

Abstract
Squamates include all lizards and snakes, and display some of the most diverse and extreme morphological adaptations among vertebrates. However, compared with birds and mammals, relatively few resources exist for comparative genomic analyses of squamates, hampering efforts to understand the molecular bases of phenotypic diversification in such a speciose clade. In particular, the ∼400 species of anole lizard represent an extensive squamate radiation. Here, we sequence and assemble the draft genomes of three anole species—Anolis frenatus, Anolis auratus, and Anolis apletophallus—for comparison with the available reference genome of Anolis carolinensis. Comparative analyses reveal a rapid background rate of molecular evolution consistent with a model of punctuated equilibrium, and strong purifying selection on functional genomic elements in anoles. We find evidence for accelerated evolution in genes involved in behavior, sensory perception, and reproduction, as well as in genes regulating limb bud development and hindlimb specification. Morphometric analyses of anole fore and hindlimbs corroborated these findings. We detect signatures of positive selection across several genes related to the development and regulation of the forebrain, hormones, and the iguanian lizard dewlap, suggesting molecular changes underlying behavioral adaptations known to reinforce species boundaries were a key component in the diversification of anole lizards.
Keywords: Anolis, genomes, molecular evolution, substitution rates, adaptation, phenotypic evolution
Introduction
The field of amniote comparative genomics has benefited from an influx of whole genome assemblies, due to efforts across multiple collaborative research groups (Genome 10K Community of Scientists 2009; Lindblad-Toh et al. 2011; Green et al. 2014; Zhang et al. 2014). This has resulted in a better phylogenetic sampling of genomes across the amniote tree of life and a vastly improved ability to understand the molecular mechanisms underlying the radiation of amniotes into terrestrial, aerial, freshwater, and marine habitats over ∼300 Myr of evolution. For instance, the eutherian mammals share a time to most recent common ancestor (TMRCA) of ∼100 Myr during the Cretaceous Period (Hedges et al. 2015), and extensively diversified during the Cenozoic Era, which began ∼66 Ma. Each eutherian mammalian order is represented by at least one whole genome sequence (Speir et al. 2016), enabling investigations into the genomic changes leading to both divergent (Kim et al. 2016) and convergent (Foote et al. 2015) phenotypes. Similarly, modern birds also share a Cretaceous TMRCA (Donoghue and Benton 2007; Hedges et al. 2015), and the Avian Genome Consortium has begun to investigate the extensive avian evolution that occurred throughout the Cenozoic at the genomic scale (Jarvis et al. 2014; Zhang et al. 2014). Although currently not as far along, numerous individual genome projects have been launched for squamate reptiles including iguanian lizards (Alföldi et al. 2011; Georges et al. 2015), snakes (Castoe et al. 2013; Vonk et al. 2013; Gilbert et al. 2014), anguimorphs (Song et al. 2015), and geckos (Liu et al. 2015). To better understand the mechanisms underlying phenotypic diversity in squamates—one of the largest groups of vertebrates—and provide meaningful comparisons with the other major groups of amniotes (i.e., mammals and birds), more robust genomic sampling across the squamate lineages is sorely needed.
Increased genomic sampling of speciose, phenotypically diverse clades such as squamates can provide better estimates of the number of mutational changes that occurred during lineage diversification and the effects of those changes, if any, on phenotypes. This can help determine whether or not underlying shifts in molecular evolution are related to morphological rates of change and species diversity, which has been a subject of debate (Bromham et al. 2002; Davies and Savolainen 2006). For instance, species are thought to diverge either by gradual change or by episodic bursts of rapid diversification such as adaptive radiations (Eldredge and Gould 1972). Punctuated episodes of speciation can cause dramatic changes in the rate in molecular evolution, possibly providing a “mutational engine” for new phenotypes (Pagel et al. 2006)—although there has been considerable debate over this issue (Lanfear et al. 2010). Genome annotations provide information about the boundaries between functional and nonfunctional loci, and DNA substitution rates can be estimated for genomic regions exposed to natural selection as well as for unconstrained regions that are expected to accumulate mutations at the neutral rate. Thus, comparative genomics creates the opportunity to understand the genetic basis of species divergence, and untangle the links between molecular evolution and morphological change (Lanfear et al. 2010).
One group of squamates with extensive diversification within the Cenozoic is the anoles, which comprise almost 400 species of mostly arboreal Neotropical lizards classified in the genus Anolis (Squamata: Dactyloidae). The morphological, ecological, and behavioral disparity among anoles makes this one of the most diverse squamates clades (Losos and Miles 2002), long serving as an ecological and evolutionary model with multiple examples of phenotypic convergence on the islands of the Caribbean and in Central and South America (Losos 2009). Anoles are an ideal group with which to test hypotheses about the relationship between rates of molecular and phenotypic evolution, as well as the genomic bases of many adaptive traits, as there is a robust reference genome and annotation for the green anole (Anolis carolinensis) (Alföldi et al. 2011; Eckalbar et al. 2013). However, despite their ongoing importance for the study of adaptive radiation and species diversification, and existing genomic resources, there has not yet been a comparative genomic investigation that seeks to identify the loci underlying adaptive divergence during Anolis evolution. For instance, body size and relative limb lengths have strong adaptive significance in anoles, as sympatric species interact differently with their environmental substrates and occupy separate areas of morphospace (Losos 2009). Although genetic control of limb development is not well studied in anoles, extensive work with model organisms has identified potential regions of interest. For instance, Hox cluster paralogs control limb element patterning during development (Pineault and Wellik 2014) and the T-box transcription factor 5 (tbx5) controls limb bud outgrowth (Rallis et al. 2003). A comparative genomic approach to understanding phenotypic diversity in anoles would provide a powerful way to scan for anole-specific mutations in candidate regions such as these, place these genetic changes in the context of what is expected under the background DNA substitution rate, and provide insights into how anoles have responded to selective pressures at the molecular level.
Here, we investigate molecular mechanisms of anole lizard diversification using complete genome assemblies from four species. Our goals were 3-fold: 1) to study patterns of genomic divergence in anoles with respect to other amniote lineages, 2) to understand how the rate of genomic evolution relates to functional genomic features and, in turn, phenotypic diversity in anoles, and 3) to identify putative adaptively evolving genomic regions that contributed to the well-studied ecological and morphological changes that occurred during the diversification of anoles. We focus our genome sequencing and assembly efforts on the following species with diverse morphologies and ecological niches: 1) the Central American giant anole (Anolis frenatus), which is a relatively large-bodied species that resides high on tree trunks (Losos et al. 1991); 2) the grass anole (Anolis auratus), which occupies grassy and bushy vegetation with narrow perches (Fleishman 1988); and 3) the slender anole (Anolis apletophallus), which is primarily found lower on tree trunks and on the ground (Köhler and Sunyer 2008). Combined with the reference genome for A. carolinensis, which resides in crowns and high trunks of trees in the southeastern United States, these species are distributed across the anole lizard phylogeny (Guyer and Savage 1986; Poe 2004), which may have originated between 120 and 45 Ma (Nicholson et al. 2012; Prates et al. 2015). The phylogenetic and ecological diversity of these species provides an ideal opportunity to study the genomic underpinnings of Anolis diversification, adaptive radiations of tetrapods in general, and how evolution has shaped genomes and phenotypes during the history of land-dwelling vertebrates.
Materials and Methods
Animal Processing, Genome Sequencing, and Assembly
Adult A. apletophallus, A. auratus, and A. frenatus were collected at field sites under permits SE/A-33-11 and SC/A-21-12 issued by the Republic of Panama Autoridad Nacional de Ambiente (ANAM; to K.K.) under Institutional Animal Care and Use Committee (IACUC) guidelines at the Smithsonian Tropical Research Institute (Protocol 2011-0616-2014-07 to K.K.). Specimens were exported under ANAM permits SEX/A-81-11 and SEX/A-71-12 (to K.K.) following regulation 50 CFR Part 14 of the United States Fish & Wildlife Service. Lizards were maintained under IACUC guidelines at Arizona State University (Protocols 09-1053 R, 12-1274 R, and 15-1416 R; supplementary table 1, Supplementary Material online). Specimens were euthanized according to ASU IACUC protocols 10-1053 R and 12-1274 R (K.K.) via intracoelemic injection of a solution of sodium pentobarbital/sodium phenytoin and secondary confirmation by thoracotomy. Twenty-four adult specimens were used for osteological analyses (13 A. apletophallus; 7 A. auratus; 4 A. frenatus) together with 10 adult A. carolinensis specimens obtained from Charles D. Sullivan Co., Inc. (Nashville, TN) and Marcus Cantos Reptiles (Fort Myers, FL). Given the lack of published studies comparing the limb morphology of A. apletophallus, A. auratus, A. frenatus, and A. carolinensis, we completed a morphometric analysis of the forelimb and hindlimb stylopod (humerus and femur), zeugopod (radius/ulna and tibia/fibula), and autopod (manus and pes) (supplementary methods and supplementary table 1, Supplementary Material online). In addition, for A. apletophallus, A. auratus, and A. frenatus, one specimen per species was selected for genomic study; we dissected skeletal muscle tissue from A. apletophallus and liver from A. frenatus and A. auratus, and extracted genomic DNA using the Qiagen DNeasy Blood & Tissue Kit standard protocol. Specimen identifiers and geographic coordinates are listed in supplementary table 1, Supplementary Material online.
Paired-end DNA libraries were constructed with the TruSeq Library Prep kit for target sizes of 180, 300, 500 bp, and 1 kb at the Translational Genomics Research Institute (TGen; Phoenix, Arizona). Mate pair libraries of target size 3 kb were constructed using the Nextera v1 Mate Pair Library Kit (Illumina) at TGen and additional mate pair libraries of target size 5 kb were constructed with the Nextera Mate Pair Library kit at the University of Arizona Genetics Core (UAGC; Tucson, Arizona). Genomic DNA was sequenced on both Illumina HiSeq 2000 and 2500 platforms at TGen and UAGC, respectively. We trimmed raw reads for nucleotide biases, adaptors, and a quality score cutoff of ≥28 with Trimmomatic v0.33 (Bolger et al. 2014). We corrected errors in the trimmed sequences using the module SOAPec in SOAPdenovo2 (Luo et al. 2012). Overlapping reads were joined to form single-end reads using FLASH v1.2.8 (Magoč and Salzberg 2011). We compared the outputs of different genome assembly pipelines including ABySS versions 1.3.7 and 1.5.2 (Simpson et al. 2009), SOAPdenovo2 (Luo et al. 2012), Platanus v1.2.1 (Kajitani et al. 2014) for both contig and scaffold assembly, and SSPACE v3.0 (Boetzer et al. 2011) for scaffold assembly. We selected assemblies with the longest contig and scaffold N50 values for further analysis. Candidate assemblies were also evaluated based on expected gene content using the Core Eukaryotic Gene Mapping Approach (CEGMA v2.5; Parra et al. 2009), which aligns genome assemblies to a database of 248 highly conserved eukaryotic proteins and reports the completeness of detected orthologs, and Benchmarking Universal Single Copy Orthologs (BUSCO v1.2; Simão et al. 2015), which scans assemblies for a vertebrate-specific database of 3,023 conserved genes.
Annotation and Analysis of Repeat Elements
To analyze the repetitive landscapes of the three de novo anole lizard genomes, we first ran RepeatMasker v4.0.5 (http://www.repeatmasker.org) (Smit et al. 2013–2015) on each assembly using a library of known Anolis repeat family consensus sequences from RepBase (Jurka et al. 2005). After this initial masking of reference repeats (i.e., those known from A. carolinensis and shared ancestrally across vertebrates), we scanned the unmasked portions of each genome for novel elements using RepeatModeler v1.0.8 (http://www.repeatmasker.org) (Smit et al. 2008–2015; Smit et al. 2013–2015). This step allowed us to identify de novo repetitive elements that may be unique to each species and thus would not have been detected by RepeatMasker alone, which relies on nucleotide similarity to previously archived elements. To estimate the amount of evolutionary divergence within repeat families and characterize the repetitive landscape of each genome, we generated repeat family-specific alignments and calculated the average Kimura 2-parameter (K2P) distance from consensus within each repeat family. We modified the weight of two transition mutations as 1% of a single transition, in order to correct for high mutation rates at CpG sites. This was done for the A. auratus, A. apletophallus, and A. frenatus assemblies using the calcDivergenceFromAlign.pl tool contained within the RepeatMasker package. We accessed the repeat landscape for A. carolinensis at http://www.repeatmasker.org/species/anoCar.html (last accessed December 2016).
Gene Annotations
We created gene models for the A. frenatus, A. auratus, and A. apletophallus genomes based on homology with protein-coding sequences in A. carolinensis (AnoCar2.0) and the UniProt/SwissProt database (UniProt Consortium 2015), combined with ab initio predictions from SNAP (November 29, 2013 release; Korf 2004) using multiple iterations of MAKER v2.31.5 (Holt and Yandell 2011) for each species assembly. The first iteration of MAKER aligned the protein sequences to the assembly to produce draft gene models. Ab initio gene predictors benefit from the training of their Hidden Markov Models (HMM), and we trained SNAP by running MAKER a second time with SNAP using the species-specific HMMs generated from the CEGMA analysis described earlier (for A. apletophallus, we used the CEGMA gene models from A. auratus, which were more complete—see Results). Using the gene models from this output, we then generated an improved HMM for SNAP in a third MAKER iteration. A fourth and final run of MAKER was then performed, which incorporated the final SNAP HMM and the aligned protein data, resulting in the final set of gene model predictions. For each species, the final genes were functionally annotated with two methods: 1) a BLASTp of MAKER2-predicted proteins to known proteins in UniProt, and 2) the scanning of MAKER-predicted proteins for the presence of functional domains using InterProScan 5 (Jones et al. 2014; Mitchell et al. 2015). Functional annotations were incorporated using scripts provided with the MAKER2 package.
Whole Genome Alignments
As the basis for our comparative genomic study of amniotes, we generated a whole genome alignment of 31 sarcopterygian vertebrates, featuring representatives of the major groups of amniotes (mammals, squamates, turtles, crocodilians, and birds) and ten available squamate genomes including the three de novo anole lizard genomes (supplementary table 2, Supplementary Material online). First, we generated pairwise syntenic alignments of each species’ genome to the A. carolinensis genome (AnoCar2.0) using LASTZ v1.02 (Harris 2007), followed by chaining to form gapless blocks and netting to rank the highest scoring chains (Kent et al. 2003). We used the pairwise nets to construct a multiple sequence alignment (MSA) with MULTIZ v11.2 (Blanchette et al. 2004) and used A. carolinensis as the reference species and a guide phylogenetic tree (see Supplementary Material online). The MSA was filtered for the presence of aligned blocks from at least 26 out of the 31 species (84% complete).
Phylogenetic Analyses
To reconstruct the amniote phylogeny and estimate neutral substitution rates across lineages, we relied on 4-fold degenerate (4 D) sites, which are positions in coding regions where all mutations are synonymous at the amino acid level and can be used as a proxy of the neutral rate of evolution (Kumar and Subramanian 2002). We extracted 4 D sites from the MSA, excluding sites from the mitochondrion, based on the A. carolinensis protein-coding sequence (CDS) annotations (Anocar2.0, Ensembl v86, last accessed December 2016) using msa_view from the Phylogenetic Analysis with Space/Time Models (PHAST) v1.3 package (Hubisz et al. 2011). Phylogenetic reconstruction of the 31 included vertebrates using the 4D site data was performed with RAxML v8.2.3 (Stamatakis 2014). We generated 20 maximum likelihood (ML) trees under the GTRCAT substitution model, setting Latimeria, a lobe-finned fish, as the outgroup, and conducted 500 bootstrap replicates, placing the bipartitions from the bootstrap analysis on the ML tree with the highest likelihood to assess node support.
Rates of Molecular Evolution
We estimated absolute rates of molecular evolution in terms of substitutions per site per million years across the best ML tree, and estimated the divergence times of the major amniote lineages, including the TMRCA of anole lizards, using the semiparametric method based on penalized likelihood (PL) in r8s v1.8 (Sanderson 2002, 2003). We used two sets of criteria to constrain minimum, maximum, or fixed node ages. The first set of criteria was based on constraints from the paleontological literature; specifically, the minimum age of Diapsida (i.e., the TMRCA of chicken and Anolis) was set at 255.9 Myr, the minimum age of Archosauria (i.e., the TMRCA of chicken and alligator) was set at 247.1 Myr, the minimum and maximum ages for Mammalia (i.e., the TMRCA of platypus and human) were set at 164.9–201.5 Myr, and the minimum and maximum ages for Neognathae (i.e., the TMRCA of chicken and zebra finch) were set at 66–86.8 Myr (Benton et al. 2015). The second analysis used comparatively fewer constraints, using the node age estimate for Archosauria of 247–250 Myr obtained from TimeTree (www.timetree.org; Hedges et al. 2015). We set the minimum and maximum ages for Crocodilia at 77.8–83.6 Myr (Green et al. 2014) and fixed the root age (i.e., the TMRCA of coelacanth and human) at 415 Myr in both analyses (Donoghue and Benton 2007). The PL method estimates a different substitution rate on each branch and implements a penalty when rates differ greatly between branches, quantified by a smoothing parameter for which larger values indicate a clock-like model (Sanderson 2002). We used cross-validation to optimize this smoothing parameter, allowing values to range on a log10 scale starting from 10° with the exponent increasing 0.3 for a total of ten steps, and reran the analysis with the optimal value. We used the gradient check implemented in r8s to ensure the signs of any active constraints were correct (i.e., negative if a minimum constraint was used).
As the evolution of anole lizards constitutes several radiations resulting in ∼400 species across two continents and several islands, we predicted that estimated substitution rates in this lineage should be faster than the phylogenetic average for amniotes and that this could be explained by either punctuated evolution (Pagel et al. 2006) or ecological opportunity (Mahler et al. 2010). In a punctuated scenario, the total genetic distance between the root and tip of a phylogeny (i.e., the path length) will be associated with the number of speciation events in a lineage (Webster et al. 2003; Pagel et al. 2006). Alternatively, in the case of ecological opportunity, the rate of evolution is correlated with the rate of speciation (Mahler et al. 2010). In either case, path lengths and speciation events inferred from molecular data will be positively correlated (Rabosky 2012). To test this prediction, we first searched for evidence of punctuated molecular evolution across the best ML phylogeny using a phylogenetic generalized least squares (PGLS) regression in BayesTraits v3.0 (Pagel et al. 2004), which incorporates a species phylogeny and weights data points according to shared ancestry. We removed the nonamniote outgroup and applied a continuous model to the phylogenetic path length (i.e., the total number of substitutions per site) and the number of nodes (i.e., speciation events) leading to each of the 30 terminal taxa as dependent and independent variables, respectively. We then estimated 1) the proportion of evolution across the tree attributed to speciation and 2) the deviation from the molecular clock due to punctuational events, using the test for punctuational evolution available at http://www.evolution.rdg.ac.uk/pe/index.html (last accessed April 2017), testing for a node-density artifact which can lead to the underestimation of branch lengths due to regions of the phylogeny with fewer taxa (Webster et al. 2003; Venditti et al. 2006). Finally, we generated a continuous regression model using the path lengths and number of speciation events for each taxon, and used it to predict the path lengths for A. frenatus, A. carolinensis, A. auratus, and A. apletophallus using BayesTraits. All MCMC analyses were run for an initial 10,000 generations for burn-in, followed by 1,000,000 iterations with sampling every 1,000 generations. We used Tracer v1.6 (http://tree.bio.ed.ac.uk/software/tracer/; last accessed December 2016) to monitor MCMC convergence, including non auto-correlation, by ensuring an effective sample size (ESS) of ≥ 200 for all parameters.
Detecting Nonneutral Substitution Rates and Identification of Anole-Accelerated Genomic Regions
In order to identify genomic regions underlying phenotypic variation in anoles, we aimed to detect loci in our aligned sequences that depart from neutral expectations, using phyloP (Pollard et al. 2010) to estimate nonneutral substitution rates both across the amniote phylogeny and specifically across the anole lineage. PhyloP computes 1) a null distribution for the total number of substitutions at a locus given a nonconserved (neutral) phylogenetic model, 2) an estimate of the observed number of substitutions in the alignment, and 3) the P value from a comparison of this estimate to the null distribution calculated with a likelihood ratio test (LRT). First, we estimated the neutral model by fitting a time-reversible substitution model (REV) to the best ML phylogeny obtained from the 4D site data and transforming branch lengths in terms of substitutions per site using phyloFit in PHAST. Second, we used phyloP to produce a conservation-acceleration (CONACC) score for every site in the alignment, which is a two-sided LRT-based P value converted into either a negative value to indicate accelerated evolution or positive value to indicate conservation. We analyzed the distributions of CONACC scores for different annotation features: CDS, introns, 5′-UTR, 3′-UTR, intergenic regions, and ancestral repeats (AR), under the expectation that AR should evolve under relatively weaker purifying selection (Mouse Genome Sequencing Consortium et al. 2002) and therefore possess lower CONACC scores. We conducted two types of tests: 1) an “all branches” test that examines rate variation across every branch in the phylogeny, and 2) multiple “subtree” tests which separately examine rate variation within a particular clade relative to the rest of the phylogeny. As suggested by Pollard et al. (2010), we computed CONACC scores for subtrees based on 10-bp windows. We compared subtree test results for anoles and snakes. As a test of the null hypothesis that substitution rates between a subtree and the rest of the phylogeny are equal, we compared the subtree CONACC score distributions for each annotation feature using the Mann–Whitney U-test in R v3.3.0 (R Core Team 2016). Finally, to determine which protein-coding genes contain anole-accelerated regions, we used phyloP to compute one-sided P values to indicate accelerated evolution (ACC). We then collected ACC regions with a P value of ≤ 0.001 which overlap with Ensembl v86 AnoCar2.0 whole gene annotations using bedtools intersect (Quinlan and Hall 2010). We tested for statistical enrichment of overrepresented GO terms associated with the genes overlapping accelerated regions using the PANTHER overrepresentation analysis tool available on the Gene Ontology Consortium website (GO Ontology database Released April 24, 2017, www.geneontology.org, last accessed May 2017; Gene Ontology Consortium 2015), applying a Bonferroni correction to account for multiple hypothesis testing. Genes overlapping accelerated regions were compared with the reference set of 18,527 A. carolinensis genes. We also collected overrepresented GO terms from ACC analyses for the snakes subtree in order to compare functional category enrichment between squamate clades.
Selection on Protein-Coding Genes
To more directly measure selective pressures acting on protein coding genes during Anolis diversification and identify genes related to ecologically relevant adaptations, we estimated the ratio of nonsynonymous to synonymous substitutions (dN/dS) in a series of analyses. Genes with pairwise dN/dS > 1 harbor an excess of nonsynonymous and potentially functionally relevant changes at the amino acid level and are candidates for inferring positive selection (Fay and Wu 2003). First, we downloaded Ensembl gene annotations for the A. carolinensis genome (AnoCar2.0) from the UCSC Genome Browser (Speir et al. 2016) and assembled exons by gene based on the pairwise syntenic nets between A. carolinensis and A. apletophallus, A. auratus, and A. frenatus, respectively (see genome alignment methods above), using the “stitch gene blocks” tool in Galaxy (Blankenberg et al. 2011). Orthologous gene alignments of each species to A. carolinensis were filtered for quality through a custom pipeline (https://github.com/WilsonSayresLab/Anole_expression/tree/master/Scripts), which deleted frame-shift mutations and replaced internal stop codons with gaps. We calculated substitution rates at nonsynonymous (dN) and synonymous sites (dS) and estimated dN/dS for each de novo species compared with A. carolinensis using KaKs_calculator v2.0 (Wang et al. 2010). We applied a Bonferroni adjustment for multiple hypothesis testing to identify positively selected genes with highly significant dN/dS > 1. To link genes with dN/dS > 1 to potential phenotypes, we used the Ensembl gene IDs to collect GO terms in Biomart (Kinsella et al. 2011). We also examined the distribution of dN/dS for genes belonging to GO categories expected a priori to be relevant to anole lizard diversification based on the literature (Losos 2009): Reproduction (GO: 0000003), pigmentation (GO: 0043473), somitogenesis (GO: 0001756), visual perception (GO: 0007601), perception of smell (GO: 0007608), and limb development (GO: 0060173) (Gene Ontology Consortium 2015).
To analyze substitution rates in protein-coding genes across sauropsids and identify positively selected genes in the four anole lizard genomes, we tested codon-based models of molecular evolution with codeml in PAML v4.9 (Yang 2007). We filtered the MSA to include only the 22 sauropsids (including representative squamates, crocodilians, birds, and turtles), stitched exons for each gene with Galaxy based on the Ensembl v86 AnoCar2.0 annotations and quality filtered for frame shifts and stop codons as above, and retained only gene alignments for which all species were present. To determine if genome-wide dN/dS in protein-coding genes is related to the background rate of molecular evolution in anoles, we concatenated three sets of 30 randomly selected genes and estimated dN/dS (the parameter ω) on all terminal branches of the sauropsid phylogeny using branch tests in codeml (model = 2, Nsites = 0). To identify specific genes undergoing positive selection in each anole lizard genome, we performed the branch-site test for each gene in codeml (model = 2, Nsites = 2). The gene phylogeny was inferred independently for each locus using PhyML v3.0 (Guindon et al. 2010). The custom pipeline used to perform these analyses is available at https://github.com/WilsonSayresLab/AlignmentProcessor. We performed independent tests with each anole lizard species as the foreground branch, comparing the likelihood of every branch-site model to a null model in which ω was fixed at 1, and the likelihood ratio test (LRT) statistic (2ΔLnl) to the chi-square distribution with a degree of freedom equal to 1. Genes with a P value < 0.01 after a Bonferroni adjustment for multiple testing and a ω > 1 were identified as positively selected. When analyzing sequence data including species which diverged over 200 Ma, saturation at synonymous sites is a potential concern; however, studies have shown that the branch-site test is quite robust to this issue. In fact, in this scenario false negatives are far more likely than false positives (Gharib and Robinson-Rechavi 2013), thus making our analysis more conservative. To link positively selected genes in each species to potential phenotypes, we examined statistically enriched GO categories as above, and tested for enrichment of mouse phenotypes using the gene IDs of the human orthologs in MamPhEA (Weng and Liao 2010).
In anoles, limb morphology is related to locomotor differences and ecological niche adaptation among sympatric species (Losos 2009); therefore, we predict strong selective pressure on limb structure and function during anole lizard diversification will result in larger dN/dS values for limb-related genes. We selected 30 protein-coding genes annotated in GO processes for limb development (GO: 0060173), limb morphogenesis (GO: 0035108), and limb bud formation (GO: 0060174). Orthologous sequences for each gene were collected from human (Homo sapiens), mouse (Mus musculus), cow (Bos taurus), African elephant (Loxodonta africanus), American opossum (Monodelphis domesticus), western clawed frog (Xenopus tropicalis), chicken (Gallus gallus), wild turkey (Meleagris gallopavo), mallard duck (Anas platyrhyncos), zebra finch (Taenopygia guttata), medium ground finch (Geospiza fortis), budgerigar (Melopsittacus undulatus), painted turtle (Chrysemys picta), Chinese softshell turtle (Pelodiscus sinensis), American alligator (Alligator mississippiensis), Burmese python (Python bivittatus), and green anole (Anolis carolinensis). For the three de novo anole lizard genomes, we generated predicted transcript sequence using genomic scaffold sequences aligning to A. carolinensis exons by BLASTn and genome annotation data. Homologous gene sequences were aligned by codon using MUSCLE (Edgar 2004) and edited by eye to remove ambiguously aligning regions and stop codons, with care taken to maintain the integrity of the amino acid sequence by frequent BLAST searches to NCBI. We analyzed the codon alignments of limb genes using branch tests in codeml. For each gene, we assumed the species tree topology and compared three models which allow ω to vary along different branches in the phylogeny: 1) the null model (M0; model = 0, Nsites = 0) in which a single ω is estimated for all branches on the phylogeny, 2) an alternative model (M2a; model = 2, Nsites = 0) that allows one ω for the Anolis branches and another for all other branches, and 3) a second alternative model (M2b; model = 2, Nsites = 0) that allows ω to vary among Anolis, python, alligator, turtles, birds, eutherian mammals, and all remaining branches. We compared each alternative model to the null model by comparing the LRT statistic to the chi square distribution given the degrees of freedom determined by the difference in the number of model parameters. Where the null was rejected, we then compared the two alternative models using the LRT. To compare relative selective pressure on limb development genes among taxa, we observed the plus or minus differential in ω between Anolis and eutherian mammals and birds, respectively.
Conservation of Regulatory Elements
Much of the phenotypic diversity within and between species is due to cis-regulatory sequences that control gene expression, including enhancers (Wittkopp and Kalay 2012). In order to determine if mutations in enhancer elements affected evolutionary change during the evolution of anole lizards, we annotated mouse enhancer regions identified from brain, liver, limb, and heart tissues and compared their conservation in A. carolinensis, A. auratus, A. apletophallus, and A. frenatus (supplementary methods, Supplementary Material online).
Results
Genome Assemblies and Gene Annotations
We carried out Illumina platform-based whole genome sequencing of three anole species representing both Dactyloa and Norops clades, using combinations of high coverage paired-end sequencing plus “jumping” mate pair libraries of varying sizes (table 1). We sequenced the Dactyloa group species A. frenatus at 116× coverage, and the two Norops group species, A. auratus and A. apletophallus, at 141× coverage and 56× coverage, respectively. The lower coverage for A. apletophallus is attributable to technical failures in the sequencing of some of the ∼300-bp insert libraries, the downstream effects of which we attempted to ameliorate by adding additional coverage using ∼500-bp insert libraries. Anolis frenatus and A. auratus contigs were assembled with ABySS v1.3.7 and scaffolded with SOAPdenovo2; A. apletophallus contigs and scaffolds were assembled using ABySS (supplementary tables 3–5, Supplementary Material online). Scaffold N50 statistics calculated were 37, 49, and 9.5 kb for A. frenatus, A. auratus, and A. apletophallus, respectively. All three de novo draft assemblies were ∼2 Gb in length, comparable in size to the estimated 2.2-Gb A. carolinensis genome and 1.78-Gb AnoCar2.0 genome assembly (Alföldi et al. 2011). Genome assembly statistics are summarized in table 2.
Table 1.
Whole Genome Sequencing Data Obtained from Three Anole Lizard Species
| Libraries |
Anolis frenatus |
Anolis auratus |
Anolis apletophallus |
|||
|---|---|---|---|---|---|---|
| Total Data (Mb) | Coverage | Total Data (Mb) | Coverage | Total Data (Mb) | Coverage | |
| 200-bp paired-end | 1,400 | 70× | 1,058 | 43× | 359 | 17× |
| 300-bp paired-end | 502 | 22× | 1,454 | 63× | 328 | 11× |
| 500-bp paired-end | – | – | – | – | 154 | 7× |
| 1-kb paired-end | 94 | 9× | 515 | 23× | 244 | 11× |
| 3-kb mate pair | 628 | 11× | 452 | 8× | 404 | 7× |
| 5-kb mate pair | 90 | 4× | 81 | 4× | 85 | 4× |
| Total | 2,714 | 116× | 3,560 | 141× | 1,574 | 56× |
Table 2.
Descriptive Statistics for Four Anole Lizard Genome Assemblies Included in This Study
|
Anolis carolinensis (AnoCar2.0) |
Anolis frenatus (Afre1.0) |
Anolis auratus (Aaur1.0) |
Anolis apletophallus (Aapl1.0) |
|||||
|---|---|---|---|---|---|---|---|---|
| Contigs | Scaffolds | Contigs | Scaffolds | Contigs | Scaffolds | Contigs | Scaffolds | |
| N50 (bp) | 79,867 | 150,641,573 | 23,240 | 36,910 | 19,858 | 48,995 | 2,534 | 9,520 |
| L50 (Number) | 6,217 | 5 | 21,942 | 14,499 | 26,569 | 11,107 | 206,073 | 53,667 |
| Longest (bp) | 582,046 | 263,920,458 | 448,330 | 649,467 | 304,182 | 563,800 | 110,998 | 217,008 |
| Total length (Gb) | 1.70 | 1.79 | 1.92 | 2.03 | 1.94 | 2.02 | 1.89 | 2.18 |
| Number ≥1 kb | 41,987 | 6,457 | 176,895 | 649,467 | 230,878 | 141,345 | 659,833 | 454,194 |
| Number ≥100 kb | 4,247 | 852 | 809 | 2,134 | 392 | 3,038 | 1 | 103 |
| Percent gaps | 5% | 6% | 4% | 14% | ||||
| BUSCOs | C: 73%[D: 1.4%], F: 15%, M: 12% | C: 50%[D: 0.7%], F: 20%, M: 29% | C: 68%,[D: 1.1%], F: 16%, M: 15% | C: 28%[D: 2.1%], F: 19%, M: 53% | ||||
| Number of annotated genes | 22,962 | 19,923 | 19,838 | 12,816 | ||||
| Percent repeats | 33% | 37% | 39% | 29% | ||||
Note.—BUSCO, Benchmarking Single Copy Orthologs; C, Complete; D, Duplicated; F, Fragmented; M, Missing.
The four anole genome assemblies differed in terms of estimated gene content, with the assemblies of A. frenatus and A. auratus approaching similar completeness to that of A. carolinensis (table 2 and fig. 1A). In the A. frenatus assembly, 86% of core eukaryotic genes (CEGs) were at least partially represented, whereas A. auratus had 97% at least partially represented CEGs; and both are similar to A. carolinensis (AnoCar2.0), which contains 91% of CEGs. In the case of A. apletophallus, 65% of CEGs were represented. BUSCO analyses estimate slightly lower gene representation than CEGMA across all anole genomes, including 71% for A. frenatus, 85% for A. auratus, and 47% for A. apletophallus, compared with 88% BUSCO representation in A. carolinensis. The relatively low estimated gene content in the A. apletophallus assembly likely reflects high fragmentation due to the lower sequencing coverage and high heterozygosity in the sequenced A. apletophallus specimen. Indeed, analysis of the kmer frequency distribution generated from the A. apletophallus Illumina reads was highly bimodal (supplementary fig. 1, Supplementary Material online) and suggestive of a high proportion of heterozygous sites, which can hinder genome assembly efforts (Kajitani et al. 2014). Analysis of the distribution of GC content across the three de novo anole lizard genomes reveals similar distributions which lie within the peaks of both A. carolinensis and chicken (Gallus gallus, fig. 1B), albeit with potentially more GC-bias in A. frenatus.
Fig. 1.

—(A) Proportion of conserved orthologs in either complete or fragmented form detected in four anole lizard genomes. Proportions are reported out of 3,023 BUSCO (Benchmarking Universal Single Copy Orthologs) and 248 CEGMA (Conserved Eukaryotic Gene Mapping Approach) orthologs. (B) Distribution of GC content in the genomes of four anole lizards and chicken, calculated in 5-kb windows.
Using both homology-based and ab initio methods for the final gene annotations for each de novo anole lizard assembly, we approached the number of predicted protein-coding genes in the A. carolinensis genome (22,962 genes predicted; Eckalbar et al. 2013, table 2). Large proportions of annotated genes consist of BLASTp matches to the SwissProt-UniProtKB database and contain known protein domains; thus, all three de novo anole genomes presented here contain genes with clear orthology in other species. The A. auratus assembly contains 19,838 annotated protein-coding genes, with 87% (17,210 genes) having significant BLASTp matches, and 88% (17,394 genes) with known protein domains. We annotated 19,923 protein-coding genes in the A. frenatus assembly; 89% (17,771 genes) of these contain a significant BLASTp match and 92% (18,353 genes) contain protein domains. Meanwhile, we were able to annotate 12,816 genes for A. apletophallus; 82% of these produced both significant BLASTp hits (10,483 genes) and protein domains (10,534 genes).
Repetitive elements make up similar proportions of each sequenced Anolis lizard genome (A. carolinensis = 33%, A. frenatus = 37%, A. auratus = 39%, A. apletophallus = 29%; supplementary tables 6–9, Supplementary Material online). We found evidence for the differential accumulation and loss of transposable elements across anole lizard genomes, such as R2 non-LTR retrotransposons which occupy between 0.92% and 1.37% of the genomes of A. carolinensis, A. auratus, and A. apletophallus genomes yet are scarcely present in A. frenatus at 0.04%. Other differences lie in the abundance of SINEs, which are three times more abundant in A. carolinensis than in A. frenatus. The starkest difference between the repetitive landscapes of the four Anolis genomes is the evidence of recent activity coupled with the loss of ancient elements in A. carolinensis, which stands out in comparison with the other three assemblies (supplementary fig. 2, Supplementary Material online). For instance, the divergence profile of interspersed repeats in A. carolinensis is characterized by a relative lack of transposable elements beyond 5–10% K2P divergence from their family consensus and is indicative of their recent insertion in the genome. In contrast, the A. auratus, A. apletophallus, and A. frenatus assemblies harbor a greater proportion of their elements far beyond this level of divergence (i.e., up to ∼45% K2P) and contain, for example, the signatures of ancient L1 and L2 retrotransposon expansions. The lack of SINEs found at <5% K2P divergence in A. frenatus, coupled with the low number of copies we detected in the genome, suggest this element family may be near extinction in the Dactyloan lineage of anoles.
Phylogenetic Results and Support for a Cenozoic Origin of Anoles
We reconstructed the ML phylogeny of 31 sarcopterygian vertebrates, including four anole lizards, using a DNA alignment of 4 D sites that contained 763,131 distinct patterns with 5.17% gaps or undetermined characters. All branches of the highest scoring ML tree had full support from the bootstrap replicates, and the topology was highly congruent with other phylogenomic studies of amniotes (fig. 2 and supplementary fig. 3 and table 10, Supplementary Material online). This includes several previously controversial nodes in the amniote phylogeny, most notably a monophyletic Atlantogenata (including elephant and armadillo) in the mammalian subtree (Tarver et al. 2016) and full support for a turtle-archosaur clade (Archelosauria) (Crawford et al. 2012, 2015; Green et al. 2014). Within the included squamates, the gecko forms the basal lineage, snakes are monophyletic and the four anole lizards and Pogona support a monophyletic Iguania, consistent with several studies (Wiens et al. 2012; Pyron et al. 2013). We obtained full support for the Dactyloan A. frenatus belonging to the basal lineage of anoles, followed by the divergence of A. carolinensis and monophyletic Norops species A. auratus and A. apletophallus, consistent with other phylogenetic studies of anoles (Guyer and Savage 1986; Nicholson et al. 2012; Del Rosario Castañeda and De Queiroz 2013; Prates et al. 2015).
Fig. 2.

—Phylogeny of 31 vertebrates from 763,131 4-fold degenerate sites reconstructed using maximum likelihood in RAxML v8.2.3 (Stamatakis 2014). All branches received full support from 500 bootstrap replicates. Representative images of species are not drawn to scale.
The fossil-based node constraints we implemented in r8s passed all gradient checks, and our estimates for the origin of amniotes at 303 Ma, the origin of sauropsids at 278 Ma, and the origin of placental mammals at 99 Ma are consistent with previously published estimates (Donoghue and Benton 2007; supplementary table 10, Supplementary Material online). Regarding the lineage leading to squamates including anoles, we estimated that the gecko lineage split from other squamates 175 Ma, and the TMRCA for iguanian lizards was 136 Myr. The TMRCA of the anoles was estimated to be 45 Myr, suggesting a Cenozoic origin, specifically during the Eocene which lasted between 56 and 34 Ma. The TMRCA of Anolis carolinensis, which is native to southeastern North America and originated from a Cuban radiation (Glor et al. 2005), and the Central American A. auratus and A. apletophallus was estimated to be 35 Myr, suggesting that the recolonization of the mainland by Caribbean anoles of the Norops lineage (Nicholson et al. 2005) occurred after the late Eocene. The r8s analysis using fewer constraints resulted in some slightly different node age estimates, particularly in mammals; however the TMRCA for anoles did not differ significantly. We also used a read-mapping approach to reconstruct the mitochondrial genomes of A. frenatus and A. auratus, obtained orthologous mitogenomic data from iguanian lizards available on NCBI (supplementary table 11, Supplementary Material Online), and used an internal fossil calibration to estimate divergence times with a partitioned data set using BEAST v1.8 (Drummond et al. 2012) (supplementary methods, Supplementary Material Online). This mitochondrial analysis was done prior to obtaining the sequence data for A. apletophallus and thus this species was not included. The results from the BEAST analysis were consistent with those from the 4D site analysis, placing the 95% highest posterior density (HPD) of the TMRCA for iguanian lizards between 195 and 113 Myr, the TMRCA of A. frenatus and A. carolinensis between 67 and 41 Myr (mean 53 Myr), and the TMRCA of A. carolinensis and A. auratus between 52 and 27 Myr (mean 40 Myr) (supplementary fig. 4, Supplementary Material Online). Thus, both 4 D sites and mitogenomic data support a hypothesis in which anole lizard evolution occurred within the Cenozoic Era, and that the Norops colonization of Central and South America began after the late Eocene.
Accelerated Molecular Evolution in Anoles
A comparison of the branch lengths of the phylogeny in figure 2 suggests rapid evolution at 4 D sites along the squamate branches, including the ancestral anole branch, as well as the common ancestor branches of birds and mammals, respectively. Using the PL method, estimated rates of neutral divergence at 4 D sites varied greatly across amniotes, and differed significantly among phylogenetic groups (fig. 3A). The optimal value for the smoothing parameter was 1, suggesting significant deviation from the molecular clock. We observed that, among the sauropsids included in this study, anoles and snakes have diverged at the fastest evolutionary rates in terms of DNA substitutions per site per million years. Consistent with previous studies, we estimated that the turtle lineage evolved at a far slower rate (Shaffer et al. 2013; Wang et al. 2013), as has the crocodilian lineage (Green et al. 2014). We also found that birds have departed significantly from crocodilians in terms of DNA substitution rates at 4 D sites since the split from their shared archosaurian ancestor ∼247 Ma. Finally, DNA substitution rates at 4 D sites in mammals surpass those of all other amniotes.
Fig. 3.
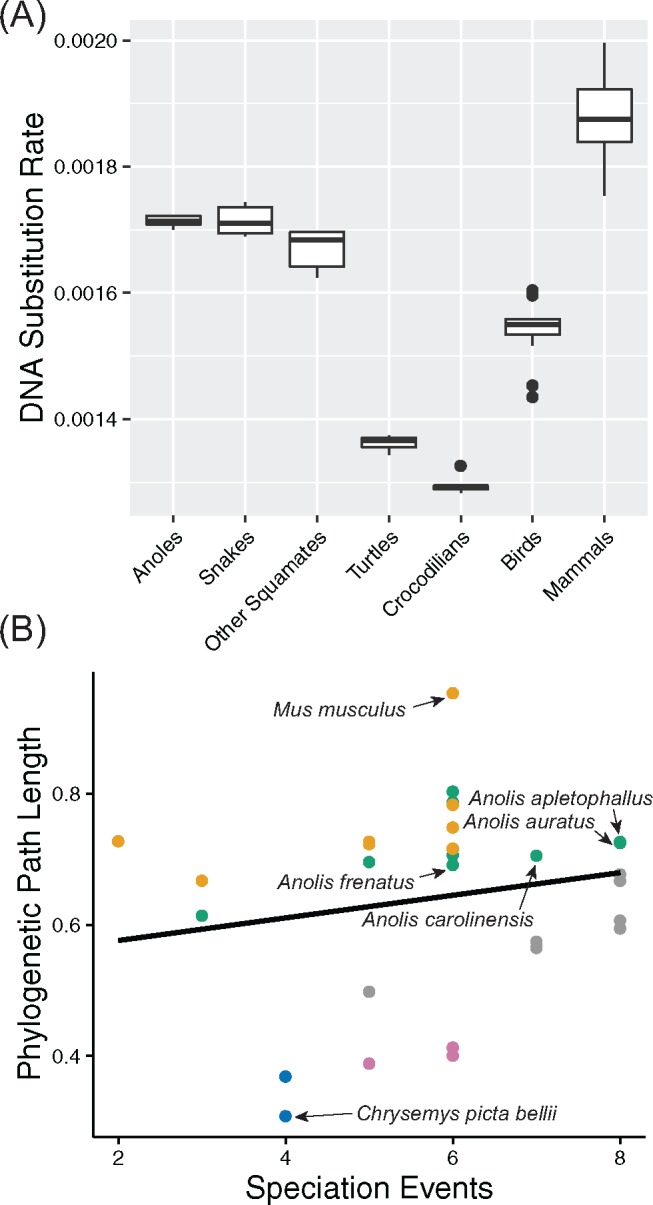
—(A) Comparison of the estimated DNA substitution rates (in terms of substitutions per site per million years) on terminal and internal branches of the amniote phylogeny. (B) Scatterplot showing the positive relationship (R2 = 0.17, P = 0.02) between the total phylogenetic path length (i.e., the sum of substitutions per site from the root to the terminal taxon) and the number of speciation events (i.e., the number of nodes leading to the terminal taxon). Values for the four anole lizards studied here and two outlier taxa (house mouse, Mus musculus and western painted turtle, Chrysemys picta bellii) are shown for comparison. Color legend is as follows: mammals = orange, squamates = green, turtles = blue, crocodilians = violet, birds = gray.
We found support for punctuated evolution across amniotes at 4 D sites, with the slope of the regression between path lengths and speciation events significantly >0 (R2 = 0.171, P < 0.02, 95% HPD = 0.170–0.174, fig. 3B). In addition, 26% of evolution across the amniote phylogeny was attributed to speciation events, with 17% of deviation from the molecular clock caused by punctuational evolution. We found no evidence for the node-density artifact. Consistent with the PL analysis, we estimated from the regression that the turtle, crocodilian, and avian lineages have evolved at comparatively much slower rates that are below the trend line for punctuated evolution. In contrast, mammals and squamates were above this trend line. Convergence was reached in all MCMC analyses, with ESS for all parameters ≫ 200. The regression model accurately predicted the estimated path lengths for A. frenatus (median 0.69 substitutions per site both predicted and observed, 95% HPD 0.50–0.90), A. carolinensis (median 0.72 predicted vs. 0.71 observed, 95% HPD 0.53–0.93), A. auratus (median 0.75 predicted vs. 0.73 observed, 95% HPD 0.53–0.96), and A. apletophallus (median 0.76 predicted vs. 0.73 observed, 95% HPD 0.57–0.99). These results suggest that rapid molecular evolution in anoles is linked to rapid speciation rates during lineage diversification.
Pervasive Conservation at the DNA Level for Functional Elements in Anoles
We used CONACC scores from phyloP to assess departures from neutrality in CDS, 5′-UTR, 3′-UTR, introns, intergenic regions, and AR, first across all branches of the vertebrate phylogeny and then in subtree analyses of anoles and snakes (fig. 4). The distribution of CONACC scores showed clear differences by genomic feature in the all branches test. For instance, CDS sequences were by far the most conserved of the analyzed functional elements, with the highest levels of acceleration in AR, consistent with expectations that evolution in coding regions is constrained with respect to nonfunctional repetitive regions. In both subtree analyses, CONACC scores for AR were significantly different from other annotation features (Mann–Whitney U-test, P = 0). CONACC scores for the anole subtree revealed conservation for several functional annotation features with respect to AR, suggesting that substitution rates in these genomic regions have diverged in the anole clade with respect to the rest of the vertebrate family tree and in the direction of stronger purifying selection. In contrast, the snakes subtree analysis resulted in higher conservation scores for AR and a shift toward more accelerated scores for intergenic regions and introns. These results suggest that the evolution of each clade was accompanied by lineage-specific changes in substitution rates in functional regions, particularly stronger conservation at the DNA level for anoles.
Fig. 4.

—Distributions of conservation-acceleration (CONACC) scores for three analyses using phyloP (Pollard et al. 2010): all branches analysis including 31 vertebrates, anoles subtree analysis, and snakes subtree analysis. CDS, protein coding sequence; UTR5, 5′-untranslated region; UTR3, 3′-untranslated region;, AR, ancestral repeats.
We detected 3,830 genes overlapping accelerated regions in the anole subtree analysis, compared with 5,247 genes in the snake subtree analysis. Of the 1,393 genes overlapping accelerated regions that are unique to anoles, 1,229 IDs were successfully mapped, resulting in enrichment of several GO categories. The category with the highest fold enrichment was diencephalon development (GO: 0021536, 4.37-fold enrichment, P = 4.26 × 10−2), which was represented by 14 genes (vs. 3.2 expected), followed by endocrine system development (GO: 0035270, 3.69-fold enrichment, P = 4.20 × 10−2), which was represented by 17 genes (vs. 4.60 expected). We detected 2,810 genes overlapping accelerated regions unique to snakes; GO terms with the highest fold enrichment in snakes were artery morphogenesis (GO: 0048844, 3-fold enrichment, 27 observed vs. 9 expected, P = 5.78 × 10−3) and artery development (GO: 0035904, 3-fold enrichment, 23 observed vs. 7.8 expected, P = 4.51 × 10−2).
Accelerated Evolution and Positive Selection Acting on Protein-Coding Genes during Anolis Diversification
We computed dN/dS through pairwise comparisons of 10,991, 13,783, and 12,246 filtered orthologous genes between A. carolinensis and A. frenatus, A. auratus, and A. apletophallus, respectively. Overall, we found high levels of protein-coding sequence conservation, with 88–93% of pairwise dN/dS estimates for genes in each comparison <0.5 (supplementary tables 12–14, Supplementary Material online), suggesting that the evolution for a majority of genes can be characterized by strong purifying selection (supplementary fig. 5, Supplementary Material online). Pairwise dN/dS was >1 for 53 genes in the A. carolinensis–A. frenatus comparison, including genes associated with specific GO terms for biological processes such as locomotion (GO: 0040011), cellular response to insulin-like growth factor stimulus (GO: 1990314), execution phase of apoptosis (GO: 0097194), and regulation of reactive oxygen species (GO: 1903426), possibly reflecting adaptive differences in adult body size, metabolic rate, and locomotor performance between the two species. The 30 accelerated genes in the A. carolinensis–A. auratus comparison were associated with more general GO terms such as metabolic process (GO: 0008152) and methylation (GO: 0032259). Finally, the A. carolinensis–A. apletophallus comparison yielded 42 accelerated genes associated with cell adhesion (GO: 0007155), circadian rhythm (GO: 0007623), fertilization (GO: 0009566), reproductive process (GO: 0022414), response to gamma radiation (GO: 001032), and urate homeostasis (GO: 1903118).
Out of all the pairwise-compared orthologs, putative positively selected genes with a pairwise dN/dS significantly >1 after a Bonferroni correction for multiple testing can highlight the molecular basis of adaptations unique to each species (supplementary table 15, Supplementary Material online). In A. apletophallus, these genes included GO annotations such as sensory perception of sound, and fertilization (sptbn4); muscle contraction (tln1); response to oxidative stress (dgkk); DNA methylation (tet3); and signal transduction and protein transport (tumor suppressor candidate rhbdb2). The only gene with a pairwise dN/dS consistent with positive selection in A. auratus was myo18a, which is involved in metabolic processes and negative regulation of apoptosis. Lastly, genes with a significantly elevated pairwise dN/dS in A. frenatus included GO terms for cellular trafficking (cog4) and calcium homeostasis (slc9a6).
Pairwise dN/dS for genes belonging to GO terms expected a priori to be relevant to Anolis adaptive radiations were highly conserved and consistent with strong purifying selection (fig. 5): For instance, the GO categories for pigmentation (GO: 0043473), somitogenesis (GO: 0001756), and visual perception (GO: 0007601) each consisted of genes with pairwise dN/dS < 1 in all species comparisons. However, the distributions of pairwise dN/dS for each a priori GO category were significantly different (Kruskal–Wallis rank sum test), suggesting different levels of functional constraint, and within each pairwise species comparison the mean dN/dS was highest for perception of smell (GO: 0007608), followed by either reproduction (GO: 0000003) or limb development (GO: 0060173). These three categories include genes with pairwise dN/dS > 1, and suggest accelerated evolution in pathways controlling ecologically relevant traits. For instance, one, 10, and 11 genes have undergone accelerated evolution in A. apletophallus, A. frenatus, and A. auratus, respectively, including the spermatogenesis-associated gene ddx25 in A. frenatus. This is consistent with an analysis that concluded reproductive genes have evolved more rapidly in the anole lineage than in other vertebrates (Grassa and Kulathinal 2011). Another outlier with high pairwise dN/dS was the gene cnga4 involved in perception of smell in A. apletophallus. Finally, the analysis of limb development genes in the pairwise comparison with A. frenatus revealed dN/dS > 1 in hoxa11 and hoxd13 (see Discussion).
Fig. 5.

—Distribution of dN/dS calculated in pairwise comparisons between the newly sequenced anole genomes and A. carolinensis for genes in adaptive radiation-relevant GO categories.
After filtering for quality and complete taxon sampling, we obtained 3,525 orthologous genes for 22 sauropsids from the MULTIZ alignment. The dN/dS estimates from the branch tests on randomized concatenated genes did not reveal a higher genome-wide dN/dS in anoles than the other species (Kruskal–Wallis rank sum test, supplementary fig. 6, Supplementary Material online), consistent with both the pairwise dN/dS and the CONACC results and suggesting widespread conservation in protein-coding genes across anole lizard evolution. Nonetheless, using the branch-site tests we found ample evidence for genes under positive selection in each species that are linked to relevant phenotypes important in anole lizard evolution. For instance, 19 genes were consistent with positive selection according to the branch-site test for A. frenatus. These include plxna4, which controls cranial nerve morphogenesis and chemorepulsion of branchiomotor axons, affecting the pharyngeal arches and the hyoid, which is the basis of the iguanian lizard dewlap (Losos 2009). Of the positively selected genes detected in the A. auratus genome, cyp26a1 controls retinoic acid metabolism necessary for inducing Hox gene expression and anterior–posterior axis specification and is associated with an absent tail phenotype in the mouse—consistent with the divergent tail morphology found in the species (Irschick et al. 1997). Twenty-seven genes were found to be consistent with positive selection according to the branch-site test for A. apletophallus, including hoxb6, which is involved in embryonic skeletal morphogenesis and anterior–posterior pattern specification. In addition, we estimate that fzd5 in A. frenatus and ror1 in both A. frenatus and A. auratus evolved under positive selection. These genes affect beta-Catenin-dependent Wnt signaling, suggesting episodic positive selection acting on different parts of the Wnt signaling pathway during the evolution of anole lizards.
Evolution of Limb Development Genes and Anole Limb Adaptations
We found 12 limb development genes for which anoles have a significantly higher dN/dS than other amniotes (supplementary tables 16 and 17, Supplementary Material online). When dN/dS values for other amniote lineages are taken into account, the anole branches have a higher dN/dS for most of the examined limb genes compared with eutherian mammals (64%) and birds (68%) (fig. 6A and supplementary table 18, Supplementary Material online). The 12 genes with higher dN/dS in anoles play key roles in limb development of vertebrates (see gene expression diagrams from chick homologues shown in fig. 6B). For instance, hoxa10, hoxd11, and hoxd12 displayed significantly higher dN/dS in anoles; Hox genes specify limb element identity along both the proximal–distal and cranial–caudal axes (Zuniga 2015). Interactions between FGF8 expression from the apical ectodermal ridge (AER) and SHH in the caudal zone of polarizing activity (ZPA) lead to prepatterning of the limb mesenchyme and later distal limb outgrowth; hand2, grem1, bmp4, and ptch1 genes are members of this regulatory network (Zuniga 2015). The en1 gene is expressed in the ventral limb bud and is involved in specification of the dorsal–ventral axis. Finally, dN/dS was higher in anoles for tbx5, which is required to specify forelimb identity, for the transcription factor pitx1 that is required for the initiation of hindlimb outgrowth, and for zinc finger protein sall4 that is required for cranial–caudal specification, particularly in the hindlimb (Infante et al. 2013; Akiyama et al. 2015; Zuniga 2015).
Fig. 6.

—(A) Barplot shows the differential in the estimated dN/dS (the parameter ω) for Anolis when compared with birds and eutherian mammals, respectively, from a codon model incorporating dN/dS values for the major clades of amniotes (M2b). Asterisks indicate genes for which M2b is favored over both M0 (the null hypothesis of a single ω for the entire phylogeny) and M2a (two ω model with Anolis branches as foreground); cross indicates M2a is favored over M0 and M2b using the likelihood ratio test (LRT). (B) Representations of expression pattern of limb development genes in chick embryonic limb bud, based on the Gallus Expression In Situ Hybridization Analysis (GEISHA) database (geisha.arizona.edu/geisha). (C) Replicates of Anolis auratus, A. apletophallus, A. carolinensis, and A. frenatus. Limb elements from the proximal stylopod (humerus or femur), middle zeugopod (radius + ulna or tibia + fibula), and distal autopod (carpus + metacarpal IV + digit IV phalanges or tarsus + metatarsal IV + digit IV phalanges) were measured in forelimb and hindlimb osteological preparations, respectively. Osteological preparations are shown to scale (2-mm bar). Additional details are in Supplementary Material online.
The morphometric analyses demonstrated that, among the four anole lizard species analyzed, A. frenatus was notable in having an elongated forelimb stylopod and zeugopod in relation to its body size (fig. 6C and supplementary fig. 7, Supplementary Material online). In addition, compared with A. carolinensis, the hindlimbs of A. apletophallus, A. auratus, and A. frenatus all exhibit elongation of the stylopod, zeugopod, and autopod. Digit 4 is especially elongated in the hindlimbs of all four species. Altogether, the morphological data are consistent with the molecular evolutionary results, suggesting that selective pressure acting on limb phenotypes has driven functional changes in genes specifying the proximal–distal axis and the hindlimb during anole lizard diversification.
Discussion
Here, we report a study of the genomic basis of adaptation during the evolution of anole lizards using complete genome sequences of multiple species. Our goals were: 1) to compare rates of genomic divergence in anoles to other amniotes, 2) to understand patterns of sequence conservation in functionally annotated features in anole lizard genomes, and 3) to identify putative adaptively evolving genomic regions that contributed to the well-studied diversification of anoles. We found that the three de novo anole lizard genomes differ in terms of scaffold contiguity and expected gene content, and contain largely similar transposable element profiles (although with some differences—see below). We estimated a ∼50-Myr divergence time for anoles and a background rate of neutral molecular evolution that is greater than many other sauropsid groups, with strong conservation in functionally annotated regions compared with other vertebrates. Finally, although the vast majority of protein-coding genes are highly conserved in anole lizard genomes, we found evidence for accelerated evolution and positive selection in genes associated with forebrain development, sensory perception, reproduction, and morphogenesis. We discuss these findings below.
Our estimates of substitution rates at 4 D sites and the relative lack of conservation at ancestral repeats suggests that neutral molecular evolution in anoles has been faster than in many other sauropsid lineages, especially turtles and archosaurs. Anoles typically mate during their first year (Losos 2009), whereas the other sequenced squamates, and even more so the turtles and crocodilians, take much longer to reach sexual maturity (Tacutu et al. 2013). The results are therefore consistent with the negative relationship between generation time and rates of molecular evolution that has been found across reptiles (Bromham 2002) as well as in mammals (Wilson Sayres et al. 2011). However, in contrast to the rapid molecular evolution in neutral regions, we identified strong conservation in the anole genomes at functional genomic loci, based on the phyloP results for CDS, 5′-UTR, 3′-UTR and the pervasively low dN/dS values across protein-coding genes. Therefore, rates of molecular evolution in anoles are widely different between functional and nonfunctional regions of the genome, which can be explained by relatively strong purifying selection during anole lizard diversification. It may be that anoles have large effective population sizes, which are less prone to fixation of alleles by genetic drift and more efficient at purging deleterious alleles. The few estimates of effective population sizes available for anoles suggest they may number in the hundreds of thousands or millions (Tollis and Boissinot 2014; Manthey et al. 2016), which is considerably greater than what has been estimated for human populations, where deleterious alleles can reach high frequencies (Lohmueller et al. 2008). Although some studies have concluded more rapid turnover of deleterious retrotransposons in larger populations of A. carolinensis (Tollis and Boissinot 2013; Ruggiero et al. 2017), additional population genomic studies of anoles will reveal the degree to which the fate of other classes of mutation is controlled by purifying selection.
Although the global repeat profiles of each anole lizard genome are largely similar, there are some notable differences in their composition, particularly in the R2/R4 non-LTR retrotransposon clade, which is almost completely absent from A. frenatus. In addition, the lack of SINEs found at <5% K2P divergence in A. frenatus coupled with the small number of copies detected in the genome suggests this element family may be near extinction in Dactyloan anoles. The almost total lack of ancient repeat elements in A. carolinensis compared with A. frenatus, A. auratus, and A. apletophallus suggests that very different evolutionary forces may have shaped the repetitive landscapes of anole lizard genomes. One explanation for this may be differences in each species’ demographic history, which can have profound effects on genome structure (Lynch and Conery 2003). Another explanation for lineage-specific differences in transposable element diversity and abundance is an interaction between rates of transposition and DNA loss (Kapusta et al. 2017). More information about the recent evolutionary histories of A. frenatus, A. auratus, and A. apletophallus and comprehensive study of their repetitive DNA contents will be required to distinguish between which of these models of genomic repeat evolution better explain the relative diversity, abundance, and age of transposable elements across anole lizard genomes.
Despite the high degree of conservation across the genome in anoles, we were able to identify numerous examples of accelerated evolution in protein-coding regions, particularly those associated with forebrain development and hormonal regulation. Anoles as a group are known to have diverse reproductive display behaviors (Losos 2009) with sex-specific seasonal changes in hormonal responses (Kerver and Wade 2013), and three regions of the anole forebrain have been linked to reproductive display behaviors, including the preoptic region, amygdala, and hypothalamus (Wade 2012). In addition, we found evidence for positive selection on at least one gene related to the anole dewlap, plxna4 (see Results), which during embryogenesis is expressed in the rhombomere 4 neural crest migration group that contributes to the second pharyngeal arch (Lumb et al. 2017). This second arch component includes the hyoid apparatus that controls the dewlap (Bels 1990), which features prominently in anole lizard reproductive behavior (Wade 2012) and aids in species recognition and territorial displays (Losos 2009). Rapid evolution in genes controlling the development and regulation of these traits may underlie interspecific differences in sexual displays, as well as other behaviors that can drive prezygotic reproductive isolation and therefore sympatric species boundaries in anoles (Ng et al. 2013).
The functional and behavioral importance of limb lengths in anoles is also well understood, and sympatric species occupying different microhabitats often occupy vastly different areas of limb morphospace (Losos 2009). Morphometric analyses of the anole lizards carried out in this study revealed elongation of the forelimb stylopod and zeugopod in A. frenatus and elongation of all hindlimb elements in A. frenatus, A. apletophallus, and A. auratus. Fittingly, we compared substitution patterns in limb development (GO: 0060173) genes in the pairwise comparison between A. carolinensis and A. frenatus and found that hoxa11 and hoxd13, which specify limb elements along the proximal–distal and cranial–caudal axes, and tbx5, which specifies forelimb identity (Zuniga 2015), were outliers with pairwise dN/dS > 1. The outgrowth and specification of the limb buds are regulated by interactions between AER, ZPA, and limb mesenchyme involving SHH and FGF regulatory network genes hand2, grem1, bmp4, and ptch1 (Zuniga 2015), which have significantly higher dN/dS in anoles compared with other amniotes. The pitx1 and sall4 genes also have higher dN/dS in anoles and are required for hindlimb bud initial outgrowth and specification, respectively (Infante et al. 2013; Akiyama et al. 2015; Zuniga 2015). The morphometric data support elongation of the stylopod, zeugopod, and autopod in A. apletophallus, A. auratus, and A. frenatus hindlimbs compared with A. carolinensis, suggesting that adaptive evolution on phenotypes during Anolis diversification may be controlled by changes in limb development genes. Although the hindlimb gene pitx1 is rapidly evolving in anoles, we did not detect accelerated evolution in tbx4 nor hoxc10, even though the expression of both genes is regulated by pitx1 binding (Infante et al. 2013). Pitx1 has also been shown to be strongly associated with limb enhancers (Infante et al. 2013). We identified 756 brain, 112 liver, 109 heart, and 289 limb enhancers in A. carolinensis, which aligned to A. auratus, A. apletophallus, and A. frenatus with 88% through 99% identity (supplementary fig. 8 and table 19, Supplementary Material online). This high alignability across anoles of enhancers from four tissues mirrors the widespread conservation in protein coding genes, and suggests that regulatory changes to limb development are modulated by a few specific mutations in enhancer motifs.
By generating de novo annotated genomes for three anole species, A. apletophallus, A. auratus, and A. frenatus, we were able to carry out a comparative genomic analysis with the A. carolinensis reference that is novel in scope for squamates. We found that the rate of molecular evolution in anoles and snakes was very rapid relative to other vertebrates, especially turtles and archosaurs, which may be a factor contributing to the successful adaptive radiation of anoles across the Neotropics resulting in ∼400 extant species. Despite this rapid background rate of evolution, strong purifying selection in the anole lineage has kept the vast majority of protein-coding genes highly conserved, even relative to other vertebrates. However, we did identify evidence for accelerated evolution in genes involved in behavior, sensory perception, and reproduction, as well as in genes regulating limb development. Morphometric analyses of anole limbs corroborated these findings, highlighting the importance of genes involved with specification of limb outgrowth and hindlimb specification. Further investigations could help to elucidate the genomic bases of evolutionary adaptations in anole lizards and provide great insight for vertebrate evolution in general.
Supplementary Material
Supplementary data are available at Genome Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Glenn Markov and Peter Marting for their help in the field and Matthew J. Huentelman, John Cornelius, Shannon Gilpin, and Elise Kulik for technical assistance. We also thank Fiona McCarthy for helpful discussion. We thank April Allen and the Collaborative Sequencing Center at the Translational Genomics Research Institute for sequencing assistance. Support for I.M. and E.L. was provided by the School of Life Sciences Undergraduate Research (SOLUR) Program at Arizona State University. This work was supported by funding from a seed grant from Arizona State University (ASU) and the Smithsonian Tropical Research Institute and funding from the College of Liberal Arts and Sciences at ASU to K.K. Additional support to D.B.M. was provided by the National Science Foundation (CAREER IOS1149453). Computational analysis was supported by allocations from the Arizona State University Research Computing and startup from the School of Life Sciences and the Biodesign Institute to M.A.W.S.
Literature Cited
- Akiyama R, et al. 2015. Sall4-Gli3 system in early limb progenitors is essential for the development of limb skeletal elements. Proc Natl Acad Sci U S A. 112(16):5075–5080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alföldi J, et al. 2011. The genome of the green anole lizard and a comparative analysis with birds and mammals. Nature 477(7366):587–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bels VL. 1990. The mechanism of dewlap extension in Anolis carolinensis (Reptilia: iguanidae) with histological analysis of the hyoid apparatus. J Morphol. 206(2):225–244. [DOI] [PubMed] [Google Scholar]
- Benton MJ, et al. 2015. Constraints on the timescale of animal evolutionary history. Palaeontol Electron 18:1–106. [Google Scholar]
- Blanchette M, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 14(4):708–715.http://dx.doi.org/10.1101/gr.1933104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenberg D, Taylor J, Nekrutenko A, Galaxy Team. 2011. Making whole genome multiple alignments usable for biologists. Bioinformatics 27(17):2426–2428.http://dx.doi.org/10.1093/bioinformatics/btr398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W.. 2011. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27(4):578–579. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B.. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30(15):2114–2120.http://dx.doi.org/10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromham L. 2002. Molecular clocks in reptiles: life history influences rate of molecular evolution. Mol Biol Evol. 19(3):302–309.http://dx.doi.org/10.1093/oxfordjournals.molbev.a004083 [DOI] [PubMed] [Google Scholar]
- Bromham L, Woolfit M, Lee MSY, Rambaut A.. 2002. Testing the relationship between morphological and molecular rates of change along phylogenies. Evolution 56:1921–1930.http://dx.doi.org/10.1111/j.0014-3820.2002.tb00118.x [DOI] [PubMed] [Google Scholar]
- Castoe TA, et al. 2013. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc Natl Acad Sci U S A. 110(51):20645–20650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GO. 2015. Gene ontology consortium: going forward. Nucleic Acids Res. 43(D1):D1049–D1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford NG, et al. 2012. More than 1000 ultraconserved elements provide evidence that turtles are the sister group of archosaurs. Biol Lett. 8(5):783–786.http://dx.doi.org/10.1098/rsbl.2012.0331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford et al. 2015. A phylogenomic analysis of turtles. Mol Phylogenet Evol. 83(2015):250–257. [DOI] [PubMed] [Google Scholar]
- Davies TJ, Savolainen V.. 2006. Neutral theory, phylogenies, and the relationship between phenotypic change and evolutionary rates. Evolution 60(3):476–483. [PubMed] [Google Scholar]
- Del Rosario Castañeda M, De Queiroz K.. 2013. Phylogeny of the Dactyloa clade of Anolis lizards: new insights from combining morphological and molecular data. Bull Mus Comp Zool. 160(7):345–398. [Google Scholar]
- Donoghue PCJ, Benton MJ.. 2007. Rocks and clocks: calibrating the Tree of Life using fossils and molecules. Trends Ecol Evol. 22(8):424–431.http://dx.doi.org/10.1016/j.tree.2007.05.005 [DOI] [PubMed] [Google Scholar]
- Drummond AJ, Suchard MA, Xie D, Rambaut A.. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 29(8):1969–1973.http://dx.doi.org/10.1093/molbev/mss075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckalbar WL, et al. 2013. Genome reannotation of the lizard Anolis carolinensis based on 14 adult and embryonic deep transcriptomes. BMC Genomics 14:49.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5:113.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldredge N, Gould J.. 1972. Punctuated equilibria: an alternative to phyletic gradualism In: Schopf TJM, editor. Models of paleobiology. San Francisco: Freeman, Cooper & Co; p. 82–115. [Google Scholar]
- Fay JC, Wu CI.. 2003. Sequence divergence, functional constraint, and selection in protein evolution. Annu Rev Genomics Hum Genet. 4:213–235.http://dx.doi.org/10.1146/annurev.genom.4.020303.162528 [DOI] [PubMed] [Google Scholar]
- Fleishman LJ. 1988. The social behavior of Anolis auratus, a grass anole from Panama. J Herpetol. 22:13. [Google Scholar]
- Foote AD, et al. 2015. Convergent evolution of the genomes of marine mammals. Nat Genet. 47(3):272–275.http://dx.doi.org/10.1038/ng.3198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genome 10K Community of Scientists 2009. Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Hered. 100(6):659–674.http://dx.doi.org/10.1093/jhered/esp086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georges A, et al. 2015. High-coverage sequencing and annotated assembly of the genome of the Australian dragon lizard Pogona vitticeps. Gigascience 4:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharib WH, Robinson-Rechavi M.. 2013. The branch-site test of positive selection is surprisingly robust but lacks power under synonymous substitution saturation and variation in GC. Mol Biol Evol. 30(7):1675–1686.http://dx.doi.org/10.1093/molbev/mst062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert C, et al. 2014. Endogenous hepadnaviruses, bornaviruses and circoviruses in snakes. Proc R Soc B. 281(1791):20141122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glor RE, Losos JB, Larson A.. 2005. Out of Cuba: overwater dispersal and speciation among lizards in the Anolis carolinensis subgroup. Mol Ecol. 14(8):2419–2432.http://dx.doi.org/10.1111/j.1365-294X.2005.02550.x [DOI] [PubMed] [Google Scholar]
- Grassa CJ, Kulathinal RJ.. 2011. Elevated evolutionary rates among functionally diverged reproductive genes across deep vertebrate lineages. Int J Evol Biol. 2011:1–9.http://dx.doi.org/10.4061/2011/274975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RE, et al. 2014. Three crocodilian genomes reveal ancestral patterns of evolution among archosaurs. Science 346(6215):1254449.http://dx.doi.org/10.1126/science.1254449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, et al. 2010. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 59(3):307–321.http://dx.doi.org/10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Guyer C, Savage JM.. 1986. Cladistic relationships among anoles (Sauria: iguanidae). Syst Biol. 35(4):509–531. [Google Scholar]
- Harris RS. 2007. Improved pairwise alignment of genomic DNA [Ph.D. thesis]. The Pennsylvania State University.
- Hedges SB, Marin J, Suleski M, Paymer M, Kumar S.. 2015. Tree of life reveals clock-like speciation and diversification. Mol Biol Evol. 32(4):835–845.http://dx.doi.org/10.1093/molbev/msv037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt C, Yandell M.. 2011. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinformatics 12:491.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubisz MJ, Pollard KS, Siepel A.. 2011. PHAST and RPHAST: phylogenetic analysis with space/time models. Brief Bioinformatics 12(1):41–51.http://dx.doi.org/10.1093/bib/bbq072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Infante CR, Park S, Mihala AG, Kingsley DM, Menke DB.. 2013. Pitx1 broadly associates with limb enhancers and is enriched on hindlimb cis-regulatory elements. Dev Biol. 374(1):234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irschick DJ, Vitt LJ, Zani PA, Losos JB.. 1997. A comparison of evolutionary radiations in mainland and caribbean Anolis lizards. Ecology 78(7):2191–2203.http://dx.doi.org/10.1890/0012-9658(1997)078[2191:ACOERI]2.0.CO;2 [Google Scholar]
- Jarvis ED, et al. 2014. Whole-genome analyses resolve early branches in the tree of life of modern birds. Science 346(6215):1320–1331.http://dx.doi.org/10.1126/science.1253451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones P, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30(9):1236–1240.http://dx.doi.org/10.1093/bioinformatics/btu031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka J, et al. 2005. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110(1–4):462–467. [DOI] [PubMed] [Google Scholar]
- Kajitani R, et al. 2014. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 24(8):1384–1395.http://dx.doi.org/10.1101/gr.170720.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapusta A, Suh A, Feschotte C.. 2017. Dynamics of genome size evolution in birds and mammals. Proc Natl Acad Sci U S A. 114:E1460–E1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Baertsch R, Hinrichs A, Miller W, Haussler D.. 2003. Evolution’s cauldron: duplication, deletion, and rearrangement in the mouse and human genomes. Proc Natl Acad Sci U S A. 100(20):11484–11489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerver HN, Wade J.. 2013. Seasonal and sexual dimorphisms in expression of androgen receptor and its coactivators in brain and peripheral copulatory tissues of the green anole. Gen Comp Endocrinol. 193:56–67.http://dx.doi.org/10.1016/j.ygcen.2013.07.006 [DOI] [PubMed] [Google Scholar]
- Kim S, et al. 2016. Comparison of carnivore, omnivore, and herbivore mammalian genomes with a new leopard assembly. Genome 17(1):211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsella RJ, et al. 2011. Ensembl BioMarts: a hub for data retrieval across taxonomic space. Database (Oxford) 2011(0):bar030–bar030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler G, Sunyer J.. 2008. Two new species of anoles formerly referred to as Anolis limifrons (Squamata: polychrotidae). Herpetologica 64(1):92–108. [Google Scholar]
- Korf I. 2004. Gene finding in novel genomes. BMC Bioinformatics 5:59.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Subramanian S.. 2002. Mutation rates in mammalian genomes. Proc Natl Acad Sci U S A. 99(2):803–808.http://dx.doi.org/10.1073/pnas.022629899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanfear R, Welch JJ, Bromham L.. 2010. Watching the clock: studying variation in rates of molecular evolution between species. Trends Ecol Evol. 25(9):495–503.http://dx.doi.org/10.1016/j.tree.2010.06.007 [DOI] [PubMed] [Google Scholar]
- Lindblad-Toh K, et al. 2011. A high-resolution map of human evolutionary constraint using 29 mammals. Nature 478(7370):476–482.http://dx.doi.org/10.1038/nature10530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, et al. 2015. Gekko japonicus genome reveals evolution of adhesive toe pads and tail regeneration. Nat Commun. 6:10033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohmueller KE, et al. 2008. Proportionally more deleterious genetic variation in European than in African populations. Nature 451(7181):994–997.http://dx.doi.org/10.1038/nature06611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losos JB. 2009. Lizards in an evolutionary tree. Berkeley: University of California Press. [Google Scholar]
- Losos JB, Andrews RM, Sexton OJ, Schuler AL.. 1991. Behavior, ecology, and locomotor performance of the giant anole, Anolis frenatus. Caribb J Sci. 27:173–179. [Google Scholar]
- Losos JB, Miles DB.. 2002. Testing the hypothesis that a clade has adaptively radiated: iguanid lizard clades as a case study. Am Nat. 160(2):147–157. [DOI] [PubMed] [Google Scholar]
- Lumb R, Buckberry S, Secker G, Lawrence D, Schwarz Q. 2017. Transcriptome profiling reveals expression signatures of cranial neural crest cells arising from different axial levels. BMC Dev Biol. 17(5):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo R, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1(1):18.http://dx.doi.org/10.1186/2047-217X-1-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Conery JS.. 2003. The origins of genome complexity. Science 302(5649):1401–1404.http://dx.doi.org/10.1126/science.1089370 [DOI] [PubMed] [Google Scholar]
- Magoč T, Salzberg SL.. 2011. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27(21):2957–2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahler DL, Revell LJ, Glor RE, Losos JB.. 2010. Ecological opportunity and the rate of morphological evolution in the diversification of greater Antillean anoles. Evolution 64(9):2731–2745.http://dx.doi.org/10.1111/j.1558-5646.2010.01026.x [DOI] [PubMed] [Google Scholar]
- Manthey JD, Tollis M, Lemmon AR, Moriarty Lemmon E, Boissinot S.. 2016. Diversification in wild populations of the model organism Anolis carolinensis: a genome-wide phylogeographic investigation. Ecol Evol. 6(22):8115–8125.http://dx.doi.org/10.1002/ece3.2547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A, et al. 2015. The InterPro protein families database: the classification resource after 15 years. Nucleic Acids Res. 43(D1):D213–D221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing Consortium, et al. 2002. Initial sequencing and comparative analysis of the mouse genome. Nature 420:520–562.http://dx.doi.org/10.1038/nature01262 [DOI] [PubMed] [Google Scholar]
- Ng J, Landeen EL, Logsdon RM, Glor RE.. 2013. Correlation between Anolis lizard dewlap phenotype and environmental variation indicates adaptive divergence of a signal important to sexual selection and species recognition. Evolution 67(2):573–582.http://dx.doi.org/10.1111/j.1558-5646.2012.01795.x [DOI] [PubMed] [Google Scholar]
- Nicholson KE, Crother BI, Guyer C, Savage JM.. 2012. It is time for a new classification of anoles (Squamata: dactyloidae). Zootaxa. 3477:e59741. [Google Scholar]
- Nicholson KE, et al. 2005. Mainland colonization by island lizards. J Biogeogr. 32(6):929–938.http://dx.doi.org/10.1111/j.1365-2699.2004.01222.x [Google Scholar]
- Pagel M, Meade A, Barker D.. 2004. Bayesian estimation of ancestral character states on phylogenies. Syst Biol. 53(5):673–684.http://dx.doi.org/10.1080/10635150490522232 [DOI] [PubMed] [Google Scholar]
- Pagel M, Venditti C, Meade A.. 2006. Large punctuational contribution of speciation to evolutionary divergence at the molecular level. Science 314(5796):119–121.http://dx.doi.org/10.1126/science.1129647 [DOI] [PubMed] [Google Scholar]
- Parra G, Bradnam K, Ning Z, Keane T, Korf I.. 2009. Assessing the gene space in draft genomes. Nucleic Acids Res. 37(1):289–297.http://dx.doi.org/10.1093/nar/gkn916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineault KM, Wellik DM.. 2014. Hox genes and limb musculoskeletal development. Curr Osteoporos Rep. 12(4):420–427.http://dx.doi.org/10.1007/s11914-014-0241-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poe S. 2004. Phylogeny of anoles. Herpetol Monogr. 18:37–89.http://dx.doi.org/10.1655/0733-1347(2004)018[0037:POA]2.0.CO;2 [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A.. 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20(1):110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prates I, Rodrigues MT, Melo-Sampaio PR, Carnaval AC.. 2015. Phylogenetic relationships of Amazonian anole lizards (Dactyloa): taxonomic implications, new insights about phenotypic evolution and the timing of diversification. Mol Phylogenet Evol. 82:258–268. [DOI] [PubMed] [Google Scholar]
- Pyron RA, Burbrink FT, Wiens JJ.. 2013. A phylogeny and revised classification of Squamata, including 4161 species of lizards and snakes. BMC Evol Biol. 13:93.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM.. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26(6):841–842.http://dx.doi.org/10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2016. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing. [Google Scholar]
- Rabosky DL. 2012. Positive correlation between diversification rates and phenotypic evolvability can mimic punctuated equilibrium on molecular phylogenies. Evolution 66(8):2622–2627.http://dx.doi.org/10.1111/j.1558-5646.2012.01631.x [DOI] [PubMed] [Google Scholar]
- Rallis C, et al. 2003. Tbx5 is required for forelimb bud formation and continued outgrowth. J Embryol Exp Morphol. 130(12):2741–2751. [DOI] [PubMed] [Google Scholar]
- Ruggiero RP, Bourgeois Y, Boissinot S.. 2017. LINE insertion polymorphisms are abundant but at low frequencies across populations of Anolis carolinensis. Front Genet. 8:44.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson MJ. 2002. Estimating absolute rates of molecular evolution and divergence times: a penalized likelihood approach. Mol Biol Evol. 19(1):101–109.http://dx.doi.org/10.1093/oxfordjournals.molbev.a003974 [DOI] [PubMed] [Google Scholar]
- Sanderson MJ. 2003. r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19(2):301–302.http://dx.doi.org/10.1093/bioinformatics/19.2.301 [DOI] [PubMed] [Google Scholar]
- Shaffer HB, et al. 2013. The western painted turtle genome, a model for the evolution of extreme physiological adaptations in a slowly evolving lineage. Genome Biol. 14(3):R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM.. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31(19):3210–3212. [DOI] [PubMed] [Google Scholar]
- Simpson JT, et al. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res. 19(6):1117–1123.http://dx.doi.org/10.1101/gr.089532.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit AFA, Hubley RM, Green P. RepeatMasker Open-4.0 2013-2015. http://www.repeatmasker.org.
- Smit AFA, Hubley RM, Green P. RepeatModeler Open-1.0 2008-2015. http://www.repeatmasker.org.
- Song B, et al. 2015. A genome draft of the legless anguid lizard, Ophisaurus gracilis. Gigascience 4:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir ML, et al. 2016. The UCSC Genome Browser database: 2016 update. Nucleic Acids Res. 44(D1):D717–D725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30(9):1312–1313.http://dx.doi.org/10.1093/bioinformatics/btu033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacutu R, et al. 2013. Human Ageing Genomic Resources: integrated databases and tools for the biology and genetics of ageing. Nucleic Acids Res. 41(Database issue):D1027–D1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarver JE, et al. 2016. The interrelationships of placental mammals and the limits of phylogenetic inference. Genome Biol Evol. 8(2):330–344.http://dx.doi.org/10.1093/gbe/evv261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollis M, Boissinot S.. 2013. Lizards and LINEs: selection and demography affect the fate of L1 retrotransposons in the genome of the green anole (Anolis carolinensis). Genome Biol Evol. 5(9):1754–1768.http://dx.doi.org/10.1093/gbe/evt133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollis M, Boissinot S.. 2014. Genetic variation in the green anole lizard (Anolis carolinensis) reveals island refugia and a fragmented Florida during the quaternary. Genetica 142(1):59–72.http://dx.doi.org/10.1007/s10709-013-9754-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium 2015. UniProt: a hub for protein information. Nucleic Acids Res. 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venditti C, Meade A, Pagel M.. 2006. Detecting the node-density artifact in phylogeny reconstruction. Syst Biol. 55(4):637–643.http://dx.doi.org/10.1080/10635150600865567 [DOI] [PubMed] [Google Scholar]
- Vonk FJ, et al. 2013. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc Natl Acad Sci U S A. 110(51):20651–20656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade J. 2012. Sculpting reproductive circuits: relationships among hormones, morphology and behavior in anole lizards. Gen Comp Endocrinol. 176(3):456–460. [DOI] [PubMed] [Google Scholar]
- Wang D, Zhang Y, Zhang Z, Zhu J, Yu J.. 2010. KaKs_Calculator 2.0: a toolkit incorporating gamma-series methods and sliding window strategies. Genomics Proteomics Bioinformatics 8(1):77–80.http://dx.doi.org/10.1016/S1672-0229(10)60008-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, et al. 2013. The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan. Nat Genet. 45(6):701–706.http://dx.doi.org/10.1038/ng.2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster AJ, Payne RJH, Pagel M.. 2003. Molecular phylogenies link rates of evolution and speciation. Science 301(5632):478–478.http://dx.doi.org/10.1126/science.1083202 [DOI] [PubMed] [Google Scholar]
- Weng MP, Liao BY.. 2010. MamPhEA: a web tool for mammalian phenotype enrichment analysis. Bioinformatics 26(17):2212–2213.http://dx.doi.org/10.1093/bioinformatics/btq359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiens JJ, et al. 2012. Resolving the phylogeny of lizards and snakes (Squamata) with extensive sampling of genes and species. Biol Lett. 8(6):1043–1046.http://dx.doi.org/10.1098/rsbl.2012.0703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson Sayres MA, Venditti C, Pagel M, Makova KD.. 2011. Do variations in substitution rates and male mutation bias correlate with life-history traits? A study of 32 mammalian genomes. Evolution 65(10):2800–2815.http://dx.doi.org/10.1111/j.1558-5646.2011.01337.x [DOI] [PubMed] [Google Scholar]
- Wittkopp PJ, Kalay G.. 2012. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nature Rev Genet. 13(1):59–69.http://dx.doi.org/10.1038/nrg3095 [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24(8):1586–1591.http://dx.doi.org/10.1093/molbev/msm088 [DOI] [PubMed] [Google Scholar]
- Zhang G, et al. 2014. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346(6215):1311–1320.http://dx.doi.org/10.1126/science.1251385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga A. 2015. Next generation limb development and evolution: old questions, new perspectives. J Embryol Exp Morphol. 142(22):3810–3820. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.