

Abstract
Nucleolytic processing by nucleases can be a relevant mechanism to allow repair/restart of stalled replication forks. However, nuclease action needs to be controlled to prevent overprocessing of damaged replication forks that can be detrimental to genome stability. The checkpoint protein Rad9/53BP1 is known to limit nucleolytic degradation (resection) of DNA double‐strand breaks (DSBs) in both yeast and mammals. Here, we show that loss of the inhibition that Rad9 exerts on resection exacerbates the sensitivity to replication stress of Mec1/ATR‐defective yeast cells by exposing stalled replication forks to Dna2‐dependent degradation. This Rad9 protective function is independent of checkpoint activation and relies mainly on Rad9‐Dpb11 interaction. We propose that Rad9/53BP1 supports cell viability by protecting stalled replication forks from extensive resection when the intra‐S checkpoint is not fully functional.
Keywords: Mec1, Rad9, replication forks, resection, Sgs1
Subject Categories: DNA Replication, Repair & Recombination
Introduction
DNA replication stress is an important source of genomic instability and can be induced by a transient slowing or stalling of replication forks due to damaged DNA, unusual DNA structures, repetitive sequences, or nucleotide depletion 1, 2. Stalled replication forks generally result in the uncoupling either of leading from lagging‐strand polymerases or of polymerases from replicative helicases. These events cause generation of tracts of replication protein A (RPA)‐covered single‐stranded DNA (ssDNA) that recruits the checkpoint kinase Mec1 (ATR in mammals) 3. Once activated, Mec1/ATR propagates the checkpoint signal to the downstream checkpoint kinase Rad53 (CHK2 in mammals), whose activation requires the interaction with the mediator protein Mrc1 1, 2. Rad53 activation in turn prevents entry into mitosis, increases the intracellular dNTP pools, represses late origin firing, and prevents fork collapse through poorly identified pathways 1, 2.
Replication stress can be experimentally induced by treatment with the ribonucleotide reductase inhibitor hydroxyurea (HU), which globally blocks active replication forks by depleting the cellular pool of deoxynucleotide triphosphates (dNTPs) 1, 2. Although the molecular mechanism is still unclear, a key function of the S‐phase checkpoint in ensuring cell survival to replication stress is to maintain the ability of the replisome to resume DNA synthesis once the block to fork progression is relieved 4, 5, 6, 7. Replisome components appear to be no longer associated with the replicative sites in Mec1‐ and Rad53‐defective mutants 8, 9, 10, although the replication proteins might still remain bound to chromatin but unable to resume replication 11.
The RecQ helicase Sgs1 (BLM in mammals) acts synergistically with Mec1 in resuming DNA replication upon replication stress, possibly by promoting the resolution of recombination structures that accumulate at damaged replication forks 8, 9, 12, 13. Furthermore, it contributes to initiate the checkpoint in response to stalled replication forks by promoting the recruitment of Rad53 into close proximity of Mec1‐Ddc2 14. Finally, in both yeast and mammals, Sgs1 and some nucleases like Mre11, Sae2 (CtIP in mammals), Exo1, and Dna2 have been implicated in the nucleolytic processing of intrachromosomal DNA double‐strand breaks (DSBs). In particular, Sae2‐dependent Mre11 endonuclease activity generates a nick in the 5′‐terminated strand that provides the access for Exo1 and Dna2 nucleases that can degrade DNA in the 5′‐3′ direction 15, 16, 17, 18, 19, 20. The helicase activity of Sgs1 unwinds double‐stranded DNA and generates a substrate for Dna2 that cleaves ssDNA overhangs adjoining a duplex DNA 16, 17, 18, 19, 21. The resection activity of Sgs1‐Dna2 is thought to be inhibited by the checkpoint protein Rad9 (53BP1 in mammals) 22, 23, which provides a barrier to DNA end resection 24, 25.
The above yeast and mammalian nucleases have key roles also in the processing of replication intermediates to allow repair/restart of stalled replication forks and/or to prevent accumulation of replication‐associated DSBs 1, 2, 26. However, unrestricted nuclease access to replication forks could destroy the fork structure and prevent continued DNA synthesis, leading to genome instability. In the absence of the intra‐S‐phase checkpoint, the genome of HU‐treated yeast cells is subjected to degradation by Exo1 27, 28, 29, 30 and Sae2 30. Furthermore, replication stress in ATR‐defective Schizosaccharomyces pombe and mammalian cells results in MRE11‐ and EXO1‐dependent ssDNA accumulation 31, 32, suggesting that the checkpoint plays a role in protecting replication forks from aberrant nuclease activity. Consistent with this hypothesis, phospho‐proteomic screens have identified Exo1 as a target of Rad53, which negatively regulates Exo1 activity through phosphorylation events 33, 34. Furthermore, the fission yeast ortholog of Rad53, Cds1, phosphorylates and regulates Dna2 activity 35, which is involved in the processing and restart of reversed forks in both yeast and mammals 36, 37.
Mammalian proteins involved in homologous recombination (HR) or in the Fanconi anemia (FA) network, including FANCD2, RAD51, BRCA1, and BRCA2, have been shown to prevent excessive fork degradation by antagonizing MRE11 and DNA2 actions 38, 39, 40, 41, 42, 43, 44, 45. Furthermore, loss of the WRN exonuclease activity enhances degradation at nascent DNA strands by EXO1 and MRE11 46, 47, whereas cells depleted of BOD1L protein exhibit a DNA2‐dependent degradation of stalled/damaged replication forks 48.
Here, we show that the Saccharomyces cerevisiae checkpoint protein Rad9, ortholog of mammalian 53BP1, is important to restrain uncontrolled nucleolytic degradation of damaged replication forks when Mec1 is not fully functional. Loss of Rad9 or expression of a Sgs1 variant (Sgs1‐G1298R), which escapes Rad9‐mediated inhibition of DNA end resection, exacerbates the sensitivity to dNTP depletion of cells expressing the Mec1‐100‐defective variant. This protective function of Rad9 is independent of checkpoint activation and is mainly due to Rad9‐Dpb11 interaction. The severe HU sensitivity of rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 cells is accompanied by increased ssDNA generation at stalled replication forks and impaired DNA replication recovery upon dNTP depletion. These findings, together with the observation that Dna2 inactivation decreases the amount of ssDNA at stalled replication forks in both rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 cells, indicate a role for Rad9 in supporting viability of Mec1‐defective cells by protecting replication forks from degradation.
Results
Both Sgs1‐G1298R and the lack of Rad9 exacerbate the sensitivity to HU of mec1‐100 cells
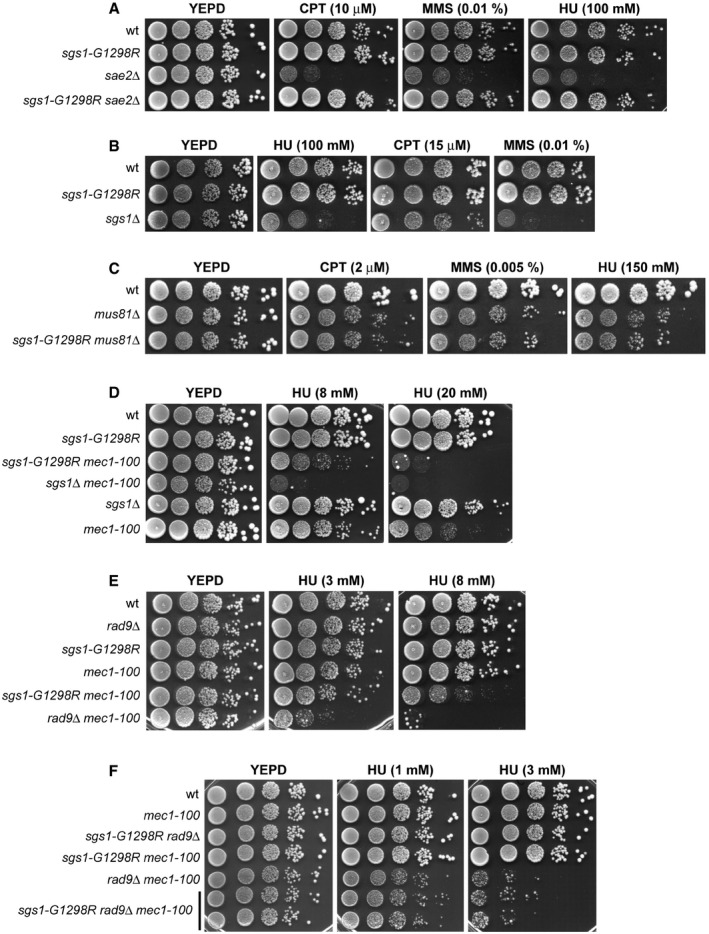
The RecQ helicase Sgs1 is involved in resection of DNA DSBs 15, 16. The lack of Sgs1 causes cell death in sae2∆ cells, and this synthetic lethality can be due to defective DSB resection, as it is suppressed by either EXO1 overexpression or elimination of the resection inhibitor Ku complex 49. We have previously described the sgs1‐G1298R allele that fully suppresses the hypersensitivity to genotoxic agents (Fig 1A) and the resection defect of sae2∆ cells 22. Unlike SGS1 deletion, the Sgs1‐G1298R variant did not cause by itself hypersensitivity to HU, camptothecin (CPT), or methyl methanesulfonate (MMS) (Fig 1B).
Figure 1. The HU sensitivity of mec1‐100 cells is exacerbated by either Sgs1‐G1298R or the lack of Rad9.
-
A–FExponentially growing cell cultures were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without CPT, MMS, or HU at the indicated concentrations.
Sgs1 is thought to work together with the recombination protein Mus81 in the processing of repair intermediates that occur at the replication forks 50. The lack of Mus81 causes cell death in a sgs1∆ background 14, presumably because Sgs1 is implicated in the resolution of, or recovery from, recombination events that arise in the absence of Mus81. We found that sgs1‐G1298R did not impair cell viability when combined with the lack of Mus81 (Fig 1C), supporting further the finding that Sgs1‐G1298R maintains most, if not all, Sgs1 functions.
In addition to its role in promoting DSB resection, Sgs1 is constitutively associated with replication forks, where it acts synergistically with Mec1 in fork maintenance under replication stress 8, 9. To understand how Sgs1 functions in DSB resection and DNA replication under stress conditions are connected to each other, we analyzed the effects of the sgs1‐G1298R allele in cells defective for the Mec1 checkpoint kinase. As mec1‐null cells (kept viable by deleting SML1) die even in the presence of very low HU doses and experience extensive fork degradation after exposure to replication stress 6, 29, 51, we took advantage of the mec1‐100 mutant allele that causes less severe sensitivity to HU than the mec1‐null allele 52. Furthermore, unlike mec1‐null cells, mec1‐100 cells are partially defective in the intra‐S checkpoint but not in the G2/M checkpoint 52 and are able to resume DNA replication after HU removal, although less efficiently than wild‐type cells 51, 53. Similar to sgs1∆ mec1‐100 cells, sgs1‐G1298R mec1‐100 double‐mutant cells were more sensitive to HU treatment than mec1‐100 single‐mutant cells (Fig 1D), indicating that Sgs1‐G1298R becomes detrimental for cell viability when the intra‐S checkpoint is not fully functional.
The resection activity of Sgs1/Dna2 is inhibited by the checkpoint protein Rad9 22, 23, which is known to limit resection of DNA DSBs 24, 25. We have previously demonstrated that Sgs1‐G1298R not only suppresses the resection defect of sae2∆ cells (Fig 1A), but it also accelerates the resection process by escaping the Rad9‐mediated inhibition of DSB resection 22. This finding prompted us to investigate the effect of deleting RAD9 in mec1‐100 cells. The lack of Rad9, which did not cause HU hypersensitivity by itself, exacerbated the sensitivity to HU of mec1‐100 cells, with mec1‐100 rad9∆ cells being more sensitive to HU than sgs1‐G1298R mec1‐100 cells (Fig 1E). The HU sensitivity of sgs1‐G1298R rad9∆ mec1‐100 triple‐mutant cells was similar to that of rad9∆ mec1‐100 double‐mutant cells (Fig 1F), indicating that the lack of Rad9 or the presence of Sgs1‐G1298R impairs viability of HU‐treated mec1‐100 cells by affecting the same mechanism.
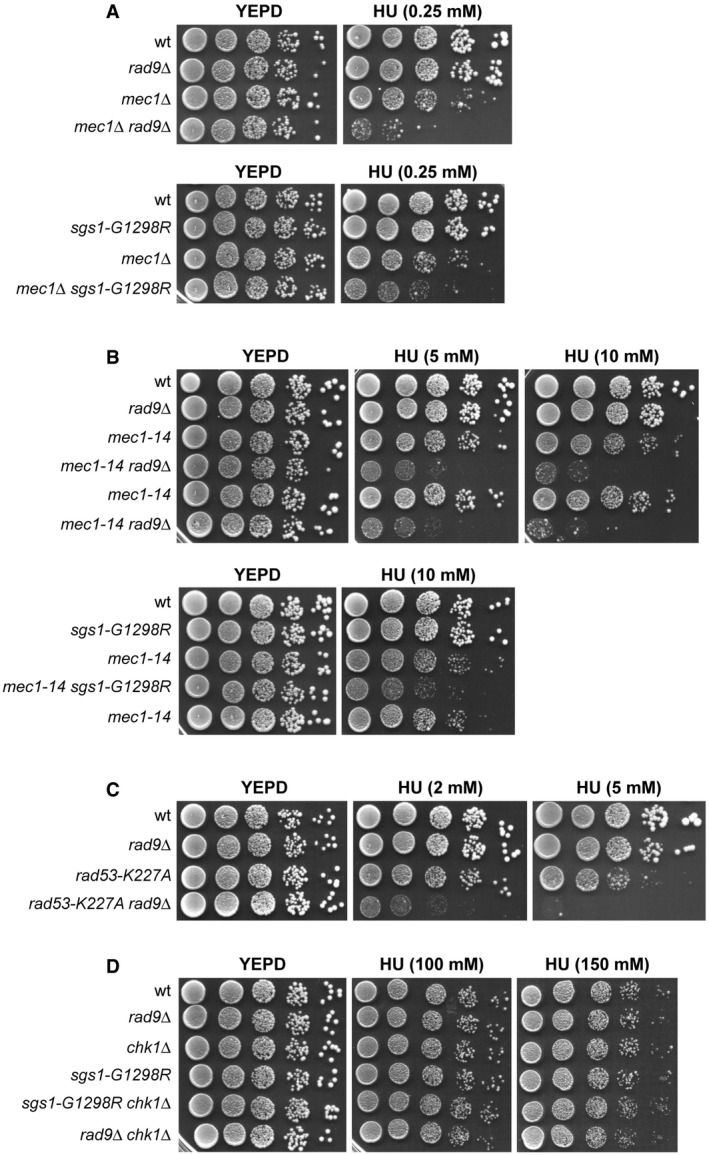
These synthetic effects on HU are not specific for a mec1‐100 background. In fact, the lack of Rad9 or the presence of Sgs1‐G1298R exacerbated the HU sensitivity of cells carrying either MEC1 deletion (kept viable by SML1 deletion) (Fig 2A) or the hypomorphic mec1‐14 allele (Fig 2B). RAD9 deletion also increased the HU sensitivity of cells carrying a Rad53 kinase‐defective variant (rad53‐K227A) (Fig 2C), whereas it had no effect on cells lacking the downstream checkpoint kinase Chk1 (Fig 2D).
Figure 2. The synthetic effects of rad9∆ and sgs1‐G129R on HU are not specific for mec1‐100 .
-
A–DExponentially growing cell cultures were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without HU at the indicated concentrations.
Both Sgs1‐G1298R and the lack of Rad9 impair the ability of mec1‐100 cells to resume DNA replication under replicative stress
We examined the effects caused by either the lack of Rad9 or the presence of Sgs1‐G1298R on the ability of mec1‐100 cells to resume DNA replication after transient HU arrest by measuring DNA content by flow cytometry. Cells were blocked in G1 with α‐factor and released into medium containing HU. After 2 h, cells were transferred to medium lacking HU but containing nocodazole to prevent passage through mitosis. Wild‐type, rad9∆ and sgs1‐G1298R cells completed DNA replication in 40–50 min after release, whereas mec1‐100 cells reached significant amounts of DNA synthesis 20–30 min later (Fig 3A). By contrast, rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 double‐mutant cells were unable to reach 2C DNA content even after 90 min (Fig 3A), indicating a severe defect in resuming DNA replication after transient HU exposure. Furthermore, both rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 cells exhibited a rapid loss of viability when synchronously released from a G1 arrest into S phase in the presence of HU (Fig 3B).
Figure 3. The ability of mec1‐100 cells to resume DNA replication under replicative stress is impaired by either Sgs1‐G1298R or the lack of Rad9.

-
ACells were arrested in G1 with α‐factor (αf) and then released in YEPD containing 0.2 M HU at time zero. After 2 h (HU), cells were transferred to medium lacking HU but containing nocodazole to prevent passage through mitosis. Aliquots of each culture were harvested at the indicated times after HU removal to determine DNA content by flow cytometry.
-
BCell viability. Cells were arrested in G1 with α‐factor and then released in YEPD containing 0.2 M HU at time zero. Cells taken at the indicated time points after release in HU were tested for colony‐forming units on YEPD plates. Plotted values are the mean values with error bars denoting s.d. (n = 3).
-
C, DImmunodetection of BrdU‐pulsed DNA. Cells were arrested in G1 with α‐factor (αf) and released into YEPD containing 0.2 M HU + 25 μM BrdU. After 1 h (HU), cells were chased with 2 mM thymidine into fresh medium and samples were taken at the indicated times after chase (−HU). DNA content during the time course was measured by flow cytometry (C). BrdU‐labeled DNA was detected with anti‐BrdU antibodies (D). High molecular weight DNA molecules are indicated by an arrow.
-
EChIP analysis. Cells were arrested in G1 with α‐factor and then released in YEPD containing 0.2 M HU at time zero. Relative fold enrichment of Myc‐tagged DNA Polε at ARS607 and ARS305 replication origins was determined after ChIP with anti‐Myc antibodies and subsequent qPCR analysis. Plotted values are the mean values with error bars denoting s.d. (n = 3). *P < 0.05 (Student's t‐test).
-
FCells were arrested in G1 with α‐factor (αf) and then released in YEPD containing 0.2 M HU at time zero, followed by Western blot analysis of protein extracts with anti‐Rad53 antibodies.
Source data are available online for this figure.
To follow the fate of stalled replication forks in a more direct way, we used bromodeoxyuridine (BrdU) pulse–chase experiments to label nascent strands during DNA replication in HU. We used strains that can incorporate BrdU into DNA because they express both the nucleoside transporter hENT and a thymidine kinase from herpes simplex virus. Cells were synchronized in G1 with α‐factor and released into medium containing HU and BrdU (Fig 3C). After the nascent DNA was labeled, the BrdU was chased by transferring cells to medium lacking both HU and BrdU and containing thymidine (Fig 3C). As previously reported 29, labeled nascent DNA replication intermediates detected by using anti‐BrdU antibodies appeared as a smear in all HU‐treated cells (Fig 3D). After release, most of the incorporated BrdU had been chased into high molecular weight within 20 and 40 min in wild‐type and mec1‐100 cells, respectively (Fig 3D). By contrast, the majority of nascent DNA in both rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 cells remained at the same position even after 60 min after release (Fig 3D), indicating a failure to resume DNA replication after HU‐induced fork stalling.
Next, we measured the association with the early ARS607 and ARS305 replication origins of Myc‐tagged DNA Polε by chromatin immunoprecipitation (ChIP) and quantitative PCR (qPCR) in cells synchronously released from a G1 arrest into S phase in the presence of HU. DNA Polε in wild‐type cells was efficiently bound to ARS607 and ARS305 about 20 min after release in HU (Fig 3E). By contrast, both ARS607‐ and ARS305‐Polε association diminished in mec1‐100 cells compared with wild‐type cells and decreased further in both sgs1‐G1298R mec1‐100 and rad9∆ mec1‐100 cells (Fig 3E).
To understand the possible impact of the sgs1‐G1298R and rad9∆ mutations on checkpoint activation, we monitored Rad53 phosphorylation in cells synchronously released from a G1 arrest into S phase in the presence of HU. According to the finding that Mec1‐100 does not completely abolish checkpoint activation 52, mec1‐100 cells showed a delay in Rad53 phosphorylation compared with wild‐type cells, and residual Rad53 phosphorylation was under detection level in rad9∆ mec1‐100 cells (Fig 3F). By contrast, the presence of the sgs1‐G1298R allele did not decrease the amount of Rad53‐phosphorylated forms in mec1‐100 cells (Fig 3F), indicating that the increased HU hypersensitivity of sgs1‐G1298R mec1‐100 double‐mutant cells cannot be ascribed to impaired checkpoint activation.
Dna2 dysfunction is epistatic to rad9∆ and sgs1‐G1298R with respect to the HU sensitivity of mec1‐100 cells
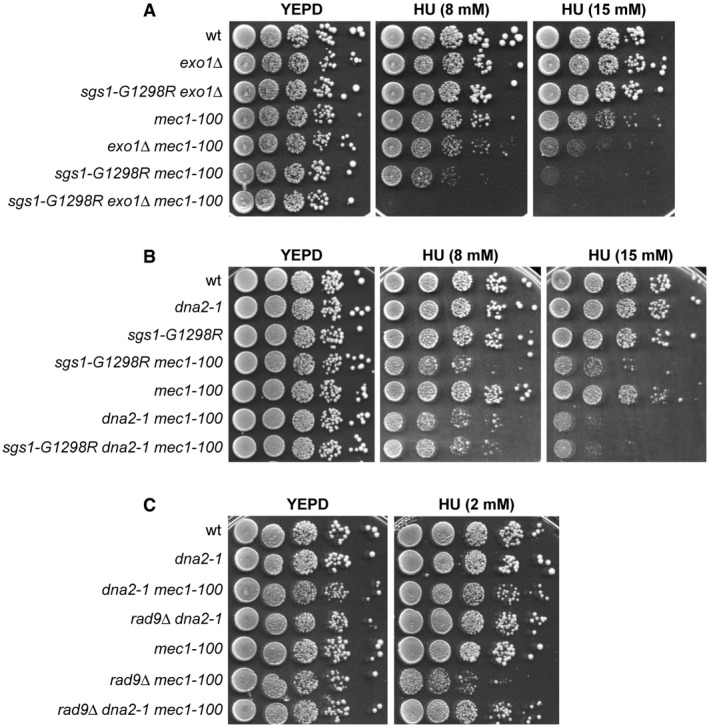
Extensive DSB resection can be carried out by either of two pathways dependent on the enzymatic activities of the nucleases Exo1 and Dna2, respectively 15, 16, 17, 18, 19. While Exo1 does not require a helicase activity to resect DNA ends, Sgs1 helicase is known to support the nuclease activity of Dna2 that degrades DNA endonucleolytically 16, 17, 18, 19, 21. Thus, we analyzed the consequences of inactivating Exo1 or Dna2 on the HU sensitivity of rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 double‐mutant cells. As expected 29, EXO1 deletion exacerbated the HU sensitivity of mec1‐100 cells and the presence of sgs1‐G1298R increased further the HU hypersensitivity of exo1∆ mec1‐100 cells (Fig 4A), indicating that sgs1‐G1298R and exo1∆ increase the HU sensitivity of mec1‐100 cells by altering two different pathways.
Figure 4. Epistatic relationships between rad9∆, sgs1‐G1298R, exo1∆, and dna2‐1 with respect to the HU sensitivity of mec1‐100 cells.
-
A–CExponentially growing cell cultures were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without HU at the indicated concentrations.
As Dna2 is essential for cell viability 54, we used the hypomorphic dna2‐1 allele, which increased the HU sensitivity of mec1‐100 cells possibly due to defects in DNA replication (Fig 4B). The presence of the dna2‐1 allele was epistatic to both sgs1‐G1298R and rad9∆ with respect to the HU sensitivity of mec1‐100 cells. In fact, the HU sensitivity of sgs1‐G1298R dna2‐1 mec1‐100 cells was similar to that of dna2‐1 mec1‐100 (Fig 4B), suggesting that Sgs1‐G1298R and Dna2‐1 increase the HU sensitivity of mec1‐100 by altering the same pathway. Furthermore, the HU sensitivity of rad9∆ dna2‐1 mec1‐100 cells was similar to that of dna2‐1 mec1‐100 cells and less severe than that of rad9∆ mec1‐100 cells (Fig 4C), suggesting that the lack of Rad9 requires Dna2 to exacerbate the HU sensitivity of mec1‐100 cells.
Dpb11‐mediated recruitment of Rad9 plays the major role in supporting mec1‐100 survival to replicative stress
We investigated whether Rad9 is recruited at stalled replication forks by ChIP and qPCR in cells synchronously released from a G1 arrest into S phase in the presence of HU. Transient Rad9 association with ARS607 replication origin was detected in both wild‐type and mec1‐100 cells about 30 min after release in the presence of HU, with mec1‐100 cells showing a stronger Rad9 association (Fig 5A).
Figure 5. The lack of Rad9‐Dbp11 interaction exacerbates HU sensitivity of mec1‐100 cells.
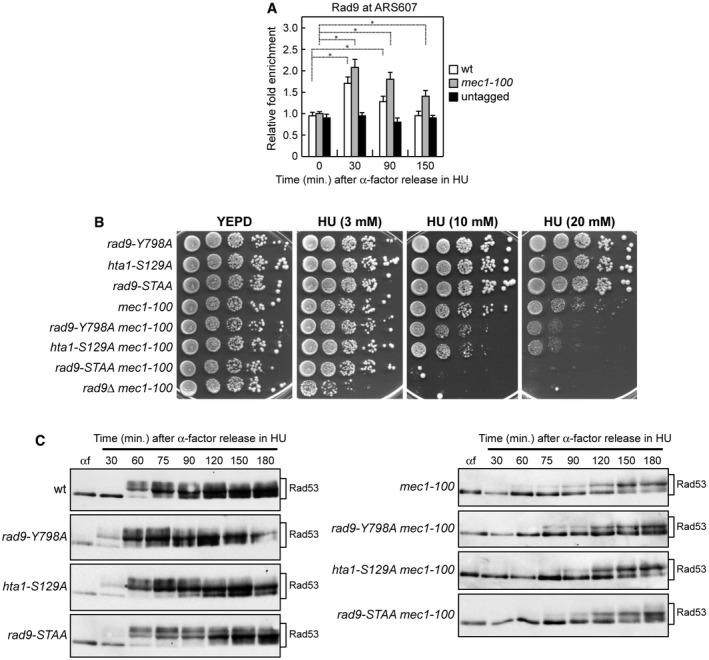
- ChIP analysis. Cells were arrested in G1 with α‐factor and then released in YEPD containing 0.2 M HU at time zero. Relative fold enrichment of Flag‐tagged Rad9 at ARS607 replication origin was determined after ChIP with anti‐Flag antibodies and subsequent qPCR analysis. Plotted values are the mean values with error bars denoting s.d. (n = 3). *P < 0.05 (Student's t‐test).
- Exponentially growing cell cultures were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without HU at the indicated concentrations. The hta1‐S129A strains also carry HTA2 deletion.
- Cells were arrested in G1 with α‐factor (αf) and then released in YEPD containing 0.2 M HU at time zero, followed by Western blot analysis of protein extracts with anti‐Rad53 antibodies.
Source data are available online for this figure.
Recruitment of Rad9 to chromatin involves multiple pathways. Rad9 is constitutively bound to chromatin even in the absence of DNA damage through the interaction between its Tudor domain and histone H3 methylated at K79 (H3‐K79me) 55, 56, 57, 58, 59. In addition, Rad9 binding to the damaged sites is further strengthened through the interaction of its BRCT domain with histone H2A phosphorylated at S129 (γH2A) by Mec1 and Tel1 checkpoint kinases 60, 61, 62. Finally, phosphorylation of the S462 and T474 Rad9 residues by cyclin‐dependent kinase (Cdk1) leads to Rad9 interaction with the multi‐BRCT domain protein Dpb11 (TopBP1 in mammals), which is a replication factor that mediates histone‐independent Rad9 recruitment to damaged sites 63, 64.
To investigate which of the above pathways could mediate Rad9 function in supporting viability of mec1‐100 cells under replication stress, we analyzed the HU sensitivity of mec1‐100 cells that were defective in Rad9 binding to H3‐K79me, γH2A, or Dpb11. The HU sensitivity of mec1‐100 cells was only slightly increased by expression of either the rad9‐Y798A or the hta1‐S129A allele (Fig 5B), which abolishes Rad9 association with H3‐K79me and γH2A generation, respectively 57, 58, 59, 60, 61, 62, 63. By contrast, the HU sensitivity of mec1‐100 cells was dramatically increased by expression of the rad9‐S462A‐T474A allele (rad9‐STAA) (Fig 5B), which lacks the S462 and T474 Cdk1‐dependent phosphorylation sites that mediate Rad9‐Dpb11 interaction 64. This finding indicates that Dpb11‐dependent recruitment of Rad9 plays the major role in supporting mec1‐100 resistance to replicative stress.
While the lack of Rad9 impairs checkpoint activation in response to DNA damage in G1 and G2, the Rad9‐STAA mutant variant is fully able to activate both the G1/S and the G2/M checkpoints 64. Moreover, the lack of either γH2A or H3‐K79me affects only activation of the G1/S checkpoint 57, 58, 59, 60, 61, 62, 63. The increased HU sensitivity of mec1‐100 cells lacking histone‐dependent or histone‐independent Rad9 association with DNA cannot be attributed to impaired activation of the downstream kinase Rad53. In fact, unlike RAD9 deletion that reduces Rad53 phosphorylation in HU‐treated mec1‐100 cells (Fig 3F), Rad53 phosphorylation in rad9‐Y798A mec1‐100, hta1‐S129A mec1‐100, and rad9‐STAA mec1‐100 cell cultures synchronously released from a G1 arrest into S phase in the presence of HU was similar to that of mec1‐100 cells (Fig 5C). The finding that rad9∆ mec1‐100 cells, which displayed an undetectable Rad53 phosphorylation upon replicative stress (Fig 3F), were more sensitive to HU than sgs1‐G1298R mec1‐100 cells (Fig 1E) and rad9‐STAA mec1‐100 cells (Fig 5B) suggests that part of the rad9∆ mec1‐100 HU hypersensitivity is due to a defect in checkpoint activation.
The lack of Fun30 or Slx4 suppresses the HU hypersensitivity of mec1‐100 cells
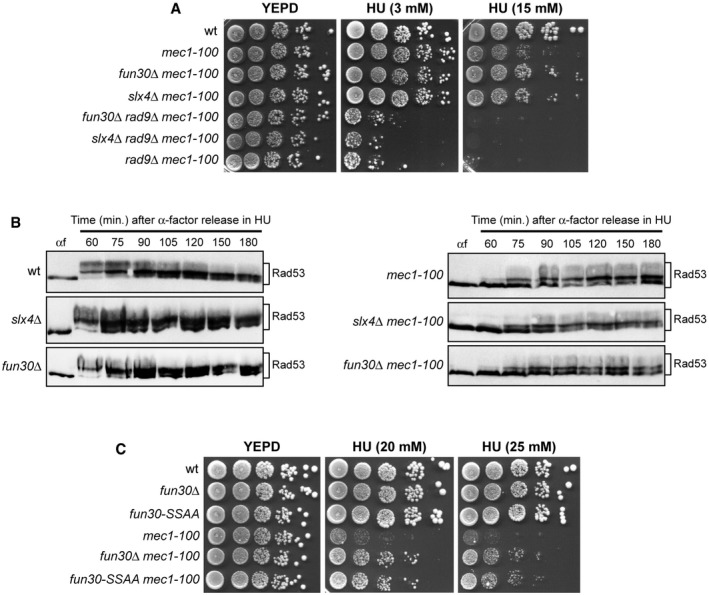
The function of Rad9 in inhibition of DSB resection is counteracted by the Swr1‐like family remodeler Fun30 (SMARCAD1 in mammals) 65, 66, 67, 68 and the scaffold protein complex Slx4‐Rtt107 69, 70, both of which promote DSB resection by limiting Rad9 accumulation to DNA DSBs 65, 69. Thus, we reasoned that if Rad9 maintains viability of HU‐treated mec1‐100 cells by limiting ssDNA generation, then the lack of Fun30 or Slx4‐Rtt107 might suppress the HU hypersensitivity of mec1‐100 cells by increasing Rad9‐mediated inhibition of resection. Indeed, both fun30∆ mec1‐100 and slx4∆ mec1‐100 double mutants were less sensitive to HU than mec1‐100 cells (Fig 6A). Moreover, the lack of Fun30 or Slx4 did not suppress the HU sensitivity of rad9∆ mec1‐100 cells (Fig 6A), suggesting that their suppression effect on mec1‐100 requires Rad9.
Figure 6. The lack of Slx4 or Fun30 suppresses the HU hypersensitivity of mec1‐100 cells.
-
A–C(A, C) Exponentially growing cell cultures were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without HU at the indicated concentrations. (B) Cells were arrested in G1 with α‐factor (αf) and then released in YEPD containing 0.2 M HU at time zero, followed by Western blot analysis of protein extracts with anti‐Rad53 antibodies.
Slx4 also counteracts the function of Rad9 in allowing Rad53 activation in response to MMS treatment 71. Suppression of the HU hypersensitivity of mec1‐100 cells by SLX4 deletion is not due to a more efficient checkpoint activation, as the kinetics of Rad53 phosphorylation in HU‐treated slx4∆ mec1‐100 cells was similar to that of mec1‐100 cells (Fig 6B).
The interaction between Slx4 and Dpb11 is strongly induced by Mec1‐dependent Slx4 phosphorylation in response to MMS treatment 70, 72. The dependency on Rad9 for survival of HU‐treated mec1‐100 cells is likely not due to decreased Slx4 phosphorylation, as HU treatment did not result in changes of Slx4 electrophoretic mobility as it did MMS treatment 73. Furthermore, the lack of Rad9 exacerbated also the HU sensitivity of cells defective for the Rad53 checkpoint kinase (Fig 2C), which is not involved in Slx4 phosphorylation 73.
Fun30 not only promotes DNA end resection by counteracting the resection block imposed by Rad9, but also it participates in chromatin organization even in the absence of DNA lesions 74. Fun30 is phosphorylated by Cdk1, and these phosphorylation events generate a binding site for Dpb11 that targets Fun30 to DSBs 68. The Fun30‐S20A‐S28A mutant variant (Fun30‐SSAA) cannot be phosphorylated by Cdk1 and is defective in DSB resection but not in silencing 68, indicating that Cdk1‐mediated Fun30 phosphorylation is required for Fun30 function in DSB resection but not for its function in chromatin organization. Thus, to rule out the possibility that general changes in chromatin organization could be responsible for suppression of the HU sensitivity of mec1‐100 cells by the lack of Fun30, we asked whether Fun30‐SSAA still suppressed the HU sensitivity of mec1‐100 cells. Indeed, fun30‐SSAA mec1‐100 cells, similar to fun30∆ mec1‐100 cells, were less sensitive to HU than mec1‐100 cells (Fig 6C), indicating that this suppression effect depends on the lack of Fun30 function in DSB resection.
Both Sgs1‐G1298R and the lack of Rad9 increase ssDNA generation at stalled replication forks in a Dna2‐dependent manner
As loss of the inhibition that Rad9 exerts on resection is sufficient to reduce survival of mec1‐100 cells to replication stress, the increased HU hypersensitivity of sgs1‐G1298R mec1‐100 and rad9∆ mec1‐100 cells might be due to uncontrolled degradation of stalled replication forks. To evaluate directly the presence of ssDNA at stalled replication forks, we took advantage of a qPCR assay that was previously used to detect ssDNA at DSBs 75 and at terminally arrested replication forks 76. This assay is based on ssDNA being refractory to digestion by the restriction enzyme SspI, which cleaves double‐stranded DNA (dsDNA) but not ssDNA. SspI‐digested and mock‐digested DNAs were amplified by qPCR using primers surrounding SspI restriction sites, and the resulting amplification products were then normalized to an amplicon on chromosome XI.
Cells arrested in G1 with α‐factor were released into medium containing HU, and ssDNA was analyzed by qPCR at different distances from the early efficient ARS607 replication origin. It has been previously reported that replication forks in untreated wild‐type cells show short gaps of ~220 nt, which likely represent the regions engaged by the replisome during replication 27. In the presence of HU, the size of these gaps increases by ~100 nt asymmetrically, possibly because of uncoupling events 27. Consistent with these findings, an increase in ssDNA above background levels (αf) was detected in HU‐treated wild‐type rad9∆, sgs1‐G1298R, and mec1‐100 cells at DNA regions closed to the replication origin (Fig 7). Both the amount and the extension of this ssDNA were dramatically increased in rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 double‐mutant cells after release in HU compared to mec1‐100 cells, with rad9∆ mec1‐100 cells showing the strongest effect (Fig 7). The ssDNA detected in rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 cells was specific to DNA regions surrounding the replication origin, as no significant differences above background levels (αf) were observed at a control locus (Fig 7). Furthermore, the amount of ssDNA decreased progressively as the distance from the replication origin increased. Strikingly, the presence of the dna2‐1 allele decreased the amount of ssDNA in both rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 cells (Fig 7), strongly suggesting that the ssDNA accumulated in the above double mutants is caused by Dna2‐mediated nucleolytic processing.
Figure 7. ssDNA generation at stalled forks in mec1‐100 cells is increased by either Sgs1‐G1298R or the lack of Rad9 in a Dna2‐dependent manner.
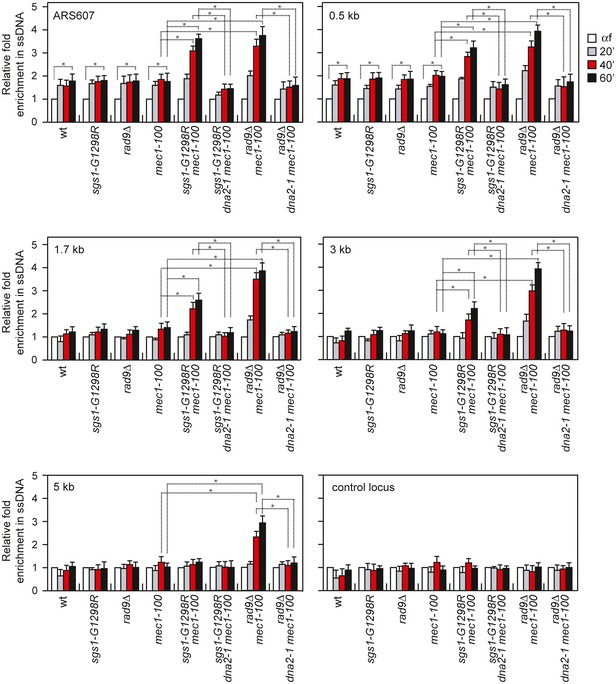
Analysis of ssDNA formation at different distances from ARS607 by qPCR. Exponentially growing YEPD cell cultures were arrested in G1 with α‐factor (αf) and then released in YEPD containing 0.2 M HU. Genomic DNA prepared at different time points after α‐factor release was either digested or mock‐digested with SspI and used as template in qPCR. The value of SspI‐digested over non‐digested DNAs was determined for each time points after normalization to an amplicon on chromosome XI that does not contain SspI sites. The data shown are expressed as fold enrichments in ssDNA at different time points after α‐factor release in HU relative to the α‐factor (αf) (set to 1.0). A locus containing SspI sites on chromosome XI is used as a control (control locus). The mean values are represented with error bars denoting s.d. (n = 3). *P < 0.05 (Student's t‐test).
As ssDNA is rapidly coated by the RPA complex that coordinates DNA damage signaling 77, we also evaluated the association of Rpa1 at stalled replication forks by ChIP and qPCR in cells synchronously released from a G1 arrest into S phase in the presence of HU. Rpa1 association at both early efficient ARS607 and ARS305 replication origins increased in all cell cultures about 20 min after release in the presence of HU and decreased about 40 min later, with sgs1‐G1298R mec1‐100 and rad9∆ mec1‐100 double‐mutant cells showing a more persistent RPA binding compared with both wild type and each single mutant (Fig 8A). Interestingly, while Rpa1 association at stalled replication forks in wild‐type, rad9∆, sgs1‐G1298R, and mec1‐100 cells paralleled that of DNA Polε and showed a pick of association at 20 min after release in HU (compare Figs 3E and 8A), the amount of ssDNA detected directly by qPCR remained constant at 20, 40, and 60 min after release (Fig 7). As the ssDNA molecules could re‐anneal to each other upon DNA extraction and deproteinization, the signal detected by qPCR could represent preferentially ssDNA gaps generated asymmetrically (which therefore cannot re‐anneal), rather than ssDNA regions covered by RPA that are engaged by the replisome during replication.
Figure 8. Rpa1, Rad51, and Rad52 association with stalled replication forks is increased by either Sgs1‐G1298R or the lack of Rad9.
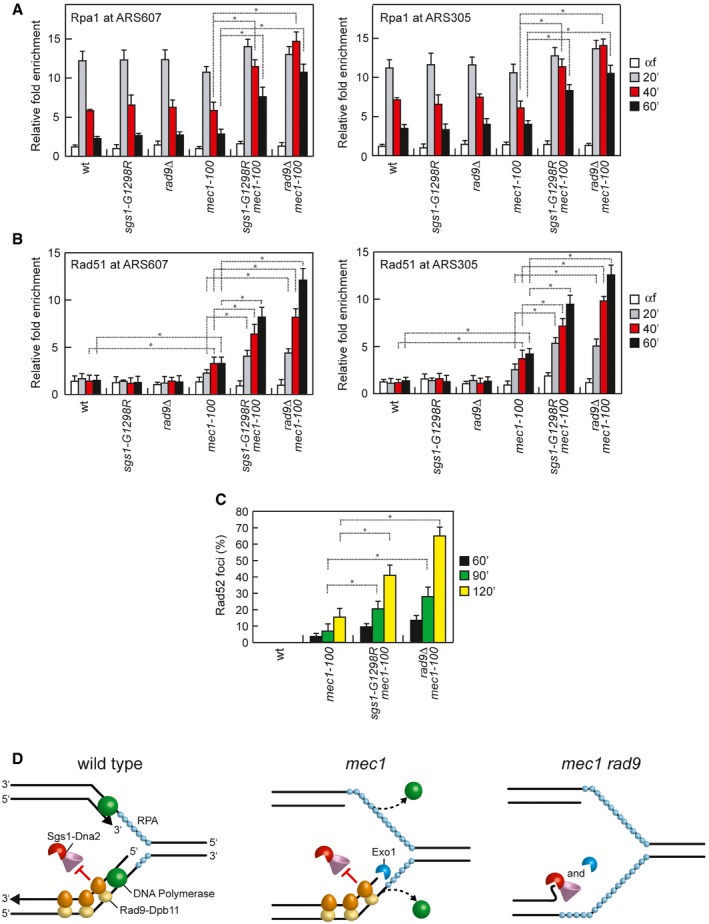
-
A, BChIP analysis. Cells were arrested in G1 with α‐factor and then released in YEPD containing 0.2 M HU at time zero. Relative fold enrichment of Rpa1 and Rad51 at ARS607 and ARS305 replication origins was determined after ChIP with anti‐Myc (A) or anti‐Rad51 (B) antibodies and subsequent qPCR analysis. Plotted values are the mean values with error bars denoting s.d. (n = 3). *P < 0.05 (Student's t‐test).
-
CRad52 foci at the indicated times after release from a G1 arrest in 0.2 M HU. Plotted values are the mean values with error bars denoting s.d. (n = 3). *P < 0.05 (Student's t‐test).
-
DA model for the role of Rad9 in protecting stalled replication forks from degradation. (Left) In wild‐type cells, activation of the checkpoint in response to inhibition of DNA replication maintains replisome integrity, couples leading‐ and lagging‐strand synthesis, and limits Exo1‐mediated degradation. Rad9 limits the resection activity of Sgs1‐Dna2 to degrade nascent lagging strands. (Middle) Checkpoint dysfunction leads to the dislocation of the replisome from sites of DNA synthesis and to the exposure of newly synthesized DNA to Exo1‐mediated degradation. Rad9 still inhibits the resection activity of Sgs1‐Dna2. (Right) When both the checkpoint and Rad9 are dysfunctional, inhibition of both Exo1 and Dna2 activity is relieved, leading to uncontrolled DNA degradation.
Replication protein A is subsequently displaced by the recombinase Rad51 to generate Rad51 nucleoprotein filaments that initiate homologous recombination 77. Consistent with an increase in ssDNA generation, the decrease in Rpa1 binding at the replication origins in sgs1‐G1298R mec1‐100 and rad9∆ mec1‐100 double‐mutant cells was concomitant with an increased accumulation of Rad51, whose association and persistence were higher in both sgs1‐G1298R mec1‐100 and rad9∆ mec1‐100 cells than in mec1‐100 cells (Fig 8B). Interestingly, although we could not detect any increase in ssDNA generation at the replication origin in HU‐treated mec1‐100 cells (Fig 7) 9, Rad51 association appeared to be increased in mec1‐100 cells compared with wild‐type cells (Fig 8B). This finding is consistent with a possible role of Mec1 in inhibiting Rad51 association with DNA that can be partially defective in mec1‐100 cells 78.
Finally, as the recombination protein Rad52 stimulates DNA annealing and Rad51‐catalyzed strand invasion reactions to allow recombination‐mediated fork restart 77, we analyzed the formation of Rad52 recombination foci. Rad52 foci were not detectable in HU‐treated wild‐type cells, while their frequency increased dramatically after HU treatment in both sgs1‐G1298R mec1‐100 and rad9∆ mec1‐100 double‐mutant cells compared with mec1‐100 cells (Fig 8C). Collectively, these results show that both sgs1‐G1298R and the lack of Rad9 increase ssDNA generation at the replication forks when the checkpoint is dysfunctional, pointing to a role for Rad9 in restricting resection at arrested replication forks.
Discussion
Controlled degradation of replication forks by nucleases can be a relevant mechanism to recover replication fork blockage by promoting HR repair and/or by processing specific stalled replication fork structures. However, unscheduled nuclease action could destroy the fork structure and prevent continued DNA synthesis, leading to genome instability. Structural analysis of DNA replication forks in S. cerevisiae has shown that rad53 mutant cells treated with HU accumulate replication forks with extended ssDNA gaps that appear to be localized on only one of the two newly synthesized strands 27. On the one hand, formation of these ssDNA gaps is partly dependent on the Exo1 nuclease 28, 29, which turns out to be a target of Mec1/ATR 31, 33, 34, suggesting that the intra‐S checkpoint suppresses Exo1‐mediated processing of stalled replication forks. On the other hand, Rad53 checkpoint kinase appears to limit ssDNA generation at stalled replication forks by ensuring the coupling of leading‐ and lagging‐strand synthesis possibly through inhibition of excessive template unwinding and upregulation of dNTP levels 79.
In both yeast and mammals, the checkpoint protein Rad9/53BP1 is known to inhibit resection of intrachromosomal DSBs 24, 25, 80, 81 by counteracting the resection activity of Sgs1‐Dna2 22, 23. Here, we describe a previously undisclosed function of Rad9 in maintaining viability of Mec1‐defective cells upon dNTP depletion by protecting stalled replication forks from detrimental nucleolytic processing. In particular, we show that the sensitivity to HU of cells either lacking Mec1 or expressing mec1 hypomorphic alleles is exacerbated by loss of Rad9 or expression of an Sgs1 variant (Sgs1‐G1298R) that escapes Rad9‐mediated inhibition of DSB resection. Furthermore, the HU hypersensitivity of mec1‐100 cells is suppressed in a Rad9‐dependent manner by elimination of Slx4 or Fun30, which are known to counteract the inhibition that Rad9 exerts on DSB resection 65, 66, 67, 68, 69, 70. Finally, both the rad9∆ and sgs1‐G1298R mutations dramatically increase the generation of ssDNA at the replication forks in HU‐treated mec1‐100 cells in a Dna2‐dependent manner. These findings, together with the observation that Dna2 deficiency is epistatic to rad9∆ and sgs1‐G1298R with respect to HU sensitivity of mec1‐100 cells, indicate a role for Rad9 in supporting viability of Mec1‐deficient cells by protecting replication forks from Dna2‐mediated degradation. This Rad9 protective function relies mainly on the interaction of Rad9 with Dpb11, which is recruited to stressed replication origins 82 and forms nuclear foci in response to replication stress 83. Altogether, our data support a model whereby survival to replication stress of Mec1‐defective cells is dependent on the Rad9‐Dpb11 complex that restrains uncontrolled Dna2‐mediated nucleolytic processing of stalled replication forks.
Whether the increased Dna2‐dependent ssDNA generation in rad9∆ mec1‐100 and sgs1‐G1298R mec1‐100 cells arise upon nucleolytic degradation of nascent DNA strands and/or of DSBs that are generated by the action of endonucleases at unprotected stalled replication forks remains to be determined. In any case, the lack of the Mus81 endonuclease, which cleaves branched structures that can be generated at stalled replication forks 84, 85, did not suppress the HU hypersensitivity of rad9∆ mec1‐100 (Fig EV1), suggesting that Mus81 is not responsible for these possible DNA cleavage events. Given the role of Dna2 in Okazaki fragment maturation 86, 21, 87, we favor the hypothesis that Rad9 prevents Dna2 activity in degrading 5′ ends generated at nascent lagging strands.
Figure EV1. The lack of Mus81 does not suppress the HU hypersensitivity of mec1‐100 rad9∆ cells.

Exponentially growing cultures were serially diluted (1:10), and each dilution was spotted out onto YEPD plates with or without HU at the indicated concentrations.
Altogether, these findings suggest a working model (Fig 8D), in which the intra‐S checkpoint prevents the generation of excessive ssDNA under replication stress both by coordinating DNA unwinding with leading‐ and lagging‐strand synthesis 79 and by limiting exposure of nascent DNA strands to Exo1‐mediated degradation 28, 29. Rad9, in turn, limits nucleolytic degradation of nascent lagging strands by inhibiting Sgs1‐Dna2 resection activity. Inactivation of the checkpoint can cause the dislocation of the replisome from sites of DNA synthesis and the exposure of newly synthesized DNA to Exo1‐mediated degradation. Under this condition, the lack of Rad9 relieves the inhibition of Sgs1‐Dna2 activity and this leads to uncontrolled Dna2‐dependent degradation of nascent lagging strands that, together with Exo1‐mediated resection, destroys the fork structure and prevents continued DNA synthesis.
Notably, the Dpb11‐Rad9 interaction appears to be conserved in human cells, where TOPBP1, the Dbp11 human ortholog, stabilizes 53BP1 to the sites of damage to exert its inhibitory function on DSB resection 70, 88. As replication stress underlies a significant proportion of the genomic instability observed in cancer cells 89, understanding whether a similar 53BP1‐mediated resection block is active also at mammalian damaged replication forks and supports survival to replicative stress of ATR‐deficient cells can be important to improve the use of ATR inhibitors in cancer therapy.
Materials and Methods
Yeast strains
Yeast strains used for this work are haploid derivatives of W303 and are listed in Table EV1. Gene disruptions were generated by one‐step PCR disruption method. Cells were grown in YEP medium (1% yeast extract, 2% bactopeptone) supplemented with 2% glucose (YEPD). The strain expressing Rad52‐YFP was provided by M. Lisby (University of Copenhagen, Denmark). The rad9‐Y798A and rad9‐STAA alleles were kindly provided by S.P. Jackson (University of Cambridge, UK) and J.F.X. Diffley (The Francis Crick Institute, UK), respectively. The fun30‐SSAA allele was kindly provided by B. Pfander (Max Planck Institute, Germany). Strains expressing both the nucleoside transporter hENT1 and the herpes simplex virus thymidine kinase (HSV‐TK) to allow BrdU incorporation were constructed by transforming cells with p306‐BrdU‐Inc plasmid kindly provided by O. Aparicio (University of Southern California, USA) 90.
Analysis of cell cycle progression and Western blotting
Exponentially growing cultures were synchronized in G1 by addition of 5 μg/ml α‐factor. G1‐arrested cell cultures were then transferred to fresh YEPD and released into S phase in the presence or in the absence of hydroxyurea 0.2 M. To detect Rad53, trichloroacetic acid protein extracts were separated on 10% polyacrylamide gels and Rad53 detection was carried out using anti‐Rad53 polyclonal antibodies (ab104232) from Abcam.
Drug sensitivity assays
Overnight‐grown saturated cultures of the indicated strains were serially diluted (10‐fold) in water; 10‐μl drops of each dilution were deposited on each plate. Images were scanned 2–3 days after plating and growth at 28°C. Each experiment was repeated at least twice.
ChIP analysis
ChIP analysis was performed as previously described 91. Input and immunoprecipitated DNA were purified and analyzed by qPCR using a Bio‐Rad MiniOpticon. Data are expressed as fold enrichment at ARS607 and ARS305 over that at a region located 14 kb from ARS607, after normalization of each ChIP signals to the corresponding input for each time point. Rad51 immunoprecipitation was carried out with anti‐Rad51 antibodies from Abcam (Ab63798). Rad9‐Flag immunoprecipitation was performed with anti‐Flag antibodies from Sigma (F1804). Rfa1‐Myc and Polε‐Myc were immunoprecipitated with anti‐Myc antibodies from Abcam (Ab32). Primers are listed in Table EV2.
Quantification of ssDNA by qPCR
Genomic DNA was digested or not with the restriction enzyme SspI, which cuts dsDNA within the PCR amplicon. Digested and mock‐digested DNAs were subjected to amplification by qPCR (iQ SYBR Green Supermix, Bio‐Rad, 1708882) using primers annealing on either side of the SspI restriction site. qPCR was performed with a Bio‐Rad MiniOpticon. We quantified ssDNA as previously described 75, using the formula: , in which ∆C t is the difference between the threshold cycles of digested and undigested DNA of a given time point. A control locus on chromosome XI with no SspI restriction sites, for which the C t values for digested and undigested DNA would be expected to be similar, was used to correct the ∆C t values of other primers and to normalize the results relative to the amount of DNA initially loaded onto the plate. The control locus is located 20 and 27 kb from ARS1103 and ARS1102, respectively, on chromosome XI. Primers are listed in Table EV2.
Other techniques
Yeast cells were grown and processed for fluorescence microscopy as previously described 92. Fluorophore was a yellow fluorescent protein (YFP) that was visualized on a Nikon Eclipse 600 equipped with a 100× 0.5–1.3 PlanFluor oil objective (Nikon). Pulse–chase BrdU experiments and detection of BrdU‐labeled DNA were performed as described 29 with anti‐BrdU antibodies from GE Healthcare (RPN202).
Author contributions
MPL conceived the project. MV, DB, and MPL designed the experiments. MV, DB, and MC performed the experiments. MPL wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Source Data for Figure 3
Source Data for Figure 5
Acknowledgements
We are grateful to O. Aparicio, J.F.X. Diffley, M. Lisby, B. Pfander, and S.P. Jackson for plasmids and yeast strains. We thank C. Cassani for technical help with fluorescence microscope and G. Lucchini and M. Clerici for critical reading of the manuscript. This work was supported by the Associazione Italiana per la Ricerca sul Cancro (AIRC) (IG 19783) and Ministero dell'Istruzione, dell'Università e della Ricerca (Progetti di Ricerca di Interesse Nazionale‐PRIN 2015) to M.P.L.
EMBO Reports (2018) 19: 351–367
References
- 1. Berti M, Vindigni A (2016) Replication stress: getting back on track. Nat Struct Mol Biol 23: 103–109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Giannattasio M, Branzei D (2017) S‐phase checkpoint regulations that preserve replication and chromosome integrity upon dNTP depletion. Cell Mol Life Sci 74: 2361–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zou L, Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science 300: 1542–1548 [DOI] [PubMed] [Google Scholar]
- 4. Desany BA, Alcasabas AA, Bachant JB, Elledge SJ (1998) Recovery from DNA replicational stress is the essential function of the S‐phase checkpoint pathway. Genes Dev 12: 2956–2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lopes M, Cotta‐Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi‐Falconi M, Newlon CS, Foiani M (2001) The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557–561 [DOI] [PubMed] [Google Scholar]
- 6. Tercero JA, Diffley JF (2001) Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 412: 553–557 [DOI] [PubMed] [Google Scholar]
- 7. Sabatinos SA, Green MD, Forsburg SL (2012) Continued DNA synthesis in replication checkpoint mutants leads to fork collapse. Mol Cell Biol 32: 4986–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cobb JA, Bjergbaek L, Shimada K, Frei C, Gasser SM (2003) DNA polymerase stabilization at stalled replication forks requires Mec1 and the RecQ helicase Sgs1. EMBO J 22: 4325–4336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, Gasser SM (2005) Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev 19: 3055–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lucca C, Vanoli F, Cotta‐Ramusino C, Pellicioli A, Liberi G, Haber J, Foiani M (2004) Checkpoint‐mediated control of replisome‐fork association and signalling in response to replication pausing. Oncogene 23: 1206–1213 [DOI] [PubMed] [Google Scholar]
- 11. De Piccoli G, Katou Y, Itoh T, Nakato R, Shirahige K, Labib K (2012) Replisome stability at defective DNA replication forks is independent of S phase checkpoint kinases. Mol Cell 45: 696–704 [DOI] [PubMed] [Google Scholar]
- 12. Bernstein KA, Gangloff S, Rothstein R (2010) The RecQ DNA helicases in DNA repair. Annu Rev Genet 44: 393–417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Liberi G, Maffioletti G, Lucca C, Chiolo I, Baryshnikova A, Cotta‐Ramusino C, Lopes M, Pellicioli A, Haber JE, Foiani M (2005) Rad51‐dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 19: 339–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hegnauer AM, Hustedt N, Shimada K, Pike BL, Vogel M, Amsler P, Rubin SM, van Leeuwen F, Guénolé A, van Attikum H et al (2012) An N‐terminal acidic region of Sgs1 interacts with Rpa70 and recruits Rad53 kinase to stalled forks. EMBO J 31: 3768–3783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mimitou EP, Symington LS (2008) Sae2, Exo1 and Sgs1 collaborate in DNA double‐strand break processing. Nature 455: 770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu Z, Chung WH, Shim EY, Lee SE, Ira G (2008) Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double‐strand break ends. Cell 134: 981–994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cejka P, Cannavo E, Polaczek P, Masuda‐Sasa T, Pokharel S, Campbell JL, Kowalczykowski SC (2010) DNA end resection by Dna2‐Sgs1‐RPA and its stimulation by Top3‐Rmi1 and Mre11‐Rad50‐Xrs2. Nature 467: 112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Niu H, Chung WH, Zhu Z, Kwon Y, Zhao W, Chi P, Prakash R, Seong C, Liu D, Lu L et al (2010) Mechanism of the ATP‐dependent DNA end‐resection machinery from Saccharomyces cerevisiae . Nature 467: 108–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nimonkar AV, Genschel J, Kinoshita E, Polaczek P, Campbell JL, Wyman C, Modrich P, Kowalczykowski SC (2011) BLM‐DNA2‐RPA‐MRN and EXO1‐BLM‐RPA‐MRN constitute two DNA end resection machineries for human DNA break repair. Genes Dev 25: 350–362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cannavo E, Cejka P (2014) Sae2 promotes dsDNA endonuclease activity within Mre11‐Rad50‐Xrs2 to resect DNA breaks. Nature 514: 122–125 [DOI] [PubMed] [Google Scholar]
- 21. Kao HI, Campbell JL, Bambara RA (2004) Dna2p helicase/nuclease is a tracking protein, like FEN1, for flap cleavage during Okazaki fragment maturation. J Biol Chem 279: 50840–50849 [DOI] [PubMed] [Google Scholar]
- 22. Bonetti D, Villa M, Gobbini E, Cassani C, Tedeschi G, Longhese MP (2015) Escape of Sgs1 from Rad9 inhibition reduces the requirement for Sae2 and functional MRX in DNA end resection. EMBO Rep 16: 351–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ferrari M, Dibitetto D, De Gregorio G, Eapen VV, Rawal CC, Lazzaro F, Tsabar M, Marini F, Haber JE, Pellicioli A (2015) Functional interplay between the 53BP1‐ortholog Rad9 and the Mre11 complex regulates resection, end‐tethering and repair of a double‐strand break. PLoS Genet 11: e1004928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lydall D, Weinert T (1995) Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science 270: 1488–1491 [DOI] [PubMed] [Google Scholar]
- 25. Lazzaro F, Sapountzi V, Granata M, Pellicioli A, Vaze M, Haber JE, Plevani P, Lydall D, Muzi‐Falconi M (2008) Histone methyltransferase Dot1 and Rad9 inhibit single‐stranded DNA accumulation at DSBs and uncapped telomeres. EMBO J 27: 1502–1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Peng G, Dai H, Zhang W, Hsieh HJ, Pan MR, Park YY, Tsai RY, Bedrosian I, Lee JS, Ira G et al (2012) Human nuclease/helicase DNA2 alleviates replication stress by promoting DNA end resection. Cancer Res 72: 2802–2813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sogo JM, Lopes M, Foiani M (2002) Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science 297: 599–602 [DOI] [PubMed] [Google Scholar]
- 28. Cotta‐Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M (2005) Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint‐defective cells. Mol Cell 17: 153–159 [DOI] [PubMed] [Google Scholar]
- 29. Segurado M, Diffley JF (2008) Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev 22: 1816–1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Colosio A, Frattini C, Pellicanò G, Villa‐Hernández S, Bermejo R (2016) Nucleolytic processing of aberrant replication intermediates by an Exo1‐Dna2‐Sae2 axis counteracts fork collapse‐driven chromosome instability. Nucleic Acids Res 44: 10676–10690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsang E, Miyabe I, Iraqui I, Zheng J, Lambert SA, Carr AM (2014) The extent of error‐prone replication restart by homologous recombination is controlled by Exo1 and checkpoint proteins. J Cell Sci 127: 2983–2994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koundrioukoff S, Carignon S, Técher H, Letessier A, Brison O, Debatisse M (2013) Stepwise activation of the ATR signaling pathway upon increasing replication stress impacts fragile site integrity. PLoS Genet 9: e1003643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Smolka MB, Albuquerque CP, Chen SH, Zhou H (2007) Proteome‐wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci USA 104: 10364–10369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morin I, Ngo HP, Greenall A, Zubko MK, Morrice N, Lydall D (2008) Checkpoint‐dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J 27: 2400–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hu J, Sun L, Shen F, Chen Y, Hua Y, Liu Y, Zhang M, Hu Y, Wang Q, Xu W et al (2012) The intra‐S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell 149: 1221–1232 [DOI] [PubMed] [Google Scholar]
- 36. Duxin JP, Moore HR, Sidorova J, Karanja K, Honaker Y, Dao B, Piwnica‐Worms H, Campbell JL, Monnat RJ Jr, Stewart SA (2012) Okazaki fragment processing‐independent role for human Dna2 enzyme during DNA replication. J Biol Chem 287: 21980–21991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Thangavel S, Berti M, Levikova M, Pinto C, Gomathinayagam S, Vujanovic M, Zellweger R, Moore H, Lee EH, Hendrickson EA et al (2015) DNA2 drives processing and restart of reversed replication forks in human cells. J Cell Biol 208: 545–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Howlett NG, Taniguchi T, Durkin SG, D'Andrea AD, Glover TW (2005) The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum Mol Genet 14: 693–701 [DOI] [PubMed] [Google Scholar]
- 39. Hashimoto Y, Ray Chaudhuri A, Lopes M, Costanzo V (2010) Rad51 protects nascent DNA from Mre11‐dependent degradation and promotes continuous DNA synthesis. Nat Struct Mol Biol 17: 1305–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair‐independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schlacher K, Wu H, Jasin M (2012) A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51‐BRCA1/2. Cancer Cell 22: 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ying S, Hamdy FC, Helleday T (2012) Mre11‐dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72: 2814–2821 [DOI] [PubMed] [Google Scholar]
- 43. Unno J, Itaya A, Taoka M, Sato K, Tomida J, Sakai W, Sugasawa K, Ishiai M, Ikura T, Isobe T et al (2014) FANCD2 binds CtIP and regulates DNA‐end resection during DNA interstrand crosslink repair. Cell Rep 7: 1039–1047 [DOI] [PubMed] [Google Scholar]
- 44. Karanja KK, Lee EH, Hendrickson EA, Campbell JL (2014) Preventing over‐resection by DNA2 helicase/nuclease suppresses repair defects in Fanconi anemia cells. Cell Cycle 13: 1540–1550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chen X, Bosques L, Sung P, Kupfer GM (2016) A novel role for non‐ubiquitinated FANCD2 in response to hydroxyurea‐induced DNA damage. Oncogene 35: 22–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Su F, Mukherjee S, Yang Y, Mori E, Bhattacharya S, Kobayashi J, Yannone SM, Chen DJ, Asaithamby A (2014) Nonenzymatic role for WRN in preserving nascent DNA strands after replication stress. Cell Rep 9: 1387–1401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Iannascoli C, Palermo V, Murfuni I, Franchitto A, Pichierri P (2015) The WRN exonuclease domain protects nascent strands from pathological MRE11/EXO1‐dependent degradation. Nucleic Acids Res 43: 9788–9803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Higgs MR, Reynolds JJ, Winczura A, Blackford AN, Borel V, Miller ES, Zlatanou A, Nieminuszczy J, Ryan EL, Davies NJ et al (2015) BOD1L is required to suppress deleterious resection of stressed replication forks. Mol Cell 59: 462–477 [DOI] [PubMed] [Google Scholar]
- 49. Mimitou EP, Symington LS (2010) Ku prevents Exo1 and Sgs1‐dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 29: 3358–3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hickson ID, Mankouri HW (2011) Processing of homologous recombination repair intermediates by the Sgs1‐Top3‐Rmi1 and Mus81‐Mms4 complexes. Cell Cycle 10: 3078–3085 [DOI] [PubMed] [Google Scholar]
- 51. Rodriguez J, Tsukiyama T (2013) ATR‐like kinase Mec1 facilitates both chromatin accessibility at DNA replication forks and replication fork progression during replication stress. Genes Dev 27: 74–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Paciotti V, Clerici M, Scotti M, Lucchini G, Longhese MP (2001) Characterization of mec1 kinase‐deficient mutants and of new hypomorphic mec1 alleles impairing subsets of the DNA damage response pathway. Mol Cell Biol 21: 3913–3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tercero JA, Longhese MP, Diffley JF (2003) A central role for DNA replication forks in checkpoint activation and response. Mol Cell 11: 1323–1336 [DOI] [PubMed] [Google Scholar]
- 54. Budd ME, Campbell JL (1997) A yeast replicative helicase, Dna2 helicase, interacts with yeast FEN‐1 nuclease in carrying out its essential function. Mol Cell Biol 17: 2136–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van Leeuwen F, Gafken PR, Gottschling DE (2002) Dot1p modulates silencing in yeast by methylation of the nucleosome core. Cell 109: 745–756 [DOI] [PubMed] [Google Scholar]
- 56. Huyen Y, Zgheib O, Ditullio RA Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD (2004) Methylated lysine 79 of histone H3 targets 53BP1 to DNA double‐strand breaks. Nature 432: 406–411 [DOI] [PubMed] [Google Scholar]
- 57. Giannattasio M, Lazzaro F, Plevani P, Muzi‐Falconi M (2005) The DNA damage checkpoint response requires histone H2B ubiquitination by Rad6‐Bre1 and H3 methylation by Dot1. J Biol Chem 280: 9879–9886 [DOI] [PubMed] [Google Scholar]
- 58. Wysocki R, Javaheri A, Allard S, Sha F, Côté J, Kron SJ (2005) Role of Dot1‐dependent histone H3 methylation in G1 and S phase DNA damage checkpoint functions of Rad9. Mol Cell Biol 25: 8430–8443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Grenon M, Costelloe T, Jimeno S, O'Shaughnessy A, Fitzgerald J, Zgheib O, Degerth L, Lowndes NF (2007) Docking onto chromatin via the Saccharomyces cerevisiae Rad9 Tudor domain. Yeast 24: 105–119 [DOI] [PubMed] [Google Scholar]
- 60. Javaheri A, Wysocki R, Jobin‐Robitaille O, Altaf M, Côté J, Kron SJ (2006) Yeast G1 DNA damage checkpoint regulation by H2A phosphorylation is independent of chromatin remodeling. Proc Natl Acad Sci USA 103: 13771–13776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Toh GW, O'Shaughnessy AM, Jimeno S, Dobbie IM, Grenon M, Maffini S, O'Rorke A, Lowndes NF (2006) Histone H2A phosphorylation and H3 methylation are required for a novel Rad9 DSB repair function following checkpoint activation. DNA Repair 5: 693–703 [DOI] [PubMed] [Google Scholar]
- 62. Hammet A, Magill C, Heierhorst J, Jackson SP (2007) Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep 8: 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Granata M, Lazzaro F, Novarina D, Panigada D, Puddu F, Abreu CM, Kumar R, Grenon M, Lowndes NF, Plevani P et al (2010) Dynamics of Rad9 chromatin binding and checkpoint function are mediated by its dimerization and are cell cycle‐regulated by CDK1 activity. PLoS Genet 6: e1001047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pfander B, Diffley JF (2011) Dpb11 coordinates Mec1 kinase activation with cell cycle‐regulated Rad9 recruitment. EMBO J 30: 4897–4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen X, Cui D, Papusha A, Zhang X, Chu CD, Tang J, Chen K, Pan X, Ira G (2012) The Fun30 nucleosome remodeller promotes resection of DNA double‐strand break ends. Nature 489: 576–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Costelloe T, Louge R, Tomimatsu N, Mukherjee B, Martini E, Khadaroo B, Dubois K, Wiegant WW, Thierry A, Burma S et al (2012) The yeast Fun30 and human SMARCAD1 chromatin remodellers promote DNA end resection. Nature 489: 581–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Eapen VV, Sugawara N, Tsabar M, Wu WH, Haber JE (2012) The Saccharomyces cerevisiae chromatin remodeler Fun30 regulates DNA end resection and checkpoint deactivation. Mol Cell Biol 32: 4727–4740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bantele SC, Ferreira P, Gritenaite D, Boos D, Pfander B (2017) Targeting of the Fun30 nucleosome remodeller by the Dpb11 scaffold facilitates cell cycle‐regulated DNA end resection. Elife 6: pii: e21687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dibitetto D, Ferrari M, Rawal CC, Balint A, Kim T, Zhang Z, Smolka MB, Brown GW, Marini F, Pellicioli A (2016) Slx4 and Rtt107 control checkpoint signalling and DNA resection at double‐strand breaks. Nucleic Acids Res 44: 669–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Liu Y, Cussiol JR, Dibitetto D, Sims JR, Twayana S, Weiss RS, Freire R, Marini F, Pellicioli A, Smolka MB (2017) TOPBP1Dpb11 plays a conserved role in homologous recombination DNA repair through the coordinated recruitment of 53BP1Rad9 . J Cell Biol 216: 623–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ohouo PY, Bastos de Oliveira FM, Liu Y, Ma CJ, Smolka MB (2013) DNA‐repair scaffolds dampen checkpoint signalling by counteracting the adaptor Rad9. Nature 493: 120–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Ohouo PY, Bastos de Oliveira FM, Almeida BS, Smolka MB (2010) DNA damage signaling recruits the Rtt107‐Slx4 scaffolds via Dpb11 to mediate replication stress response. Mol Cell 39: 300–306 [DOI] [PubMed] [Google Scholar]
- 73. Flott S, Rouse J (2005) Slx4 becomes phosphorylated after DNA damage in a Mec1/TEL1‐dependent manner and is required for repair of DNA alkylation damage. Biochem J 391: 325–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Neves‐Costa A, Will WR, Vetter AT, Miller JR, Varga‐Weisz P (2009) The SNF2‐family member Fun30 promotes gene silencing in heterochromatic loci. PLoS One 4: e8111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zierhut C, Diffley JF (2008) Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J 27: 1875–1885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Ait Saada A, Teixeira‐Silva A, Iraqui I, Costes A, Hardy J, Paoletti G, Fréon K, Lambert SAE (2017) Unprotected replication forks are converted into mitotic sister chromatid bridges. Mol Cell 66: 398–410 [DOI] [PubMed] [Google Scholar]
- 77. San Filippo J, Sung P, Klein H (2008) Mechanism of eukaryotic homologous recombination. Annu Rev Biochem 77: 229–257 [DOI] [PubMed] [Google Scholar]
- 78. Flott S, Kwon Y, Pigli YZ, Rice PA, Sung P, Jackson SP (2011) Regulation of Rad51 function by phosphorylation. EMBO Rep 12: 833–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Gan H, Yu C, Devbhandari S, Sharma S, Han J, Chabes A, Remus D, Zhang Z (2017) Checkpoint kinase Rad53 couples leading‐ and lagging‐strand DNA synthesis under replication stress. Mol Cell 68: 446–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez‐Capetillo O, Cao L et al (2010) 53BP1 inhibits homologous recombination in Brca1‐deficient cells by blocking resection of DNA breaks. Cell 141: 243–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E et al (2011) Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell 42: 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Balint A, Kim T, Gallo D, Cussiol JR, Bastos de Oliveira FM, Yimit A, Ou J, Nakato R, Gurevich A, Shirahige K et al (2015) Assembly of Slx4 signaling complexes behind DNA replication forks. EMBO J 34: 2182–2197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Germann SM, Oestergaard VH, Haas C, Salis P, Motegi A, Lisby M (2011) Dpb11/TopBP1 plays distinct roles in DNA replication, checkpoint response and homologous recombination. DNA Repair 10: 210–224 [DOI] [PubMed] [Google Scholar]
- 84. Fricke WM, Bastin‐Shanower SA, Brill SJ (2005) Substrate specificity of the Saccharomyces cerevisiae Mus81‐Mms4 endonuclease. DNA Repair 4: 243–251 [DOI] [PubMed] [Google Scholar]
- 85. Hanada K, Budzowska M, Davies SL, van Drunen E, Onizawa H, Beverloo HB, Maas A, Essers J, Hickson ID, Kanaar R (2007) The structure‐specific endonuclease Mus81 contributes to replication restart by generating double‐strand DNA breaks. Nat Struct Mol Biol 14: 1096–1104 [DOI] [PubMed] [Google Scholar]
- 86. Bae SH, Seo YS (2000) Characterization of the enzymatic properties of the yeast dna2 Helicase/endonuclease suggests a new model for Okazaki fragment processing. J Biol Chem 275: 38022–38031 [DOI] [PubMed] [Google Scholar]
- 87. Levikova M, Cejka P (2015) The Saccharomyces cerevisiae Dna2 can function as a sole nuclease in the processing of Okazaki fragments in DNA replication. Nucleic Acids Res 43: 7888–7897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cescutti R, Negrini S, Kohzaki M, Halazonetis TD (2010) TopBP1 functions with 53BP1 in the G1 DNA damage checkpoint. EMBO J 29: 3723–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hsieh HJ, Peng G (2017) Cellular responses to replication stress: implications in cancer biology and therapy. DNA Repair 49: 9–20 [DOI] [PubMed] [Google Scholar]
- 90. Viggiani CJ, Aparicio OM (2006) New vectors for simplified construction of BrdU‐Incorporating strains of Saccharomyces cerevisiae . Yeast 23: 1045–1051 [DOI] [PubMed] [Google Scholar]
- 91. Cassani C, Gobbini E, Wang W, Niu H, Clerici M, Sung P, Longhese MP (2016) Tel1 and Rif2 regulate MRX functions in end‐tethering and repair of DNA double‐strand breaks. PLoS Biol 14: e100238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Eckert‐Boulet N, Rothstein R, Lisby M (2011) Cell biology of homologous recombination in yeast. Methods Mol Biol 745: 523–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Review Process File
Source Data for Figure 3
Source Data for Figure 5