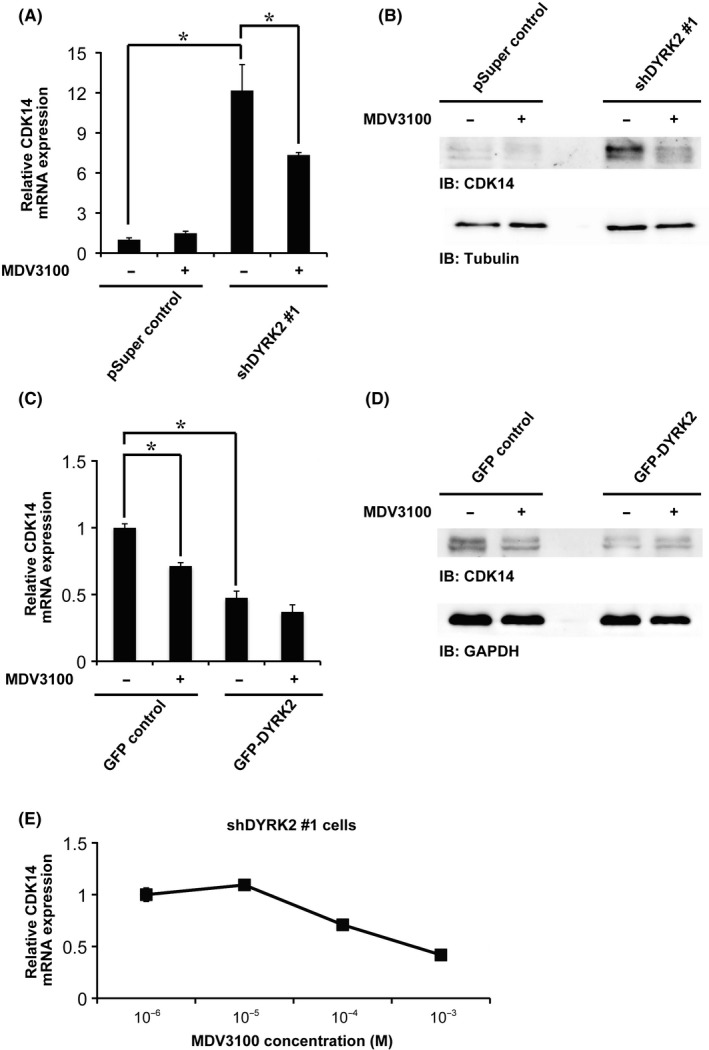
Androgen receptor (AR) is a dual‐specificity tyrosine‐regulated kinase 2 (DYRK2)‐dependent transcriptional activator of CDK14. (A) Relative cyclin‐dependent kinase 14 (CDK14) mRNA expression in pSuper control or shDYRK2 #1 cells with or without treatment with the AR inhibitor MDV3100 (10 μmol/L, 24 h) was quantitated by real‐time RT‐PCR. Data are presented as the mean ± SD (n = 3; *P < .05). (B) Lysates from pSuper control or shDYRK2 #1 cells with or without treatment with the AR inhibitor MDV3100 (10 μmol/L, 24 h) were analyzed by immunoblotting with the indicated antibodies. (C) Relative CDK14 mRNA expression in GFP control or GFP‐DYRK2 cells with or without treatment with the AR inhibitor MDV3100 (10 μmol/L, 24 h), was quantitated by real‐time RT‐PCR. Data are presented as the mean ± SD (n = 3; *P < .05). (D) Lysates from GFP control or GFP‐DYRK2 cells with or without treatment with the AR inhibitor MDV3100 (10 μmol/L, 24 h) were analyzed by immunoblotting with the indicated antibodies. (E) Dose‐response curve of relative CDK14 mRNA expression in shDYRK2 #1 cells treated with the indicated concentration of the AR inhibitor MDV3100 (24 h). Data are presented as the mean ± SD (n = 3; *P < .05)
3.5. Clinical relevance of cyclin‐dependent kinase 14 and dual‐specificity tyrosine‐regulated kinase 2 to breast cancer
To determine the clinical relevance of DYRK2 and CDK14, we examined breast cancer specimens (Figure 5A; Figure S3 and Table S2). Both CDK14 and DYRK2 were detected immunohistochemically in the cytoplasm of cancer cells. We analyzed specimens from 60 patients with invasive ductal carcinomas. We uncovered an inverse correlation between the expression levels of DYRK2 and CDK14 (Figure 5B). Taken together, these findings indicate that DYRK2 suppresses tumor cell proliferation and invasion by inhibiting CDK14 transcription. In contrast, reduced DYRK2 expression in invasive ductal carcinoma likely promotes tumor cell proliferation and invasion by modulating CDK14 expression via AR (Figure 5C).
Figure 5.
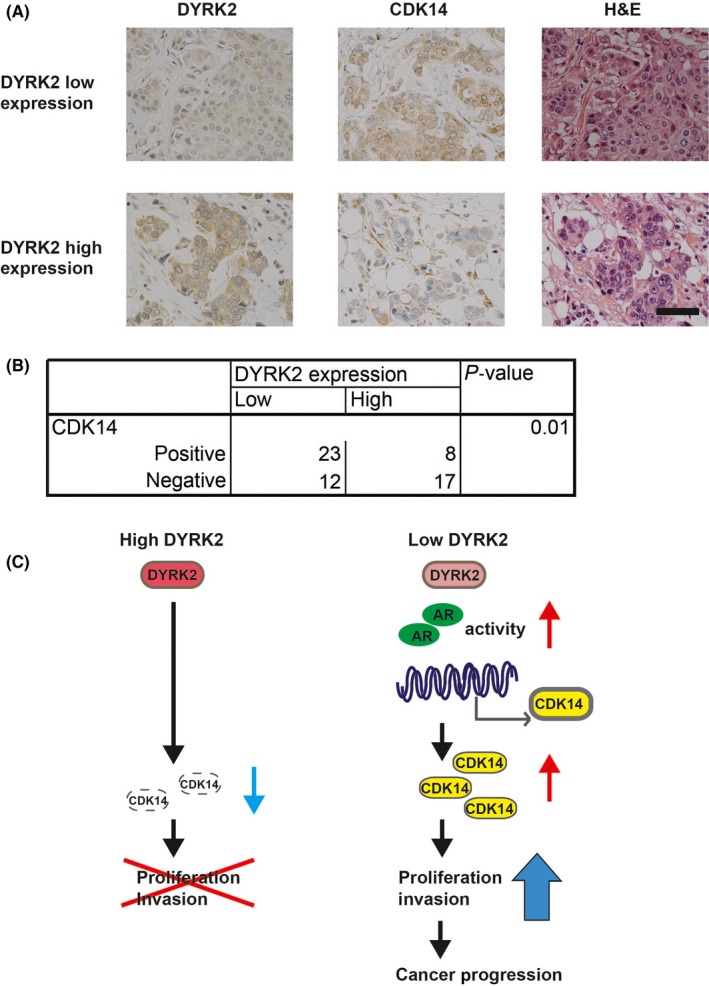
Clinical relevance of cyclin‐dependent kinase 14 (CDK14) and DYRK2 in breast cancer. (A) Breast cancer tissues were immunostained with anti‐DYRK2 or anti‐CDK14 antibodies. Images were obtained at 400 × magnification. Scale bar represents 50 μm. (B) The correlation between DYRK2 and CDK14 expression was assessed. The P‐value was calculated using the χ2‐test. (C) DYRK2 suppresses tumor cell proliferation and invasion by inhibiting CDK14 transcription, and reduced DYRK2 expression in invasive ductal carcinoma promotes tumor cell proliferation and invasion through CDK14 expression via transcriptional regulation by AR
4. DISCUSSION
In this study, we demonstrated that DYRK2 is a regulator of breast cancer progression. DYRK2 suppresses cancer cell proliferation and invasion by inhibiting the transcription of CDK14. The following evidence supports this conclusion. First, CDK14 mRNA and protein levels increased in DYRK2‐depleted cells (Figure 1). Second, low DYRK2 expression was associated with increased proliferation in breast cancer cell lines. In contrast, high DYRK2 expression was associated with decreased cell proliferation (Figure 2). Third, cancer cell proliferation and invasion were suppressed by inhibition of CDK14 expression in DYRK2‐depleted cells (Figure 3). Fourth, it is conceivable that AR could promote transcription of CDK14 in DYRK2‐depleted cells (Figure 4). Finally, CDK14 and DYRK2 expression were inversely correlated in human breast cancer tissues (Figure 5).
Using a microarray, we identified CDK14 as a target of DYRK2, which is, to the best of our knowledge, the first time this has been demonstrated. Previous studies showed that CDK14 is an oncogene.11, 12, 13, 14 It has been reported that upregulated expression of CDK14 promotes tumor cell proliferation, migration and invasion through Wnt/β‐catenin signaling pathway in breast cancer.11 Our results demonstrated that DYRK2 suppresses cancer cell proliferation and invasion through CDK14.
Several studies have demonstrated that DYRK2 expression is significantly reduced in breast cancer, ovarian serous carcinoma, non‐small cell lung cancer, bladder cancer, colorectal cancer, hepatocellular carcinoma and non‐Hodgkin's lymphoma.5, 6, 17, 18, 19, 20, 21, 22, 23 More importantly, diminished DYRK2 expression was correlated with cancer progression and chemotherapy resistance in these cancers. Although DYRK2 has been suggested to have both pro‐tumor and anti‐tumor activities,4, 5, 6, 8, 24, 25 our data clearly demonstrate its role as a tumor suppressor that modulates CDK14 in breast cancer.
Previous studies have also described the knockdown phenotype and downstream targets of CDK14.11, 12, 13, 15 However, our study demonstrated that DYRK2 is an upstream regulator of CDK14. We demonstrate the possibility that AR acts as a transcriptional activator of CDK14, and it is dependent on DYRK2 expression. AR inhibitor partially reduced CDK14 transcription, suggesting that other molecules would be also involved. We previously reported that AR acts as a transcriptional activator of KLF4, and it is also dependent on DYRK2.8 The mechanism underlying DYRK2‐mediated AR promoter binding remains unclear. Further research is needed to uncover this mechanism.
The AR inhibitor MDV3100 affects the androgen receptor signaling pathway in multiple steps: (i) inhibiting binding of androgens to AR; (ii) inhibiting nuclear translocation of AR; and (iii) inhibiting association of AR with DNA.26 When AR is bound to MDV3100, it adopts a conformation incapable of DNA binding or of coactivator recruitment.27 Overall, AR is expressed in approximately 70‐80% of breast cancers.28, 29 A recent review article reported that androgen receptor‐targeted therapies have shown promising preliminary results in breast cancer.30
There was a limitation in the current study. DYRK2 regulates c‐Jun, c‐Myc and Snail expression by promoting ubiquitination and proteasomal degradation.4, 5 These are key molecules of tumor cell proliferation and invasion, and are overexpressed in low DYRK2‐expressing cancer cells. Whereas the effect of CDK14 on tumor cell proliferation and invasion is robust and essential, the influence of c‐Jun, c‐Myc and Snail in DYRK2‐depleted cells cannot be excluded. The combination of these transcriptional factors could accelerate tumor cell proliferation and invasion in DYRK2‐depleted cells.
Our immunohistochemical and clinicopathological analysis was limited mainly due to heterogeneity of patients. Whereas immunohistochemical staining uncovered an inverse correlation between the expression levels of DYRK2 and CDK14, DYRK2 expression was not statistically correlated with several clinical parameters such as lymph invasion or tumor size in this study. The reason could be that the initial treatment of the 60 patients with invasive ductal carcinomas was not only surgery but also chemotherapy.
In conclusion, our results delineate a novel mechanism for cancer cell proliferation and invasion through the DYRK2–AR–CDK14 axis. Restoration of the expression and function of DYRK2 is a potential therapeutic strategy for treating breast cancer.
DISCLOSURE STATEMENT
The authors have no conflict of interest to declare.
Supporting information
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
The authors are grateful to all patients who provided the clinical samples for the study. We thank Naoe T Nihira for microarray analysis, Naoko Tago and Katsuhiko Aoki for in vitro experiments, and Mamiko Owada for immunohistochemistry.
Imawari Y, Mimoto R, Hirooka S, Morikawa T, Takeyama H, Yoshida K. Downregulation of dual‐specificity tyrosine‐regulated kinase 2 promotes tumor cell proliferation and invasion by enhancing cyclin‐dependent kinase 14 expression in breast cancer. Cancer Sci. 2018;109:363–372. https://doi.org/10.1111/cas.13459
Funding information
JSPS KAKENHI Grant Numbers JP26290041, JP17H03584 and JP26861056; Takeda Science Foundation; Vehicle Racing Commemorative Foundation; Research Grant of the Princess Takamatsu Cancer Research Fund; Jikei University Research Fund for Graduate Students.
REFERENCES
- 1. Forouzanfar MH, Foreman KJ, Delossantos AM, et al. Breast and cervical cancer in 187 countries between 1980 and 2010: a systematic analysis. Lancet. 2011;378:1461‐1484. [DOI] [PubMed] [Google Scholar]
- 2. Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell. 2007;25:725‐738. [DOI] [PubMed] [Google Scholar]
- 3. Yoshida K, Miki Y. The cell death machinery governed by the p53 tumor suppressor in response to DNA damage. Cancer Sci. 2010;101:831‐835. [DOI] [PubMed] [Google Scholar]
- 4. Taira N, Mimoto R, Kurata M, et al. DYRK2 priming phosphorylation of c‐Jun and c‐Myc modulates cell cycle progression in human cancer cells. J Clin Invest. 2012;122:859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mimoto R, Taira N, Takahashi H, et al. DYRK2 controls the epithelial–mesenchymal transition in breast cancer by degrading Snail. Cancer Lett. 2013;339:214‐225. [DOI] [PubMed] [Google Scholar]
- 6. Yamaguchi N, Mimoto R, Yanaihara N, et al. DYRK2 regulates epithelial–mesenchymal‐transition and chemosensitivity through Snail degradation in ovarian serous adenocarcinoma. Tumour Biol. 2015;36:5913‐5923. [DOI] [PubMed] [Google Scholar]
- 7. Mimoto R, Nihira NT, Hirooka S, Takeyama H, Yoshida K. Diminished DYRK2 sensitizes hormone receptor‐positive breast cancer to everolimus by the escape from degrading mTOR. Cancer Lett. 2017;384:27‐38. [DOI] [PubMed] [Google Scholar]
- 8. Mimoto R, Imawari Y, Hirooka S, Takeyama H, Yoshida K. Impairment of DYRK2 augments stem‐like traits by promoting KLF4 expression in breast cancer. Oncogene. 2017;36:1862‐1872. [DOI] [PubMed] [Google Scholar]
- 9. Shu F, Lv S, Qin Y, et al. Functional characterization of human PFTK1 as a cyclin‐dependent kinase. Proc Natl Acad Sci U S A. 2007;104:9248‐9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang T, Chen JY. Identification and cellular localization of human PFTAIRE1. Gene. 2001;267:165‐172. [DOI] [PubMed] [Google Scholar]
- 11. Gu X, Wang Y, Wang H, et al. Upregulated PFTK1 promotes tumor cell proliferation, migration, and invasion in breast cancer. Med Oncol. 2015;32:195. [DOI] [PubMed] [Google Scholar]
- 12. Yang L, Zhu J, Huang H, et al. PFTK1 promotes gastric cancer progression by regulating proliferation, migration and invasion. PLoS One. 2015;10:e0140451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang W, Liu R, Tang C, et al. PFTK1 regulates cell proliferation, migration and invasion in epithelial ovarian cancer. Int J Biol Macromol. 2016;85:405‐416. [DOI] [PubMed] [Google Scholar]
- 14. Miyagaki H, Yamasaki M, Miyata H, et al. Overexpression of PFTK1 predicts resistance to chemotherapy in patients with oesophageal squamous cell carcinoma. Br J Cancer. 2012;106:947‐954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Leung WK, Ching AK, Chan AW, et al. A novel interplay between oncogenic PFTK1 protein kinase and tumor suppressor TAGLN2 in the control of liver cancer cell motility. Oncogene. 2011;30:4464‐4475. [DOI] [PubMed] [Google Scholar]
- 16. Riva A. The MAPPER2 Database: a multi‐genome catalog of putative transcription factor binding sites. Nucleic Acids Res. 2012;40:D155‐D161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Enomoto Y, Yamashita S, Yoshinaga Y, et al. Downregulation of DYRK2 can be a predictor of recurrence in early stage breast cancer. Tumour Biol. 2014;35:11021‐11025. [DOI] [PubMed] [Google Scholar]
- 18. Yamashita S, Chujo M, Moroga T, et al. DYRK2 expression may be a predictive marker for chemotherapy in non‐small cell lung cancer. Anticancer Res. 2009;29:2753‐2757. [PubMed] [Google Scholar]
- 19. Nomura S, Suzuki Y, Takahashi R, et al. Dual‐specificity tyrosine phosphorylation‐regulated kinase 2 (DYRK2) as a novel marker in T1 high‐grade and T2 bladder cancer patients receiving neoadjuvant chemotherapy. BMC Urol. 2015;15:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yan H, Hu K, Wu W, et al. Low expression of DYRK2 (Dual Specificity Tyrosine Phosphorylation Regulated Kinase 2) correlates with poor prognosis in colorectal cancer. PLoS One. 2016;11:e0159954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang X, Xu P, Ni W, et al. Downregulated DYRK2 expression is associated with poor prognosis and Oxaliplatin resistance in hepatocellular carcinoma. Pathol Res Pract. 2016;212:162‐170. [DOI] [PubMed] [Google Scholar]
- 22. Wang Y, Wu Y, Miao X, et al. Silencing of DYRK2 increases cell proliferation but reverses CAM‐DR in Non‐Hodgkin's Lymphoma. Int J Biol Macromol. 2015;81:809‐817. [DOI] [PubMed] [Google Scholar]
- 23. Ito D, Yogosawa S, Mimoto R, et al. Dual‐specificity tyrosine‐regulated kinase 2 is a suppressor and potential prognostic marker for liver metastasis of colorectal cancer. Cancer Sci. 2017;108:1565‐1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller CT, Aggarwal S, Lin TK, et al. Amplification and overexpression of the dual‐specificity tyrosine‐(Y)‐phosphorylation regulated kinase 2 (DYRK2) gene in esophageal and lung adenocarcinomas. Cancer Res. 2003;63:4136‐4143. [PubMed] [Google Scholar]
- 25. Guo X, Wang X, Wang Z, et al. Site‐specific proteasome phosphorylation controls cell proliferation and tumorigenesis. Nat Cell Biol. 2016;18:202‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen Y, Clegg NJ, Scher HI. Anti‐androgens and androgen‐depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10:981‐991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tran C, Ouk S, Clegg NJ, et al. Development of a second‐generation antiandrogen for treatment of advanced prostate cancer. Science. 2009;324:787‐790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Park S, Koo J, Park HS, et al. Expression of androgen receptors in primary breast cancer. Ann Oncol. 2010;21:488‐492. [DOI] [PubMed] [Google Scholar]
- 29. Collins LC, Cole KS, Marotti JD, Hu R, Schnitt SJ, Tamimi RM. Androgen receptor expression in breast cancer in relation to molecular phenotype: results from the Nurses' Health Study. Mod Pathol. 2011;24:924‐931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kono M, Fujii T, Lim B, Karuturi MS, Tripathy D, Ueno NT. Androgen receptor function and androgen receptor‐targeted therapies in breast cancer: a review. JAMA Oncol. 2017;3:1266‐1273. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials