

Abstract
Background
Advanced glycation end products (AGEs) have been related to the pathogenesis of cardiovascular diseases (CVD), chronic kidney disease (CKD) and diabetes mellitus. We sought to investigate the binding capacity of sevelamer to both AGEs and uremic serum in vitro and then test this pharmaceutical effect as a potential vascular anti-inflammatory strategy.
Methods
AGEs were prepared by albumin glycation and characterized by absorbance and electrophoresis. Human endothelial cells were incubated in culture media containing AGEs and uremic serum with or without sevelamer. Receptor for advanced glycation end product (RAGE) expression was evaluated through immunocytochemistry and western blot to explore the interactions between AGEs and the endothelium. Inflammatory and endothelial dysfunction biomarkers, such as interleukin 6 (IL-6) and IL-8, monocyte chemoattractant protein-1 (MCP-1), plasminogen activator inhibitor-1 (PAI-1) and serum amyloid A (SAA) were also measured in cell supernatant. The chemotactic property of the supernatant was evaluated.
Results
AGEs significantly induced the expression of RAGE, inflammatory and endothelial activation biomarkers [IL-6, (P < 0.005); IL-8, MCP-1, PAI-1 and SAA (P < 0.001)] and monocyte chemotaxis as compared with controls. In addition, AGEs increased the levels of inflammatory biomarkers, which were observed after 6 h of endothelial cell incubation with uremic serum [IL-6 (P < 0.001) IL-8, MCP-1 and PAI-1 (P < 0.05)]. On the other hand, after 6 h of endothelial cell treatment with sevelamer, RAGE expression (P < 0.05) and levels of inflammatory biomarkers [IL-6 and IL-8 (P < 0.001), MCP-1 (P < 0.01), PAI-1 and SAA (P < 0.005)] significantly decreased compared with the AGEs/uremic serum treatment alone.
Conclusions
Sevelamer decreased both endothelial expression of RAGE and endothelial dysfunction biomarkers, induced by AGEs, and uremic serum. Further studies are necessary for a better understanding of the potential protective role of sevelamer on uremic serum and AGEs-mediated endothelial dysfunction.
Keywords: AGEs, chronic kidney disease, endothelial dysfunction, inflammation, sevelamer, uremic serum
Introduction
Advanced glycation end-products (AGEs) are a heterogeneous group of molecules such as sugars, lipids and nucleic acids, formed by nonenzymatic glycosylation reactions through a complex sequence of reactions referred to as the Maillard reactions [1]. The most prevalent AGE in vivo is carboxymethyl-lysine, better known as CML [2]. AGEs have a high renal clearance in the body and the main form of excretion is through urine [3]. They are considered medium-sized uremic toxins that accumulate in the bloodstream of patients with chronic kidney disease (CKD). Their its origin is endogenous, occurring mainly when the body is exposed to high sugar levels, such as in diabetes [4].
The excessive intake of dietary AGEs has been linked to endothelial dysfunction, insulin resistance and the progression of atherosclerosis [5, 6]. Furthermore, recent studies have suggested that the increased intake of AGEs leads to their accumulation in the brain of diabetic and obese patients [7, 8] and thus it contributes to cognitive decline in these patients [9]. To exert their cellular effect, AGEs bind to cell surface receptors, such as AGER1 or, most importantly, to the receptor for advanced glycation end products (RAGE) [10, 11]. The RAGE–AGEs linkage stimulates several pro-inflammatory cytokines, including tumour necrosis factor alpha (TNF-α), interleukin (IL)-6 and IL-8, as well as profibrotic cytokines such as transforming growth factor beta (TGF-β) [12] through activation of the transcription factor nuclear factor kappa B (NF-κB), which in turn leads to gene expression and release of pro-inflammatory molecules as well as the release of reactive oxygen species (ROS) [13]. Furthermore, studies led by our group showed that endothelial cell exposure to a uremic environment increases monocyte chemoattractant protein-1 (MCP-1), IL-8 and vascular adhesion molecule-1 (VCAM-1) expression, which suggests a relationship between vascular injury, systemic inflammation and uremic toxicity [14].
Sevelamer is a phosphate binder used in CKD patients to control hyperphosphatemia. In addition to its effects on phosphate levels, sevelamer seems to have pleiotropic effects that include a reduction of lipid levels and clearance of some uremic toxins in the bloodstream [15, 16]. Clinical studies enrolling CKD patients have shown that sevelamer may reduce serum levels of some low-molecular-weight uremic toxins [17]. We have previously demonstrated that sevelamer reduces systemic inflammatory response both in an animal model of CKD and in hemodialysis (HD) patients [18, 19]. Other studies have also demonstrated that sevelamer may decrease systemic inflammation in patients with CKD, as it reduces circulating levels of uremic toxins such as AGEs [20] as well as indoxyl sulfate, and p-cresol [17, 21]. Moreover, sevelamer can reduce the expression of adhesion molecules intercellular adhesion molecule-1 (ICAM-1) and VCAM-1 (markers of vascular injury) consequently it reduces circulating and cellular levels of AGEs, increases antioxidant defense, and decreases circulating levels of pro-oxidants molecules [22–24]. Taken together, these studies indicate that sevelamer may help remove uremic toxins via nondialytic process.
Thus, the present study aimed to investigate the potential protective endothelial effects of sevelamer through its binding capacity on other uremic toxins beyond phosphate, particularly AGEs. The potential chelation effect of sevelamer on uremic serum of patients with CKD was also investigated. Human endothelial cells and monocytes were used to mimic the early model of endothelial dysfunction. Hence we suggest a novel approach for the use of sevelamer, which could hypothetically contribute to the decrease in inflammatory cellular response and, consequently, avoid the development of CVD in AGEs-elevated pathologies such as CKD and diabetes.
Materials and methods
Patient selection
The patients were selected among 104 CKD patients on HD from a single dialysis center in Curitiba, Brazil. After applying the inclusion and exclusion criteria listed below, 26 patients were enrolled in the study. The inclusion criteria were that all patients had to be on chronic HD three times a week (3.5–4 h per session), using polysulphone dialysis membranes and dialysate with bicarbonate and calcium concentrations of 32 mEq/L and 3.5 mEq/L, respectively. The exclusion criteria were all patients (i) undergoing HD sessions through a central venous catheter as vascular access, (ii) with the presence of infectious disease or severe chronic inflammation or malignancies, (iii) with active liver disease, (iv) with autoimmune diseases, (v) using immunosuppressive or anti-inflammatory agents within the last 3 months prior to enrollment in the study and (vi) who have had a cardiovascular event (i.e. myocardial infarction, unstable angina, stroke or myocardial revascularization) within 3 months before the study began. All patients signed an informed consent agreeing to participate in the study. The protocol was approved by the Ethics Committee in Human Research of the Universidade Federal do Paraná (CEP/SD: 974.079.10.07). For comparison purposes, serum samples from healthy volunteers (N = 10) were used as controls.
Clinical data collection
Clinical and demographic data were collected through interviews and physical examinations performed on the initial assessment day and analysis of the medical records. The following data were collected: age, gender, race, comorbidities, CKD etiology, time on dialysis and use of statins, aspirin or cholecalciferol.
Collection of laboratory data
Blood samples were collected immediately before the first HD session of the week, centrifuged and stored at −80°C. Serum levels of total cholesterol, low-density lipoprotein (LDL) and high-density lipoprotein (HDL) cholesterol fractions, triglycerides, hemoglobin, albumin, calcium, phosphorus, parathyroid hormone (PTH), alkaline phosphatase and C-reactive protein (CRP) were measured in all patients.
In vitro experiments
Endothelial cell culture
An immortalized human endothelial cell line EA.hy926 (CRL-2922; ATCC, Manassas, VA) was used. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (Life Technologies, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA) and 10 mg/mL of penicillin/streptomycin (Gibco) and maintained at 37°C in a humidified atmosphere containing 5% CO2.
U-937 cell culture
U-937 cells (CRL-1593.2; ATCC) were purchased from a commercial tumor cell line. These cells are accepted as proxies for circulating monocytes, which are difficult to obtain in satisfactory amounts from donors’ total blood. The U-937 cells were cultured in RPMI 1640 (Gibco) supplemented with 10% FBS (Gibco), 100 U/mL of penicillin and 50 mg/mL of streptomycin (Gibco). As the cells grew in suspension, a ratio of 105 monocytes/mL was maintained in the culture medium for 3 days. The cells were kept in culture flasks and incubated at 37°C in a 5% CO2 atmosphere.
Synthesis and characterization of AGEs
AGEs and bovine serum albumin (BSA) (Sigma-Aldrich, St Louis, MO, USA) were prepared as previously described [25]. In short, AGEs and BSA were characterized by spectrophotometry (Tecan, Männedorf, Switzerland) by comparing the absorbance at 330, 360, 400 and 420 nm, as previously described [26, 27]. For cell treatment, AGEs and BSA were applied to the culture media at 0.2 mg/mL. AGEs and BSA were also characterized by basic protein polyacrylamide gel electrophoresis (PAGE). Then, 15 µg of the sample was electrophoresed in 10% resolving gel at 100 V for 4 h at room temperature with inverted poles and the bands were visualized after Coomassie blue staining.
Endotoxin assay
To exclude endotoxin contamination of AGEs and BSA, a limulus amebocyte lysate (LAL) assay test (Thermo Fisher Scientific, Rockford, IL, USA) was performed according to the manufacturer’s recommendations. The absorbance was measured at 410 nm (Tecan) and all measurements were taken in duplicate.
Cell viability assay
Cell viability was assessed using the 3-[4,5-dimethyl-thiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) (Sigma-Aldrich) assay according to Mosmann [28]. The endothelial cells were plated in 96-wells culture plates at a density of 104 cells/well. After 24 h of incubation, the medium was replaced and cells were treated with BSA and AGEs with or without 3% sevelamer for 24 h. All analyses were performed in triplicate.
Treatment of endothelial cells with AGEs, uremic pool and sevelamer
Endothelial cells were cultured at 106 cells/well in 96-well plates as described and incubated at 37°C in a 5% CO2 atmosphere overnight. For cell treatment, AGEs and BSA were diluted at a concentration of 0.2 mg/mL in DMEM with or without 3% sevelamer. To prepare the uremic medium, serum from patients was pooled. For this purpose, equal volumes of serum from all patients (N = 26) were mixed to form a single uremic pool. Subsequently the endothelial cells were incubated with uremic medium (10% uremic pool) with or without 3% sevelamer. Finally, the endothelial cell supernatants were recovered at 0 and 6 h by simple aspiration of the contents of the wells and stored at −20°C for subsequent quantification of IL-6, IL-8, MCP-1, plasminogen activator inhibitor-1 (PAI-1) and serum amyloid A (SAA) as well as for the chemotaxis assay. Three experiments were performed in triplicate for all cell culture models used.
Nuclear magnetic resonance
Spectra were obtained with a 1D31P nuclear magnetic resonance (NMR) spectrometer (Avance III; Bruker, Billerica, MA, USA) operating at 14.1 Tesla (600 MHz for 1H and 242 MHz for 31P) equipped with a reverse pentanuclear observation probe of 5 mm field gradient z (QXI) axis. We evaluated the phosphate concentration in samples containing phosphate (4 mM), with or without 3% sevelamer, to validate in vitro the chelating effect of sevelamer to phosphate. The samples were solubilized in 600 μL of a solution of D2O:H2O (1:9 v/v) at 30°C. The 31P decoupled spectra were obtained with a 30° angle, with a magnetization window of 400 ppm and an acquisition time of 0.17 s. The relaxation time of 0.200 and 1024 transients was processed with the help of the TopSpin program (Bruker, Karlsruhe, Germany) applying exponential multiplication on the acquired free induction decays (FIDs). All analyses were performed in triplicate.
RAGE immunostaining
RAGE expression in cells treated with AGEs was as previously described by Rempel et al. [27]. The images were captured using an AxioImager Z2 light microscope (Carl Zeiss, Jena, Germany) equipped with an automated scanner slide (Metasystems, Altlussheim, Germany). All analyses were performed in triplicate.
Western blot analysis
Growth-arrested cells cultured in 100-mm dishes were stimulated for 24 h with BSA and AGEs at 0.2 mg/mL, with or without 3% sevelamer, as previously described [27]. RAGE antibody (1:1000; Santa Cruz Biotechnology, Dallas, TX, USA) and horseradish peroxidase-conjugated sheep anti-rabbit IgG polyclonal antibody (1:10000) (Sigma-Aldrich) were used as primary and secondary antibodies, respectively. The reaction was visualized by enhanced chemiluminescence western blotting reagents (GE Health Care Life Sciences, Little Chalfont, UK). Band intensity was analyzed using Image Studio Lite 5.0 (LI-COR Biotechnology, Lincoln, NE, USA). All analyses were performed in triplicate.
Biomarker assays (IL-6, IL-8, MCP-1, PAI-1 and SAA)
Endothelial supernatant levels of IL-6, IL-8, MCP-1, PAI-1 and SAA were measured by the Luminex methodology in combination with the xPONENT 3.1 software package (Luminex, Austin, TX, USA). The MAGPIX system (Darmstadt, Germany) with 14 markers was used following the manufacturer’s instructions (adipokine human magnetic 14-plex panel for Luminex platform). Results were expressed through a standard curve (in pg/mL). All samples were analyzed at the same time under standardized experimental conditions and measurements were performed in triplicate.
Monocyte migration assay
Cell migration assays were performed using modified Boyden chambers with a 6.5-mm diameter, 10-µm thickness, porous (8.0 µm) polystyrene membrane separating the two chambers (GE Healthcare Bio-Sciences, Westborough, MA, USA). Briefly, the endothelial cells were treated with AGEs, with or without 3% sevelamer, and the cell supernatant was added to the lower compartment of the chamber to evaluate the chemoattractant effect of this supernatant on the monocytes. Subsequently, 106 U-937 cells/mL were added to the top of the chamber over the membrane. The cells were incubated at 37°C and 5% CO2 for 90 min. The chemoattractant ability of the supernatant (migration) was measured by counting the number of cells that were in the lower chamber compartment with a Neubauer chamber.
Data analysis
Statistical analyses were performed using the statistical packages JMP (version 8.0; SAS Institute, Cary, NC) and SigmaStat (version 3.5; Systat Software, Erkrath, Germany). The determination of significant differences was performed by either Student’s t-test or analysis of variance (ANOVA) for parametric data and Mann–Whitney and ANOVA on ranks for nonparametric data. The values were expressed as mean ± standard error of the mean (SEM) of three or five independent experiments. P < 0.05 was considered statistically significant.
Results
The main clinical and laboratory characteristics of the 26 patients enrolled in the study are described in Tables 1 and 2, respectively. The mean age was 52 ± 2 years and 38% were male. Hypertensive nephrosclerosis was the main cause of CKD and all patients in the sample were hypertensive. Only 11% of the patients had diabetes mellitus as an associated disease. The patients were treated with statins, aspirin and antihypertensive drugs in 30%, 43% and 100% of cases, respectively. The median values found for total calcium, PTH, Kt/V, albumin and total cholesterol were within the reference range for patients in stage 5 CKD.
Table 1.
Main clinical characteristics of the study population
| Parameters | |
| Number of patients | 26 |
| Age (years) | 52 ± 2 |
| Gender (% men) | 38 |
| Ethnicity (% Caucasian) | 81 |
| Comorbitidies (%) | |
| Diabetes mellitus | 11 |
| Cardiovascuar diseases | 15 |
| Hypertension | 100 |
| Etiology of CKD (%) | |
| Hypertensive nephrosclerosis | 30 |
| Diabetic nephropathy | 11 |
| Chronic glomerulonephritis | 50 |
| Other | 9 |
| Vitamin D (% used) | 45 |
| Statins (% used) | 30 |
| Aspirin (% used) | 43 |
| Antihypertensive agents (% used) | 100 |
| Time on dialysis (months) | 17 ± 3 |
Values expressed as mean ± SD.
Table 2.
Main laboratory characteristics of the study population
| Cholesterol (mg/dL) | 180 (108–248) |
| Low-density lipoprotein cholesterol (mg/dL) | 106 (42–176) |
| High-density lipoprotein cholesterol (mg/dL) | 44 (21–80) |
| Triglycerides (mg/dL) | 150 (73–240) |
| Hemoglobin (g/dL) | 11.5 (10.8–12.0) |
| Albumin (g/dL) | 3.9 (3.3–4.7) |
| Calcium (mg/dL) | 9.0 (7.6–10.3) |
| Phosphate (mg/dL) | 6.7 (4.1–9.6) |
| PTH (pg/ml) | 446 (11–1666) |
| Alkaline phosphatase (UI/L) | 149 (65–602) |
| Kt/V | 1.5 (1.1–1.8) |
| CRP (mg/ml) | 4.9 ± 4.8 |
| IL-6 (pg/ml) | 6.7 ± 8.1 |
| IL-8 (pg/ml) | 128.2 ± 206.2 |
| SDF-1 (pg/ml) | 2625.9 ± 1288.6 |
Values expressed as mean ± SEM or median (25th-75th percentiles). SDF-1, stromal cell-derived factor-1.
AGEs characterization
To verify AGE formation after albumin glycation, AGEs were characterized by ultraviolet (UV) absorption (Figure 1A), as previously described elsewhere [26, 27]. Figure 1B shows the electrophoretic migration of AGEs by PAGE.
Fig. 1.

To characterize AGEs, the samples were subjected to electrophoresis and absorbance reading. (A) Absorption spectra of AGEs and BSA at 330, 360, 400 and 420 nm. Data are expressed as mean ± SEM of three independent experiments. (B) Electrophoretic migration of AGEs by PAGE. Samples were electrophoresed using 10% PAGE for 4 h at 100 V. Gel represents two separate experiments.
AGEs endotoxin levels
An LAL assay (Thermo Fisher Scientific) was performed to exclude the possibility that AGE and BSA were contaminated by endotoxins. The levels of endotoxins in BSA and AGEs were 1.65 and 1.04 EU/mL, respectively, which are not considered relevant.
Cell viability assay
Endothelial cell viability was assessed by MTT. Endothelial cells were cultured with BSA and AGEs at 0.2 mg/mL, with or without 3% sevelamer. Neither AGEs nor sevelamer had a significant effect on cell viability (Figure 2).
Fig. 2.

The effect of AGEs and sevelamer on endothelial cell viability. Endothelial cells were cultured with BSA and AGEs with or without sevelamer for 24 h and then treated with MTT for 3 h. The cell viability was determined by measuring the absorbance at 570 nm. The viability of untreated control cells was taken as 100%. Data are expressed as mean ± SEM of three independent experiments.
NMR
To verify if the binding capacity of sevelamer was preserved in vitro, the phosphate concentration on media samples containing phosphate (4 mM) was evaluated with and without sevelamer (3%). A reduction in phosphate concentration in samples treated with sevelamer was noted, confirming that its in vitro chelating action was preserved (Figure 3).
Fig. 3.
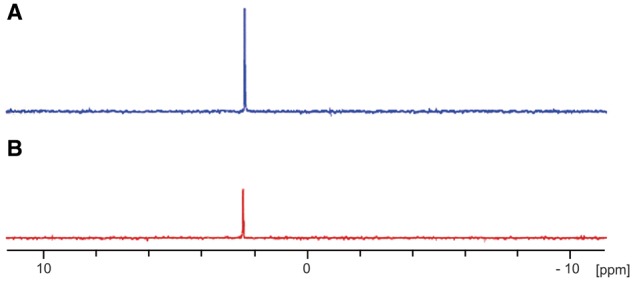
The effect of sevelamer (Sev) in samples with a phosphate (Pi) concentration of 4 mM with or without sevelamer. (A) Solution with 4 mM Pi; (B) solution containing 4 mM Pi + Sev (3%). Three independent experiments were performed.
Effect of AGEs on RAGE immunostaining and protein expression
Endothelial cells were treated with AGEs and specifically stained to detect this receptor. As shown in Figure 4, positive staining was visualized in AGE-treated cells (Figure 4D). Endothelial cells treated with either media alone (Figure 4A) or BSA alone (Figure 4C) were used as negative controls (Figure 4B). The staining was RAGE specific because the secondary antibody alone did not produce any coloration (Figure 4B). Additionally, to confirm and quantify the RAGE expression, we examined whether AGEs induced an increase in RAGE protein levels by western blot analysis. AGEs increased RAGE protein levels after 24 h of incubation (Figure 4E). However, a decrease in RAGE expression was observed when endothelial cells were incubated with AGEs and sevelamer.
Fig. 4.

The effect of AGEs on RAGE expression in endothelial cells. Endothelial cells were treated with BSA and AGEs (0.2 mg/mL) or untreated (control) for 24 h and stained for RAGE by immunocytochemistry. Panels show (A) control cells (untreated cells), (B) secondary antibody alone, (C) BSA and (D) AGEs. Magnification: 200x. (E) Endothelial cells were stimulated with AGEs or BSA (0.2 mg/mL), with or without sevelamer (Sev.) (3%) for 24 h. Immunoblotting for β-actin was used as the protein-loading control. RAGE levels were quantified as the ratio to β-actin by densitometry. Data are expressed as mean ± SEM of four independent experiments. *P < 0.05, AGEs versus AGEs + Sev.; ***P < 0.005, AGEs versus controls.
Effect of AGEs, uremic serum and sevelamer on biomarkers expression
IL-6, IL-8, MCP-1, PAI-1 and SAA expression increased significantly in a time-dependent manner when the endothelial cells were exposed to AGEs (Figure 5). However, when the cells were exposed to the uremic serum pool for 0 and 6 h, only SAA did not have a significant increase (Figure 6). In contrast, a significant time-dependent decrease in the expression of these biomarkers was observed in cells incubated with AGEs or the uremic serum pool in the presence of sevelamer. Nonetheless, sevelamer was not effective in reducing PAI-1 levels after uremic serum treatment.
Fig. 5.

The effect of AGEs on systemic inflammatory biomarker production (IL-6 and SAA) and vascular inflammatory biomarker (IL-8, MCP-1 and PAI-1) production by endothelial cells. Endothelial cells were incubated with AGEs or BSA (0.2 mg/mL), with or without sevelamer (Sev.) (3%), for 0 and 6 h. Data are expressed as mean ± SEM of three independent experiments. (A) IL-6: ***P < 0.005, AGEs 0 h versus AGEs 6 h; ****P < 0.001, AGEs 6 h versus AGEs + Sev. 6 h. (B) SAA: *P < 0.05, BSA 0 h versus BSA 6 h; ***P < 0.005, AGEs 6 h versus AGEs + Sev. 6 h; ****P < 0.001, AGEs 0 h versus AGEs 6 h. (C) IL-8: *P < 0.05, control 0 versus control 6; ***P < 0.005, BSA 0 h versus BSA 6 h; ****P < 0.001, AGEs 6 h versus AGEs + Sev. 6 h, AGEs 0 h versus AGEs 6 h. (D) MCP-1: *P < 0.05, BSA 6 versus BSA + Sev. 6 h; **P < 0.01, AGEs 6 h versus AGEs + Sev. 6 h; ***P < 0.005, control 6 h versus control 0 h, BSA 0 h versus BSA 6 h; ****P < 0.001, AGEs 6 h versus AGEs 0 h, AGEs 6 h versus control. (E) PAI: ***P < 0.005, AGEs 6 h versus AGEs + Sev. 6 h; ****P < 0.001, control 0 h versus control 6 h, BSA 0 h versus BSA 6 h, AGEs 0 h versus AGEs 6 h. ### P < 0.005, AGEs 6 h versus control 0 h.
Fig. 6.

The effect of uremic serum on systemic (IL-6 and SAA) and vascular (IL-8 and MCP-1) inflammatory biomarker production by endothelial cells. Endothelial cells were incubated with uremic serum, with or without sevelamer (Sev.) (3%), for 0 and 6 h. (A) IL-6: *P < 0.05, SU 0 h versus SU + Sev. 0 h; ****P < 0.001, SU 0 h versus SU 6 h, SU 6 h versus SU + Sev. 6 h. (B) IL-8: *P < 0.05, SU 0 h versus SU 6 h, SU 6 h versus SU + Sev. 6 h; **P < 0.01, SU 0 h versus SU + Sev. 0 h. (C) SAA: *P < 0.05, SU 6 h versus SU + Sev. 6 h. (D) MCP-1: *P < 0.05, SU 0 h versus SU 6 h, SU 6 h versus SU + Sev. 6 h, SU 0 h versus SU + Sev. 0 h.
Monocyte migration assay
Figure 7 illustrates the effect of BSA and AGEs, with or without sevelamer, on monocyte migration. Media alone (DMEM) was used as a negative control to evaluate the spontaneous movement of monocytes. MCP-1, IL-8 and MCP-1 with IL-8, known chemoattractant molecules, were used as positive controls at 50 ng/mL. After 90 min of incubation, a significant increase in cell migration was noted in the media enriched with MCP-1 (254.2 ± 46.8%; P < 0.05) and in the supernatant of the endothelial cells treated with AGEs (402.3 ± 163.8%; P < 0.01) compared with the negative controls (DMEM alone, taken as 100%). Conversely, the sevelamer-enriched supernatant led to a significant decrease in cell migration induced by AGE-enriched media (402.3 ± 163.8 versus 33.3 ± 7.4%; P < 0.005) and BSA-enriched media (277.7 ± 70.87 versus 62.5 ± 4.1%; P < 0.05).
Fig. 7.

Effect of endothelial cell supernatants on monocyte migration after treatment with AGEs and BSA 0.2 mg/mL, with and without sevelamer (Sev.) 3%, after 6 h. Data are expressed as mean ± SEM of six independent experiments. *P < 0.05, MCP-1 versus control, BSA versus BSA + Sev.; **P < 0.01, control versus AGEs; ***P < 0.005, AGEs versus AGEs + Sev.
Discussion
Vascular inflammation plays a crucial role in the development of CVD complications and comorbidities in CKD [29, 30]. AGEs are a heterogeneous group of compounds responsible for the activation of inflammatory responses, causing deleterious effects on vascular tissue [31]. Moreover, AGEs may accumulate in arteries, thus increasing the risk of endothelial dysfunction and atherosclerosis progression [5, 32].
Sevelamer is a phosphate binder with potential pleiotropic effects used in CKD patients for hyperphosphatemia treatment [33]. In addition, sevelamer may decrease the circulating levels of uremic toxins [24], which reduces the possibility of uremic-related cardiovascular complications such as atherosclerosis [34]. In this context, Nikolov etal. [35] demonstrated that sevelamer combined with lanthanum carbonate leads to a decrease in serum phosphate, vascular calcification and atherosclerosis in mice with CKD.
The administration of sevelamer for 3 weeks reduced the circulating levels of CML, methylglyoxal (MG), phosphatemia and lipid levels in diabetic HD patients, which resulted in a reduction in inflammatory biomarkers and oxidative stress [36]. Interestingly, some studies on HD and pre-dialysis patients and recent meta-analysis studies have suggested that sevelamer use was associated with lower all-cause mortality compared with calcium-based binders [37–39], suggesting a potential advantage of the former binder over the latter.
To the best of our knowledge, this is the first in vitro demonstration using endothelial cells in which sevelamer reduced systemic and vascular inflammation. The reduction of inflammation was evaluated through the decreased expression of RAGE, inflammatory biomarkers (IL-6, IL-8, MCP-1 PAI-1 and SAA) and monocyte migration induced by AGEs. Taken together, these findings suggest a possible role of sevelamer in attenuating inflammatory pathways stimulated by uremic serum and AGEs. Exposure of the endothelium to uremic toxins leads to a series of changes in vascular homeostasis and its functions [39]. We demonstrated previously—using this same model—that uremic plasma activates inflammatory pathways by inducing the production of chemokines and adhesion molecules, usually present in early events of atherosclerosis in CKD patients [14, 40]. We also observed, using endothelial cells and monocytes treated with AGEs, that both cells were activated by AGEs via RAGE through protein kinase C beta pathway, producing MCP-1 and VCAM-1 [27]. In addition, AGEs impaired the production of nitric oxide (NO), in part by NO synthase downregulation by increasing both mRNA degradation [41] and nicotinamide adenine dinucleotide phosphate oxidase expression, which suggests a link between AGEs and endothelial dysfunction [42]. Thus there is growing interest among the scientific community in developing ways to minimize the deleterious effects of uremic toxins, such as AGEs.
The involvement of RAGE on endothelial cell activation after AGE exposure was previously demonstrated [13, 27]. In contrast, we found a significant decrease in RAGE expression in the presence of sevelamer, which indirectly suggests that this chelator can act by binding in vitro to AGEs, therefore reducing the expression of RAGE. This finding was also observed in a study by Yubero-Serrano et al. [24] in patients with type 2 diabetes mellitus in which sevelamer reduced the circulating levels of AGEs. Other studies have also demonstrated the pleiotropic binding capacity of sevelamer in the reduction of circulatory levels of p-cresol and AGEs [18, 20].
Our study demonstrated that the longer the exposure of endothelial cells to both AGEs and uremic serum, the more systemic (IL-6 and SAA) and vascular (IL-8, MCP-1 and PAI-1) biomarkers are expressed. These results further confirm the link between vascular activation, systemic inflammation and uremic toxicity [2, 14]. Recent studies have implicated a causal role of SAA as a pro-inflammatory and pro-thrombotic mediator in the pathogenesis of atherosclerosis [43]. Moreover, others have shown that SAA is a potent pro-atherogenic chemokine on the endothelium, inducing the expression of cytokines and adhesion molecules such as IL-6, IL-8, MCP-1, ICAM-1, VCAM-1 and tissue factor [40, 44, 45]. Furthermore, AGEs increase the endothelial cell levels of mRNA coding for PAI-1 mediated by RAGE [46]. Our results demonstrated that sevelamer led to a significant decrease in the expression of IL-6, IL-8, MCP-1, PAI-1 and SAA, molecules related to systemic and vascular inflammation and induced by AGEs and uremic serum, suggesting a potential protective role of sevelamer on endothelium dysfunction beyond its classic effect via phosphate binding. On the other hand, sevelamer was not effective in reducing PAI-1 levels after uremic serum treatment, suggesting that the high serum levels of PAI-1 found in CKD patients [47] may be due to activation of other pathways different from the ones activated by phosphate and AGEs.
Finally, the cell migration assay was performed to mimic the early events of atherosclerosis development. Our findings reveal that the binding capacity of sevelamer to AGEs reduced monocyte migration, suggesting a possible early effect of sevelamer in preventing monocyte recruitment and attachment to the endothelium (thus all the activation of pathways) and finally plaque formation. Taken together, these findings suggest that sevelamer has a protective effect by binding nonspecifically to uremic serum and AGEs and therefore reduces the expression of inflammatory biomarkers present in the atherosclerosis process.
One of the limitations of our project is that it was not possible to measure the AGE levels in vitro by the NMR technique because it lacks the sensitivity and specificity to detect which specific molecule of the heterogeneous group of AGEs was bound to sevelamer. To overcome this limitation, the measurements of RAGE and inflammatory biomarker expression were used to indirectly assess sevelamer's action on reducing inflammation mediated by AGEs. Additionally, even though U-937 cells are commonly used as a model of monocytes, it would also be interesting to investigate the effect of AGEs and sevelamer in other cell lines, such as THP-1, or in monocytes extracted from the peripheral blood of CKD patients.
Conclusion
In conclusion, this study demonstrates, in vitro, that the anti-inflammatory effects of sevelamer on endothelial cells and monocytes may be due not only to its classic effect on phosphate, but also to its binding capacity to AGEs and possibly other uremic toxins. Sevelamer might be considered a therapeutic option to reduce the high AGE levels observed in pathological conditions such as CKD and perhaps diabetes, regardless of the presence of hyperphosphatemia. In the future, clinical studies should be designed to validate the potential capacity of sevelamer to decrease systemic AGEs and uremic serum concentrations. Thus it is hypothesized that this clinical treatment could provide a beneficial effect on vascular function, yielding relevant clinical outcomes.
Acknowledgments
The authors would like to thank the Academic Publishing Advisory Center [Centro de Assessoria de Publicação Acadêmica (CAPA), www.capa.ufpr.br] of the Universidade Federal do Paraná and Rafaella Tami Shima for assistance with English-language editing, Elaine Doff Sotta for assistance with Luminex analysis and Arquimedes Santana for assistance with NMR analysis. This work was supported by Demanda Social and Apoio à Pós-Graduação from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior.
Funding
Brazilian PhD. Scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes) to Alessandra Becker-Finco. Brazilian MSc. Scholarship from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes) to Paulo C. Gregório, Giane Favretto and Regiane S. Cunha. Research Grant from Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (Capes, PROAP). Research grant scholarship received by Professor Roberto Pecoits-Filho from Conselho Nacional de Pesquisa (CNPq).
Conflict of interest statement
None declared.
References
- 1. Bodiga VL, Eda SR, Bodiga S.. Advanced glycation end products: role in pathology of diabetic cardiomyopathy. Heart Fail Rev 2014; 19: 49. [DOI] [PubMed] [Google Scholar]
- 2. Hegab Z. Role of advanced glycation end products in cardiovascular disease. World J Cardiol 2012; 4: 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Thornalley PJ, Rabbani N.. Highlights and hotspots of protein glycation in end-stage renal disease. Semin Dial 2009; 22: 400–404 [DOI] [PubMed] [Google Scholar]
- 4. Bucala R. Diabetes, aging, and their tissue complications. J Clin Invest 2014; 124: 1887–1888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Stinghen AEM, Massy ZA, Vlassara H. et al. Uremic toxicity of advanced glycation end products in CKD. 2016; 27: 354–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang W, Xie D, Anderson AH, et al. Association of kidney disease outcomes with risk factors for CKD: findings from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis 2014; 63: 236–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. West RK, Moshier E, Lubitz I. et al. Dietary advanced glycation end products are associated with decline in memory in young elderly. Mech Ageing Dev 2014; 140: 10–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai W, Ramdas M, Zhu L. et al. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin1. Proc Natl Acad Sci USA 2012; 109: 15888–15893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yaffe K, Lindquist K, Schwartz AV. et al. Advanced glycation end product level, diabetes, and accelerated cognitive aging. Neurology 2011; 77: 1351–1356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu J, Huang K, Cai G-Y. et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal 2013; 26: 110–121 [DOI] [PubMed] [Google Scholar]
- 11. Chawla D, Bansal S, Banerjee BD. et al. Role of advanced glycation end product (AGE)-induced receptor (RAGE) expression in diabetic vascular complications. Microvasc Res 2014; 95: 1–6 [DOI] [PubMed] [Google Scholar]
- 12. Miyata T, Hori O, Zhang J. et al. Rapid publication the receptor for advanced glycation end products (RAGE) is a central mediator of the interaction of AGE-beta2microglobulin with human. J Clin Invest 1996; 98: 1088–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Quehenberger P, Bierhaus A, Fasching P. et al. Endothelin 1 transcription is controlled by nuclear factor-kappaB in AGE-stimulated cultured endothelial cells. Diabetes 2000; 49: 1561–1570 [DOI] [PubMed] [Google Scholar]
- 14. Stinghen AEM, Gonçalves SM, Martines EG. et al. Increased plasma and endothelial cell expression of chemokines and adhesion molecules in chronic kidney disease. Nephron Clin Pract 2009; 111: 117–126 [DOI] [PubMed] [Google Scholar]
- 15. Meng L, Fu B. Practical use of sevelamer in chronic kidney disease patients on dialysis in People’s Republic of China. Ther Clin Risk Manag 2015; 11: 705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nikolov IG, Joki N, Maizel J. et al. Pleiotropic effects of the non-calcium phosphate binder sevelamer. Kidney Int 2006; 70: S16–S23 [DOI] [PubMed] [Google Scholar]
- 17. Garg JP, Chasan-Taber S, Blair A. et al. Effects of sevelamer and calcium-based phosphate binders on uric acid concentrations in patients undergoing hemodialysis: a randomized clinical trial. Arthritis Rheum 2005; 52: 290–295 [DOI] [PubMed] [Google Scholar]
- 18. Hauser AB, Azevedo IRF, Gonçalves S. et al. Sevelamer carbonate reduces inflammation and endotoxemia in an animal model of uremia. Blood Purif 2010; 30: 153–158 [DOI] [PubMed] [Google Scholar]
- 19. Stinghen a. EM, Gonçalves SM, Bucharles S. et al. Sevelamer decreases systemic inflammation in parallel to a reduction in endotoxemia. Blood Purif 2010; 29: 352–356 [DOI] [PubMed] [Google Scholar]
- 20. Guida B, Cataldi M, Riccio E, et al. Plasma p-cresol lowering effect of sevelamer in peritoneal dialysis patients: evidence from a cross-sectional observational study. PLoS One 2013; 8: e73558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rodríguez-Osorio L, Zambrano DP, Gracia-Iguacel C. et al. Uso del sevelamer en la enfermedad renal crónica. Más allá del control del fósforo. Nefrología 2015; 35: 207–217 [DOI] [PubMed] [Google Scholar]
- 22. Maizel J, Six I, Dupont S. et al. Effects of sevelamer treatment on cardiovascular abnormalities in mice with chronic renal failure. Kidney Int 2013; 84: 491–500 [DOI] [PubMed] [Google Scholar]
- 23. Six I, Maizel J, Barreto FC, et al. Effects of phosphate on vascular function under normal conditions and influence of the uraemic state. Cardiovasc Res 2012; 96: 130–139 [DOI] [PubMed] [Google Scholar]
- 24. Yubero-Serrano EM, Woodward M, Poretsky L. et al. Effects of sevelamer carbonate on advanced glycation end products and antioxidant/pro-oxidant status in patients with diabetic kidney disease. Clin J Am Soc Nephrol 2015; 10: 759–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Iwashima Y, Eto M, Horiuchi S, Sano H.. Advanced glycation end product-induced peroxisome proliferator-activated receptor gamma gene expression in the cultured mesangial cells. Biochem Biophys Res Commun 1999; 264: 441–448 [DOI] [PubMed] [Google Scholar]
- 26. Pongor S, Ulrich PC, Bencsath FA. et al. Aging of proteins: isolation and identification of a fluorescent chromophore from the reaction of polypeptides with glucose. Proc Natl Acad Sci USA 1984; 81: 2684–2688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rempel LCT, Finco AB, Maciel RAP. et al. Effect of PKC-β signaling pathway on expression of MCP-1 and VCAM-1 in different cell models in response to advanced glycation end products (AGEs). Toxins (Basel) 2015; 7: 1722–1737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 1983; 65: 55–63 [DOI] [PubMed] [Google Scholar]
- 29. Goligorsky MS. Pathogenesis of endothelial cell dysfunction in chronic kidney disease: a retrospective and what the future may hold. Kidney Res Clin Pract 2015; 34: 76–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barreto DV, Barreto FC, Liabeuf S. et al. Plasma interleukin-6 is independently associated with mortality in both hemodialysis and pre-dialysis patients with chronic kidney disease. Kidney Int 2010; 77: 550–556 [DOI] [PubMed] [Google Scholar]
- 31. Vlassara H, Cai W, Chen X. et al. Managing chronic inflammation in the aging diabetic patient with CKD by diet or sevelamer carbonate: A modern paradigm shift. J Gerontol Ser A Biol Sci Med Sci 2012; 67: 1410–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang C, Wang Y, Wang G. et al. Skin autofluorescence is associated with endothelial dysfunction in uremic subjects on hemodialysis. PLoS One 2016; 11: e0147771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Block GA, Wheeler DC, Persky MS, et al. Effects of phosphate binders in moderate CKD. J Am Soc Nephrol 2012; 23: 1407–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Phan O, Ivanovski O, Nguyen-Khoa T, et al. Sevelamer prevents uremia-enhanced atherosclerosis progression in apolipoprotein E-deficient mice. Circulation 2005; 112: 2875–2882 [DOI] [PubMed] [Google Scholar]
- 35. Nikolov IG, Joki N, Nguyen-Khoa T, et al. Lanthanum carbonate, like sevelamer-HCl, retards the progression of vascular calcification and atherosclerosis in uremic apolipoprotein E-deficient mice. Nephrol Dial Transplant 2012; 27: 505–513 [DOI] [PubMed] [Google Scholar]
- 36. Vlassara H, Uribarri J, Cai W. et al. Effects of sevelamer on HbA1c, inflammation, and advanced glycation end products in diabetic kidney disease. Clin J Am Soc Nephrol 2012; 7: 934–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Di Iorio B, Bellasi A, Russo D.. Mortality in kidney disease patients treated with phosphate binders: a randomized study. Clin J Am Soc Nephrol 2012; 7: 487–493 [DOI] [PubMed] [Google Scholar]
- 38. Di Iorio B, Molony D, Bell C. et al. Sevelamer versus calcium carbonate in incident hemodialysis patients: results of an open-label 24-month randomized clinical trial. Am J Kidney Dis 2013; 62: 771–778 [DOI] [PubMed] [Google Scholar]
- 39. Günthner T, Jankowski V, Kretschmer A. et al. Endothelium and vascular smooth muscle cells in the context of uremia. Semin Dial 2009; 22: 428–432 [DOI] [PubMed] [Google Scholar]
- 40. Ribeiro V, Bosquetti B, Gonçalves SM. et al. Uremic serum inhibits in vitro expression of chemokine SDF-1: impact of uremic toxicity on endothelial injury. J Bras Nefrol 2014; 36: 123–131 [DOI] [PubMed] [Google Scholar]
- 41. Rojas A, Romay S, González D. et al. Regulation of endothelial nitric oxide synthase expression by albumin-derived advanced glycosylation end products. Circ Res 2000; 86: e50–e54 [DOI] [PubMed] [Google Scholar]
- 42. Jourde-Chiche N, Dou L, Cerini C. et al. Vascular incompetence in dialysis patients-protein-bound uremic toxins and endothelial dysfunction. Semin Dial 2011; 24: 327–337 [DOI] [PubMed] [Google Scholar]
- 43. Cai H, Song C, Endoh I. et al. Serum amyloid A induces monocyte tissue factor. J Immunol 2007; 178: 1852–1860 [DOI] [PubMed] [Google Scholar]
- 44. Chami B, Barrie N, Cai X. et al. Serum amyloid a receptor blockade and incorporation into high-density lipoprotein modulates its pro-inflammatory and pro-thrombotic activities on vascular endothelial cells. Int J Mol Sci 2015; 16: 11101–11124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nedić O, Rattan SIS, Grune T. et al. Molecular effects of advanced glycation end products on cell signalling pathways, ageing and pathophysiology. Free Radic Res 2013; 47(Suppl 1): 28–38 [DOI] [PubMed] [Google Scholar]
- 46. Yamagishi S, Fujimori H, Yonekura H. et al. Advanced glycation endproducts inhibit prostacyclin production and induce plasminogen activator inhibitor-1 in human microvascular endothelial cells. Diabetologia 1998; 41: 1435–1441 [DOI] [PubMed] [Google Scholar]
- 47. Jeong BY, Uddin J, Park JH. et al. Novel plasminogen activator inhibitor-1 inhibitors prevent diabetic kidney injury in a mouse model. PLoS One 2016; 11: e0157012 [DOI] [PMC free article] [PubMed] [Google Scholar]