

Abstract
Although several studies have exploited the effects of PB1-F2 in swine influenza viruses, its contribution to the pathogenicity of swine influenza viruses remains unclear. Herein, we investigated the effects of PB1-F2 on the pathogenicity of influenza virus using a virulent H1N1 A/swine/Kansas/77778/2007 (KS07) virus, which expresses a full-length PB1-F2, in mice and pigs. Using reverse genetics, we generated the wild-type KS07 (KS07_WT), a PB1-F2 knockout mutant (KS07_K/O) and its N66S variant (KS07_N66S). KS07_K/O showed similar pathogenicity in mice to the KS07_WT, whereas KS07_N66S displayed enhanced virulence when compared to the other two viruses. KS07_WT exhibited more efficient replication in lungs and nasal shedding in infected pigs than the other two viruses. Pigs infected with the KS07_WT had higher pulmonary levels of granulocyte-macrophage colony-stimulating factor, IFN-γ, IL-6 and IL-8 at 3 and 5 days post-infection, as well as lower levels of IL-2, IL-4 and IL-12 at 1 day post-infection compared to those infected with the KS07_K/O. These results indicate that PB1-F2 modulates KS07 H1N1 virus replication, pathogenicity and innate immune responses in pigs and the single substitution at position 66 (N/S) in the PB1-F2 plays a critical role in virulence in mice. Taken together, our results provide new insights into the effects of PB1-F2 on the virulence of influenza virus in swine and support PB1-F2 as a virulence factor of influenza A virus in a strain- and host-dependent manner.
Keywords: PB1-F2, swine influenza H1N1 virus, pathogenicity, mouse and pig
Introduction
Influenza A virus (IAV) is a single-stranded negative-sense RNA virus of the family Orthomyxoviridae that infects a wide variety of host species, from wild and domestic birds to several mammalian species including humans. It causes annual epidemics and occasional pandemics in humans, which leads to significant public health and economic burdens worldwide. Swine influenza is caused by IAV and is an important acute respiratory disease to the swine industry and has also become a considerable zoonotic agent that threatens public health [1, 2]. This fact is exemplified by the 2009 pandemic H1N1 influenza virus, which is considered to be a swine-origin virus [3].
The first swine influenza virus (SIV) isolated from pigs was the H1N1 subtype in 1930 [4], also known as the classical H1N1 virus (cH1N1), which circulated predominantly in North American swine herds until 1998. Then, a novel triple-reassortant (TR) H3N2 SIV emerged in 1998 and successfully became established and widespread in North American pig populations [5, 6]. The TR H3N2 virus contains the haemagglutinin, neuraminidase (NA) and PB1 genes from human IAVs; the PB2 and PA genes from avian IAVs; and the M, NP and NS genes from the cH1N1 virus [5, 6]. Subsequently, TR H3N2 viruses reassorted with cH1N1 SIVs and human seasonal H1N1 viruses, resulting in novel reassortants such as H1N2, reassortant H1N1 (rH1N1) and H3N2 SIVs, which have become endemic in North American swine herds [7–10]. These reassortant viruses have a similar composition of six internal genes of human origin (PB1), avian origin (PB2 and PA) and classical swine origin (M, NS and NP), which has been called the triple-reassortant internal gene cassette (TRIG) [10, 11]. The TRIG cassette is readily able to accept different haemagglutinin and NA combinations [10]. With the introduction of the 2009 pandemic H1N1 virus into pigs, the TRIG M gene was replaced by the Eurasian swine M gene [12, 13].
IAV contains eight viral RNA segments, which encode 10–17 viral proteins depending on the strain. The PB1-F2 protein discovered in 2001 is a small non-structural protein that is encoded by alternative translation from the PB1 genome [14]. Although it can have various lengths from 8 to 101 amino acids, it generally consists of 90 amino acids. The PB1-F2 protein is known to be an important virulence factor with diverse functions. It is localized into the mitochondria and induces apoptosis by interacting with the mitochondrial-dependent apoptotic pathway [14, 15]. In most avian IAVs, PB1-F2 proteins are expressed in their full-length form (over 78 amino acid residues), while human and swine influenza viruses commonly express truncated PB1-F2 proteins. This suggests that truncated PB1-F2 proteins could be beneficial for adaptation of the virus to the mammalian host [16, 17]. Overall, the PB1-F2 protein is associated with the survival advantage of IAV in different hosts and modulates virus pathogenicity in a strain- and host-dependent manner. A previous study has shown that restoring the PB1-F2 ORF in the context of the 2009 pandemic H1N1 virus has minimal effects in swine [18]. Although numerous studies that investigate the effects of PB1-F2 on virus pathogenicity and transmissibility of different IAVs in different animal models have been published [19–23], the contribution of the PB1-F2 protein to the viral pathogenicity of IAVs in natural hosts such as birds and pigs remains unclear.
In this study, we used a virulent TR H1N1 SIV, A/swine/Kansas/77778/2007 (KS07), which has a human-origin PB1 gene and expresses a full-length PB1-F2 protein, to investigate the effects of PB1-F2 on viral pathogenicity in a mouse model and in a natural host, i.e. pigs.
Results
Generation and characterization of wild-type KS07 and its mutated viruses in vitro
KS07_WT and its mutated viruses including both KS07_K/O and KS07_N66S were generated by reverse genetics and confirmed by sequencing. Since the N66S substitution in PB1-F2 protein is associated with enhanced virulence in mice as shown previously [24], we generated the mutant KS07_N66S virus to determine whether the effect of this mutation can be applied to the swine-adapted KS07 virus either in mice or in pigs. The rescued three viruses replicated efficiently in Madin–Darby canine kidney (MDCK) cells, and amplified viruses were used for further characterization. The plaque assay showed that KS07_WT as well as both mutated KS07_K/O and KS07_N66S viruses formed similar small pinpoint plaques in MDCK cells (Fig. 1a). KS07_WT and its two mutated viruses grew efficiently in both MDCK and PK-15 cells, and their titres reached approximately 108 TCID50 ml−1; however, no significant difference was observed among the three viruses in their growth kinetics (Fig. 1b). In MDCK cells, all three viruses reached their peak virus titre at 24 hours post-infection (p.i.), while in PK-15 cells, they showed their peak virus titre at 48 h p.i. (Fig. 1b). These results indicate that both PB1-F2 knockout and single substitution N66S in the PB1-F2 do not affect virus replication and phenotype in tested cells.
Fig. 1.

Plaque morphology and growth kinetics of the recombinant influenza viruses and alignment of C-terminal sequences of PB1-F2 from selected IAVs. (a) Plaque sizes formed by recombinant viruses in MDCK cells at 3 days p.i. (b) Growth kinetics of indicated recombinant viruses in MDCK or PK-15 cells infected at an m.o.i. of 0.001. Each data point on the curve indicates the mean of the results in triplicate, and the error bars indicate sem. (c) Amino acid sequences of the C-terminal region in KS07 PB1-F2 protein were aligned to those of the last-century pandemic strains (H1N1 1918, H2N2 1957 and H3N2 1968), a previously characterized strain (PR8 H1N1) and one H5N1 highly pathogenic avian influenza A virus. Amino acid residues with bold letters have been identified as inflammatory residues (L62, R75, R79 and L82) or cytotoxic residues (I68, L69 and V70), respectively. Numbers with asterisks indicate amino acid positions according to PB1-F2 numbering.
PB1-F2 in KS07 does not affect virus replication and pathogenicity in mice, while KS07_N66S enhances virulence in mice
As the KS07 H1N1 SIV is not a mouse-adapted virus, we infected BALB/c mice with a high dose (1.5×106 TCID50/mouse) of each virus to evaluate the effect of PB1-F2 on viral pathogenicity. All infected mice showed clinical symptoms such as depression, decreased activities, ruffled fur and obvious weight loss starting at 2 p.i. when compared to the control mice. In contrast, KS07_N66S induced more severe body weight loss than both KS07_WT and KS07_K/O viruses, resulting in 100 % mortality by 8 days p.i. (Fig. 2a, b). Both KS07_WT and KS07_K/O caused 75 % mortality in infected mice (Fig. 2b); however, surviving mice infected with KS07_K/O started to recover 2 days earlier (6 days p.i.) compared to those infected with KS07_WT (8 days p.i.) (Fig. 2a). Interestingly, KS07_K/O replicated to a significantly higher virus titre in mouse lungs than the KS07_N66S at 3 days p.i., whereas virus titres in the mouse lungs were similar among three infection groups at 7 days p.i. (Fig. 3a). Histopathological analysis showed that all three infection groups exhibited more severe lung damage at 7 days p.i. when compared to that at 3 days p.i. despite no significant differences of histopathological scores observed between different groups at each time point (Fig. 3b). Most mice had varying degrees of neutrophilic airway inflammation, airway epithelial degeneration and necrosis, peribronchiolar lymphocytic cuffing, interstitial pneumonia and epithelial hyperplasia (Fig. 4). A small number of mice (n=6) also had hyaline membranes lining alveoli (Fig. 4). NP antigen deposition of IAV was detected in lung tissues from each mouse in all three infection groups at 3 and 7 days p.i. by immunohistochemistry (IHC) staining (Fig. 4).
Fig. 2.

Contribution of PB1-F2 to pathogenicity of recombinant KS07 in BALB/c mice. (a) Average body weights of surviving mice in each group up to 14 days p.i. are represented as percentages of the original weight on day 0. Letters a and/or b on the value point of each day address significant differences (P<0.05) between infected groups (a, KS07_WT and KS07_N66S; b, KS07_K/O and KS07_N66S). The error bars indicate sem. (b) Survival rates of mice infected with indicated viruses.
Fig. 3.

Lung virus replication and histopathological scores of mouse study. (a) Lung virus titres [10% (w/v) lung homogenate] of infected mice were determined at 3 and 7 days p.i. by calculating the TCID50 ml−1 in MDCK cells. The error bars represent sem. (b) Microscopic lung scores are presented as average scores±sem of three mice in each group at 3 and 7 days p.i. The asterisks (*) indicate a statistically significant difference between groups (P<0.05).
Fig. 4.
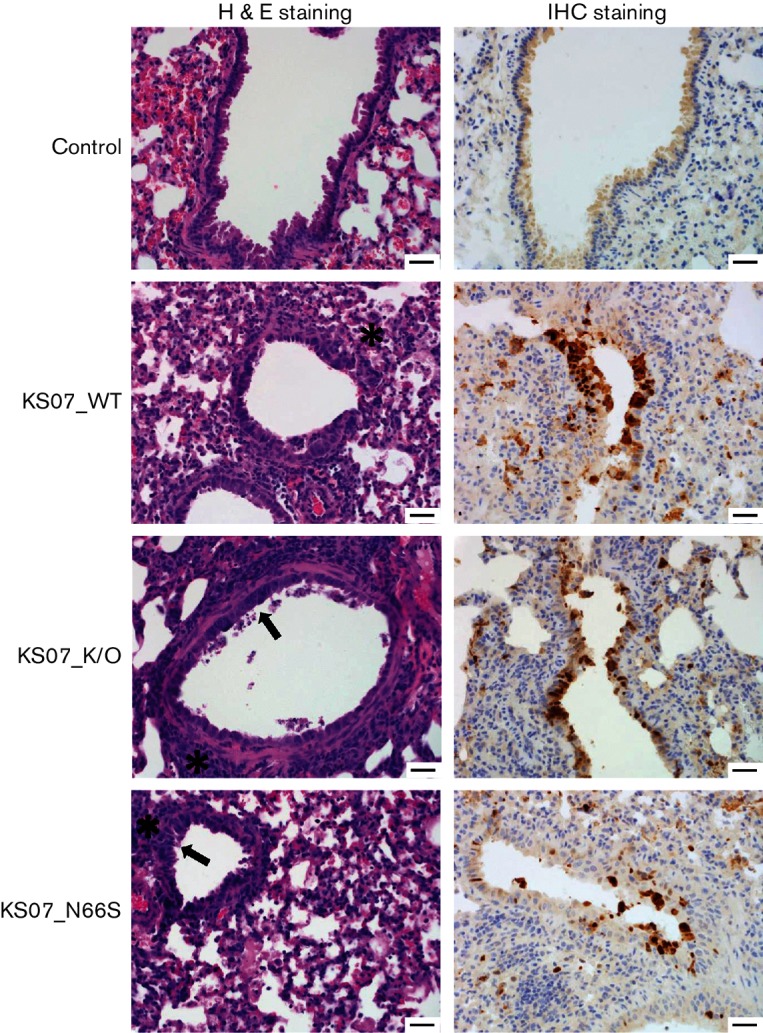
Haematoxylin and eosin and IHC staining of mouse lung sections at 7 days p.i. Haematoxylin-and-eosin-stained sections of mouse lungs infected with the indicated viruses at 7 days p.i. showed typical influenza pneumonia. Pictures are representative sections. In the control, no lesions are present, and there is no antigen deposition in the airway epithelium. In KS07_WT, there are areas of multifocal mild interstitial pneumonia (asterisk), and there is cuffing of bronchioles and vessels by lymphocytes and plasma cells. In KS07_K/O and KS07_N66S, there is mild epithelial hyperplasia (arrow), mild lymphocytic peribronchiolar cuffing (asterisk) and small amounts of fibrin and increased alveolar macrophages in alveolar lumina. IHC staining of lung sections was also conducted to detect influenza virus antigen using an anti-influenza A NP mAb (brown staining). All three treatment groups have positive antigen labelling for influenza NP in the nucleus of airway epithelial cells. Bars, 100 µm. H&E, Haematoxylin and eosin.
PB1-F2 has moderate effects on viral pathogenicity in pigs
A previous study showed that minimal effects were observed in pigs inoculated with a high dose (105 TCID50 per pig) of the 2009 pandemic H1N1 virus expressing either full-length or non-functional PB1-F2 protein [18], and our former studies also demonstrated that the 2009 pandemic H1N1 is virulent and causes disease in pigs [25]. As the KS07 virus is virulent in pigs, we infected pigs with a low dose (103 TCID50 per pig) of each virus to investigate the effect of PB1-F2 and single substitution N66S in PB1-F2 on viral pathogenicity. None of the infected pigs presented obvious respiratory symptoms such as nasal secretion, coughing or sneezing. Several pigs in each infection group (5/9 pigs in KS_WT group, 6/9 pigs in KS07_K/O or in KS07_N66S group) had mild fever starting at 1 day p.i. and lasting 1–2 days; only one pig in the KS07_N66S group had a fever lasting for 5 days. During necropsy, no gross lung lesions were observed in any of the three pigs infected with either the KS07_WT or KS07_K/O virus at 1 day p.i., whereas two out of three KS07_N66S-infected pigs exhibited minimal gross lung lesions (averaged less than 1 %) (Fig. 5a). Interestingly, KS07_K/O induced more macroscopic lesions in infected pigs which spread to five or six lung lobes at 3 days p.i. when compared to the other two viruses. At 5 days p.i., lung lesions were present in each lung lobe of each infected pig of three groups, but fewer lesions were observed in the pigs infected with the KS07_N66S virus (Fig. 5a).
Fig. 5.

Contribution of PB1-F2 to pathogenicity of recombinant KS07 in pigs. (a) Macroscopic lung lesions of infected pigs are shown as the mean percentage±sem of gross lesions of three pigs in each group at 1, 3 and 5 days p.i. Mean of virus titres in bronchoalveolar lavage fluid (BALF) (b) and in nasal swabs (c) of infected pigs on the days indicated. The number of pigs with positive virus isolation out of the total number of tested pigs is shown above each bar in (b, c).
All three recombinant viruses replicated in the lungs of infected pigs. Virus was detected from bronchoalveolar lavage fluid (BALF) samples collected at 1, 3 and 5 days p.i. from all infected pigs with KS07_WT (Fig. 5b). In contrast, virus was detected in BALF from only two of three pigs at 1 and 3 days p.i. in the KS07_K/O-infected group and from all three pigs at 5 days p.i. On the other hand, virus was detected in BALF from two of three pigs in the KS07_N66S-infected group at 1 and 5 days p.i. and from all three infected pigs at 3 days p.i. (Fig. 5b). Although virus titres were variable between different groups, there were no significant differences of virus titres between the three infected groups. No virus was detected in nasal swab samples collected from infected pigs of each group at 1 day p.i. (Fig. 5c). Virus nasal shedding was detected from five of six pigs infected with the KS07_WT virus and from four out of six pigs infected with either the KS07_K/O or the KS07_N66S virus at 3 days p.i. Virus was found in nasal swabs collected from all three infected pigs of each group at 5 days p.i. (Fig. 5c). Virus titre of nasal shedding of the KS07_WT group was higher than those of the other two infection groups despite no significant differences in virus titres observed among infected groups.
Nasal turbinate, trachea and lungs collected from pigs necropsied at 1, 3 and 5 days p.i. were examined for histopathological analysis (Table 1, Fig. 6). Microscopic lesions were not found in these three tissues of control pigs at the three time points. All influenza-infected pigs showed pulmonary lesions characterized by varying degrees of interstitial pneumonia, bronchiolar epithelial necrosis and bronchiolitis with peribronchiolar lymphocytic cuffing and airway epithelial hyperplasia and had different degrees of tracheal (Fig. 6) and nasal cavity inflammation and necrosis. Microscopic lesions were found in the lung tissues of all three pigs infected with the KS07_WT virus at 1 day p.i.; however, none of the three pigs infected with the KS07_N66S had lung lesions, and two of three pigs infected with the KS07_K/O displayed lesions in lung tissues (Table 1). Microscopic lesions were observed in trachea tissues of all three pigs infected with the KS07_WT virus at 1 day p.i., but only one of three pigs infected with either the KS07_N66S or the KS07_K/O had lesions in the trachea (Table 1). Lung lesions were found in three or two pigs infected with the KS07_K/O at 3 and 5 days p.i., respectively; one or no pigs in the other two infection groups showed microscopic lung lesions at 3 and 5 days p.i. One or two pigs from each infected group showed lesions in nasal turbinate tissues at 1 and 3 days p.i., while no pigs had lesions in this tissue at 5 days p.i. (Table 1). In contrast to both KS07_WT and KS07_K/O viruses, the KS07_N66S virus induced less histopathological lung and trachea damage in pigs (Table 1).
Table 1. Microscopic lesions of respiratory tract and lungs in infected and control pigs at 1, 3, and 5 days p.i.
Results are presented as the number of pigs with microscopic lesions out of the total number of pigs on the indicated days. Three different tissues in the respiratory tract were evaluated. Numbers in parentheses indicate the mean score of microscopic lesions±sem. NT, Nasal turbinate.
| Virus | No. of pigs with microscopic lesions/total no. of pigs euthanized on the indicated day (microscopic lesion scores) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 day p.i. | 3 days p.i. | 5 days p.i. | |||||||
| Lung | Trachea | NT | Lung | Trachea | NT | Lung | Trachea | NT | |
| KS07_WT | 3/3 (0.80±0.23) | 3/3 (0.50±0.00) | 1/3 (0.50) | 1/3 (1.60) | 2/3 (1.25±0.20) | 1/3 (0.50) | 1/3 (1.60) | 0/3 | 0/3 |
| KS07_K/O | 2/3 (1.70±0.90) | 1/3 (1.00) | 2/3 (0.50±0.00) | 3/3 (1.33±0.29) | 1/3 (0.50) | 1/3 (0.50) | 2/3 (0.7±0.41) | 1/3 (1.00) | 0/3 |
| KS07_N66S | 0/3 | 1/3 (0.50) | 2/3 (0.50±0.00) | 1/3 (1.60) | 1/3 (0.50) | 1/3 (0.50) | 0/3 | 0/3 | 0/3 |
| Control | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/3 | 0/2 | 0/2 | 0/2 |
Fig. 6.
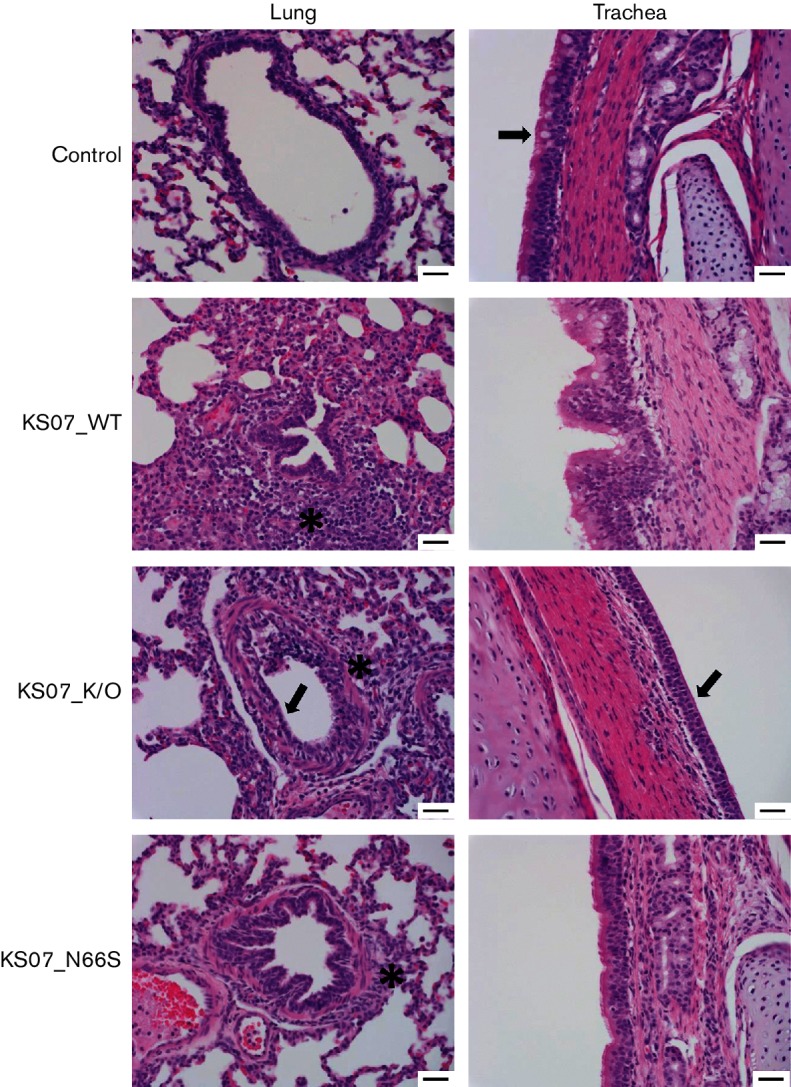
Haematoxylin and eosin staining of pig lung and trachea sections at 3 days p.i. Lung and trachea sections of pigs infected with indicated viruses at 3 days p.i. were stained with haematoxylin and eosin. Pictures are representative sections. In the control, the lung is normal, which is demonstrated by normal airway epithelium and alveoli, and tracheal epithelium has goblet cells and cilia (arrow). In KS07_WT, the bronchiole is surrounded by lymphocytes and plasma cells, which extend into the alveolar septa (interstitial pneumonia) (asterisk). In KS07_K/O, there is flattening of airway epithelium (arrow) and cuffing by peribronchiolar cuffing by lymphocytes (asterisk). The trachea in this pig has diffuse loss of cilia and goblet cells consistent with early degeneration due to influenza (arrow). In KS07_N66S, there is cuffing of bronchioles and vessels by lymphocytes and plasma cells (asterisk), and the trachea lacks goblet cells but has a normal layer of cilia. Bars, 100 µm.
PB1-F2 modulates host immune responses in pigs
To investigate the effects of PB1-F2 expression in KS07 on the host immune response, we next assessed the pulmonary levels of cytokines and chemokines in BALF collected from each pig at 1, 3 and 5 days p.i. (Fig. 7). All measured chemokine and cytokine levels in BALF from KS07_N66S-infected pigs were similar to those of control pigs except for IL-1β and IL-6 at 5 days p.i. All tested cytokine/chemokines except TNF-α in KS07_WT-infected pigs and six cytokines/chemokines including IL-1β, IL-2, IL-4, IL-6, IL-10 and IL-12 in KS07_K/O-infected pigs were significantly higher compared to those detected in control pigs at 3 or 5 days p.i. The KS07_WT-infected group showed lower levels of all tested cytokines/chemokines including significantly lower expression of IL-2, IL-4 and IL-12 than the KS07_K/O-infected group at 1 day p.i. In contrast, KS07_WT-infected pigs had higher levels of all tested cytokines/chemokines including significantly higher levels of granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-γ, IL-6 and IL-8 than those in the KS07_K/O-infected pigs at 3 or 5 days p.i. Collectively, N66S substitution in PB1-F2 protein of the KS07 virus had a minor effect on innate immune responses in pigs. On the other hand, PB1-F2 protein expression suppressed innate immune responses at early time points, while PB1-F2 protein upregulated cytokine/chemokine expression at later time points in pigs. These data indicate that PB1-F2 modulates immune responses in pigs during viral infection with the KS07 virus.
Fig. 7.

Cytokine/chemokine levels in BALF of infected and control pigs. The expressions of the indicated 12 porcine cytokine/chemokines in BALF were quantified using the Luminex technology. Data represent the average values±sem of three pigs in each group on the days indicated. The hash symbol (#) represents a statistically significant difference with the control group (P<0.05). The asterisks (*) indicate a statistically significant difference between virus-infected groups (*P<0.05, **P<0.01 and ***P<0.001).
Discussion
The PB1-F2 protein is an accessory protein encoded from the +1 ORF in PB1 gene through leaky ribosomal scanning. It has been shown to play important roles in IAV replication, which include cell-type- and virus-strain-specific proapoptotic functions [14, 15, 26, 27] and regulation of polymerase activity by co-localization as well as interacting with PB1 [28, 29]. Furthermore, the PB1-F2 protein modulates the innate immune response and pro-inflammatory reactions, which exacerbate or attenuate IAV pathogenicity [20, 30]. It also facilitates secondary bacterial infection in a mouse model [31, 32]. It has been shown that the PB1-F2 protein has effects on viral pathogenicity in a strain- and host-dependent manner. In H5N1 highly pathogenic avian influenza A viruses (HPAIVs), the PB1-F2 protein has shown different contributions to viral pathogenicity in different hosts such as mice, ducks and chickens [19, 20]. In addition, restoring PB1-F2 in the 2009 pandemic H1N1 influenza virus has minimal effects in pigs [18]. In TR H3N2 SIVs, PB1-F2 expression has strain-dependent effects in swine and altered viral pathogenicity and transmission in turkeys [21, 22].
Although previous studies have shown the effects of PB1-F2 in low-virulent SIVs in pigs [21, 22] and a former study has suggested a very low level of PB1-F2 expression in SIV-infected cells [33], the effect of PB1-F2 protein in a highly virulent SIV on viral pathogenicity in different animal models remains unclear. Therefore, in this study, we selected a highly virulent H1N1 TR SIV that causes approximately 10 % mortality in finishing pigs [34] and expresses a full-length PB1-F2 protein. Our results showed that the expression of PB1-F2 and the presence of the N66S mutation in the KS07 virus had no effect on virus replication in both MDCK and PK-15 cells as well as on the formation of plaques in MDCK cells (Fig. 1). These results are in agreement with a previous study that showed that PB1-F2 expression in TR H3N2 SIVs did not impact virus replication in A549 and 3D4/31 cells [21]. In contrast, Pena et al. described that PB1-F2 expression in two different TR H3N2 SIV backgrounds had a different impact on virus replication kinetics in porcine alveolar macrophages and porcine respiratory explants [22]. These findings support the hypothesis that the effects of PB1-F2 are cell type and virus strain specific in vitro.
In mice, PB1-F2 expression in 1918 H1N1 virus enhanced viral pathogenicity, and the N66S mutation in PB1-F2 protein of this virus contributed to increased virulence [24, 31]. Another previous study found that expression of full-length or truncated PB1-F2 protein or its N66S substitution in the 2009 pandemic H1N1 virus does not have a significant impact on virulence in both DBA/2 and BALB/c mice [35]. In addition, the N66S substitution in the PB1-F2 protein of H5N1 HPAIV showed increased replication and virulence, while deletion of the PB1-F2 protein had no effect on viral pathogenicity in mice [19]. In our mouse study, the PB1-F2 knockout mutant did not alter viral virulence when compared to the KS07_WT virus; however, the single substitution N66S in PB1-F2 resulted in enhanced virulence in mice, evidenced by induced severe disease and 100 % mortality in infected mice when compared to either KS07_WT or KS07_K/O virus (Fig. 2). In full-length PB1-F2, the N66S mutation has been shown to contribute to viral pathogenicity and delaying type I IFN response [19, 24]. The type I IFN suppression activity of PB1-F2 is due to inhibition of the early IFN-stimulated genes as well as decreasing mitochondrial membrane potential via interacting with the mitochondrial antiviral signalling protein [24, 36, 37]. Consistent with previous studies, it could be possible that the N66S substitution in KS07 virus leads to enhanced type I IFN antagonist activity of the PB1-F2, thereby increasing viral virulence.
In pigs, previous studies using low-virulent TR H3N2 SIVs have revealed that deletion of the PB1-F2 ORF or incorporation of the N66S mutation in the PB1-F2 ORF does not influence virus replication, shedding or pathogenicity in pigs [21, 22]. In contrast, the deletion of the PB1-F2 protein or N66S substitution in virulent KS07 H1N1 virus has some degree of influence on pathogenicity in swine, i.e. both mutated viruses showed less efficient virus replication in lungs at 1 day p.i. and nasal shedding at 3 and 5 days p.i. (Fig. 5b, c), and induced histopathologic lesions in both lung and trachea tissues from less infected pigs at 1 day p.i. when compared to the KS07_WT virus (Table 1). One recent study demonstrated that swine IAVs express a very low level of PB1-F2 protein relative to human isolates [33]. In particular, Buehler et al. [33] showed that A/swine/Ohio/511445/2007 (H1N1), which has the highest nucleotide sequence identity (99.7–99.9 %) to the virus genome of KS07 virus [34], expressed PB1-F2 protein from fewer than 1 % of the infected cells and PB1-F2 could not be detected in virus-infected cells, suggesting that carrying a full-length PB1-F2 ORF does not serve as a predictor for sufficient PB1-F2 protein expression. This could explain why no significantly different phenotypes were observed between wild-type and PB1-F2 knockout viruses in pigs in this study or in former studies. The effect of PB1-F2 protein on virus replication and host immune response needs to be investigated in future studies when it is sufficiently expressed in SIV-infected cells.
The pro-inflammatory cytokine response is crucial for recruiting effector cells to the site of infection to clear virus. The PB1-F2 protein has been shown to influence viral pathogenicity by modulating the host innate immune response, which helps to promote lung inflammation [30, 32]. Our pig study showed that decreased levels of all cytokines/chemokines as well as significantly lower levels of IL-2, IL-4 and IL-12 at early time points (1 day p.i.) and significantly increased levels of GM-CSF, IFN-γ, IL-6 and IL-8 at later time points (3 or 5 days p.i.) were found in lungs of pigs infected with the KS07_WT compared to what were observed for the KS07_K/O (Fig. 7). Downregulated and upregulated cytokines/chemokines at early and later time points seem to correlate with observed severity of microscopic lesions in the lung and trachea tissues in both KS07_WT and KS07_K/O groups (Fig. 6, Table 1). Our results suggest that PB1-F2 modulates the host immune response in swine throughout the course of infection. These results are in disagreement with the findings of a former study which showed that PB1-F2 modulation of host immune responses only occurs shortly after infection (24 h p.i.) [18]. This discrepancy might be due to the difference in the virus strain used in the studies. In our study, we used a virulent North American TR H1N1 virus, rather than the 2009 pandemic H1N1 virus which contains Eurasian swine-origin NA and M genes.
The presence of specific amino acid residues, known as inflammatory residues or cytotoxic residues, at the C-terminal region of the PB1-F2 protein is linked to increased pathogenicity and secondary bacterial infection [24, 32, 37, 38]. The PB1-F2 sequence of KS07 contains only two pro-inflammatory residues (L62 and L82) and none of the cytotoxic motifs, whereas H5N1 HPAIVs and all three pandemic IAVs from the last century (H1N1 1918, H2N2 1957 and H3N2 1968) include three or more of the virulence genetic markers (Fig. 1c). In addition, the C-terminal part of the PB1-F2 protein contains a mitochondrial targeting sequence, which allows the PB1-F2 protein to localize within the mitochondria [39, 40]. This localization allows PB1-F2 to interact with mitochondrial membrane-related proteins followed by mitochondrial permeability alteration, resulting in the induction of apoptosis [26, 41]. One recent report revealed that a translocated full-length PB1-F2 protein in the mitochondria attenuated the mitochondrial membrane potential, which suppressed mitochondrial-mediated innate immunity, such as retinoic-acid-inducible gene 1 signalling pathway, and also inhibited the activation of NLRP3 inflammasomes, while a truncated PB1-F2 protein that lacks a C-terminal region was diffused in cytoplasm and did not impact mitochondrial functions [42]. Consistent with these findings, all cytokines/chemokines at 1 day p.i. in lungs of pigs infected with the KS07_WT in our studies showed lower levels than those of pigs infected with the KS07_K/O (Fig. 7). It is possible that full-length PB1-F2 expression in KS07_WT negatively regulated mitochondrial-mediated innate immunity and that may affect the secretion of cytokines/chemokines in virus-infected pigs at early post-infection time points.
Another previous study demonstrated that different secondary structural forms were observed at the C-terminal region of the PB1-F2 protein based on amino acid sequences under membranous solution conditions. One continuous C-terminal α-helix was seen in less-virulent strains, while a divided C-terminal α-helix was located in the PB1-F2 proteins of HPAIVs [43]. These structural differences at the C-terminal region of PB1-F2 may allow it to serve as different danger signals and induce different immune reactions in the host. It remains to be determined whether the KS07 PB1-F2 has the C-terminal α-helix structure and how that structure affects immune responses in different hosts.
In summary, our data indicate that PB1-F2 expression in virulent H1N1 KS07 SIV has effects on virus replication and pathogenicity in the natural host, pigs, but not in mice. In addition, PB1-F2 expression modulates host immune responses in pigs, and the substitution N66S in PB1-F2 plays a critical role in virulence in mice, while no effect was found in pigs. Our results provide new insights into the impact of PB1-F2 on virulence of IAVs in swine and support PB1-F2 as a virulence factor of IAV in a strain- and host-dependent manner.
Methods
Cells and virus strain
Human embryonic kidney (293T) cells were maintained in Opti-modified minimal essential medium (MEM) containing 10 % (v/v) FBS (Hyclone) and 1 % (v/v) antibiotic/antimycotic (Gibco 100X). MDCK cells were cultured in MEM with 5 % (v/v) FBS, 1 % (v/v) antibiotic/antimycotic, 2 mM l-glutamine (Gibco) and 1× MEM vitamin solution (Gibco). Porcine kidney (PK-15) cells were grown in Dulbecco's modified Eagle medium supplemented with 5 % (v/v) FBS, 2 mM l-glutamine, 1× MEM vitamin solution and 1 % (v/v) antibiotic/antimycotic. MEM infecting media containing 0.3 % (w/v) BSA (Sigma-Aldrich), 1 % (w/v) antibiotic/antimycotic and 1 µg ml−1 TPCK-treated trypsin (Sigma-Aldrich) were used for virus infection of cells. The TR H1N1 SIV A/swine/Kansas/77778/2007 (KS07) (GenBank accession number: GQ484355-GQ484362) was isolated from diseased pigs as previously described [34] and was used for this study.
Plasmid constructions and rescue of recombinant viruses
Eight-gene segments of KS07 were amplified and cloned into a pHW2000 vector as described previously [44, 45]. To generate the KS07 PB1-F2 knockout gene, the PB1-F2 expression initiation start codon and three possible downstream start codons were mutated from ATG to ACG (T120C, T234C, T255C and T270C), while one stop codon was mutated from TAA to TTA (A390T) using a GeneArt site-directed mutagenesis kit (Invitrogen) according to the manufacturer’s instructions. A single amino acid substitution at position 66 of the PB1-F2 (N66S) was introduced, which has been shown to be associated with increased virulence in mice [24]. None of the mutations altered the PB1 ORF as silent mutation. The sequences of the constructed plasmids and introduced mutations were confirmed by sequencing.
Wild-type (KS07_WT), PB1-F2 knocked out (KS07_K/O) and N66S single mutant (KS07_N66S) viruses were generated by reverse genetics as described previously [45]. Briefly, co-cultured MDCK and 293T cells were transfected with eight constructed pHW2000 plasmids encoding viral genomic RNA segments using Lipofectamine 2000 (Invitrogen). After 48 h transfection, supernatants were collected and passaged three times on MDCK cells. The rescued viruses were confirmed by sequencing.
Growth kinetics
To evaluate the replication kinetics of recombinant viruses, MDCK and PK-15 cells were grown in 12-well plates and infected with each virus at a m.o.i. of 0.001 in triplicate. Cell culture supernatants were collected at 12, 24, 36 and 48 h p.i. Virus titres were determined by calculating the 50 % TCID50 ml−1 in MDCK cells using the Reed and Muench method. The plaque assay was performed to compare the size of plaques formed by each recombinant virus on MDCK cells.
Pathogenicity study in mice
Six-week-old female BALB/c mice were used for the pathogenicity study. A total of 56 mice were randomly divided into four groups (14 mice per group). Mice from each group were inoculated intranasally with 1.5×106 TCID50 of each virus in a volume of 50 µl under slight anaesthesia with isoflurane. For the control group, mice were mock infected with 50 µl virus-free MEM. Mice were monitored daily for clinical signs and weighed daily until 14 days p.i. If mice lost more than 25 % of their original body weight, they were humanely euthanized. Three mice from each group were euthanized at 3 and 7 days p.i., and lungs were collected from each mouse to assess virus replication and to perform histopathological analysis. For virus titration, 10 % lung homogenates were made using fresh MEM with 1 % (v/v) antibiotic/antimycotic, and virus titres in the lung homogenates were determined on MDCK cells.
Pathogenicity study in pigs
Thirty-five 3- to 4-week-old pigs, which were confirmed to be seronegative for porcine reproductive and respiratory syndrome virus and SIVs, were used in this study. Each infection group contained nine pigs, while the control group contained eight pigs, and each group was housed in separate isolation rooms. Pigs were intratracheally infected with 1×103 TCID50 of each recombinant virus or with virus-free MEM as controls. Body temperature and clinical symptoms were monitored daily. At 0, 1, 3 and 5 days p.i., nasal swabs were collected from each pig and processed for virus titration on MDCK cells. Three pigs from each group were necropsied at 1 and 3 days p.i.; three pigs from each infection group and two pigs of the control group were necropsied at 5 days p.i. At necropsy, lungs were removed in toto, and the percentage of gross lesions on each lobe (each lung lobe is considered as 100 %) was assessed by a single experienced veterinarian. The mean of gross lung lesions of seven lung lobes was calculated and presented for the average lung lesions of each pig. BALF samples were collected by flushing each lung with 50 ml fresh MEM, and viral titres were determined on MDCK cells by calculating the TCID50 ml−1.
Histopathology and IHC
Tissue samples (right lungs from mice and nasal turbinates, trachea and right cardiac lung lobes from pigs) were fixed in 10 % (v/v) neutral buffered formalin and processed by the histopathology and IHC laboratory of the Kansas State Veterinary Diagnostic Laboratory. A board-certified veterinary pathologist evaluated histopathological lesions of haematoxylin-and-eosin-stained tissues. IHC staining for mouse lungs was conducted to detect influenza virus antigen in tissues using an anti-influenza A NP mAb. The primary antibody was detected with the Leica Bond Polymer Refine Detection kit on the Leica Bond-Max. For the lung microscopic lesions in mice, lungs were graded on the following seven criteria: subjective percentage of lung involved in the histological section examined (4 point scale of 1–4); airway epithelial necrosis, neutrophilic airway inflammation, peribronchiolar lymphocyte cuffing, interstitial pneumonia, airway epithelial hyperplasia (all on a 3 point scale of 1–3); and lastly the presence or absence of hyaline membranes (2 point scale, 0=absent and 1=present). In pigs, microscopic lesions were graded on a 0–3 or 0–4 scale for percentage of airways with epithelial necrosis or inflammation (0–4), peribronchiolar lymphocyte cuffing (0–3), interstitial pneumonia (0–4), airway epithelial hyperplasia (0–4), respiratory epithelial degeneration and necrosis in nasal turbinates and trachea (0–4) and degree of inflammation in trachea and nasal turbinates (0–3) similar to that described previously.
Cytokine and chemokine levels in BALF
Expressions of 12 porcine cytokines/chemokines (GM-CSF, IFN-γ, IL-1α, IL-1β, IL-2, IL-4, IL-6, IL-8, IL-10, IL-12, IL-18 and TNF-α) in BALF were quantified by MILLIPLEX MAP Porcine Cytokine/Chemokine Magnetic Bead Panel using the Luminex technology following the manufacturer’s instructions (Millipore). The data was read and analysed by a Bio-Plex 200 Suspension Array Luminex System (Bio-Rad).
Statistical analysis
For statistical analysis among groups, ANOVA test was used in GraphPad Prism version 5.0 (GraphPad Software). A P value of 0.05 or less was considered statistically significant.
Funding information
This work was partially supported by a Kansas State University Start-Up Fund (SRO# 001) and by a National Institute of Allergy and Infectious Diseases-funded Center of Excellence for Influenza Research and Surveillance, under contract number HHSN266200700006C.
Acknowledgements
We would like to thank Hui He, Haixia Liu, Nan Cao and Chester McDowell from Kansas State University for helping with the animal studies. The authors thank Russell Ransburgh from Kansas State University for helping with Luminex assay. We thank the staff from the Comparative Medicine Group at Kansas State University for supporting the animal studies and Jennifer Hill from the Kansas State Veterinary Diagnostic Lab for technical assistance with haematoxylin and eosin and IHC staining.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical statement
All animal studies including the mouse study (IACUC no. 3339) and the pig study (IACUC no. 3146) were reviewed and approved by the Institutional Animal Care and Use Committee at Kansas State University and were performed in Biosafety Level 2 or 2+ animal facilities under guidance from the Comparative Medicine Group at Kansas State University.
Footnotes
Abbreviations: BALF, bronchoalveolar lavage fluid; HPAIV, highly pathogenic avian influenza A virus; GM-CSF, granulocyte-macrophage colony-stimulating factor; IAV, influenza A virus; IHC, immunohistochemistry; MDCK, Madin–Darby canine kidney; NA, neuraminidase; p.i., post-infection; SIV, swine influenza virus; TR, triple-reassortant; TRIG, triple-reassortant internal gene cassette.
References
- 1.Dowdle WR, Hattwick MA. Swine influenza virus infections in humans. J Infect Dis. 1977;136:S386–S389. doi: 10.1093/infdis/136.supplement_3.s386. [DOI] [PubMed] [Google Scholar]
- 2.Myers KP, Olsen CW, Gray GC. Cases of swine influenza in humans: a review of the literature. Clin Infect Dis. 2007;44:1084–1088. doi: 10.1086/512813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peiris JS, Poon LL, Guan Y. Emergence of a novel swine-origin influenza A virus (S-OIV) H1N1 virus in humans. J Clin Virol. 2009;45:169–173. doi: 10.1016/j.jcv.2009.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shope RE. The etiology of swine influenza. Science. 1931;73:214–215. doi: 10.1126/science.73.1886.214. [DOI] [PubMed] [Google Scholar]
- 5.Zhou NN, Senne DA, Landgraf JS, Swenson SL, Erickson G, et al. Emergence of H3N2 reassortant influenza A viruses in North American pigs. Vet Microbiol. 2000;74:47–58. doi: 10.1016/S0378-1135(00)00165-6. [DOI] [PubMed] [Google Scholar]
- 6.Webby RJ, Swenson SL, Krauss SL, Gerrish PJ, Goyal SM, et al. Evolution of swine H3N2 influenza viruses in the United States. J Virol. 2000;74:8243–8251. doi: 10.1128/JVI.74.18.8243-8251.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karasin AI, Olsen CW, Anderson GA. Genetic characterization of an H1N2 influenza virus isolated from a pig in Indiana. J Clin Microbiol. 2000;38:2453–2456. doi: 10.1128/jcm.38.6.2453-2456.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Webby RJ, Rossow K, Erickson G, Sims Y, Webster R. Multiple lineages of antigenically and genetically diverse influenza A virus co-circulate in the United States swine population. Virus Res. 2004;103:67–73. doi: 10.1016/j.virusres.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 9.Lekcharoensuk P, Lager KM, Vemulapalli R, Woodruff M, Vincent AL, et al. Novel swine influenza virus subtype H3N1, United States. Emerg Infect Dis. 2006;12:787–794. doi: 10.3201/eid1205.051060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vincent AL, Ma W, Lager KM, Janke BH, Richt JA. Swine influenza viruses: a North American perspective. Adv Virus Res. 2008;72:127–154. doi: 10.1016/S0065-3527(08)00403-X. [DOI] [PubMed] [Google Scholar]
- 11.Ma W, Lager KM, Vincent AL, Janke BH, Gramer MR, et al. The role of swine in the generation of novel influenza viruses. Zoonoses Public Health. 2009;56:326–337. doi: 10.1111/j.1863-2378.2008.01217.x. [DOI] [PubMed] [Google Scholar]
- 12.Nelson MI, Stratton J, Killian ML, Janas-Martindale A, Vincent AL. Continual reintroduction of human pandemic H1N1 influenza A viruses into swine in the United States, 2009 to 2014. J Virol. 2015;89:6218–6226. doi: 10.1128/JVI.00459-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson MI, Vincent AL, Kitikoon P, Holmes EC, Gramer MR. Evolution of novel reassortant A/H3N2 influenza viruses in North American swine and humans, 2009–2011. J Virol. 2012;86:8872–8878. doi: 10.1128/JVI.00259-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen W, Calvo PA, Malide D, Gibbs J, Schubert U, et al. A novel influenza A virus mitochondrial protein that induces cell death. Nat Med. 2001;7:1306–1312. doi: 10.1038/nm1201-1306. [DOI] [PubMed] [Google Scholar]
- 15.Lowy RJ. Influenza virus induction of apoptosis by intrinsic and extrinsic mechanisms. Int Rev Immunol. 2003;22:425–449. doi: 10.1080/08830180305216. [DOI] [PubMed] [Google Scholar]
- 16.Zell R, Krumbholz A, Eitner A, Krieg R, Halbhuber KJ, et al. Prevalence of PB1-F2 of influenza A viruses. J Gen Virol. 2007;88:536–546. doi: 10.1099/vir.0.82378-0. [DOI] [PubMed] [Google Scholar]
- 17.Pasricha G, Mishra AC, Chakrabarti AK. Comprehensive global amino acid sequence analysis of PB1F2 protein of influenza A H5N1 viruses and the influenza A virus subtypes responsible for the 20th-century pandemics. Influenza Other Respir Viruses. 2013;7:497–505. doi: 10.1111/j.1750-2659.2012.00400.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pena L, Vincent AL, Loving CL, Henningson JN, Lager KM, et al. Restored PB1-F2 in the 2009 pandemic H1N1 influenza virus has minimal effects in swine. J Virol. 2012;86:5523–5532. doi: 10.1128/JVI.00134-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schmolke M, Manicassamy B, Pena L, Sutton T, Hai R, et al. Differential contribution of PB1-F2 to the virulence of highly pathogenic H5N1 influenza A virus in mammalian and avian species. PLoS Pathog. 2011;7:e1002186. doi: 10.1371/journal.ppat.1002186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leymarie O, Embury-Hyatt C, Chevalier C, Jouneau L, Moroldo M, et al. PB1-F2 attenuates virulence of highly pathogenic avian H5N1 influenza virus in chickens. PLoS One. 2014;9:e100679. doi: 10.1371/journal.pone.0100679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deventhiran J, Kumar SR, Raghunath S, Leroith T, Elankumaran S. PB1-F2 protein does not impact the virulence of triple-reassortant H3N2 swine influenza virus in pigs but alters pathogenicity and transmission in turkeys. J Virol. 2015;90:222–231. doi: 10.1128/JVI.01551-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pena L, Vincent AL, Loving CL, Henningson JN, Lager KM, et al. Strain-dependent effects of PB1-F2 of triple-reassortant H3N2 influenza viruses in swine. J Gen Virol. 2012;93:2204–2214. doi: 10.1099/vir.0.045005-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zamarin D, Ortigoza MB, Palese P. Influenza A virus PB1-F2 protein contributes to viral pathogenesis in mice. J Virol. 2006;80:7976–7983. doi: 10.1128/JVI.00415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Conenello GM, Zamarin D, Perrone LA, Tumpey T, Palese P. A single mutation in the PB1-F2 of H5N1 (HK/97) and 1918 influenza A viruses contributes to increased virulence. PLoS Pathog. 2007;3:1414–1421. doi: 10.1371/journal.ppat.0030141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma W, Belisle SE, Mosier D, Li X, Stigger-Rosser E, et al. 2009 pandemic H1N1 influenza virus causes disease and upregulation of genes related to inflammatory and immune responses, cell death, and lipid metabolism in pigs. J Virol. 2011;85:11626–11637. doi: 10.1128/JVI.05705-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zamarin D, García-Sastre A, Xiao X, Wang R, Palese P. Influenza virus PB1-F2 protein induces cell death through mitochondrial ANT3 and VDAC1. PLoS Pathog. 2005;1:e4. doi: 10.1371/journal.ppat.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen CJ, Chen GW, Wang CH, Huang CH, Wang YC, et al. Differential localization and function of PB1-F2 derived from different strains of influenza A virus. J Virol. 2010;84:10051–10062. doi: 10.1128/JVI.00592-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mazur I, Anhlan D, Mitzner D, Wixler L, Schubert U, et al. The proapoptotic influenza A virus protein PB1-F2 regulates viral polymerase activity by interaction with the PB1 protein. Cell Microbiol. 2008;10:1140–1152. doi: 10.1111/j.1462-5822.2008.01116.x. [DOI] [PubMed] [Google Scholar]
- 29.McAuley JL, Zhang K, Mccullers JA. The effects of influenza A virus PB1-F2 protein on polymerase activity are strain specific and do not impact pathogenesis. J Virol. 2010;84:558–564. doi: 10.1128/JVI.01785-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Le Goffic R, Bouguyon E, Chevalier C, Vidic J, da Costa B, et al. Influenza A virus protein PB1-F2 exacerbates IFN-β expression of human respiratory epithelial cells. J Immunol. 2010;185:4812–4823. doi: 10.4049/jimmunol.0903952. [DOI] [PubMed] [Google Scholar]
- 31.McAuley JL, Hornung F, Boyd KL, Smith AM, Mckeon R, et al. Expression of the 1918 influenza A virus PB1-F2 enhances the pathogenesis of viral and secondary bacterial pneumonia. Cell Host Microbe. 2007;2:240–249. doi: 10.1016/j.chom.2007.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alymova IV, Samarasinghe A, Vogel P, Green AM, Weinlich R, et al. A novel cytotoxic sequence contributes to influenza A viral protein PB1-F2 pathogenicity and predisposition to secondary bacterial infection. J Virol. 2014;88:503–515. doi: 10.1128/JVI.01373-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Buehler J, Navi D, Lorusso A, Vincent A, Lager K, et al. Influenza A virus PB1-F2 protein expression is regulated in a strain-specific manner by sequences located downstream of the PB1-F2 initiation codon. J Virol. 2013;87:10687–10699. doi: 10.1128/JVI.01520-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma W, Vincent AL, Lager KM, Janke BH, Henry SC, et al. Identification and characterization of a highly virulent triple reassortant H1N1 swine influenza virus in the United States. Virus Genes. 2010;40:28–36. doi: 10.1007/s11262-009-0413-7. [DOI] [PubMed] [Google Scholar]
- 35.Hai R, Schmolke M, Varga ZT, Manicassamy B, Wang TT, et al. PB1-F2 expression by the 2009 pandemic H1N1 influenza virus has minimal impact on virulence in animal models. J Virol. 2010;84:4442–4450. doi: 10.1128/JVI.02717-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varga ZT, Grant A, Manicassamy B, Palese P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J Virol. 2012;86:8359–8366. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Conenello GM, Tisoncik JR, Rosenzweig E, Varga ZT, Palese P, et al. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. J Virol. 2011;85:652–662. doi: 10.1128/JVI.01987-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alymova IV, Green AM, van de Velde N, Mcauley JL, Boyd KL, et al. Immunopathogenic and antibacterial effects of H3N2 influenza A virus PB1-F2 map to amino acid residues 62, 75, 79, and 82. J Virol. 2011;85:12324–12333. doi: 10.1128/JVI.05872-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibbs JS, Malide D, Hornung F, Bennink JR, Yewdell JW. The influenza A virus PB1-F2 protein targets the inner mitochondrial membrane via a predicted basic amphipathic helix that disrupts mitochondrial function. J Virol. 2003;77:7214–7224. doi: 10.1128/JVI.77.13.7214-7224.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamada H, Chounan R, Higashi Y, Kurihara N, Kido H. Mitochondrial targeting sequence of the influenza A virus PB1-F2 protein and its function in mitochondria. FEBS Lett. 2004;578:331–336. doi: 10.1016/j.febslet.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 41.Chanturiya AN, Basañez G, Schubert U, Henklein P, Yewdell JW, et al. PB1-F2, an influenza A virus-encoded proapoptotic mitochondrial protein, creates variably sized pores in planar lipid membranes. J Virol. 2004;78:6304–6312. doi: 10.1128/JVI.78.12.6304-6312.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshizumi T, Ichinohe T, Sasaki O, Otera H, Kawabata S, et al. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat Commun. 2014;5:4713. doi: 10.1038/ncomms5713. [DOI] [PubMed] [Google Scholar]
- 43.Solbak SM, Sharma A, Bruns K, Röder R, Mitzner D, et al. Influenza A virus protein PB1-F2 from different strains shows distinct structural signatures. Biochim Biophys Acta. 2013;1834:568–582. doi: 10.1016/j.bbapap.2012.11.009. [DOI] [PubMed] [Google Scholar]
- 44.Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. Universal primer set for the full-length amplification of all influenza A viruses. Arch Virol. 2001;146:2275–2289. doi: 10.1007/s007050170002. [DOI] [PubMed] [Google Scholar]
- 45.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci USA. 2000;97:6108–6113. doi: 10.1073/pnas.100133697. [DOI] [PMC free article] [PubMed] [Google Scholar]