

Abstract
Accurate control of macromolecule transport between nucleus and cytoplasm underlines several essential biological processes, including gene expression. According to the canonical model, nuclear import of soluble proteins is based on nuclear localization signals and transport factors. We challenge this view by showing that nuclear localization of the actin‐dependent motor protein Myosin‐1C (Myo1C) resembles the diffusion–retention mechanism utilized by inner nuclear membrane proteins. We show that Myo1C constantly shuttles in and out of the nucleus and that its nuclear localization does not require soluble factors, but is dependent on phosphoinositide binding. Nuclear import of Myo1C is preceded by its interaction with the endoplasmic reticulum, and phosphoinositide binding is specifically required for nuclear import, but not nuclear retention, of Myo1C. Our results therefore demonstrate, for the first time, that membrane association and binding to nuclear partners is sufficient to drive nuclear localization of also soluble proteins, opening new perspectives to evolution of cellular protein sorting mechanisms.
Keywords: myosin, nuclear import, nuclear transport, phosphoinositide
Subject Categories: Membrane & Intracellular Transport
Introduction
The high degree of compartmentalization, and especially the segregation of genomic DNA from the cytoplasm by means of the nuclear envelope, poses a logistical challenge for the cell. Since different compartments have to preserve their special identity, reflected in their protein repertoire, proper protein targeting and delivery are indispensable for well‐being of the cell. This is especially critical in the case of nucleo‐cytoplasmic transport, since expression of multiple genes is often triggered by a certain protein factor, which needs to be imported from the cytoplasm in a selective and timely manner. NFAT signaling 1 and MKL1‐dependent activation of SRF‐dependent genes 2, 3 represent typical examples of gene expression reliance on nuclear import of transcriptional regulators.
Mechanistically, nuclear import of soluble cargo occurs through nuclear pore complexes (NPC), composed of up to 30 different nucleoporins (Nups) that create a semipermeable channel through the nuclear envelope (NE) 4. Water, small molecules, and proteins can pass freely through the NPC by passive diffusion, while larger proteins require an energy‐dependent import mechanism, with the cutoff traditionally thought to be around 40 kDa 5. However, microscopy experiments have revealed that the yeast NPC is a rather poor barrier to most proteins and can leak proteins as large as 150 kDa 6. In line with this, quantitative analysis of frog oocyte nuclear proteome has revealed that assembly of complexes and passive retention contribute more to the maintenance of nuclear and cytoplasmic protein distribution than active nuclear transport 7. Hence, the mechanism of nuclear transport cannot be directly inferred from the size of the protein. In a canonical sequence of active nuclear transport, soluble import receptors (karyopherins) recognize specific nuclear localization sequence (NLS) in the cargo protein. This complex then passes through the central channel of the NPC that is lined with phenylalanine–glycine (FG)‐rich Nups. Inside the nucleus, cargo protein is liberated from the complex with karyopherins by binding to the small GTPase Ran in its GTP‐bound form and can perform its nuclear functions. In the cytoplasm, Ran is mostly GDP‐bound, which favors karyopherin–cargo complex formation, and thus, the Ran‐gradient determines the directionality of transport 5.
Also less conventional mechanisms of nuclear import exist. For instance, recent progress in the field has revealed that nuclear import of inner nuclear membrane (INM) proteins, such as Lap2β, LBR, and SUN2, differs drastically from soluble cargos 8, 9. In particular, their nuclear translocation is independent of transport receptors. Translocation through the NPC is the rate‐limiting step and most probably occurs through peripheral channels of the NPC scaffold and not through the central channel. According to the currently favored diffusion–retention model, INM proteins become embedded in the membrane of endoplasmic reticulum (ER) upon their synthesis and then reach the INM by means of unrestricted diffusion in the 2D membrane space. At the destination site in the nucleus, these proteins interact with chromatin, which decreases their diffusion rate and leads to accumulation at the INM.
In this study, we focus on actin‐based molecular motor protein Myosin‐1C (Myo1C), which is present in both cytoplasm and the nucleus of the cell 10, 11. Myo1C has a very typical domain organization for class 1 myosins: N‐terminal motor (“head”) domain occupies the largest portion of the molecule and generates force upon actin binding, while the relatively short and positively charged C‐terminal “tail” domain directly interacts with cell membranes through a pleckstrin homology (PH)‐like lipid‐binding motif. The head and tail domains are connected by a “neck” domain, which consists of three calmodulin‐binding IQ motifs and plays a crucial role in force transduction between the head and the tail domains 12. In mammalian cells, MYO1C gene encodes three isoforms (termed A, B, and C), which only differ in short (up to 35 aa) N‐terminal peptides and originate through alternative splicing and the use of alternative promoters 13. Being able to interact with both lipids and actin, Myo1C physically connects and generates mechanical force between actin cytoskeleton and cell membranes, which has a plethora of applications in cytoplasmic processes as diverse as exocytosis 14, regulation of membrane tension in stereocilia of the inner ear 15, and integrin‐mediated cellular migration and spreading 16. In addition, Myo1C has been linked to actin‐dependent maintenance of ER sheets 17. In the nucleus, Myo1C isoform B, also known as nuclear myosin 1 (NM1), has been implicated in chromatin remodeling as a part of B‐WICH complex 18, 19, in transcription 20 and in long‐range directional movements of chromosomal loci 21.
Originally, the unique N‐terminal sequences in Myo1C isoforms A and B were postulated to confer the nuclear localization of these proteins 10, 13. Subsequently, Myo1C has been suggested to have an NLS‐like sequence in the neck domain 22, shared by all of three Myo1C isoforms, suggesting that all of them can localize to the nucleus. Instead of nuclear import, the N‐terminal sequences in Myo1C isoforms may regulate their specific intranuclear distributions to, for example, the nucleolus in the case of isoform B 13. The NLS‐like sequence consists of six positively charged amino acid residues (lysines and arginines) overlapping with the second IQ motif and meets two formal requirements for an NLS sequence: Mutations in it abrogate nuclear import and, when fused to a cytosolic protein, the NLS‐like sequence promotes its nuclear accumulation. Importins 5, 7, and β1 were shown to interact with the NLS‐like sequence. Moreover, it was suggested that reversible calcium‐dependent calmodulin binding to the second IQ motif could be a regulatory factor in Myo1C nuclear import 22. Indeed, experimental elevation of intracellular calcium concentration induces nuclear accumulation of Myo1C isoforms 23. Overall, according to this model, nuclear import of Myo1C uses an NLS‐dependent active transport pathway typical for soluble cargos.
In the present study, we further address the determinants of Myo1C nuclear import. Surprisingly, our data suggest that the diffusion–retention model devised for the import of INM proteins would more accurately describe Myo1C nuclear import than the previously proposed import receptor‐based transport mechanism. This opens a new perspective for our understanding of protein sorting mechanisms, showing that membrane association and binding to nuclear partners is sufficient to drive nuclear localization of also soluble proteins.
Results
Myo1C shuttles between cytoplasm and nucleus
Although Myo1C has been suggested to utilize a typical NLS/importin‐dependent mechanism, its dynamics in live cells have remained unclear, which prompted us to address this experimentally. In most of our assays, we used Myo1C isoform B (NM1), but for the sake of simplicity, we refer to it as Myo1C. In order to investigate the dynamic properties of Myo1C, we used photobleaching‐based techniques that have earlier been successfully applied in studies on nucleo‐cytoplasmic shuttling of actin as well as other proteins 3, 24, 25. To understand the nuclear import dynamics of Myo1C, we took advantage of a fluorescence recovery after photobleaching (FRAP) assay based on high‐power laser‐assisted bleaching of the whole nucleus of human osteosarcoma (U2OS) cells stably expressing GFP‐tagged Myo1C, and monitored the time course of fluorescence recovery as the proteins are imported from the unbleached cytoplasm. Figure 1A shows an averaged FRAP curve for GFP‐Myo1C in comparison with those for GFP and GFP‐actin. Due to its small size (27 kDa), GFP travels in and out of the nucleus by means of passive diffusion, while actin uses an active import mechanism dependent on cofilin and importin 9 24. Like for GFP and GFP‐actin, the nuclear fluorescence of GFP‐Myo1C recovered after photobleaching, indicating that myosin indeed undergoes constant import into the nucleus. Comparison of nuclear import rates of GFP‐Myo1C and GFP‐actin (Fig 1B), derived from respective FRAP curves 24, 25, shows that nuclear import of Myo1C is substantially slower than that of actin. Since active transport is insensitive to molecular size, this difference can hardly be attributed to molecular weights of GFP‐Myo1C and GFP‐actin (~150 kDa vs. ~70 kDa). More likely, it suggests that Myo1C uses a different import pathway than actin, and the kinetics of this pathway is much slower, due to, for example, lower abundance of import factors. Alternatively, only a limited fraction of Myo1C could be available for import. The GFP‐Myo1C used in this study was not degraded in cells (Fig 1E).
Figure 1. Myo1C constantly shuttles between the nucleus and cytoplasm.
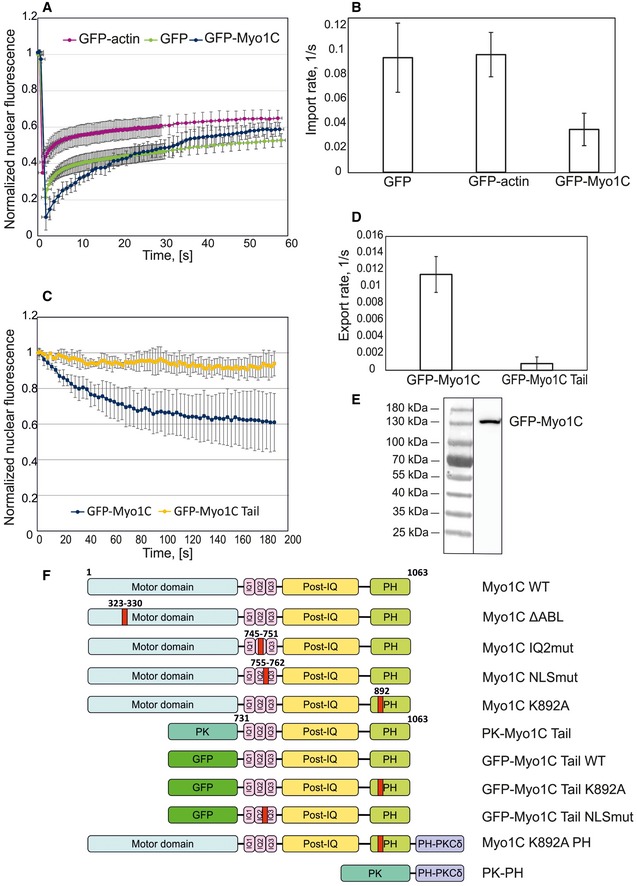
- Fluorescence recovery after photobleaching experiments with GFP‐Myo1C, GFP‐actin, and GFP; data represent nuclear fluorescence levels normalized to the value before bleaching and are the mean ± SD (GFP n = 10; GFP‐actin n = 8; GFP‐Myo1C n = 11).
- Nuclear import rate derived from the FRAP curves; data represent mean rates from individual experiments ± SD (n as in A).
- FLIP experiments with GFP‐Myo1C and GFP‐Myo1C tail WT; data represent normalized nuclear fluorescence levels and are the mean ± SD (n = 8).
- Nuclear export rate quantified from FLIP curves; data represent mean rates from individual experiments ± SD (n = 8).
- Western blot showing that GFP‐Myo1C is expressed as a full‐size protein with no signs of degradation.
- Schematic of Myo1C constructs used in the present study. Pleckstrin homology domain of Myo1C (PH), chicken pyruvate kinase (PK), and pleckstrin homology domain of phospholipase C, isoform δ (PH‐PKCδ), are indicated in the chart. HA‐tag was in the N‐terminus of the constructs. Note that “tail” construct contains also the neck region of Myo1C.
Nuclear export of GFP‐tagged Myo1C was studied by means of fluorescence loss in photobleaching (FLIP). Here, the cytoplasm is repeatedly bleached with a high‐power laser and loss of nuclear fluorescence due to protein export is observed 24, 25. Figure 1C shows typical FLIP curves for GFP‐Myo1C and its truncated construct representing the GFP‐labeled tail portion (GFP‐Myo1C tail; see Fig 1F for the constructs used in this study. Note also that tail constructs contain both neck and tail regions of Myo1C, but are called only “tail” for simplicity). Nuclear fluorescence of GFP‐Myo1C decreases upon cytoplasmic bleaching, indicating that Myo1C is constantly exported from the nucleus. However, a significant fraction of the GFP‐Myo1C is export‐incompetent and cannot be bleached out of the nucleus. This suggests that a substantial pool of nuclear Myo1C molecules (up to 50%) is tightly bound to chromatin or other nuclear structures, which likely renders them inaccessible for export factors. Surprisingly, GFP‐Myo1C tail did not show any export from the nucleus in contrast to the full‐length construct (Fig 1C and D). This strongly suggests that the putative export signal is localized in the “head” portion (motor domain) of the protein. The absence of nuclear export in the case of GFP‐Myo1C tail helps to explain the predominantly nuclear localization of this construct (see below, Fig 2A).
Figure 2. Phosphoinositide binding is indispensable for nuclear localization of Myo1C.
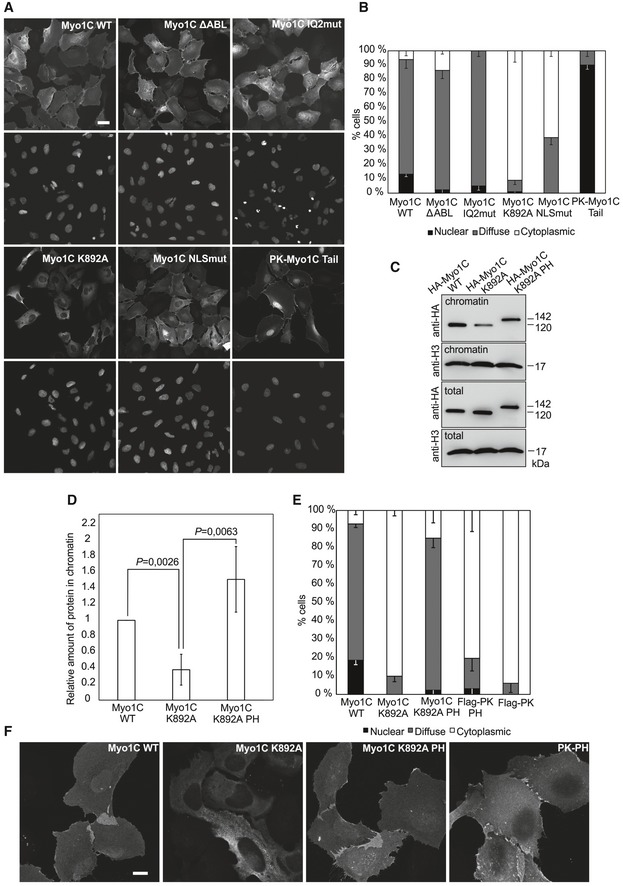
- Wide‐field fluorescence microscopy images showing localization of HA‐tagged Myo1C constructs in transiently transfected human osteosarcoma (U2OS) cells. Scale bar, 10 μm.
- Quantification of the percentage of U2OS cells showing nuclear, diffuse, or cytoplasmic localization of HA‐tagged Myo1C constructs in three independent experiments ± SD (n = 90 cells quantified per construct).
- Western blot analysis of chromatin fractions and total lysates of U2OS cells transfected with specified HA‐Myo1C constructs.
- Quantification of relative amounts of HA‐tagged Myo1C species in the chromatin fraction. Data represent mean ± SD from three independent experiments and are normalized to Myo1C WT. Statistically significant differences tested by Student's t‐test.
- Quantification of the HA‐tagged Myo1C constructs localizations as in (B), data are the mean from three independent experiments ± SD (n = 90 cells quantified per construct).
- Confocal microscopy images of U2OS cells transiently transfected with indicated HA‐tagged Myo1C constructs. Scale bar, 10 μm.
Overall, Myo1C shows a dynamic behavior in the cell, actively traveling between nucleus and cytoplasm, and a substantial portion of Myo1C is stably bound to nuclear constituents.
Phosphoinositide binding is crucial for nuclear localization of Myo1C
To gain further insight into the import mechanism of Myo1C, we set out to target various functional properties in this protein (Fig 1F). Among those, we chose actin binding, which we disrupted by replacing the so‐called myopathy loop in Myo1C motor domain with AGA tripeptide 26 (Myo1C ΔABL). We also mutated the positively charged amino acid residues of the previously identified NLS‐like sequence 22 into small neutral alanines and serines to disrupt possible interactions with import factors. Furthermore, we introduced mutations into calmodulin‐binding IQ2 motif, adjacent to the NLS‐like sequence, to study the hypothesized competition of calmodulin with import factors for the binding site 22. Finally, we introduced a point mutation (K892A) into the putative pleckstrin homology (PH) domain of Myo1C. This substitution abolishes PI binding as measured by co‐sedimentation of recombinant Myo1C with PIP2‐containing phospholipid vesicles 27. Hemagglutinin (HA)‐tagged versions of these constructs were used for transient transfection of U2OS cells, and protein localization was studied by means of wide‐field fluorescence microscopy (Fig 2A). To characterize the construct localization in a quantitative way, observed localization patterns were categorized as “cytoplasmic”, “nuclear”, and “diffuse” and the fraction of cells exhibiting corresponding phenotype was counted (Fig 2B). We also quantified the ratios of nuclear to cytoplasmic fluorescence (N/C ratios) in U2OS cells transfected with above‐mentioned Myo1C constructs (Fig EV1A) and ensured their equal expression levels (Fig EV1B). The wild‐type protein (Myo1C WT) exhibits predominantly diffuse localization (Fig 2A and B), with no clear accumulation of the protein in either nucleus or cytoplasm, which corresponds to N/C ratio of 1 (Fig EV1A). The same pattern was observed for ΔABL and IQ2mut constructs, which suggests that neither actin nor calmodulin binding is crucial for nucleo‐cytoplasmic transport of Myo1C. On the contrary, the Myo1C construct with mutated NLS‐like motif showed substantially decreased, albeit not fully abolished nuclear localization, which was expected in light of previous data 22. In agreement with photobleaching data (Fig 1C), a fusion of Myo1C tail domain with chicken pyruvate kinase (a cytosolic protein with no nuclear localization or export sequences 28) showed an exclusively nuclear localization (Fig 2A and B). Fusion to PK was used to ensure that the size of the construct remains well above diffusion limit of the NPC. Mutations in the putative NLS sequence did not affect nuclear localization of Myo1C tail (Fig EV1E). Although these data suggest that the nuclear export signal is located in the head region of Myo1C, it does not seem to depend on actin binding. The most surprising behavior was demonstrated by the construct with impaired PI binding (Myo1C K892A), which showed, against all expectations, a very clear cytoplasmic localization (Figs 2A and B, and EV1A). This indicates that interaction with PI could play an essential role in Myo1C nuclear targeting.
Figure EV1. Cellular localization of Myosin‐1C constructs.
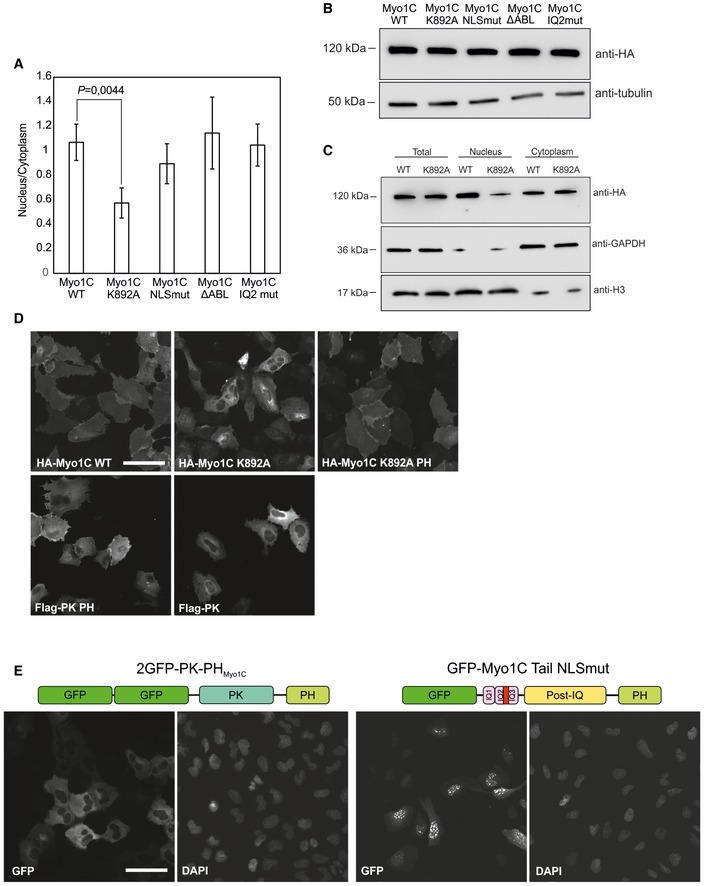
- Quantification of nuclear to cytoplasmic ratios of fluorescence signal in cells transfected with HA‐tagged Myo1C constructs. Human osteosarcoma cells (U2OS) were imaged with confocal microscope and fluorescence intensities of randomly chosen cells (40–50 cells/construct) were calculated using Microscopy image browser software. Data are mean ± SD. Student's t‐test for statistical significance.
- Western blot showing expression levels of HA‐Myo1C constructs in the stable cell lines used in Fig 2A and B.
- Western blot analysis of Myo1C WT and K892 in nuclear and cytoplasmic fractions obtained by cell fractionation in detergent‐free conditions (see Materials and Methods for more details).
- Addition of PH domain to Myo1C K892A rescues its nuclear localization. Figure shows wide‐field fluorescence microscopy images of human osteosarcoma (U2OS) cells transiently transfected with specified HA‐tagged Myo1C constructs; localizations are quantified in Fig 2E. Scale bar, 40 μm.
- Immunofluorescence images showing localization of the PH domain of Myo1C fused with chicken pyruvate kinase and that of Myo1C tail with mutated NLS sequence in human osteosarcoma cells (U2OS). Scale bar, 40 μm.
To confirm the peculiar localization pattern of Myo1C K892A, we used subcellular fractionation as a complementary biochemical approach. Indeed, HA‐tagged Myo1C WT was tightly bound to chromatin, whereas Myo1C K892A was clearly less abundant in this fraction (Fig 2C and D), corroborating the imaging data (Fig 2A and B). As a method of choice, chromatin isolation was preferred over purification of whole intact nuclei, since the presence of Myo1C associated with the nuclear envelope through PI could have distorted the true picture of myosin abundance inside the nucleus. Nevertheless, also fractionation with hypotonic buffer demonstrates decreased nuclear abundance of Myo1C K892A (Fig EV1C).
To confirm the requirement for PI binding in Myo1C nuclear localization, and to rule out possible secondary effects of the K892A substitution, we designed a rescue construct consisting of the PH domain of phospholipase C isoform δ (PLC δ) known for its high affinity for PIP2 29 fused to the C‐terminus of full‐length Myo1C carrying K892A substitution (Fig 1F). Addition of the PH domain to Myo1C K892A efficiently rescues its nuclear localization (Figs 2E and EV1D), confirming the importance of PI binding by the Myo1C tail for this process. Importantly, confocal microscopy shows that similar to the WT protein, the rescue construct localizes to the nuclear interior and not simply to the membrane components of the nucleus (Fig 2F). In addition, biochemical fractionation confirmed the nuclear localization of the rescue construct (Fig 2C and D). Noteworthy, addition of the PH domain, from either PLC δ or Myo1C, to pyruvate kinase did not promote its nuclear localization (Figs 2E, and EV1D and E), demonstrating that PH domain is required, but not sufficient, for nuclear localization of Myo1C.
Of note, also Myo1C isoform C, which lacks the 16‐amino acid N‐terminal extension, fully recapitulated the localization pattern of Myo1C isoform B and the effect of K892A substitution on it (Fig EV2A). This indicates that PI binding is a critical determinant of nuclear localization of all Myo1C isoforms. Thus, our data lead to the conclusion that PI binding by the PH‐like domain, localized in the tail region of Myo1C, is indispensable for nuclear localization of the protein.
Figure EV2. Myosin‐1C constructs NLSmut and K892A show decreased nuclear localization.

- Isoform C of Myo1C recapitulates the effects of K892A and NLSmut mutations on the nuclear localization of the protein. Figure shows wide‐field fluorescence microscopy images of human osteosarcoma (U2OS) cells transiently transfected with specified HA‐tagged Myo1C constructs. Scale bar, 20 μm.
- GFP‐Myo1C tail WT and K892A show different kinetics of accumulation in the nucleus after cell division. Cell cycle of human osteosarcoma cells (U2OS) stably expressing GFP‐Myo1C tail WT and K892A was monitored by means of wide‐field fluorescence microscopy; see Fig 3C and D for other representation of the data. Figure shows initial segments of averaged fluorescence recovery curves, whose slope reflects the rate of recovery. Data are mean from 15 cells ± SD. Measurements of nuclear fluorescence were only possibly as early as 30 min after cytokinesis, since then nuclei could be distinguished from the cytoplasm.
Phosphoinositide binding is required for nuclear import of Myo1C
Having established the importance of PI binding for the nuclear localization of Myo1C, we sought to uncover its molecular mechanism. Two alternative scenarios, nuclear retention and import, could explain our observations. The first one involves an interaction of Myo1C with nuclear PIs, which would lead to its retention in the nucleus. PIs have been identified as components of such nuclear compartments as interchromatin granules and nucleolus 30, 31, 32 as well as nuclear speckles 33, where they were proposed to play a role in polyadenylation of pre‐mRNA 34. In the other scenario, the PI interaction is required for the actual nuclear import process of Myo1C. To distinguish between nuclear retention and import, we first compared the nuclear import rates of Myo1C WT and Myo1C K892A. Unfortunately, FRAP measurements with the K892A mutant were impossible due to its low nuclear abundance. To circumvent this issue, we took advantage of the truncated version of Myo1C lacking the head domain (Myo1C tail, Fig 1F), which is export‐incompetent and thus nuclear (Fig 1C), allowing us to analyze nuclear import without the need of taking export into account. GFP‐Myo1C tail WT shows a uniformly nuclear localization, while a careful examination of cells expressing GFP‐Myo1C tail K892A yields an uneven localization pattern: While there are cells with a very clear nuclear signal, a certain subpopulation shows varying degrees of cytoplasmic localization (Fig 3A). Noteworthy, these latter cells are often found close to each other, as if they have just divided. These observations suggest that GFP‐Myo1C tail K892A might show delayed kinetics of nuclear accumulation after the re‐formation of nuclear envelope after mitosis, which could be explained in terms of import differences between WT and K892A constructs. To test this possibility, we imaged U2OS cells stably expressing GFP‐Myo1C tail WT and GFP‐Myo1C tail K892A at roughly the same levels (Fig 3B) in the course of 12–14 h to capture cell divisions (Fig 3C). Analysis of videos showed that nuclear accumulation of K892A mutant construct after mitosis is dramatically slower than that of the WT protein (Figs 3D and EV2B), indicating that PI binding indeed contributes to the nuclear import of Myo1C.
Figure 3. PI binding is required for nuclear import of Myo1C.
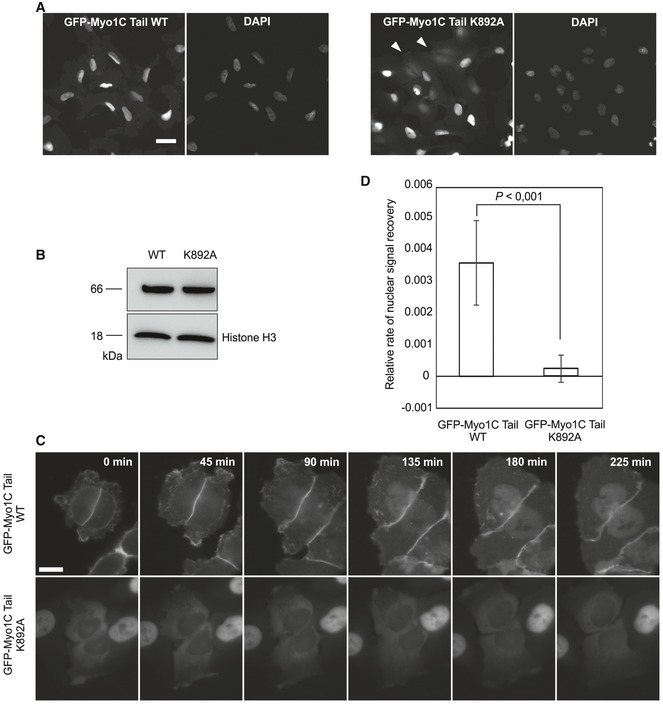
- Localization of GFP‐Myo1C tail WT and K892A constructs in inducible stable U2OS cell lines by wide‐field fluorescence microscopy. Scale bar, 40 μm.
- Western blot with antibody against GFP, showing equal expression of GFP‐Myo1C tail WT and K892A in respective cell lines.
- Representative time‐lapse images of U2OS cells stably expressing GFP‐Myo1C tail WT and K892A. Time point “0 min” corresponds to the earliest detected moment after cytokinesis. Scale bar, 20 μm.
- Relative nuclear fluorescence recovery rates after cytokinesis in cells expressing GFP‐Myo1C tail WT and K892A. Data are mean ± SD (n = 22 cells from three independent experiments). Rate was approximated with straight lines from the fluorescence recovery curves (see Fig EV2B), and statistically significant differences were tested by Student's t‐test.
PIs are membrane lipids, and with the notable and peculiar exception of nuclear PIs 35, they do not exist beyond lipid bilayers, implying that Myo1C interaction with membrane might be at the core of its nuclear import mechanism. A good candidate organelle for this would be endoplasmic reticulum (ER), since it is enriched in phosphatidylcholine and PIs 36. Second, ER forms a continuous membrane structure with the nuclear envelope, and third, proteins of the INM have been shown to use ER for their nuclear import 37. Indeed, confocal microscopy revealed that Myo1C partially colocalized with the ER marker calreticulin (Fig EV3A) and cell fractionation experiments demonstrated the presence of both endogenously (Fig EV3C) and exogenously expressed Myo1C in the ER membrane fraction, while Myo1C K892A mutant was less abundant in ER fraction (Fig EV3B). To further address the nuclear import mechanism of Myo1C, we used a well‐established in vitro import system based on selective permeabilization of plasma membrane in HeLa cells with digitonin 38, 39. Semipermeabilized cells were then incubated with the import substrates, HeLa lysate as a source of transport factors and energy‐regenerating mix. As a result, we observed clear nuclear accumulation of both the model import substrate (NLS‐GST‐GFP) and recombinant GFP‐Myo1C tail/calmodulin complex (Fig 4). Remarkably, the presence of soluble import factors did not seem to be crucial for Myo1C nuclear import, since the GFP‐Myo1C tail/calmodulin complex showed significant nuclear accumulation even in the absence of HeLa lysate, while import of NLS‐GST‐GFP was highly inefficient under these conditions (Figs 4A and B, and EV3C). In line with this, transport of GFP‐Myo1C was insensitive to addition of RanQ69L. Interestingly, addition of wheat germ agglutinin (WGA) halted the transport of both constructs (Fig 4). Finally, removal of the energy source prevented nuclear localization of also Myo1C (Fig 4).
Figure EV3. Localization of Myosin‐1C to the ER.
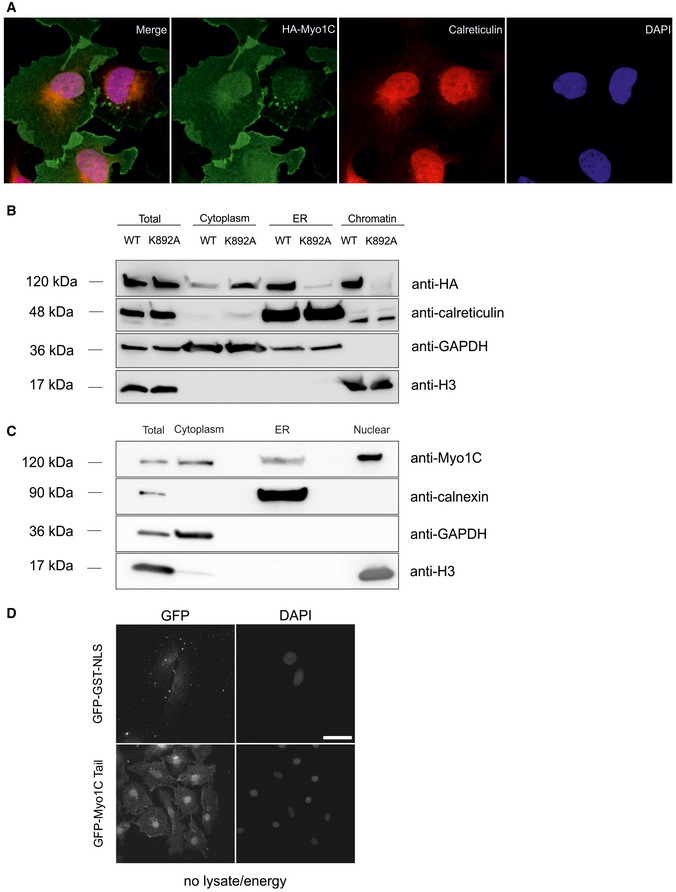
- Confocal immunofluorescence images showing colocalization of HA‐tagged Myo1C WT with the ER marker calreticulin in U2OS cells.
- Western blotting analyses of subcellular fractions showing that HA‐Myo1C WT is present in the ER fraction, while HA‐Myo1C K892A is not (see Materials and Methods for more details).
- Western blotting analysis showing that endogenous Myosin‐1C localizes in the endoplasmic reticulum.
- 63× magnification images of GFP‐GST‐NLS and GFP‐Myo1C tail under condition “− lysate/+ energy” in in vitro import assay; for smaller magnification, see Fig 4.
Figure 4. In vitro import assay shows no requirement for soluble transport factors in Myo1C nuclear import.
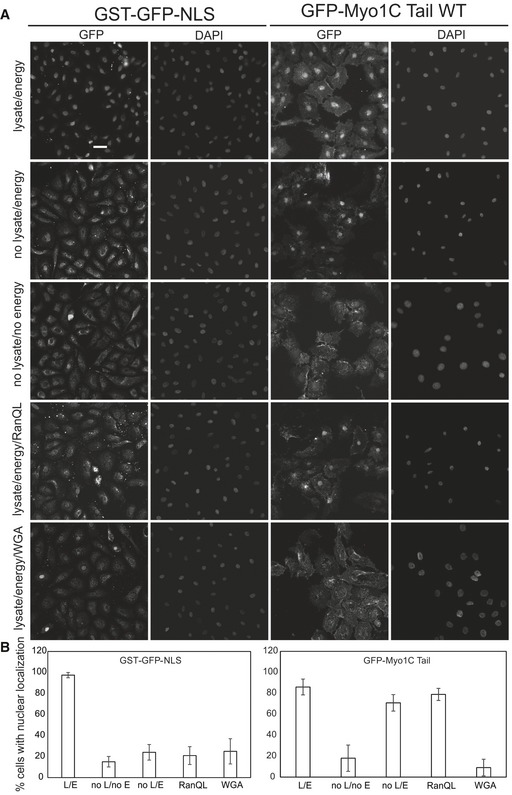
- Nuclear import of recombinant GFP‐Myo1C tail WT complexes with calmodulin was analyzed in semipermeablized HeLa cells. NLS‐GST‐GFP expressed in Escherichia coli was used as control import substrate. Where indicated, HeLa lysate and/or energy regeneration mix was omitted and import inhibitors were added. Scale bar, 20 μm.
- Quantification of in vitro import experiments with percentage of cells exhibiting nuclear accumulation of substrate plotted. Data are mean from three fields of view (each containing 40–60 cells) ± SD. L, lysate; E, energy; RanQL, RanQ69L; WGA, wheat germ agglutinin.
To further probe the assumption that soluble transport factors are not required for nuclear import of Myo1C, we analyzed the effect of importin knockdown on Myo1C localization. Based on the literature data, we targeted those importins, which were earlier shown to co‐precipitate with Myo1C tail constructs in pull‐down experiments, namely importin 5 (Ipo5), importin 7 (Ipo7), and importin β (Ipoβ) 22. Neither individual depletion of these importins, nor their pairwise depletions, however, led to any significant effect on nuclear localization of stably expressed HA‐tagged Myo1C in U2OS cells (Fig EV4A and B), although the levels of knockdown were confirmed to be sufficient (Fig EV4C). Thus, at least above‐mentioned importins do not seem to mediate Myo1C nuclear import. Finally, to analyze how much of Myo1C is present in the soluble form in U2OS cells, and hence accessible for soluble cytoplasmic import factors, we fused HA‐tagged Myo1C WT with the NLS sequence of SV40 Large T antigen known to exploit importin α/β‐dependent import pathway 28. Addition of the NLS sequence did not affect the localization of Myo1C WT, while it efficiently drove the Myo1C K892A protein into the nucleus (Fig EV5C). This shows that majority of cytoplasmic Myo1C is most likely bound to membranes and is simply inaccessible for soluble transport factors. At the same time, the K892A mutant is more soluble than the wild‐type protein and, therefore, could interact with cytosolic import factors. Yet, the localization of this mutant is almost exclusively cytoplasmic (Fig 2), which once again suggests that Myo1C uses a less conventional import pathway based on its association with the ER membrane through PI binding.
Figure EV4. Knockdown of importins 5, 7, and β does not affect cellular localization of HA‐tagged Myo1C.
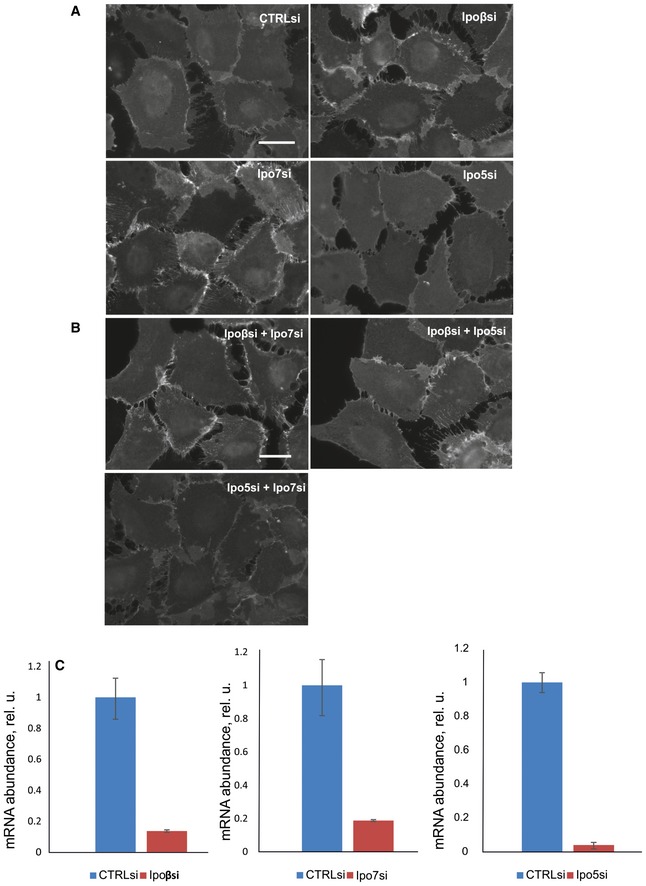
-
A, BU2OS cells stably expressing HA‐tagged Myo1C WT were depleted of single importins (A) or their combinations (B) using commercially available siRNAs (Sigma). After knockdown, cells were fixed and stained with HA antibody (HA‐7, Sigma) to visualize cellular localization of the myosin construct. As a control, a commercially available mix of random siRNAs was used (CTRLsi). Scale bars, 20 μm.
-
CEfficiency of knockdown of importins 5, 7, and β. After siRNA transfection, cells were grown for 4 days, after which total RNA was purified and converted into DNA by means of reverse transcription, and levels of specific mRNAs were determined by qPCR analysis using GAPDH as a housekeeping reference gene. Data are mean ± SD from three biological replicates.
Figure EV5. Effects of NLS on nuclear localization of Myo1C constructs.
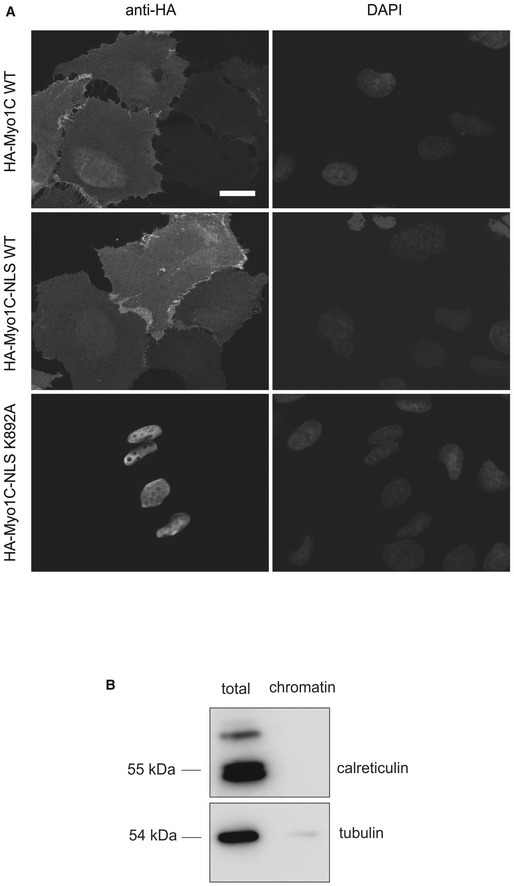
- Addition of SV40 NLS sequence to HA‐tagged Myo1C WT does not drive it to the nucleus, while HA‐Myo1C K892A NLS fusion localized exclusively to the nucleus. Wide‐field immunofluorescence images of U2OS transfected with corresponding constructs are shown. Scale bar, 20 μm.
- Purity control of purified chromatin used in chromatin precipitation assay (Fig 6). Total and chromatin fractions were immunoblotted against calreticulin (luminal protein of the endoplasmic reticulum) and tubulin (cytosolic protein).
The fact that GFP‐Myo1C tail failed to accumulate in the nucleus in the absence of energy (Fig 4A and B) and the robust binding of Myo1C to ER membranes in semipermeabilized cells (Fig 5B) allowed us to study whether ER binding precedes nuclear localization of Myo1C. We first allowed GFP‐Myo1C to bind to ER in the absence of energy, then washed away the soluble proteins and added energy. Since GFP‐Myo1C only accumulated in the nucleus upon addition of energy, at conditions where the only source of Myo1C is membrane‐bound (Fig 5A), this demonstrates that ER binding precedes nuclear localization of Myo1C.
Figure 5. In vitro import assay shows that association of Myo1C with ER precedes its nuclear import.
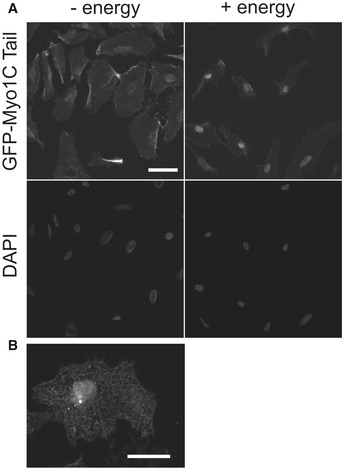
- First, cells were incubated with import substrate (GFP‐Myo1C tail WT) in the absence of energy to saturate ER with the myosin (“− energy”). Then, unbound substrate was washed off and cells were supplemented with energy mix (creatine kinase, phosphocreatine, GTP, ATP), HeLa cytosolic extract, and incubated for 30 min at 30°C. Scale bar, 20 μm.
- A higher‐magnification image showing that recombinant GFP‐Myo1C tail WT associated with the membranes of the endoplasmic reticulum in digitonin‐permeabilized HeLa cells. Scale bar, 10 μm.
Any role for the NLS‐like sequence?
If Myo1C relies primarily on ER membrane binding for its nuclear import, how can this be reconciled with the presence of an NLS‐like sequence in this molecule? As this is a polybasic region, we considered the possibility that the NLS‐like sequence might mediate Myo1C interactions with either DNA or PIs, which are both negatively charged at physiological pH. To test these two possibilities, we produced recombinant GFP‐tagged tail domain of Myo1C carrying substitutions in the NLS‐like sequence identical to those in the full‐length protein (NLSmut in Fig 1F). To study chromatin‐binding properties of the NLSmut protein, we used a co‐precipitation assay in which isolated chromatin is incubated with purified proteins, precipitated by centrifugation, washed, and analyzed for the presence of the protein in question. Of note, purity of chromatin was ensured by immunoblotting against ER lumenal protein calreticulin and cytosolic tubulin (Fig EV5B). GFP‐Myo1C tail WT and GFP‐Myo1C tail NLSmut were both found to co‐precipitate with purified chromatin at similar levels, while purified GFP alone showed virtually no binding (Fig 6A). Noteworthy, both protein species are highly soluble and do not precipitate on their own due to complex formation with calmodulin. Furthermore, subcellular fractionation of U2OS cells transfected with GFP‐tagged Myo1C tail constructs showed no difference in their abundance in the chromatin fraction (Fig 6B and C), in agreement with chromatin co‐precipitation experiments (Fig 6A). Likewise, also GFP‐Myo1C tail K892A efficiently interacted with chromatin, thus confirming that PI binding is likely required for import and not nuclear retention, as also demonstrated by our live‐cell imaging assay (Fig 3). To sum up, the NLS‐like sequence is unlikely to play crucial role in Myo1C interaction with chromatin.
Figure 6. NLS‐like sequence in Myo1C does not contribute to interaction with chromatin, but is involved in lipid binding.
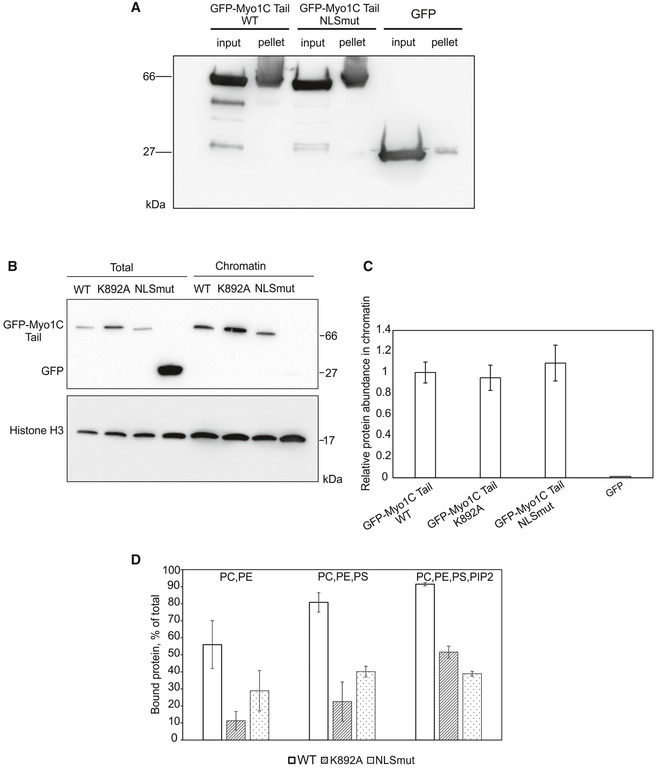
- Chromatin co‐precipitation experiment with recombinant GFP, GFP‐Myo1C tail WT in complex with calmodulin, as well as indicated mutants. Proteins co‐precipitating with chromatin were detected with GFP antibody.
- Western blotting analysis of chromatin fractions and total lysates of U2OS cells transfected with indicated GFP‐Myo1C tail constructs.
- Quantification of relative amounts of GFP‐tagged Myo1C tail constructs in the chromatin fraction. Data represent mean ± SD from three independent experiments and are normalized to GFP‐Myo1C tail WT.
- Quantification of lipid‐binding activity of recombinant GFP‐Myo1C tail WT in complex with calmodulin, as well as respective mutants from a phospholipid vesicle co‐sedimentation assay. Data are mean ± SD from three independent experiments. PC, phosphatidylcholine; PE, phosphatidylethanolamine; PS, phosphatidylserine.
Source data are available online for this figure.
To test the second possibility, lipid binding, we carried out co‐sedimentation experiments with PIP2‐containing phospholipid vesicles. Up to 90% of total GFP‐Myo1C tail WT binds to PIP2‐containing lipid vesicles, while the bound fraction for both K892A and NLSmut mutants is reduced to 30–40% (Fig 6D). These data confirm the earlier studies that implicate the PH‐like domain of Myo1C in PI binding 27, 40, but also suggest that another site, overlapping with the NLS‐like sequence, contributes to the interaction. Therefore, the effect of mutations in the NLS‐like sequence on nuclear localization can be explained in terms of impaired interaction of Myo1C with phospholipids.
Discussion
Protein Myo1C, typically deemed cytoplasmic together with other myosins, is also present in the nucleus, where it plays important roles in regulating gene expression. In this study, we analyze the molecular determinants of its localization and unexpectedly uncover that Myo1C does not follow a conventional transport pathway for soluble cargos. We show that interaction with membrane PIs is indispensable for nuclear targeting of Myo1C and that its localization to the chromatin fraction is preceded by interaction with the ER membrane. Thus, we argue that Myo1C utilizes a nuclear import route reminiscent of diffusion–retention mechanism of INM proteins, which makes it the first up to date non‐integral membrane protein to follow this pathway.
To continue the analogy with INM proteins, the import reaction of Myo1C is stalled when no energy regeneration mix is provided (Fig 4). Ungricht et al 8 showed that energy depletion affects ER structure and diffusional mobility of ER proteins due to decreased ER connectivity. Likewise, mobility of Myo1C in the ER membrane might decrease upon energy depletion, which will ultimately result in diminished nuclear accumulation. However, certain features of Myo1C import differ from INM proteins. Most significantly, there is a clear size limitation for diffusion–retention‐based nuclear import of INM proteins. Polypeptides with nucleoplasmic domains larger than 60 kDa cannot reach the INM 41, 42, which might be explained in terms of restricted NPC permeability. It has been suggested that upon transport through NPC, INM proteins do not use the central channel, but more likely pass through the so‐called lateral openings in the NPC, positioned close to the pore membrane. Myo1C physically cannot pass through such a channel, as its largest dimension of ~12 Å 43 surpasses the narrowest dimension of the lateral opening in NPC (~9 Å), 44. Therefore, it is likely that Myo1C uses another mechanism to pass through NPC. Indeed, inhibition of FG‐Nups with WGA prevented nuclear accumulation of Myo1C (Fig 4), suggesting that unlike INM proteins, Myo1C uses the central channel of NPC to enter the nucleus. Although not formally proven, it has been suggested that nuclear import of yeast INM proteins Heh1 and Heh2 would also take place through the central channel. These proteins contain canonical NLSs and require karyopherins Kap60/95 and the Ran GTPase cycle for INM targeting 45. Mechanistically, an intrinsically disordered linker in Heh2 is proposed to facilitate the access of the karyopherins to FG‐Nups 46. Although nuclear targeting of Myo1C does not require soluble factors (Fig 4), its transport might be mediated by membrane‐bound protein adaptors. Of note, importin α has been shown to interact with membranes in a co‐fractionation assay 47, and importin α16 was identified as a membrane‐bound karyopherin involved in INM targeting of certain viral proteins 48. Moreover, a number of proteins, such as β‐catenin 49 and human T lymphotropic virus type 1 protein 50 are able to directly interact with nucleoporins and thus do not require karyopherins for their import. The mechanism by which Myo1C is translocated from the ER membrane through the NPC will be an interesting subject for further studies.
Another striking difference between Myo1C and INM proteins is in the mechanism of membrane targeting. INM components are integral membrane proteins, which are tightly anchored in lipid bilayers by means of their transmembrane domains and thus can only exist within the membrane milieu. On the contrary, Myo1C represents a peripheral membrane protein, meaning it can bind membranes in a reversible manner. The mechanism of this interaction is based on specific PI binding through a PH‐like domain in the tail of Myo1C 27. Binding to PI, in particular to PIP2, appears to be the basic mechanism for Myo1C targeting to the sites of high accumulation of actin and various actin‐binding proteins, which also bind PI 51. It might also be the main mechanism of Myo1C recruitment to the sites of ongoing endocytosis, secretion, and membrane contraction 40, 52. Our results indicate that PI binding can be also essential for Myo1C targeting to the nucleus (Figs 2 and 3). Involvement of PIs provides new possibilities for the regulation of Myo1C nuclear localization. Levels of membrane PI are subject to multiple layers of regulation by dedicated phospholipases, phosphatidylinositol phosphatases, and kinases 53, which should have an impact on Myo1C interaction with the ER membrane and hence nuclear localization. The pathway we suggest would be, in principle, similar to the one described for ERM proteins (ezrin, radixin, and moesin). Their activity and membrane localization is regulated by chemokine signaling: SDF1‐induced activation of phospholipase C results in PIP2 degradation, which subsequently leads to release of ERM proteins from the membrane and their inactivation by dephosphorylation 54.
The PH domain of Myo1C is necessary, but not sufficient for nuclear localization of Myo1C (Figs 2 and EV1E). Thus, another essential part of our model is nuclear retention of Myo1C through association with certain nuclear components. Chromatin is a good candidate, since ChIP‐seq analysis has suggested co‐occupancy of Myo1C with the RNA polymerase II and active epigenetic marks across the entire genome 55. According to our results, nuclear retention is not based on Myo1C binding to nuclear PIs, since protein carrying K892A substitution interacts with isolated chromatin as efficiently as the wild‐type protein (Fig 6B). Another possibility might be direct DNA binding by Myo1C, earlier proposed by De Lanerolle et al 56. Indeed, the tail of Myo1C carries a substantial positive net charge due to high abundance of arginines and lysines, which could account for electrostatic interactions with negatively charged DNA backbone. Although PH domain of Myo1C alone cannot localize to the nucleus, addition of the IQ and post‐IQ regions, even when the putative NLS is mutated, promotes efficient nuclear accumulation of Myo1C (Figs 2 and EV1E). It has earlier been shown that GFP‐post‐IQ‐PHMyo1C shows no nuclear localization 22. This suggests that especially the IQ region, but not the NLS within this region, could play a role in nuclear retention of Myo1C. Interestingly, structural studies have shown that calcium binding to calmodulin induces drastic conformational changes in Myo1C IQ‐post‐IQ, which probably indicates that this domain is a hotspot of protein–protein interactions 57. Alternatively, the N‐terminal sequences of at least the longer isoforms A and B of Myo1C could contribute to nuclear targeting and thus retention 13. In budding yeast, the histone variant H2A.Z is required for INM targeting of the SUN domain protein Mps3 58. Detailed analysis of nuclear interaction partners for Myo1C could reveal protein factors that contribute to its nuclear localization either through retention or the actual transport.
A particularly puzzling element of Myo1C structure is an NLS‐like sequence, which was earlier suggested to function as a genuine NLS in an importin‐based import pathway 22. Our results show no particular requirement for soluble transport factors in the nuclear import of Myo1C (Figs 4 and EV4), thus implying other functions for the NLS‐like sequence. It should be noted that this is again very reminiscent of INM proteins, which also possess functionally redundant NLS‐like sequences 8. Since the NLS‐like sequence represents a stretch of six positively charged residues, a possibility of electrostatic interaction with either DNA or negatively charged phospholipids, such as PIs, arises. We show that interaction of Myo1C tail with PIP2‐containing lipid vesicles is substantially diminished in the NLSmut construct (Fig 6D), which leads us to the conclusion that Myo1C possesses in fact two PIP2‐binding sites. One of them is represented by the above‐mentioned PH‐like domain, while the second seems to overlap with calmodulin‐binding IQ2 motif. Of note, the existence of a second phospholipid‐binding site in Myo1C tail has long been discussed in the literature, and IQ motifs have been suggested to mediate phosphatidylserine binding 27, 59. However, up until now this possibility has not been tested by means of site‐directed mutagenesis.
In the course of evolution, cells have developed multiple mechanisms of nuclear protein targeting. Our research suggests that alongside with the conventional karyopherin‐based import pathways, such factors as interaction with membrane phospholipids can play a role in nuclear import. A substantial part of this mechanism would be interaction with nuclear structures, which would guarantee nuclear retention. In fact, such a mechanism would not necessarily require the presence of separate binding sites for, for example, phospholipids and DNA, as either patches of positively charged residues or more specialized domains could interact with both. For instance, interaction with acidic phospholipids competes with DNA binding in Escherichia coli protein DnaA, which is responsible for initiation of chromosomal DNA replication. In this case, the membrane‐binding domain overlaps with the domain of sequence‐independent DNA binding 60. In a similar way, direct interaction with phospholipids causes inhibition of DNA binding by mammalian DNA topoisomerase I 61. Following this line of reasoning, one can speculate that such a relationship between DNA‐ and phospholipid‐binding sites might have been beneficial at the early stages of eukaryote evolution, before the emergence of more specialized import pathways.
Materials and Methods
Plasmids and antibodies
Details of the plasmids cloned for the study are available upon request.
Isoform B of mouse Myo1C, whose amino acid sequence is identical to that of the human protein, was amplified using cDNA of mouse NIH 3T3 fibroblasts and cloned into earlier described p2HA‐C1 vector [24] or pEGFP‐C1 vector (Clontech). Mutation ΔABL (323IIAKGEEL330 replacing with AGA peptide), IQ2mut (I745N/Q746A/R750A/G751S), NLSmut (R755A/R756S/K757A/K760A/R761S/K762A), and K892A were introduced using standard PCR overlap extension protocol. SV40 NLS was added to the C‐terminus of HA‐tagged constructs. Constructs for inducible stable cell lines were prepared using pcDNA 4TO (Life Technologies) as backbone, into which GFP‐Myo1C tail WT and K892A were cloned. For protein production in E. coli, His6‐GFP‐Myo1C tail WT, K892, and NLSmut fusions were cloned into pET Duet vector (Novagen) together with human calmodulin 1 sequence. For the control import substrate, EGFP was amplified from pEGFP‐C1 using 3′ primer with NLS sequence and cloned into pGAT2 vector for fusion with GST.
Antibodies such as anti‐HA (HA7), anti‐Flag (M2), anti‐Myo1C (anti‐myosin Iβ, M 3567), anti‐Myo1C (HPA001768), anti‐histone H3 (H0164), anti‐actin (AC40), and anti‐tubulin (B‐5‐1‐2) were from Sigma. Anti‐GFP (GF28R) and anti‐calnexin (6F12BE10) were from Abcam and anti‐calreticulin (2679S) from Cell Signaling Technologies. Alexa Fluor 488‐ and HRP‐conjugated streptavidin as well as HRP‐conjugated anti‐mouse, HRP‐conjugated anti‐rabbit, Alexa Fluor 488‐conjugated anti‐mouse, Alexa Fluor 488‐conjugated anti‐rabbit, Alexa Fluor 594‐conjugated anti‐mouse were from Life Technologies.
Cell culture, transfections, immunofluorescence, and microscopy
Human osteosarcoma (U2OS) and HeLa cells (both were gifts from Lappalainen Lab, University of Helsinki) were cultured in Dulbecco's modified Eagle's medium (DMEM, Lonza) containing 10% FBS (GIBCO), 100 Units/ml penicillin, and 0.1 mg/ml streptomycin (Pen‐Strep, GIBCO). All cells were grown in humidified 95% air/5% CO2 incubator at +37°C, and regularly tested for mycoplasma contamination by PCR‐based methods and DAPI staining.
For transfections, U2OS cells were plated onto coverslips in a 24‐well tissue culture plate (~30,000 cells/well) 1 day prior to transfection. On the next day, cells were transfected with DNA constructs of interest using JetPrime transfection reagent (Polyplus transfection) according to the manufacturer's instructions (200 ng DNA per well was used). After 24 h, cells were processed for immunofluorescent staining (see below).
For microscopy, cells were fixed with 4% paraformaldehyde for 15 min, washed three times with Dulbecco's medium containing 0.2% BSA, and permeabilized for 5 min with 0.5% Triton X‐100 in PBS. For antibody staining, permeabilized cells were first incubated in Dulbecco's medium containing 0.2% BSA for 30 min and incubated with primary antibody for 1 h. Coverslips were washed and incubated with Alexa Fluor‐conjugated secondary antibody for 1 h with or without DAPI. Coverslips were washed three times with Dulbecco's medium, rinsed with MilliQ water, and mounted in DABCO‐supplemented Moviol. Cells were imaged using 63× or 20× objective of Leica DM6000B upright fluorescence wide‐field microscope equipped with Hamamatsu Orca‐Flash4.0V2 sCMOS camera. Confocal images were acquired with either Leica TCS SP5 confocal microscope, 63×/1.3 objective and LAS AF software, or Zeiss LSM 700 confocal microscope, 63×/1.30 objective and ZEN software.
Recombinant protein expression and purification
His‐tagged N‐terminal GFP fusions of Myo1C were expressed in complex with human calmodulin 1 using BL codon + E. coli strain. Protein expression was induced by addition of 0.4 mM IPTG to bacterial cultures grown in BL medium 20°C overnight. After that, cells were harvested, lysed, and Myo1C/calmodulin complexes were purified by combination of Ni affinity and size‐exclusion chromatography. GST‐GFP‐NLS was expressed in E. coli Rosetta strain and purification by means of Ni sepharose and size‐exclusion chromatography.
In vitro transport assay
HeLa cells were grown on fibronectin (10 μg/ml)‐coated glass coverslips at the density of 20,000 cells/slide. On the day of experiment, culturing medium was aspirated, and cells were washed with ice‐cold PBS once and incubated for 5 min with assay buffer (20 mM HEPES–KOH pH 7.2, 110 mM potassium acetate, 4 mM magnesium acetate, 0.5 mM EGTA) containing 25 μg/ml digitonin (Sigma). Semipermeabilization was followed by 3 × 5 min washes with cold assay buffer; 50 μl of import mixture containing transport‐competent cytoplasmic HeLa cell extract 8, energy‐regenerating system (10 mM creatine phosphate, 0.5 mM GTP, 0.5 mM ATP, 0.05 mg/ml creatine kinase in assay buffer), 2 mg/ml WGA, and recombinant GFP‐Myo1C tail WT (2 μM) were added to cells on coverslips, and import was allowed to proceed at room temperature.
Lipid‐binding Assays
1‐Palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphocholine (POPC), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phosphoethanolamine (POPE), 1‐palmitoyl‐2‐oleoyl‐sn‐glycero‐3‐phospho‐L‐serine (POPS), and porcine brain L‐α‐phosphatidylinositol‐4,5‐bisphosphate (PI(4,5)P2) were purchased from Avanti Polar Lipids (Alabaster, AL).
Lipids were mixed in desired ratios, and the organic solvents were evaporated under a stream of nitrogen. The lipid film was further dried under vacuum for at least 3 h and hydrated in 20 mM HEPES buffer, pH 7.4 containing 150 mM NaCl to yield multilamellar vesicles in a lipid concentration of 1 mM. To obtain unilamellar vesicles, vesicles were extruded through a polycarbonate membrane filter (100‐nm pore size) using a mini‐extruder (Avanti Polar Lipids Inc., Alabaster, AL). Proteins and liposomes with final concentrations of 3 and 250 μM, respectively, were incubated at room temperature for 10 min and centrifuged at 385,000 g using a Beckman rotor (TLA 100) for 30 min at 20°C. After centrifugation, equal proportions of supernatants and pellets were loaded onto SDS–PAGE and the gels were stained with Coomassie Blue. The intensities of protein bands were quantified using the Quantity One program (Bio‐Rad), and the data are shown as % of protein in the pellet fraction.
Live‐cell imaging
Stable cell lines were plated onto glass‐bottomed dishes (MatTek Corporation) induced with 1 μg/ml tetracycline and incubated for 24 h prior to live‐cell imaging. Time‐lapse images were acquired with 3I Marianas imaging system (3I Intelligent Imaging Innovations) in the wide‐field mode, equipped with appropriate filters, heated sample chamber (+37°C) with controlled CO2 and 63×/1.2 W C‐Apochromat Corr WD = 0.28 M27 objective. SlideBook 5.0 software (3I Intelligent Imaging Innovations) and sCMOS (Andor) Neo camera were used for the image acquisition. Time‐lapse images were acquired with the frequency one image in 15 min during 14 h. Further analysis of the video frames was performed with Microscopy Image Browser software 62. For quantification of nuclear fluorescence recovery, background signal was subtracted from the nuclear signal and divided by background‐adjusted cytoplasmic fluoresce. Initial segments of thus obtained fluorescence recovery curves (normally, in the interval 30–165 min) were then approximated with straight lines, whose slopes reflect the recovery rate of nuclear fluorescence after cell division.
Fluorescence recovery after photobleaching experiments were performed on Leica TCS SP5II HCS A confocal microscope using the protocol described in Ref. 25. For analysis, background was subtracted and prebleach nuclear intensity was set to one. FLIP imaging to measure nuclear export rates was performed on Zeiss LSM 700 confocal microscope equipped with motorized Axio Imager M2 upright microscope stand, W Plan‐Apochromat 40×/1.0 objective, 488 nm/10 mW laser, heated sample chamber (+37°C) with controlled CO2. In this assay, cytoplasm of a cell expressing GFP‐tagged protein is continuously bleached and loss of nuclear fluorescence is monitored as a measure of export. ZEN software was used to create a bleaching protocol consisting of 40 cycles. Every cycle consisted of a scan followed by up to five iteration of bleaching. For analysis, background was subtracted and prebleach nuclear intensity was set to one.
Biochemical fractionation and chromatin isolation
For chromatin isolation, cells were grown on 6‐cm plates and when applicable transfected with 2 μg construct of interest 48 h prior to experiment. Isolation of chromatin was carried out according to a modified protocol by Holden and Horton 63. Briefly, cells were harvested by trypsinization, washed with PBS containing “Complete” EDTA‐free protease inhibitor cocktail (Sigma), and pelleted by centrifugation (500 g, 5 min). Part of cell suspension was taken for the “total” samples prior to centrifugation. Cells were then lysed with lysis buffer containing 50 mM HEPES pH 8.0, 150 mM NaCl, and 25 μg/ml digitonin (Sigma) on ice for 10 min. Crude nuclei were pelleted (1,500 g, 5 min) and resuspended in chromatin buffer, containing 50 mM HEPES pH 8.0, 150 mM NaCl, and 1% Triton X‐100. Extraction of intracellular membranous components including endoplasmic reticulum, nuclear envelope, and mitochondria was carried out for 30 min on ice. Enrichments of this fraction were monitored by blotting against ER luminal marker calreticulin. After centrifugation (2,000 g, 5 min), resulting chromatin pellet was washed with PBS and resuspended in 1× Laemmli sample buffer. After sonication and boiling at 98°C for 5 min, samples were run on SDS polyacrylamide gradient gels (Bio‐Rad), transferred onto nitrocellulose membrane, and blotted with antibody of interest. Anti‐histone H3 and anti‐tubulin antibody were used to control equal loading of different samples. For quantification, the signals were first normalized to the respective loading control, and then, the signal in the chromatin fraction was normalized against the total signal, and the value for GFP‐Myo1C tail was set to one.
Isolation of intact nuclei was carried out as follows. Around 5 × 106 cells grown on Petri dishes were trypsinized and washed once with ice‐cold PBS, after which cells were soaked in 10 mM HEPES pH 7.9 containing 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT, and protease inhibitors (buffer A). After 30‐min incubation on ice, cells were lysed with B‐type tight pestle Dounce homogenizer. Subsequently, lysate was layered over equal volume of buffer A containing 1 M sucrose and centrifuged for 30 min at 4°C (5,000 g). Upper layer was used as cytoplasmic fraction, and nuclear pellet was washed once again with the sucrose buffer prior to being lysed with 1× Laemmli loading buffer.
For chromatin co‐precipitation experiments, chromatin isolated from 50 × 103 to 70 × 103 cells was incubated with 2 μM solution of recombinant GFP‐Myo1C tail/calmodulin complex in assay buffer (25 mM HEPES pH 7.4, 150 mM NaCl, 1.5 mM MgCl2, 0.1% Triton X‐100) for 30 min at room temperature. Chromatin was then precipitated by centrifugation (2,000 g, 5 min), washed several times with assay buffer, lysed with Laemmli sample buffer, run on SDS–PAGE, transferred on nitrocellulose membrane, and blotted with GFP antibody.
Statistical analysis
Statistical analyses were performed in Excel by two‐tailed Student's t‐test, with two‐sample unequal variance, because the data conformed to normal distribution.
Author contributions
MKV and IN designed the study with valuable contribution of HZ, IN and ES performed the experiments with help from WW, IN and MKV wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Review Process File
Source Data for Figure 6B
Acknowledgements
Imaging was carried out at the Light Microscopy Unit of the Institute of Biotechnology. Dr. Henna Moore is acknowledged for her help with live‐cell imaging. Authors are grateful to Mrs. Paula Maanselkä for excellent technical assistance, Dr. Konstantin Kogan for helpful discussions and assistance with cloning, and Dr. Ilya Belevich for assistance in image analysis. I.N. is funded by the Academy of Finland. M.K.V. is funded by the Academy of Finland, Sigrid Juselius Foundation, Biocentrum Helsinki, Finnish Cancer Foundation, Loreal for Women in Science grant, and ERC starting grant.
EMBO Reports (2018) 19: 290–304
References
- 1. Crabtree GR, Olson EN (2002) NFAT signaling: choreographing the social lives of cells. Cell 109(Suppl): S67–S79 [DOI] [PubMed] [Google Scholar]
- 2. Miralles F, Posern G, Zaromytidou AI, Treisman R (2003) Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell 113: 329–342 [DOI] [PubMed] [Google Scholar]
- 3. Vartiainen MK, Guettler S, Larijani B, Treisman R (2007) Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316: 1749–1752 [DOI] [PubMed] [Google Scholar]
- 4. Stewart M (2007) Molecular mechanism of the nuclear protein import cycle. Nat Rev Mol Cell Biol 8: 195–208 [DOI] [PubMed] [Google Scholar]
- 5. Terry LJ, Shows EB, Wente SR (2007) Crossing the nuclear envelope: hierarchical regulation of nucleocytoplasmic transport. Science 318: 1412–1416 [DOI] [PubMed] [Google Scholar]
- 6. Popken P, Ghavami A, Onck PR, Poolman B, Veenhoff LM (2015) Size‐dependent leak of soluble and membrane proteins through the yeast nuclear pore complex. Mol Biol Cell 26: 1386–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wuhr M, Guttler T, Peshkin L, McAlister GC, Sonnett M, Ishihara K, Groen AC, Presler M, Erickson BK, Mitchison TJ et al (2015) The nuclear proteome of a vertebrate. Curr Biol 25: 2663–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ungricht R, Klann M, Horvath P, Kutay U (2015) Diffusion and retention are major determinants of protein targeting to the inner nuclear membrane. J Cell Biol 209: 687–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Boni A, Politi AZ, Strnad P, Xiang W, Hossain MJ, Ellenberg J (2015) Live imaging and modeling of inner nuclear membrane targeting reveals its molecular requirements in mammalian cells. J Cell Biol 209: 705–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pestic‐Dragovich L, Stojiljkovic L, Philimonenko AA, Nowak G, Ke Y, Settlage RE, Shabanowitz J, Hunt DF, Hozak P, de Lanerolle P (2000) A myosin I isoform in the nucleus. Science 290: 337–341 [DOI] [PubMed] [Google Scholar]
- 11. Nowak G, Pestic‐Dragovich L, Hozak P, Philimonenko A, Simerly C, Schatten G, de Lanerolle P (1997) Evidence for the presence of myosin I in the nucleus. J Biol Chem 272: 17176–17181 [DOI] [PubMed] [Google Scholar]
- 12. Coluccio LM (ed.) (2008) Myosins: a superfamily of molecular motors, pp 95–124. Dordrecht, The Netherlands: Springer. [Google Scholar]
- 13. Ihnatovych I, Migocka‐Patrzalek M, Dukh M, Hofmann WA (2012) Identification and characterization of a novel myosin Ic isoform that localizes to the nucleus. Cytoskeleton (Hoboken) 69: 555–565 [DOI] [PubMed] [Google Scholar]
- 14. Bose A, Guilherme A, Robida SI, Nicoloro SM, Zhou QL, Jiang ZY, Pomerleau DP, Czech MP (2002) Glucose transporter recycling in response to insulin is facilitated by myosin Myo1c. Nature 420: 821–824 [DOI] [PubMed] [Google Scholar]
- 15. Gillespie PG, Cyr JL (2004) Myosin‐1c, the hair cell's adaptation motor. Annu Rev Physiol 66: 521–545 [DOI] [PubMed] [Google Scholar]
- 16. Brandstaetter H, Kendrick‐Jones J, Buss F (2012) Molecular roles of Myo1c function in lipid raft exocytosis. Commun Integr Biol 5: 508–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Joensuu M, Belevich I, Ramo O, Nevzorov I, Vihinen H, Puhka M, Witkos TM, Lowe M, Vartiainen MK, Jokitalo E (2014) ER sheet persistence is coupled to myosin 1c‐regulated dynamic actin filament arrays. Mol Biol Cell 25: 1111–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Percipalle P, Farrants AK (2006) Chromatin remodelling and transcription: be‐WICHed by nuclear myosin 1. Curr Opin Cell Biol 18: 267–274 [DOI] [PubMed] [Google Scholar]
- 19. Sarshad A, Sadeghifar F, Louvet E, Mori R, Bohm S, Al‐Muzzaini B, Vintermist A, Fomproix N, Ostlund AK, Percipalle P (2013) Nuclear myosin 1c facilitates the chromatin modifications required to activate rRNA gene transcription and cell cycle progression. PLoS Genet 9: e1003397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Philimonenko VV, Zhao J, Iben S, Dingova H, Kysela K, Kahle M, Zentgraf H, Hofmann WA, de Lanerolle P, Hozak P et al (2004) Nuclear actin and myosin I are required for RNA polymerase I transcription. Nat Cell Biol 6: 1165–1172 [DOI] [PubMed] [Google Scholar]
- 21. Chuang CH, Carpenter AE, Fuchsova B, Johnson T, de Lanerolle P, Belmont AS (2006) Long‐range directional movement of an interphase chromosome site. Curr Biol 16: 825–831 [DOI] [PubMed] [Google Scholar]
- 22. Dzijak R, Yildirim S, Kahle M, Novak P, Hnilicova J, Venit T, Hozak P (2012) Specific nuclear localizing sequence directs two myosin isoforms to the cell nucleus in calmodulin‐sensitive manner. PLoS One 7: e30529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maly IV, Hofmann WA (2016) Calcium‐regulated import of myosin IC into the nucleus. Cytoskeleton (Hoboken) 73: 341–350 [DOI] [PubMed] [Google Scholar]
- 24. Dopie J, Skarp KP, Rajakyla EK, Tanhuanpaa K, Vartiainen MK (2012) Active maintenance of nuclear actin by importin 9 supports transcription. Proc Natl Acad Sci USA 109: E544–E552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Skarp K‐P, Vartiainen MK (2013) Actin as a model for the study of nucleocytoplasmic shuttling and nuclear dynamics. Methods Mol Biol 1042: 245–255 [DOI] [PubMed] [Google Scholar]
- 26. Sasaki N, Asukagawa H, Yasuda R, Hiratsuka T, Sutoh K (1999) Deletion of the myopathy loop of Dictyostelium myosin II and its impact on motor functions. J Biol Chem 274: 37840–37844 [DOI] [PubMed] [Google Scholar]
- 27. Hokanson DE, Laakso JM, Lin T, Sept D, Ostap EM (2006) Myo1c binds phosphoinositides through a putative pleckstrin homology domain. Mol Biol Cell 17: 4856–4865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kalderon D, Roberts BL, Richardson WD, Smith AE (1984) A short amino acid sequence able to specify nuclear location. Cell 39: 499–509 [DOI] [PubMed] [Google Scholar]
- 29. Lemmon MA, Ferguson KM, O'Brien R, Sigler PB, Schlessinger J (1995) Specific and high‐affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc Natl Acad Sci USA 92: 10472–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Osborne SL, Thomas CL, Gschmeissner S, Schiavo G (2001) Nuclear PtdIns(4,5)P2 assembles in a mitotically regulated particle involved in pre‐mRNA splicing. J Cell Sci 114: 2501–2511 [DOI] [PubMed] [Google Scholar]
- 31. Watt SA, Kular G, Fleming IN, Downes CP, Lucocq JM (2002) Subcellular localization of phosphatidylinositol 4,5‐bisphosphate using the pleckstrin homology domain of phospholipase C delta1. Biochem J 363: 657–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yildirim S, Castano E, Sobol M, Philimonenko VV, Dzijak R, Venit T, Hozák P (2013) Involvement of phosphatidylinositol 4,5‐bisphosphate in RNA polymerase I transcription. J Cell Sci 126: 2730–2739 [DOI] [PubMed] [Google Scholar]
- 33. Boronenkov IV, Loijens JC, Umeda M, Anderson RA (1998) Phosphoinositide signaling pathways in nuclei are associated with nuclear speckles containing pre‐mRNA processing factors. Mol Biol Cell 9: 3547–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mellman DL, Gonzales ML, Song C, Barlow CA, Wang P, Kendziorski C, Anderson RA (2008) A PtdIns4,5P2‐regulated nuclear poly(A) polymerase controls expression of select mRNAs. Nature 451: 1013–1017 [DOI] [PubMed] [Google Scholar]
- 35. Shah ZH, Jones DR, Sommer L, Foulger R, Bultsma Y, D'Santos C, Divecha N (2013) Nuclear phosphoinositides and their impact on nuclear functions. FEBS J 280: 6295–6310 [DOI] [PubMed] [Google Scholar]
- 36. van Meer G, de Kroon AIPM (2011) Lipid map of the mammalian cell. J Cell Sci 124: 5–8 [DOI] [PubMed] [Google Scholar]
- 37. Ungricht R, Kutay U (2015) Establishment of NE asymmetry – targeting of membrane proteins to the inner nuclear membrane. Curr Opin Cell Biol 34: 135–141 [DOI] [PubMed] [Google Scholar]
- 38. Adam SA, Marr RS, Gerace L (1990) Nuclear protein import in permeabilized mammalian cells requires soluble cytoplasmic factors. J Cell Biol 111: 807–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ribbeck K, Gorlich D (2001) Kinetic analysis of translocation through nuclear pore complexes. EMBO J 20: 1320–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ostap EM, Maupin P, Doberstein SK, Baines IC, Korn ED, Pollard TD (2003) Dynamic localization of myosin‐I to endocytic structures in Acanthamoeba. Cell Motil Cytoskelet 54: 29–40 [DOI] [PubMed] [Google Scholar]
- 41. Soullam B, Worman HJ (1995) Signals and structural features involved in integral membrane protein targeting to the inner nuclear membrane. J Cell Biol 130: 15–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zuleger N, Kelly DA, Richardson AC, Kerr AR, Goldberg MW, Goryachev AB, Schirmer EC (2011) System analysis shows distinct mechanisms and common principles of nuclear envelope protein dynamics. J Cell Biol 193: 109–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Munnich S, Taft MH, Manstein DJ (2014) Crystal structure of human myosin 1c–the motor in GLUT4 exocytosis: implications for Ca2+ regulation and 14‐3‐3 binding. J Mol Biol 426: 2070–2081 [DOI] [PubMed] [Google Scholar]
- 44. Beck M, Lucic V, Forster F, Baumeister W, Medalia O (2007) Snapshots of nuclear pore complexes in action captured by cryo‐electron tomography. Nature 449: 611–615 [DOI] [PubMed] [Google Scholar]
- 45. King MC, Lusk CP, Blobel G (2006) Karyopherin‐mediated import of integral inner nuclear membrane proteins. Nature 442: 1003–1007 [DOI] [PubMed] [Google Scholar]
- 46. Meinema AC, Laba JK, Hapsari RA, Otten R, Mulder FA, Kralt A, van den Bogaart G, Lusk CP, Poolman B, Veenhoff LM (2011) Long unfolded linkers facilitate membrane protein import through the nuclear pore complex. Science 333: 90–93 [DOI] [PubMed] [Google Scholar]
- 47. Hachet V, Köcher T, Wilm M, Mattaj IW (2004) Importin α associates with membranes and participates in nuclear envelope assembly in vitro . EMBO J 23: 1526–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Saksena S, Summers MD, Burks JK, Johnson AE, Braunagel SC (2006) Importin‐alpha‐16 is a translocon‐associated protein involved in sorting membrane proteins to the nuclear envelope. Nat Struct Mol Biol 13: 500–508 [DOI] [PubMed] [Google Scholar]
- 49. Fagotto F, Gluck U, Gumbiner BM (1998) Nuclear localization signal‐independent and importin/karyopherin‐independent nuclear import of beta‐catenin. Curr Biol 8: 181–190 [DOI] [PubMed] [Google Scholar]
- 50. Tsuji T, Sheehy N, Gautier VW, Hayakawa H, Sawa H, Hall WW (2007) The nuclear import of the human T lymphotropic virus type I (HTLV‐1) tax protein is carrier‐ and energy‐independent. J Biol Chem 282: 13875–13883 [DOI] [PubMed] [Google Scholar]
- 51. Saarikangas J, Zhao H, Lappalainen P (2010) Regulation of the actin cytoskeleton‐plasma membrane interplay by phosphoinositides. Physiol Rev 90: 259–289 [DOI] [PubMed] [Google Scholar]
- 52. Novak KD, Peterson MD, Reedy MC, Titus MA (1995) Dictyostelium myosin I double mutants exhibit conditional defects in pinocytosis. J Cell Biol 131: 1205–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. McCrea HJ, De Camilli P (2009) Mutations in phosphoinositide metabolizing enzymes and human disease. Physiology (Bethesda) 24: 8–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hao JJ, Liu Y, Kruhlak M, Debell KE, Rellahan BL, Shaw S (2009) Phospholipase C‐mediated hydrolysis of PIP2 releases ERM proteins from lymphocyte membrane. J Cell Biol 184: 451–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Almuzzaini B, Sarshad AA, Farrants AK, Percipalle P (2015) Nuclear myosin 1 contributes to a chromatin landscape compatible with RNA polymerase II transcription activation. BMC Biol 13: 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. de Lanerolle P, Johnson T, Hofmann WA (2005) Actin and myosin I in the nucleus: what next? Nat Struct Mol Biol 12: 742–746 [DOI] [PubMed] [Google Scholar]
- 57. Lu Q, Li J, Ye F, Zhang M (2015) Structure of myosin‐1c tail bound to calmodulin provides insights into calcium‐mediated conformational coupling. Nat Struct Mol Biol 22: 81–88 [DOI] [PubMed] [Google Scholar]
- 58. Gardner JM, Smoyer CJ, Stensrud ES, Alexander R, Gogol M, Wiegraebe W, Jaspersen SL (2011) Targeting of the SUN protein Mps3 to the inner nuclear membrane by the histone variant H2A.Z. J Cell Biol 193: 489–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tang N, Lin T, Ostap EM (2002) Dynamics of myo1c (myosin‐ibeta) lipid binding and dissociation. J Biol Chem 277: 42763–42768 [DOI] [PubMed] [Google Scholar]
- 60. Makise M, Mima S, Katsu T, Tsuchiya T, Mizushima T (2002) Acidic phospholipids inhibit the DNA‐binding activity of DnaA protein, the initiator of chromosomal DNA replication in Escherichia coli . Mol Microbiol 46: 245–256 [DOI] [PubMed] [Google Scholar]
- 61. Tamura H‐o, Ikegami Y, Ono K, Sekimizu K, Andoh T (1990) Acidic phospholipids directly inhibit DNA binding of mammalian DNA topoisomerase I. FEBS Lett 261: 151–154 [DOI] [PubMed] [Google Scholar]
- 62. Belevich I, Joensuu M, Kumar D, Vihinen H, Jokitalo E (2016) Microscopy image browser: a platform for segmentation and analysis of multidimensional datasets. PLoS Biol 14: e1002340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Holden P, Horton WA (2009) Crude subcellular fractionation of cultured mammalian cell lines. BMC Res Notes 2: 243 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Review Process File
Source Data for Figure 6B