

Abstract
Troponin I (TnI) is the inhibitory subunit of the troponin complex in the sarcomeric thin filament of striated muscle and plays a central role in the calcium regulation of contraction and relaxation. Vertebrate TnI has evolved into three isoforms encoded by three homologous genes: TNNI1 for slow skeletal muscle TnI, TNNI2 for fast skeletal muscle TnI and TNNI3 for cardiac TnI, which are expressed under muscle type-specific and developmental regulations. To summarize the current knowledge on the TnI isoform genes and products, this review focuses on the evolution, gene regulation, posttranslational modifications, and structure-function relationship of TnI isoform proteins. Their physiological and medical significances are also discussed.
Keywords: troponin I, cardiac and skeletal muscle, evolution, gene regulation, isoform, posttranslational modification
Graphical Abstract
1. Introduction
Troponin I (TnI) is the inhibitory subunit of the troponin complex in striated muscle (skeletal and cardiac muscles). The basic contractile machinery of striated muscles is the sarcomere that consists of overlapping myosin thick filaments and actin thin filaments. Contraction is produced by sliding between the thick and thin filaments, a process that is powered by actin-activated myosin ATPase and regulated by cytosolic Ca2+ via the troponin complex associated with the sarcomeric thin filament (Jin et al., 2008). Troponin I functions along with the other two subunits of troponin, troponin T (TnT) (Wei and Jin, 2011) and troponin C (TnC) (Li and Hwang, 2015), to govern muscle contraction and relaxation. To summarize the current knowledge on the TnI isoform genes and products, this review focuses on the evolution, gene regulation, posttranslational modifications, and structure-function relationship of TnI isoform proteins. Their physiological and medical significances are also discussed.
2. Evolution of TnI Isoform Genes
Three homologous genes have evolved in higher vertebrates, encoding the three muscle fiber type-specific isoforms of TnI (Hastings, 1997; Perry, 1999; Chong and Jin, 2009) (Table 1). In the human chromosomal genome, TNNI1 is located at 1q31.3, encoding the slow skeletal muscle isoform of TnI (ssTnI); TNNI2 is located at 11p15.5, encoding the fast skeletal muscle isoform of TnI (fsTnI); and TNNI3 is located at 19q13.4, encoding the cardiac isoform of TnI (cTnI). The three TnI isoform genes are structurally diverged, differentially expressed under fiber type-specific transcriptional control, and regulated during embryonic and postnatal development (Jin et al., 2008).
Table 1.
Human TnI isoform genes and tissue-specific expression
| Protein isoforms | Slow Skeletal Muscle TnI |
Fast Skeletal Muscle TnI |
Cardiac TnI |
|---|---|---|---|
| Gene | TNNI1 | TNNI2 | TNNI3 |
| Chromosomal Location | 1q31.3 | 11p15.5 | 19q13.4 |
| Number of Exons | 9 | 8 | 8 |
| Number of Amino Acids | 187 | 182 | 210 |
| Molecular Weight (kDa) | 21.7 | 21.3 | 24.0 |
| Isoelectric Point | 9.59 | 8.74 | 9.87 |
| Tissue Specificity | Slow-twitch fibers Embryonic heart |
Fast-twitch fibers | Adult heart |
A comparison of the amino acid sequences of rabbit cardiac, fast and slow skeletal muscle TnI isoforms suggested that the three TnI genes arose by gene duplication (Baldwin et al., 1985). Sequence analysis and co-evolutionary relationship with TnT isoforms established via an epitope similarity and conformational modulation study indicated that the original TnI gene was fast skeletal muscle TnI-like (Chong and Jin, 2009).
Diverged from the fsTnI gene lineage, the present-day ssTnI and cTnI sequences exhibit higher degree of similarity among the three isoforms. Their co-evolutionary relationship with the closely linked TnT isoform genes (Huang and Jin, 1999) indicates that they evolved from a slow skeletal muscle TnI-like common ancestor gene (Chong and Jin, 2009). Therefore, the first gene duplication event generated a slow skeletal muscle TnI-like gene that was further duplicated into the present-day ssTnI and cTnI genes.
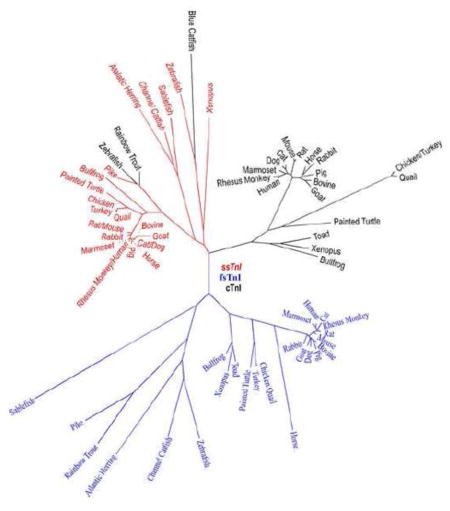
The direct evolutionary relationship between ssTnI and cTnI genes is supported by the facts that a) cTnI in lower vertebrates, such as zebra fish, aligned with the ssTnI monophyletic group (Fu et al., 2009) (Fig. 1), b) the unique N-terminal extension of mammalian and avian cTnI, which is absent in fast and slow skeletal muscle isoforms of TnI, is an adult heart specific structure added during evolution as it is absent in fish cTnI (Fig. 2), and c) ssTnI is expressed as the sole TnI isoform in embryonic hearts (Jin, 1996).
Figure 1.

A phylogenetic tree of vertebrate TnI isoform genes is derived from protein sequence alignment. ssTnI, fsTnI and cTnI are marked in red, blue and black fonts, respectively. The NCBI database accession numbers for the sequences analyzed are: Sablefish ssTnI, ACQ58112.1; Sablefish fsTnI, ACQ58096.1; Atlantic Herring slow-like TnI, XP_012674152.1; Atlantic Herring fsTnI, AAB05825.1; Zebra fish ssTnI, NP_001002101.1; Zebra fish fsTnI, NP_991138.2; Zebra fish cTnI, NP_001008613.1; Channel Catfish ssTnI, NP_001187788.1; Channel Catfish fsTnI, NP_001187493.1; Blue Catfish cTnI, ADO28353.1; Pike ssTnI, NP_001291009.1; Pike fsTnI, XP_010900246.1; Rainbow Trout fsTnI, NP_001123462.1; Rainbow Trout cTnI, NP_001171957.1; Painted Turtle ssTnI, XP_005310684.1; Painted Turtle fsTnI, XP_005307820.1; Painted Turtle cTnI, XP_008176331.1; Xenopus ssTnI, NP_001079781.1; Xenopus fsTnI, NP_001079556.1; Xenopus cTnI, NP_001088122.1; Toad fsTnI, AEZ53888.1; Toad cTnI, AAX69047.1; Bullfrog ssTnI, AAO33938.1; Bullfrog fsTnI, AAW73073.1; Bullfrog cTnI, AAO33937.1; Turkey ssTnI, XP_010722333.1; Turkey fsTnI, XP_003206336.1; Turkey cTnI, NP_001290153.1; Quail ssTnI, AAC59937.1; Quail fsTnI, AAB00122.1; Quail cTnI, AAA49513.1; Chicken ssTnI, XP_004934896.1; Chicken fsTnI, NP_990748.1; Chicken cTnI, NP_998735.1; Bovine ssTnI, NP_001290364.1; Bovine fsTnI, NP_001179023.1; Bovine cTnI, NP_001035607; Dog ssTnI, XP_003639192.1; Dog fsTnI, XP_851068.1; Dog cTnI, NP_001003041.1; Goat ssTnI, AFN20331.1; Goat fsTnI, AHK12864.1; Goat cTnI, AFN20332.1; Horse ssTnI, XP_005608116.1; Horse fsTnI, XP_005613620.1; Horse cTnI, NP_001075373; Cat ssTnI, XP_006942912.1; Cat fsTnI, XP_011285371.1; Cat cTnI, Q863B6.3; Rabbit ssTnI, XP_008266820.1; Rabbit fsTnI, NP_001076252.1; Rabbit cTnI, P02646.2; Pig ssTnI, AAP37479.1; Pig fsTnI, NP_001027530.1; Pig cTnI, NP_001092069.1; Mouse ssTnI, NP_001106173.1; Mouse fsTnI, NP_033431.1; Mouse cTnI, NP_033432.1; Rat ssTnI, NP_058880.1; Rat fsTnI, NP_058881.1; Rat cTnI, NP_058840.1; Marmoset (New World Monkey) ssTnI, NP_001230000.1; Marmoset fsTnI, JAB14438.1; Marmoset cTnI, XP_002762542.2; Rhesus monkey (Old World Monkey) ssTnI, NP_001252569.1; Rhesus monkey fsTnI, XP_001117040.1; Rhesus monkey cTnI, XP_001085820.1; Human ssTnI, NP_003272.3; Human fsTnI, NP_003273.1; Human cTnI, P19429.3.
Figure 2.

Amino acid sequence alignment of the N-terminal extension of vertebrate cTnI was performed with the MegAlign computer program (Lasergene; DNASTAR, Inc, Madison, WI) using the Clustal V method. The sequence alignment demonstrated that the N-terminal extension of mammalian cTnI is highly conserved, whereas it shows notable sequence variations in avian, reptile and amphibian species, and is absent in fish cTnI. The three arrows indicate the exon boundaries based on the structure of human TNNI3 gene. The two arrowheads indicate the two PKA phosphorylated Ser residues. The NCBI database accession numbers for the protein sequences analyzed are the same as that for Fig. 1.
Phylogenetic analysis of TnI isoform sequences also demonstrated that each of the muscle type-specific isoforms is conserved among species while the three TnI isoforms in a given species are significantly diverged (Jin et al., 1998). This observation suggests that the TnI isoform genes were diverged early on during vertebrate evolution (Chong & Jin, 2009). The phylogenetic tree in Fig. 1 further demonstrates that the divergence of cTnI genes is greater than that of fsTnI genes, whereas the ssTnI genes are the most conserved among vertebrate species. This pattern implicates different rates of the evolution of TnI isoform genes, possibly reflecting different functional demands in the three types of muscle fibers.
3. Muscle Type-Specific Expression and Developmental Regulation of TnI Isoform Genes
The expression of TnI isoform genes is under muscle-specific and developmental controls (Jin et al., 2008). ssTnI is expressed in both skeletal muscle and heart in embryos (Sasse et al., 1993). The expression of ssTnI in the heart is switched to cTnI during perinatal development (Saggin et al., 1989; Sasse et al., 1993). The presence of solely ssTnI in fetal hearts demonstrates the functional conservation and exchangeability of TnI isoforms in sustaining muscle contraction. Consistently, transgenic mice with cTnI replaced by ssTnI in the heart were viable and fertile, exhibiting no cardiac hypertrophy or failure (Fentzke et al., 1999).
fsTnI is restrictively expressed in fast twitch skeletal muscle fibers early on during myogenesis and continues expression through adulthood (Hastings and Emerson, 1982). A switch from slow to fast skeletal muscle TnI isoforms concurs with the switch of type I to type II fibers during skeletal muscle adaptation to simulated microgravity (Stevens et al., 2002). cTnI is exclusively expressed in the heart (Bodor et al., 1995), becoming predominant in postnatal cardiac muscle (Sasse et al., 1993). The adult heart expresses cTnI as the sole isoform with no change found in pathological conditions such as ischemic heart disease, dilated cardiomyopathy, and end-stage heart failure (Sasse et al., 1993).
Whereas cardiac TnT is expressed in developing fetal (Anderson et al., 1991) and regenerating adult (Bodor et al., 1997b; Rittoo et al., 2014) skeletal muscles, cTnI was exclusively expressed in cardiac muscle and not detected at protein level in skeletal muscle (Bodor et al., 1995; Rittoo et al., 2014). cTnI mRNA was controversially reported positive (Messner et al., 2000) or negative (Ricchiuti and Apple, 1999) in skeletal muscle of patients with Duchenne muscular dystrophy. No cTnI mRNA was detected in embryonic or adult skeletal muscle or in embryonic skeletal muscle cell cultures (Hastings et al., 1991).
Interestingly, ssTnI is not expressed in the embryonic hearts of Xenopus and zebra fish, while it is expressed in the somites and skeletal muscles (Warkman and Atkinson, 2002; Fu et al., 2009). In the meantime, the expression of zebra fish cTnI is not restricted to cardiac muscle and is detectable in craniofacial muscles (Fu et al., 2009). These observations indicate the high degree of similarity between lower vertebrate ssTnI and cTnI that lacks the N-terminal extension (Fig. 2). Therefore, the apparently high diversity of cTnI across vertebrate species (Fig. 1) mainly reflects the evolutionary addition of the adult heart-specific N-terminal extension in cTnI of higher vertebrates.
The different TnI isoforms expressed in different types of muscle fibers and regulated during heart and skeletal muscle development may function in fine tuning myofilament Ca2+-sensitivity, cooperativity and pH tolerance as an adaptation to the cellular environment and contractile performance (Westfall et al., 1997; Westfall et al., 1999). Over-expression of ssTnI in the cardiac muscle of adult transgenic mice altered relaxation and diastolic function by increasing myofilament Ca2+ sensitivity (Fentzke et al., 1999) and the tolerance to acidosis-induced decrease in myofilament Ca2+ sensitivity in cardiomyocytes (Westfall et al., 1997; Westfall et al., 2000). These findings indicate that ssTnI produces a higher Ca2+ affinity for the troponin complex than that of cTnI, which may sustain Ca2+ sensitivity of myofilaments at the lower pH (6.5 versus 7.0) in embryonic cardiomyocytes (Solaro et al., 1988).
Although TnI is generally considered a striated muscle-specific protein, recent studies reported that fsTnI, as well as fsTnT and fsTnC were expressed at significant levels in smooth muscle cells in mouse blood vessels (Moran et al., 2008), while fsTnT was found in smooth muscle cells in aorta, bladder and bronchus (Ju et al., 2013). Expression of fsTnI was also found in non-muscle cells, such as human corneal epithelium (Kinoshita et al., 2001) and cartilage (Moses et al., 1999; Li et al., 2003b). fsTnI was localized to the nuclei of breast cancer cells and may be a co-activator of estrogen receptor related-receptor α (Li et al., 2008).
4. Structure-Function Relationship of TnI Isoforms
TnI is a protein of 21 – 24 kDa in size with an alkaline isoelectric point (Table 1). Of the exons of the TnI genes, exons 4 – 8 are conserved among the isoforms and across species. In contrast, exons 1, 2 and 3 encode as few as 5 amino acids in fsTnI and ssTnI but as many as 30 amino acids in cTnI corresponding to the unique N-terminal extension (Fig. 2) (Jin et al., 2008). The last exon (exon 9, Table 1) of human TNNI1 gene is rather big (5,484 base pairs) and contains only a non-coding sequence (Corin et al., 1994). The last exon of mouse TNNI1 gene also contains only non-coding sequence. This additional non-coding exon is only found in TNNI1 genes and its functional significance is unknown.
Troponin I interacts with all known regulatory proteins in the thin filament: TnC, TnT, actin, and tropomyosin, reflecting its key position in the Ca2+ regulation of striated muscle contraction (Perry, 1999). Crystallography of the troponin complex determined the high resolution structure of human cTnI from amino acid 31 to 191 except for the inhibitory peptide (residues 137–148) (Takeda et al., 2003) (Fig. 3). Crystallography of chicken fast skeletal muscle troponin showed similar overall organization to that of cardiac troponin (Vinogradova et al., 2005). However, the inhibitory region of fsTnI is well ordered in skeletal muscle troponin whereas it is flexible and not visible in the crystal structure of cardiac troponin (Takeda et al., 2003) (Fig. 3).
Figure 3.

The left schematic structure illustrates the position of cTnI in the crystal structure of the troponin complex and its interactions with TnC and the partial structure of TnT in the Ca2+-saturated state (PDB 1J1E) (Takeda et al., 2003). The four α-helices are indicated: H1 (43–79), H2 (90–135), H3 (150–159), H4 (164–188). The counterpart residues in ssTnI and fsTnI were shown in the inset box to denote the helix boundaries. As there is no crystal structure available for ssTnI, the helix boundaries (in Italic font) in ssTnI are deduced from protein sequence alignment with cardiac and fast skeletal muscle TnI. No high resolution structure of the N-terminal extension, the C-terminal end segment and the inhibitory region (IR) were resolved for human cardiac troponin. The right schematic crystal structure of chicken fast skeletal troponin (PDB 1YTZ) (Vinogradova et al., 2005) showed only three α-helices (H1, H2 and H3) in fsTnI but included the inhibitory region. The C-terminal end segment of fsTnI is disordered in the crystal structure of chicken fast skeletal muscle troponin complex when Ca2+ is bound.
Based on in vitro structure-function relationship studies and interactions with other thin filament proteins, the structure of TnI can be divided into six functional segments (Li et al., 2004) (Fig 3): 1) the cardiac-specific N-terminal extension (residues 1–30 in cTnI); 2) the N-terminal conserved region (residues 42–65 in cTnI and 1–40 in fsTnI) that is the amphiphilic portion of H1 α-helix and binds the C domain of TnC; 3) the TnT-binding region (residues 66–136 in cTnI and 50–106 in fsTnI) from the C-terminal portion of H1 to H2 α-helices, which forms a coiled-coil interface with TnT; 4) the inhibitory region (residue 137–148 in cTnI and 107–115 in fsTnI) that interacts with TnC and actin–tropomyosin filament; 5) the switch or triggering region (residue 148–163 in cTnI and 115–131 in fsTnI) that is an α-helix (H3) and binds the N domain of TnC via its N-terminal segment; and 6) the C-terminal mobile domain (residue 164–210 in cTnI and 132–180 in fsTnI) that consists of a protruding α-helix (H4) (cTnI 164–188).
Recent studies demonstrated that the last 20 amino acids of the C-terminal end segment of TnI (residues 191–210 in cTnI) encoded by exon 8 with a highly conserved sequence is a Ca2+-modulated allosteric structure and interacts with tropomyosin (Zhang et al., 2011; Akhter et al., 2014). While the N-terminal extension of cTnI does not have definitive interaction with other known myofilament proteins, it plays a role in modulating the conformation of other regions of the cTnI molecule (Akhter et al., 2012).
5. Posttranslational Modifications
No alternative RNA splicing has been reported for the transcripts of TnI isoform genes. On the other hand, posttranslational modifications play a major role in regulating structure and function of TnI (Solaro et al., 2008). The mechanisms include amino acid side chain modifications and cleavages of the polypeptide chain, which induce conformational changes to modify troponin function and muscle contractility (Pi et al., 2003; Layland et al., 2005; Westfall et al., 2005; Solaro and van der Velden, 2010).
5.1. Phosphorylation
Whereas there is very limited information for the phosphorylation of ssTnI and fsTnI, phosphorylation of cTnI plays significant but sometimes controversial roles in cardiac muscle function (Solaro and Kobayashi, 2011). The 210 amino acid human cTnI polypeptide chain contains 12 serine, 8 threonine, and 3 tyrosine residues. Eighteen of these residues are predicted to be potentially phosphorylatable and 16 of them were experimentally demonstrated (Zhang et al., 2012). The known phosphorylation sites, corresponding kinases and function of these sites are summarized in Table 2.
Table 2.
Phosphorylation sites in troponin I
| Phosphorylation Site | Kinases | Function | Reference | ||
|---|---|---|---|---|---|
| cTnI | ssTnI | fsTnI | |||
| S5,S6 | NA | NA | Unknown | Unknown | (Zhang et al., 2012) |
| S23,S24 | NA | NA | PKA PKC-β PKC-ε PKD PKG |
Enhancing diastolic function | (Solaro et al., 2008) (Kobayashi et al., 2005) (Haworth et al., 2004 (Layland et al., 2002) |
| Y26 | NA | NA | Unknown | Enhancing diastolic function | (Zhang et al., 2012) (Salhi et al., 2014) |
| T31 | NA | NA | Mst1 | Increasing affinity for TnC; Decreasing affinity for TnT |
(You et al., 2009) |
| S39 | (P8) | (N8) | PKA | Decreasing affinity for TnC | (Ward et al., 2001) |
| S42,S44 | T11,S13 | (I11,A13) | PKC | Slowing cardiac relaxation | (Kooij et al., 2011) |
| T51 | S20 | S20 | Mst1 | Unknown | (You et al., 2009) |
| S77,T78 | (A46,E47) | (A46,E47) | Unknown | Unknown | (Zhang et al., 2012) |
| T129 | (K98) | (N97) | Mst1 | Unknown | (You et al., 2009) |
| T143 | (112P) | (111P) | PKC PKC-βII Mst1 |
Decreasing relaxation and contraction | (Westfall et al., 2005) (You et al., 2009) |
| S150 | S119 | S118 | Pak AMPK |
Slowing cardiac relaxation | (Buscemi et al., 2002) (Oliveira et al., 2012) (Sancho Solis et al., 2011) |
| S166 | S135 | (C134) | PKA | Decreasing affinity to TnC | (Ward et al., 2001) |
| T181 | T150 | T149 | Unknown | Unknown | (Zhang et al., 2012) |
| S199 | S169 | S169 | PKC | Decreasing affinity to actin-tropomyosin | (Wijnker et al., 2015) |
The amino acid residues in cTnI with known phosphorylation are listed. The residue numbers refer to that in human TnI isoforms with the first methionine included. NA, not applicable. Some of the residues are conserved between cardiac and skeletal muscle TnI, suggesting possible phosphorylations in skeletal muscle TnI. Some of the sites have been studied with engineered substitutions in cTnI with the counterpart amino acids in ssTnI or fsTnI (in brackets), suggesting that the phosphorylation modification of these sites in cTnI is a mechanism of tuning the function toward a skeletal muscle-like state. Phosphorylation of the sites in cTnI has been experimentally identified. Among the potential phosphorylation sites in ssTnI and fsTnI (in Italic font), only S118 in fsTnI was experimentally demonstrated.
Discussed above, cTnI of higher vertebrates differs from the two skeletal muscle TnI isoforms mainly by its N-terminal extension of ~30 amino acids, which contains a characteristic protein kinase A (PKA) substrate motif RRRSS (residues 20–24 in human cTnI) (Fig. 2). PKA phosphorylation of Ser23 and Ser24 under the regulation of adrenergic signaling cascades (Quirk et al., 1995; Solaro et al., 2008; Solaro and Kobayashi, 2011; Rao et al., 2012) enhances the diastolic function of cardiac muscle (Zhang et al., 1995a; Stelzer et al., 2007; Li et al., 2010) by reducing the Ca2+-binding affinity of the N domain regulatory site of cardiac TnC (Zhang et al., 1995b) and weakened TnC-TnI interaction in the presence of Ca2+ (Rao et al., 2014). These two serine residues have also been reported to be phosphorylated in vitro by PKC-β, PKC-ε (Kobayashi et al., 2005), PKD (previously named PKCμ) (Haworth et al., 2004; Cuello et al., 2007; Bardswell et al., 2010) and PKG (Layland et al., 2002).
Four other sites (Ser5, Ser6, Tyr26 and Thr31) in the N-terminal extension of cTnI are proposed with potential phosphorylations. Thr31 is a substrate of mammalian sterile 20-like kinase 1 (Mst1) (You et al., 2009). Tyr26 phosphorylation was shown to function similarly to that of Ser23/24 in decreasing myofilament Ca2+ sensitivity and increasing cardiac muscle relaxation (Salhi et al., 2014). Physiological significance and regulatory mechanisms of these phosphorylation sites remain to be further investigated.
The absence of the cardiac specific N-terminal extension in fish cTnI (Fig. 2) indicates its nature as an evolutionarily added regulatory structure in TnI of higher vertebrates. Troponin complex containing trout cTnI that lacks the N-terminal extension showed greater Ca2+ affinity than that containing human cTnI (Kirkpatrick et al., 2011). Although trout cTnI lacks the two N-terminal Ser residues, myofilament Ca2+ affinity decreased upon PKA treatment similar to the response of mammalian cTnI control (Kirkpatrick et al., 2011). This observation is worth further investigation.
Phosphorylation at Ser42 and Ser44 by PKC produces opposite effects to that of PKA at Ser23 and Ser24 by slowing down cardiac muscle relaxation and increasing the durations of Ca2+ transient and twitch contraction (MacGowan et al., 2001; Pi et al., 2002; Burkart et al., 2003; Westfall et al., 2005).
The TnT binding region, i.e., residues 66–136 in cTnI, contains several potentially phosphorylatable residues, Ser77, Thr78 and Thr129 (Zhang et al., 2012) with unknown function. Thr143 in the inhibitory region is a cTnI-specific phosphorylation site. In mouse heart, phosphorylation of Thr143 of cTnI by PKC-βII increased myofilament Ca2+ sensitivity (Wang et al., 2006). However, another study observed that Thr143 phosphorylation did not alter Ca2+ sensitivity, but depressed cooperative activation of the thin filaments (Lu et al., 2010). Replacing Thr143 with Pro to mimic that in ssTnI resulted in delayed relaxation of cardiomyocytes (Westfall et al., 2005). PKC phosphorylation of cTnI at Thr143 also impaired the interaction between the inhibitory region and TnC, leading to depressed actomyosin ATPase and contractility (Lindhout et al., 2002; Li et al., 2003a). The physiological function of Thr143 phosphorylation requires more investigation.
Ser150 of cTnI in the TnC binding region has been shown as a phosphorylation site with opposite effect to that of PKA phosphorylation at Ser23/24 (Nixon et al., 2014). Ser150 can be phosphorylated by P21-activated kinase (Pak) (Buscemi et al., 2002; Ke et al., 2004) and AMP-activated protein kinase (AMPK), resulting in increased Ca2+ sensitivity of cardiac myofibrils, prolonged relaxation (Oliveira et al., 2012), and increased development of adrenergic-induced myocardial hypertrophy (Taglieri et al., 2011). Equivalent to Ser150 in cTnI, Ser118 in fsTnI was also reported to be phosphorylated by AMPK (Sancho Solis et al., 2011). As AMPK is a key regulator of cellular energetics, phosphorylation of Ser150 may suggest an adaptive mechanism in energy deprivation of both cardiac and skeletal muscles.
Phosphorylation at Ser166 in cTnI reduced binding affinity for cardiac TnC (Zhang et al., 2012). Phosphorylation at Ser199 in the actin-binding region of cTnI by PKC increased Ca2+ sensitivity of troponin with decreased affinity for actin-tropomyosin thin filament (Wijnker et al., 2015). Thr181 in this region was phosphorylated with a level higher than that of Ser166 and Ser199 (Zhang et al., 2012), but the responsible kinase and physiological function are unknown.
The phosphorylation of cTnI changes in cardiomyopathy and heart failure. In human end-stage dilated cardiomyopathy, the baseline phosphorylation of cTnI was diminished with increased myofilament Ca2+ affinity (Zakhary et al., 1999). In failing human heart, PKA phosphorylation of Ser23/S24 in cTnI was decreased (Bodor et al., 1997a; Messer et al., 2007) and PKC phosphorylation of Ser42/Ser44/Thr143 increased (Zhang et al., 2012), resulting in ventricular diastolic dysfunction. In post-infarct myocardium under remodeling, the expression of PKA and PKA-mediated phosphorylation of cTnI were decreased (Van der Velden et al., 2004).
5.2. O-linked GlcNAc modification
O-Linked N-acetylglucosaminylation (O-GlcNAc) is a dynamic cytosolic and nuclear mechanism of glycosylation, which is a ubiquitous post-translational modification and plays a role in regulating protein functions. Studies of isolated cardiomyocytes suggested increased levels of O-GlcNAcylation of cardiac muscle proteins in hearts with diabetic cardiac dysfunction (Fulop et al., 2007). High O-GlcNAc level would decrease Ca2+ sensitivity and affinity of myofibrils (Hedou et al., 2007). Mass spectrometry identified Ser150 of mouse cTnI as one of the O-GlcNAcylation sites (Ramirez-Correa et al., 2008). Interestingly, Ser150 is also a phosphorylation site by Pak and AMPK (discussed above), increasing Ca2+ sensitivity. O-GlcNAc of cTnI at Ser150 may compete for this site with phosphorylation to down-regulate protein phosphorylation. Conversely, it is conceivable that decreased phosphorylation may be a result of increased O-GlcNAc levels. The balance between phosphorylation and O-GlcNAc is worth further investigation.
5.3. S-glutathionylation
Reactive oxygen and nitrogen species are generated in skeletal muscle with normal activity and also in pathological conditions, and acutely or chronically affect muscle function (Lamb and Westerblad, 2011). H2O2 treatment decreased Ca2+ sensitivity of intact muscle fibers, which was reversed by glutathione pretreatment in only fast-twitch fibers, indicating specific S-glutathionylation target proteins in fast-twitch fibers (Murphy et al., 2008). It was found that fsTnI in rodent skeletal muscle was S-glutathionylated at Cys134, which increased the Ca2+ activation of contraction. Similarly, fsTnI in human type II muscle fibers was S-glutathionylated with increased Ca2+ sensitivity in physically active individuals, which may benefit skeletal muscle performance (Mollica et al., 2012). fsTnI of chicken and toad, both of which lack cysteine 133, showed no S-glutathionylation effects when the fast-twitch fiber went through the same treatment as that for rodent muscles (Mollica et al., 2012). This observation suggests a protective role of fsTnI S-glutathionylation against oxidative stress.
5.4. Proteolytic modifications
Cardiac TnI is a substrate of intracellular proteases, with a demonstrated sensitivity to μ-calpain and m-calpain (Di Lisa et al., 1995). Its degradation by μ-calpain was regulated by phosphorylation of cTnI, in which phosphorylation by PKA reduced the sensitivity whereas phosphorylation by PKC increased the sensitivity to μ-calpain proteolysis (Di Lisa et al., 1995). Phosphorylation of cTnI by PKC at Ser199 increased myofilament Ca2+ sensitivity and cTnI’s susceptibility to calpain-mediated proteolysis (Wijnker et al., 2015).
In addition to the overall degradation, C-terminal or N-terminal truncations have been reported in cTnI with distinct effects on cardiac function.
5.4.1. C-terminal truncation
The C-terminal region of TnI binds and stabilizes tropomyosin on the actin filament in the absence of Ca2+ to inhibit muscle contraction (Galińska et al., 2010; Zhang et al., 2011). The C-terminal end segment (192–210) is the most conserved region of the TnI polypeptide chain (Jin et al., 2001) and a Ca2+-modulated allosteric structure in the troponin complex (Jin et al., 2001; Zhang et al., 2011; Wang et al., 2012a). R192H and R204H mutations in the C-terminal end segment of human cTnI cause restrictive cardiomyopathy (Mogensen et al., 2003; Gambarin et al., 2008) with alterations in the conformation and function of the TnI-TnT interface and increased binding affinity of cTnI for TnT (Akhter et al., 2014).
A truncation of the C-terminal 19 amino acids of cTnI was found during myocardial ischemia-reperfusion injury in Langendorff perfused rat hearts (McDonough et al., 1999). It was also seen in myocardial stunning in coronary bypass grafted patients (McDonough et al., 2001). Over-expression of the C-terminal truncated cTnI (cTnI1–193) in transgenic mouse heart reproduced myocardial stunning with systolic and diastolic dysfunctions (Murphy et al., 2000). Partial replacement of intact cTnI with cTnI1–192 in myofibrils in vitro and in cardiomyocytes ex vivo did not affect maximal tension development but hindered the rates of force redevelopment and relaxation (Narolska et al., 2006). Troponin complex containing cTnI1–192 exhibited increased activity of Ca2+-activated actomyosin ATPase and faster sliding velocity as compared with that of troponin containing intact cTnI (Foster et al., 2003).
However, the pathological significance of the C-terminal truncation of cTnI is controversial. No C-terminal truncated cTnI was detectable in swine hearts after in vivo regional ischemia-reperfusion (Thomas et al., 1999). Myocardial stunning in pigs induced by regional ischemia was found with dephosphorylation of phospholamban without degradation of cTnI (Kim et al., 2001). No degradation of cTnI was detected in the hearts of conscious dogs after reversible ischemia (Lüss et al., 2000; Sherman et al., 2000).
5.4.2. Restrictive N-terminal truncation
The adult heart-specific N-terminal extension of cTnI is a regulatory structure (Sheng and Jin, 2014). The N-terminal extension contains the key PKA phosphorylation sites and plays a role in modulating the overall molecular conformation and function of cTnI (Akhter et al., 2012). Different from the C-terminal truncation, a selective removal of the N-terminal extension of cTnI through restrictive proteolysis occurs as a regulatory mechanism in cardiac adaptation in physiological and pathological stress conditions.
The restrictive N-terminal truncation of cTnI is at low levels in normal hearts of all species examined including human, and is significantly increased in response to hemodynamic stress (Yu et al., 2001) and β-adrenergic deficiency-caused failing mouse hearts (Feng et al., 2008). The N-terminal truncated cardiac TnI (cTnI-ND) remains in the myofibrils with a function in increasing myocardial relaxation to improve ventricular filling, similar to the effect of PKA phosphorylation (Barbato et al., 2005).
Transgenic mouse hearts expressing cTnI-ND exhibit improved diastolic function (Feng et al., 2008) and a better preservation of cardiac function in aging (Biesiadecki et al., 2010). Co-expression of cTnI-ND corrected the diastolic dysfunction of restrictive cardiomyopathy mouse hearts caused by cTnI-R193H mutation (Li et al., 2010). Isolated cTnI-ND mouse cardiomyocytes exhibited increased diastolic and systolic functions (Wei and Jin, 2013). These findings indicate that the N-terminal extension of cTnI is a potential site for targeted treatment for the clinically challenging condition of diastolic heart failure (Zile and Brutsaert, 2002).
6. Pathogenic Mutations
6.1. Patient phenotypes
To date, no human disease has been reported with mutations in ssTnI. Mutations in the fsTnI gene have been found to cause myopathy and distal arthrogryposis (DA). A missense mutation R174Q, a nonsense mutation (premature stop codon R156X), and three in-frame deletion mutations ΔE167, ΔK175 and ΔK176 have been reported in DA patients (Sung et al., 2003; Jiang et al., 2006; Kimber et al., 2006; Robinson et al., 2007). The mutations associated with DA are all in the C-terminal actin-tropomyosin binding domain.
Many mutations of cTnI have been found to cause cardiomyopathies (Seidman and Seidman, 2001; Curila et al., 2012). Comparing with pathogenic mutations in other myofilament proteins, cTnI mutations are often associated with more severe clinical courses (Doolan et al., 2005). R21C mutation in the N-terminal extension (Wang et al., 2012b), R141Q, L144P, R145Q, R145G in the inhibition region, A157V, R162W, R162Q, R162P in the switch region (Willott et al., 2010), and S166F, ΔK177, K178del, K183E, ΔK183, R186Q, I195M, D196N, L198V, L198P, S199N, E202G, G203R, G203S, R204C, R204H and K206Q in the C-terminal region (Willott et al., 2010) were found in hypertrophic cardiomyopathy patients. A116G mutation in the α-helix in TnI at the interface with TnT was found in human dilated cardiomyopathy (Millat et al., 2011). A171T, K178E, D190G and R192H in the C-terminal region of cTnI were found in restrictive cardiomyopathy patients (Willott et al., 2010).
Most of these disease-causing single nucleotide mutations in cTnI are located in the C-terminal half of the polypeptide chain (residues 128–210) (Palpant et al., 2010), demonstrating the critical role of the C-terminal domain of TnI in muscle relaxation and diastolic function of the heart (Davis et al., 2007). This observation may indicate more stringent structure-function relationships in this region, or on the other hand reflect that this region of TnI has a high tolerance to structural variations to avoid embryonic lethality, allowing mutations to remain in the population. Both hypotheses are worth investigating.
6.2. Experimental studies
The pathophysiologic mechanism underlying the cTnI mutation-caused cardiomyopathies is a current research topic and much data have been elucidated from molecular to animal level studies.
R21C, which is the only in cTnI mutation found in the N-terminal extension, alters the PKA substrate motif and abolishes in vivo phosphorylation of Ser23 and Ser24. cTnI-R21C knock-in mice showed diastolic dysfunction with delayed Ca2+ re-sequestration (Dweck et al., 2014), a phenotype that supports the role of the N-terminal extension of cTnI in the regulation of diastolic function of the heart.
Mouse cTnI-A117G, the corresponding site of human A116G, exhibits faster mobility in SDS-PAGE as compared with wild type control, indicating a significant change in overall protein conformation (Akhter and Jin, 2015). Interestingly, an adjacent mutation K118C produces slower SDS-gel mobility than wild type control (Wei et al., 2010; Akhter and Jin, 2015), further demonstrating the potent and apparently complex effects of the TnI-TnT interface structure of TnI on molecular conformation and function. The K118C mutation decreases binding affinity of cTnI for TnC at pCa 4, which can be reversed by the restrictive N-terminal truncation (Akhter and Jin, 2015), reflecting a dominant effect of the N-terminal extension of cTnI on modulating the function of TnI-TnT interface.
R145G mutation in the inhibitory region of cTnI alters the interaction of cTnI with cardiac TnC, reduces the inhibition of actomyosin ATPase, and thus increases Ca2+ sensitivity (Kimura et al., 1997; Lindhout et al., 2002).
Different mutations of the same residue in cTnI may produce different phenotypes. For example, R204P mutation showed weakened interaction with TnT and TnC, R204C showed mild impairment of affinity for TnT and greater impairment of affinity for TnC (Cui et al., 2013), and R204W produced increased Ca2+ sensitivity.
Transgenic mouse models of cTnI mutation-caused cardiomyopathy are valuable tools to study the pathogenesis and pathophysiology of the human diseases, as well as to better understand the structure-function relationship of TnI. Some extensively characterized representative mouse models are summarized in Table 3.
Table 3.
Transgenic mouse models of cTnI mutations and truncations
| Mouse Line | Phenotype | Reference |
|---|---|---|
| R21C knock-in | Hypertrophic cardiomyopathy | (Wang et al., 2012b) |
| K118C over-expression | Diastolic dysfunction | (Wei et al., 2010) |
| R145G over-expression* | Hypertrophic cardiomyopathy | (Wen et al., 2008) |
| R145W over-expression* | Restrictive cardiomyopathy | (Wen et al., 2009) |
| K178E over-expression | Restrictive cardiomyopathy | (Jean-Charles et al., 2012) |
| K179E over-expression | Restrictive cardiomyopathy | (Jean-Charles et al., 2012) |
| K184deletion over-expression | Diastolic dysfunction | (Iorga et al., 2008) |
| R193H over-expression | Restrictive cardiomyopathy | (Du et al., 2008) |
| C-terminal truncation over-expression | Stunning heart | (Murphy et al., 2000) |
| cTnI-ND over-expression | Enhanced diastolic function | (Feng et al., 2008) |
Several gene-targeted or transgenic overexpression mouse models of cTnI are listed with their major phenotypes. The mutation sites in mouse cTnI are indicated with residue # counting from the first methionine.
The R145G and R145W mouse lines over-express human cTnI in the heart.
7. Applications in Clinical Diagnosis of Acute Myocardial Infarction and Muscle Injuries
Cardiac troponin has been widely used as an indicator for cardiac muscle injuries. Testing of plasma cardiac TnI and cardiac TnT is uniformly performed in the diagnosis of patients with ischemic heart diseases (Januzzi et al., 2012). cTnI is exclusively expressed in the adult cardiac muscle cells and is a more specific diagnostic marker for myocardial infarction (Bodor et al., 1995) over cardiac TnT that is expressed in the heart as well as in fetal and regenerating skeletal muscles with elevations in patients with neuromuscular diseases (Rittoo et al., 2014). cTnT is also a sensitive and specific marker for even minor myocardial injuries. In addition, an increased level of serum cTnI independently predicts poor prognosis of critically ill patients in the absence of acute coronary syndrome (Reynolds et al., 2012; Lee et al., 2015). When skeletal muscle diseases are ruled out, elevation of cTnI together with cTnT in plasma could predict poor prognosis due to cardiomyocyte injury secondary to non-cardiac diseases (Giannitsis et al., 2000).
Skeletal muscle TnI has been proposed as a muscle fiber specific and sensitive marker of skeletal muscle injuries (Simpson et al., 2005; Chapman et al., 2013). fsTnI concentration in serum increased more than that of ssTnI after eccentric exercise of elbow flexors, indicating more damages of fast twitch fibers (Chapman et al., 2013).
7. Summary
An essential component of the thin filament Ca2+ regulatory system of vertebrate striated muscles, TnI has evolved with three muscle fiber type-specific isoform genes and diverse posttranslational modifications. Understanding the regulation and structure-function relationships of TnI isoforms, posttranslational modifications, and pathogenic mutations has broad biological and medical significance and applications.
Based on the current knowledge, many important questions regarding the gene evolution, regulation, structure-function relationship of TnI and its interactions with other myofilament proteins remain to be answered. For example, what are the key functional differences that prevent the isoforms from substituting for each other? How does the cardiac specific N-terminal extension regulate the molecular conformation and function of cTnI? What is the molecular mechanism that governs the restrictive N-terminal truncation of cTnI in physiological adaptations? How does the phosphorylation of TnI regulate its functions? And what is the function of TnI found in smooth muscle and non-muscle cells? Much future work is needed to address these questions, which will enrich our knowledge on muscle contraction in health and diseases.
Highlights.
Human and higher vertebrate genomes contain three homologous troponin I isoform genes, TNNI1, TNNI2 and TNNI3.
The muscle fiber type-specific troponin I isoforms are evolutionarily diverged whereas each isoform is conserved across species.
The N-terminal extension of cardiac troponin I is an adult heart-specific structure, and its phosphorylation by PKA and restrictive deletion regulate cardiac function.
Mutations of troponin I cause myopathies.
Acknowledgments
This work was also supported in part by National Institutes of Health grants AR-048816 and HL-098945 to J-P Jin. Juan-Juan Sheng is a PhD candidate partially supported by a fellowship from China Scholarship Council.
This review and the corresponding Gene Wiki articles are written as part of the Gene Wiki Review series - a series resulting from a collaboration between the journal GENE and the Gene Wiki Initiative. The Gene Wiki Initiative is supported by National Institutes of Health (GM089820 and GM114833). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. The corresponding Gene Wiki entries for this review can be found here: https://en.wikipedia.org/wiki/TNNI1; https://en.wikipedia.org/wiki/TNNI2; and https://en.wikipedia.org/wiki/TNNI3.
Abbreviations
- AMPK
AMP-activated protein kinase
- cTnI
cardiac isoform of troponin I
- cTnI-ND
N-terminal truncated cardiac troponin I
- DA
distal arthrogryposis
- fsTnI
fast skeletal muscle isoform of troponin I
- Mst1
mammalian sterile 20-like kinase 1
- O-GlcNAc
O-linked β-N-acetyl-D-glucosamine
- PKA
protein kinase A
- PKC
protein kinase C
- PKD
protein kinase D
- PKG
protein kinase G
- ssTnI
slow skeletal muscle isoform of troponin I
- TnI
troponin I
- TnT
troponin T
- TnC
troponin C
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akhter S, Bueltmann K, Jr, Huang X, Jin JP. Restrictive cardiomyopathy mutations demonstrate functions of the C-terminal end-segment of troponin I. Arch Biochem Biophys. 2014;552–553:3–10. doi: 10.1016/j.abb.2013.12.001. [DOI] [PubMed] [Google Scholar]
- Akhter S, Jin JP. Distinct conformational and functional effects of two adjacent pathogenic mutations in cardiac troponin I at the interface with troponin T. FEBS Open Bio. 2015;5:64–75. doi: 10.1016/j.fob.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhter S, Zhang Z, Jin JP. The heart-specific NH2-terminal extension regulates the molecular conformation and function of cardiac troponin I. Am J Physiol Heart Circ Physiol. 2012;302:H923–33. doi: 10.1152/ajpheart.00637.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson PA, Malouf NN, Oakeley AE, Pagani ED, Allen PD. Troponin T isoform expression in humans. A comparison among normal and failing adult heart, fetal heart, and adult and fetal skeletal muscle. Circ Res. 1991;69:1226–33. doi: 10.1161/01.res.69.5.1226. [DOI] [PubMed] [Google Scholar]
- Baldwin AS, Jr, Kittler EL, Emerson CP., Jr Structure, evolution, and regulation of a fast skeletal muscle troponin I gene. Proc Natl Acad Sci U S A. 1985;82:8080–4. doi: 10.1073/pnas.82.23.8080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbato JC, Huang QQ, Hossain MM, Bond M, Jin JP. Proteolytic N-terminal truncation of cardiac troponin I enhances ventricular diastolic function. J Biol Chem. 2005;280:6602–9. doi: 10.1074/jbc.M408525200. [DOI] [PubMed] [Google Scholar]
- Bardswell SC, Cuello F, Rowland AJ, Sadayappan S, Robbins J, Gautel M, Walker JW, Kentish JC, Avkiran M. Distinct sarcomeric substrates are responsible for protein kinase D-mediated regulation of cardiac myofilament Ca2+ sensitivity and cross-bridge cycling. J Biol Chem. 2010;285:5674–5682. doi: 10.1074/jbc.M109.066456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesiadecki BJ, Tachampa K, Yuan C, Jin JP, de Tombe PP, Solaro RJ. Removal of the cardiac troponin I N-terminal extension improves cardiac function in aged mice. J Biol Chem. 2010;285:19688–98. doi: 10.1074/jbc.M109.086892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodor GS, Oakeley AE, Allen PD, Crimmins DL, Ladenson JH, Anderson PAW. Troponin I phosphorylation in the normal and failing adult human heart. Circulation. 1997a;96:1495–1500. doi: 10.1161/01.cir.96.5.1495. [DOI] [PubMed] [Google Scholar]
- Bodor GS, Porterfield D, Voss EM, Smith S, Apple FS. Cardiac troponin-I is not expressed in fetal and healthy or diseased adult human skeletal muscle tissue. Clin Chem. 1995;41:1710–5. [PubMed] [Google Scholar]
- Bodor GS, Survant L, Voss EM, Smith S, Porterfield D, Apple FS. Cardiac troponin T composition in normal and regenerating human skeletal muscle. Clin Chem. 1997b;43:476–84. [PubMed] [Google Scholar]
- Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem. 2003;278:11265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
- Buscemi N, Foster DB, Neverova I, Van Eyk JE. p21-activated kinase increases the calcium sensitivity of rat triton-skinned cardiac muscle fiber bundles via a mechanism potentially involving novel phosphorylation of troponin I. Circ Res. 2002;91:509–516. doi: 10.1161/01.res.0000035246.27856.53. [DOI] [PubMed] [Google Scholar]
- Chapman DW, Simpson JA, Iscoe S, Robins T, Nosaka K. Changes in serum fast and slow skeletal troponin I concentration following maximal eccentric contractions. J Sci Med Sport. 2013;16:82–5. doi: 10.1016/j.jsams.2012.05.006. [DOI] [PubMed] [Google Scholar]
- Chong SM, Jin JP. To investigate protein evolution by detecting suppressed epitope structures. J Mol Evol. 2009;68:448–60. doi: 10.1007/s00239-009-9202-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corin SJ, Juhasz O, Zhu L, Conley P, Kedes L, Wade R. Structure and expression of the human slow twitch skeletal muscle troponin I gene. J Biol Chem. 1994;269:10651–9. [PubMed] [Google Scholar]
- Cuello F, Bardswell SC, Haworth RS, Yin X, Lutz S, Wieland T, Mayr M, Kentish JC, Avkiran M. Protein kinase D selectively targets cardiac troponin I and regulates myofilament Ca2+ sensitivity in ventricular myocytes. Circ Res. 2007;100:864–873. doi: 10.1161/01.RES.0000260809.15393.fa. [DOI] [PubMed] [Google Scholar]
- Cui Z, Gilda J, Gomes G, Gomes AV. Effect of Amino Acid Changes in a Troponin I FHC Hotspot on Protein:Protein Binding and Calcium Sensitivity of Force Development. Biophys J. 2013;106:723a. [Google Scholar]
- Curila K, Benesova L, Penicka M, Minarik M, Zemanek D, Veselka J, Widimsky P, Gregor P. Spectrum and clinical manifestations of mutations in genes responsible for hypertrophic cardiomyopathy. Acta Cardiol. 2012;67:23–9. doi: 10.1080/ac.67.1.2146562. [DOI] [PubMed] [Google Scholar]
- Davis J, Wen H, Edwards T, Metzger JM. Thin filament disinhibition by restrictive cardiomyopathy mutant R193H troponin I induces Ca2+-independent mechanical tone and acute myocyte remodeling. Circ Res. 2007;100:1494–1502. doi: 10.1161/01.RES.0000268412.34364.50. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, De Tullio R, Salamino F, Barbato R, Melloni E, Siliprandi N, Schiaffino S, Pontremoli S. Specific degradation of troponin T and I by mu-calpain and its modulation by substrate phosphorylation. Biochem J. 1995;308:57. doi: 10.1042/bj3080057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolan A, Tebo M, Ingles J, Nguyen L, Tsoutsman T, Lam L, Chiu C, Chung J, Weintraub RG, Semsarian C. Cardiac troponin I mutations in Australian families with hypertrophic cardiomyopathy: clinical, genetic and functional consequences. J Mol Cell Cardiol. 2005;38:387–393. doi: 10.1016/j.yjmcc.2004.12.006. [DOI] [PubMed] [Google Scholar]
- Du J, Liu J, Feng HZ, Hossain MM, Gobara N, Zhang C, Li Y, Jean-Charles PY, Jin JP, Huang XP. Impaired relaxation is the main manifestation in transgenic mice expressing a restrictive cardiomyopathy mutation, R193H, in cardiac TnI. Am J Physiol Heart Circ Physiol. 2008;294:H2604–13. doi: 10.1152/ajpheart.91506.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dweck D, Sanchez-Gonzalez MA, Chang AN, Dulce RA, Badger CD, Koutnik AP, Ruiz EL, Griffin B, Liang J, Kabbaj M, Fincham FD, Hare JM, Overton JM, Pinto JR. Long term ablation of protein kinase A (PKA)-mediated cardiac troponin I phosphorylation leads to excitation-contraction uncoupling and diastolic dysfunction in a knock-in mouse model of hypertrophic cardiomyopathy. J Biol Chem. 2014;289:23097–111. doi: 10.1074/jbc.M114.561472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng HZ, Chen M, Weinstein LS, Jin JP. Removal of the N-terminal extension of cardiac troponin I as a functional compensation for impaired myocardial beta-adrenergic signaling. J Biol Chem. 2008;283:33384–93. doi: 10.1074/jbc.M803302200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fentzke RC, Buck SH, Patel JR, Lin H, Wolska BM, Stojanovic MO, Martin AF, Solaro RJ, Moss RL, Leiden JM. Impaired cardiomyocyte relaxation and diastolic function in transgenic mice expressing slow skeletal troponin I in the heart. J Physiol. 1999;517:143–157. doi: 10.1111/j.1469-7793.1999.0143z.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DB, Noguchi T, VanBuren P, Murphy AM, Van Eyk JE. C-Terminal Truncation of Cardiac Troponin I Causes Divergent Effects on ATPase and Force Implications for the Pathophysiology of Myocardial Stunning. Circ Res. 2003;93:917–924. doi: 10.1161/01.RES.0000099889.35340.6F. [DOI] [PubMed] [Google Scholar]
- Fu CY, Lee HC, Tsai HJ. The molecular structures and expression patterns of zebrafish troponin I genes. Gene Expr Patterns. 2009;9:348–56. doi: 10.1016/j.gep.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Fulop N, Mason MM, Dutta K, Wang P, Davidoff AJ, Marchase RB, Chatham JC. Impact of Type 2 diabetes and aging on cardiomyocyte function and O-linked N-acetylglucosamine levels in the heart. Am J Physiol Cell Physiol. 2007;292:C1370–8. doi: 10.1152/ajpcell.00422.2006. [DOI] [PubMed] [Google Scholar]
- Galińska A, Hatch V, Craig R, Murphy AM, Van Eyk JE, Wang CLA, Lehman W, Foster DB. The C terminus of cardiac troponin I stabilizes the Ca2+-activated state of tropomyosin on actin filaments. Circ Res. 2010;106:705–711. doi: 10.1161/CIRCRESAHA.109.210047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambarin FI, Tagliani M, Arbustini E. Pure restrictive cardiomyopathy associated with cardiac troponin I gene mutation: mismatch between the lack of hypertrophy and the presence of disarray. Heart. 2008;94:1257. doi: 10.1136/hrt.2008.154203. [DOI] [PubMed] [Google Scholar]
- Giannitsis E, Muller-Bardorff M, Kurowski V, Weidtmann B, Wiegand U, Kampmann M, Katus HA. Independent prognostic value of cardiac troponin T in patients with confirmed pulmonary embolism. Circulation. 2000;102:211–7. doi: 10.1161/01.cir.102.2.211. [DOI] [PubMed] [Google Scholar]
- Hastings K. Molecular evolution of the vertebrate troponin I gene family. Cell Struct Funct. 1997;22:205. doi: 10.1247/csf.22.205. [DOI] [PubMed] [Google Scholar]
- Hastings KE, Emerson CP., Jr cDNA clone analysis of six co-regulated mRNAs encoding skeletal muscle contractile proteins. Proc Natl Acad Sci U S A. 1982;79:1553–7. doi: 10.1073/pnas.79.5.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings KE, Koppe RI, Marmor E, Bader D, Shimada Y, Toyota N. Structure and developmental expression of troponin I isoforms.cDNA clone analysis of avian cardiac troponin I mRNA. J Biol Chem. 1991;266:19659–65. [PubMed] [Google Scholar]
- Haworth RS, Cuello F, Herron TJ, Franzen G, Kentish JC, Gautel M, Avkiran M. Protein kinase D is a novel mediator of cardiac troponin I phosphorylation and regulates myofilament function. Circ Res. 2004;95:1091–1099. doi: 10.1161/01.RES.0000149299.34793.3c. [DOI] [PubMed] [Google Scholar]
- Hedou J, Cieniewski-Bernard C, Leroy Y, Michalski JC, Mounier Y, Bastide B. O-linked N-acetylglucosaminylation is involved in the Ca2+ activation properties of rat skeletal muscle. J Biol Chem. 2007;282:10360–10369. doi: 10.1074/jbc.M606787200. [DOI] [PubMed] [Google Scholar]
- Huang QQ, Jin JP. Preserved close linkage between the genes encoding troponin I and troponin T reflecting an evolution of adapter proteins coupling the Ca2+-signaling of contractility. J Mol Evol. 1999;49:780–788. doi: 10.1007/pl00006600. [DOI] [PubMed] [Google Scholar]
- Iorga B, Blaudeck N, Solzin J, Neulen A, Stehle I, Davila AJL, Pfitzer G, Stehle R. Lys184 deletion in troponin I impairs relaxation kinetics and induces hypercontractility in murine cardiac myofibrils. Cardiovasc Res. 2008;77:676–686. doi: 10.1093/cvr/cvm113. [DOI] [PubMed] [Google Scholar]
- Januzzi JL, Jr, Filippatos G, Nieminen M, Gheorghiade M. Troponin elevation in patients with heart failure: on behalf of the third Universal Definition of Myocardial Infarction Global Task Force: Heart Failure Section. Eur Heart J. 2012;33:2265–71. doi: 10.1093/eurheartj/ehs191. [DOI] [PubMed] [Google Scholar]
- Jean-Charles PY, Li Y, Nan C, Gobara N, Huang X. Cardiac Troponin I C-Terminal Mutations (K178E and K179E) Cause Severe Heart Failure and Early Mortality in Transgenic Mice. Biophys J. 2012;102:560a. [Google Scholar]
- Jiang M, Zhao X, Han W, Bian C, Li X, Wang G, Ao Y, Li Y, Yi D, Zhe Y, Lo WH, Zhang X, Li J. A novel deletion in TNNI2 causes distal arthrogryposis in a large Chinese family with marked variability of expression. Hum Genet. 2006;120:238–42. doi: 10.1007/s00439-006-0183-4. [DOI] [PubMed] [Google Scholar]
- Jin JP. Alternative RNA splicing-generated cardiac troponin T isoform switching: a non-heart-restricted genetic programming synchronized in developing cardiac and skeletal muscles. Biochem Biophys Res Commun. 1996;225:883–9. doi: 10.1006/bbrc.1996.1267. [DOI] [PubMed] [Google Scholar]
- Jin JP, Yang FW, Yu ZB, Ruse CI, Bond M, Chen A. The highly conserved COOH terminus of troponin I forms a Ca2+-modulated allosteric domain in the troponin complex. Biochemistry. 2001;40:2623–31. doi: 10.1021/bi002423j. [DOI] [PubMed] [Google Scholar]
- Jin JP, Zhang Z, Bautista JA. Isoform diversity, regulation, and functional adaptation of troponin and calponin. Crit Rev Eukaryot Gene Expr. 2008;18:93–124. doi: 10.1615/critreveukargeneexpr.v18.i2.10. [DOI] [PubMed] [Google Scholar]
- Ju Y, Li J, Xie C, Ritchlin CT, Xing L, Hilton MJ, Schwarz EM. Troponin T3 expression in skeletal and smooth muscle is required for growth and postnatal survival: characterization of Tnnt3(tm2a(KOMP)Wtsi) mice. Genesis. 2013;51:667–75. doi: 10.1002/dvg.22407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y, Wang L, Pyle WG, de Tombe PP, Solaro RJ. Intracellular localization and functional effects of P21-activated kinase-1 (Pak1) in cardiac myocytes. Circ Res. 2004;94:194–200. doi: 10.1161/01.RES.0000111522.02730.56. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Kudej RK, Yatani A, Kim YK, Takagi G, Honda R, Colantonio DA, Van Eyk JE, Vatner DE, Rasmusson RL. A novel mechanism for myocardial stunning involving impaired Ca2+ handling. Circ Res. 2001;89:831–837. doi: 10.1161/hh2101.098547. [DOI] [PubMed] [Google Scholar]
- Kimber E, Tajsharghi H, Kroksmark AK, Oldfors A, Tulinius M. A mutation in the fast skeletal muscle troponin I gene causes myopathy and distal arthrogryposis. Neurology. 2006;67:597–601. doi: 10.1212/01.wnl.0000230168.05328.f4. [DOI] [PubMed] [Google Scholar]
- Kimura A, Harada H, Park JE, Nishi H, Satoh M, Takahashi M, Hiroi S, Sasaoka T, Ohbuchi N, Nakamura T. Mutations in the cardiac troponin I gene associated with hypertrophic cardiomyopathy. Nat Genet. 1997;16:379–382. doi: 10.1038/ng0897-379. [DOI] [PubMed] [Google Scholar]
- Kinoshita S, Adachi W, Sotozono C, Nishida K, Yokoi N, Quantock AJ, Okubo K. Characteristics of the human ocular surface epithelium. Prog Retin Eye Res. 2001;20:639–73. doi: 10.1016/s1350-9462(01)00007-6. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Yang X, Walker LA, Van Breemen RB, Solaro RJ. A non-equilibrium isoelectric focusing method to determine states of phosphorylation of cardiac troponin I: identification of Ser-23 and Ser-24 as significant sites of phosphorylation by protein kinase C. J Mol Cell Cardiol. 2005;38:213–218. doi: 10.1016/j.yjmcc.2004.10.014. [DOI] [PubMed] [Google Scholar]
- Lamb GD, Westerblad H. Acute effects of reactive oxygen and nitrogen species on the contractile function of skeletal muscle. J Physiol. 2011;589:2119–27. doi: 10.1113/jphysiol.2010.199059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland J, Li JM, Shah AM. Role of cyclic GMP-dependent protein kinase in the contractile response to exogenous nitric oxide in rat cardiac myocytes. J Physiol. 2002;540:457–67. doi: 10.1113/jphysiol.2001.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovasc Res. 2005;66:12–21. doi: 10.1016/j.cardiores.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Lee H, Park JS, Kim SJ, Cho YJ, Yoon HI, Lee JH, Lee CT. Cardiac troponin I as a prognostic factor in critically ill pneumonia patients in the absence of acute coronary syndrome. J Crit Care. 2015;30:390–4. doi: 10.1016/j.jcrc.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Li MX, Hwang PM. Structure and function of cardiac troponin C (TNNC1): Implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene. 2015;571:153–166. doi: 10.1016/j.gene.2015.07.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li MX, Wang X, Lindhout DA, Buscemi N, Van Eyk JE, Sykes BD. Phosphorylation and mutation of human cardiac troponin I deferentially destabilize the interaction of the functional regions of troponin I with troponin C. Biochemistry. 2003a;42:14460–14468. doi: 10.1021/bi035408y. [DOI] [PubMed] [Google Scholar]
- Li MX, Wang X, Sykes BD. Structural based insights into the role of troponin in cardiac muscle pathophysiology. J Muscle Res Cell Motil. 2004;25:559–79. doi: 10.1007/s10974-004-5879-2. [DOI] [PubMed] [Google Scholar]
- Li Q, Shen PY, Wu G, Chen XZ. Polycystin-2 interacts with troponin I, an angiogenesis inhibitor. Biochemistry. 2003b;42:450–7. doi: 10.1021/bi0267792. [DOI] [PubMed] [Google Scholar]
- Li Y, Charles PY, Nan C, Pinto JR, Wang Y, Liang J, Wu G, Tian J, Feng HZ, Potter JD, Jin JP, Huang X. Correcting diastolic dysfunction by Ca2+ desensitizing troponin in a transgenic mouse model of restrictive cardiomyopathy. J Mol Cell Cardiol. 2010;49:402–11. doi: 10.1016/j.yjmcc.2010.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Chen B, Chen J, Lou G, Chen S, Zhou D. Fast skeletal muscle troponin I is a co-activator of estrogen receptor-related receptor alpha. Biochem Biophys Res Commun. 2008;369:1034–40. doi: 10.1016/j.bbrc.2008.02.147. [DOI] [PubMed] [Google Scholar]
- Lindhout DA, Li MX, Schieve D, Sykes BD. Effects of T142 phosphorylation and mutation R145G on the interaction of the inhibitory region of human cardiac troponin I with the C-domain of human cardiac troponin C. Biochemistry. 2002;41:7267–7274. doi: 10.1021/bi020100c. [DOI] [PubMed] [Google Scholar]
- Lu QW, Hinken AC, Patrick SE, Solaro RJ, Kobayashi T. Phosphorylation of cardiac troponin I at protein kinase C site threonine 144 depresses cooperative activation of thin filaments. J Biol Chem. 2010;285:11810–11817. doi: 10.1074/jbc.M109.055657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüss H, Meissner A, Rolf N, Van Aken H, Bokník P, Kirchhefer U, Knapp J, Läer S, Linck B, Lüss I. Biochemical mechanism (s) of stunning in conscious dogs. Am J Physiol Heart Circ Physiol. 2000;279:H176–H184. doi: 10.1152/ajpheart.2000.279.1.H176. [DOI] [PubMed] [Google Scholar]
- MacGowan GA, Du C, Cowan DB, Stamm C, McGowan FX, Solaro RJ, Koretsky AP, Del Nido PJ. Ischemic dysfunction in transgenic mice expressing troponin I lacking protein kinase C phosphorylation sites. Am J Physiol Heart Circ Physiol. 2001;280:H835–H843. doi: 10.1152/ajpheart.2001.280.2.H835. [DOI] [PubMed] [Google Scholar]
- McDonough J, Labugger R, Pickett W, Tse M, MacKenzie S, Pang S, Atar D, Ropchan G, Van Eyk J. Cardiac troponin I is modified in the myocardium of bypass patients. Circulation. 2001;103:58–64. doi: 10.1161/01.cir.103.1.58. [DOI] [PubMed] [Google Scholar]
- McDonough JL, Arrell DK, Van Eyk JE. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ Res. 1999;84:9–20. doi: 10.1161/01.res.84.1.9. [DOI] [PubMed] [Google Scholar]
- Messer AE, Jacques AM, Marston SB. Troponin phosphorylation and regulatory function in human heart muscle: dephosphorylation of Ser23/24 on troponin I could account for the contractile defect in end-stage heart failure. J Mol Cell Cardiol. 2007;42:247–259. doi: 10.1016/j.yjmcc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Messner B, Baum H, Fischer P, Quasthoff S, Neumeier D. Expression of messenger RNA of the cardiac isoforms of troponin T and I in myopathic skeletal muscle. Am J Clin Pathol. 2000;114:544–9. [PubMed] [Google Scholar]
- Millat G, Bouvagnet P, Chevalier P, Sebbag L, Dulac A, Dauphin C, Jouk PS, Delrue MA, Thambo JB, Le Metayer P, Seronde MF, Faivre L, Eicher JC, Rousson R. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur J Med Genet. 2011;54:e570–5. doi: 10.1016/j.ejmg.2011.07.005. [DOI] [PubMed] [Google Scholar]
- Mogensen J, Kubo T, Duque M, Uribe W, Shaw A, Murphy R, Gimeno JR, Elliott P, McKenna WJ. Idiopathic restrictive cardiomyopathy is part of the clinical expression of cardiac troponin I mutations. J Clin Invest. 2003;111:209–16. doi: 10.1172/JCI16336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica JP, Dutka TL, Merry TL, Lamboley CR, McConell GK, McKenna MJ, Murphy RM, Lamb GD. S-glutathionylation of troponin I (fast) increases contractile apparatus Ca2+ sensitivity in fast-twitch muscle fibres of rats and humans. J Physiol. 2012;590:1443–63. doi: 10.1113/jphysiol.2011.224535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran CM, Garriock RJ, Miller MK, Heimark RL, Gregorio CC, Krieg PA. Expression of the fast twitch troponin complex, fTnT, fTnI and fTnC, in vascular smooth muscle. Cell Motil Cytoskelet. 2008;65:652–61. doi: 10.1002/cm.20291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses MA, Wiederschain D, Wu I, Fernandez CA, Ghazizadeh V, Lane WS, Flynn E, Sytkowski A, Tao T, Langer R. Troponin I is present in human cartilage and inhibits angiogenesis. Proc Natl Acad Sci U S A. 1999;96:2645–50. doi: 10.1073/pnas.96.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy AM, Kögler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marbán E. Transgenic Mouse Model of Stunned Myocardium. Science. 2000;287:488–491. doi: 10.1126/science.287.5452.488. [DOI] [PubMed] [Google Scholar]
- Murphy RM, Dutka TL, Lamb GD. Hydroxyl radical and glutathione interactions alter calcium sensitivity and maximum force of the contractile apparatus in rat skeletal muscle fibres. J Physiol. 2008;586:2203–16. doi: 10.1113/jphysiol.2007.150516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narolska NA, Piroddi N, Belus A, Boontje NM, Scellini B, Deppermann S, Zaremba R, Musters RJ, dos Remedios C, Jaquet K. Impaired diastolic function after exchange of endogenous troponin I with C-terminal truncated troponin I in human cardiac muscle. Circ Res. 2006;99:1012–1020. doi: 10.1161/01.RES.0000248753.30340.af. [DOI] [PubMed] [Google Scholar]
- Nixon BR, Walton SD, Zhang B, Brundage EA, Little SC, Ziolo MT, Davis JP, Biesiadecki BJ. Combined troponin I Ser-150 and Ser-23/24 phosphorylation sustains thin filament Ca(2+) sensitivity and accelerates deactivation in an acidic environment. J Mol Cell Cardiol. 2014;72:177–85. doi: 10.1016/j.yjmcc.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira SM, Zhang YH, Solis RS, Isackson H, Bellahcene M, Yavari A, Pinter K, Davies JK, Ge Y, Ashrafian H. AMP-Activated Protein Kinase Phosphorylates Cardiac Troponin I and Alters Contractility of Murine Ventricular MyocytesNovelty and Significance. Circ Res. 2012;110:1192–1201. doi: 10.1161/CIRCRESAHA.111.259952. [DOI] [PubMed] [Google Scholar]
- Palpant NJ, Houang EM, Delport W, Hastings KEM, Onufriev AV, Sham YY, Metzger JM. Pathogenic peptide deviations support a model of adaptive evolution of chordate cardiac performance by troponin mutations. Physiol Genomics. 2010;42:287–299. doi: 10.1152/physiolgenomics.00033.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry S. Troponin I: inhibitor or facilitator. Mol Cell Biochem. 1999;190:9–32. [PubMed] [Google Scholar]
- Pi YQ, Kemnitz KR, Zhang D, Kranias EG, Walker JW. Phosphorylation of Troponin I Controls Cardiac Twitch Dynamics Evidence From Phosphorylation Site Mutants Expressed on a Troponin I-Null Background in Mice. Circ Res. 2002;90:649–656. doi: 10.1161/01.res.0000014080.82861.5f. [DOI] [PubMed] [Google Scholar]
- Pi YQ, Zhang D, Kemnitz KR, Wang H, Walker JW. Protein kinase C and A sites on troponin I regulate myofilament Ca2+ sensitivity and ATPase activity in the mouse myocardium. J Physiol. 2003;552:845–857. doi: 10.1113/jphysiol.2003.045260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quirk PG, Patchell VB, Gao Y, Levine BA, Victor Perry S. Sequential phosphorylation of adjacent serine residues on the N-terminal region of cardiac troponin-I: Structure-activity implications of ordered phosphorylation. FEBS Lett. 1995;370:175–178. doi: 10.1016/0014-5793(95)00812-n. [DOI] [PubMed] [Google Scholar]
- Ramirez-Correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, Vecoli C, Hart GW, Murphy AM. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res. 2008;103:1354–8. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao V, Cheng Y, Lindert S, Wang D, Oxenford L, McCulloch AD, McCammon JA, Regnier M. PKA phosphorylation of cardiac troponin I modulates activation and relaxation kinetics of ventricular myofibrils. Biophys J. 2014;107:1196–204. doi: 10.1016/j.bpj.2014.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao VS, Korte FS, Razumova MV, Feest ER, Hsu H, Irving TC, Regnier M, Martyn DA. N - terminal phosphorylation of cardiac troponin - I reduces length dependent calcium sensitivity of contraction in cardiac muscle. The Journal of physiology. 2012;591:475–490. doi: 10.1113/jphysiol.2012.241604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds T, Cecconi M, Collinson P, Rhodes A, Grounds RM, Hamilton MA. Raised serum cardiac troponin I concentrations predict hospital mortality in intensive care unit patients. Br J Anaesth. 2012;109:219–24. doi: 10.1093/bja/aes141. [DOI] [PubMed] [Google Scholar]
- Ricchiuti V, Apple FS. RNA expression of cardiac troponin T isoforms in diseased human skeletal muscle. Clin Chem. 1999;45:2129–35. [PubMed] [Google Scholar]
- Rittoo D, Jones A, Lecky B, Neithercut D. Elevation of cardiac troponin T, but not cardiac troponin I, in patients with neuromuscular diseases: implications for the diagnosis of myocardial infarction. J Am Coll Cardiol. 2014;63:2411–20. doi: 10.1016/j.jacc.2014.03.027. [DOI] [PubMed] [Google Scholar]
- Robinson P, Lipscomb S, Preston LC, Altin E, Watkins H, Ashley CC, Redwood CS. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007;21:896–905. doi: 10.1096/fj.06-6899com. [DOI] [PubMed] [Google Scholar]
- Saggin L, Gorza L, Ausoni S, Schiaffino S. Troponin I switching in the developing heart. J Biol Chem. 1989;264:16299–16302. [PubMed] [Google Scholar]
- Salhi HE, Walton SD, Hassel NC, Brundage EA, de Tombe PP, Janssen PM, Davis JP, Biesiadecki BJ. Cardiac troponin I tyrosine 26 phosphorylation decreases myofilament Ca2+ sensitivity and accelerates deactivation. J Mol Cell Cardiol. 2014;76:257–64. doi: 10.1016/j.yjmcc.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho Solis R, Ge Y, Walker JW. A preferred AMPK phosphorylation site adjacent to the inhibitory loop of cardiac and skeletal troponin I. Protein Sci. 2011;20:894–907. doi: 10.1002/pro.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasse S, Brand N, Kyprianou P, Dhoot G, Wade R, Arai M, Periasamy M, Yacoub M, Barton P. Troponin I gene expression during human cardiac development and in end-stage heart failure. Circ Res. 1993;72:932–938. doi: 10.1161/01.res.72.5.932. [DOI] [PubMed] [Google Scholar]
- Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001;104:557–67. doi: 10.1016/s0092-8674(01)00242-2. [DOI] [PubMed] [Google Scholar]
- Sheng JJ, Jin JP. Gene regulation, alternative splicing, and posttranslational modification of troponin subunits in cardiac development and adaptation: a focused review. Front Physiol. 2014;5:165. doi: 10.3389/fphys.2014.00165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman AJ, Klocke FJ, Decker RS, Decker ML, Kozlowski KA, Harris KR, Hedjbeli S, Yaroshenko Y, Nakamura S, Parker MA. Myofibrillar disruption in hypocontractile myocardium showing perfusion-contraction matches and mismatches. Am J Physiol Heart Circ Physiol. 2000;278:H1320–H1334. doi: 10.1152/ajpheart.2000.278.4.H1320. [DOI] [PubMed] [Google Scholar]
- Simpson JA, Labugger R, Collier C, Brison RJ, Iscoe S, Van Eyk JE. Fast and slow skeletal troponin I in serum from patients with various skeletal muscle disorders: a pilot study. Clin Chem. 2005;51:966–72. doi: 10.1373/clinchem.2004.042671. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem. 2011;286:9935–9940. doi: 10.1074/jbc.R110.197731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ, Lee JA, Kentish JC, Allen DG. Effects of acidosis on ventricular muscle from adult and neonatal rats. Circ Res. 1988;63:779–787. doi: 10.1161/01.res.63.4.779. [DOI] [PubMed] [Google Scholar]
- Solaro RJ, Rosevear P, Kobayashi T. The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem Biophys Res Commun. 2008;369:82–87. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solaro RJ, van der Velden J. Why does troponin I have so many phosphorylation sites? Fact and fancy. J Mol Cell Cardiol. 2010;48:810–816. doi: 10.1016/j.yjmcc.2010.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stelzer JE, Patel JR, Walker JW, Moss RL. Differential roles of cardiac myosin-binding protein C and cardiac troponin I in the myofibrillar force responses to protein kinase A phosphorylation. Circ Res. 2007;101:503–511. doi: 10.1161/CIRCRESAHA.107.153650. [DOI] [PubMed] [Google Scholar]
- Stevens L, Bastide B, Kischel P, Pette D, Mounier Y. Time-dependent changes in expression of troponin subunit isoforms in unloaded rat soleus muscle. Am J Physiol Cell Physiol. 2002;282:C1025–30. doi: 10.1152/ajpcell.00252.2001. [DOI] [PubMed] [Google Scholar]
- Sung SS, Brassington AM, Grannatt K, Rutherford A, Whitby FG, Krakowiak PA, Jorde LB, Carey JC, Bamshad M. Mutations in genes encoding fast-twitch contractile proteins cause distal arthrogryposis syndromes. Am J Hum Genet. 2003;72:681–90. doi: 10.1086/368294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglieri DM, Monasky MM, Knezevic I, Sheehan KA, Lei M, Wang X, Chernoff J, Wolska BM, Ke Y, Solaro RJ. Ablation of p21-activated kinase-1 in mice promotes isoproterenol-induced cardiac hypertrophy in association with activation of Erk1/2 and inhibition of protein phosphatase 2A. J Mol Cell Cardiol. 2011;51:988–996. doi: 10.1016/j.yjmcc.2011.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- Thomas SA, Fallavollita JA, Lee TC, Feng J, Canty JM. Absence of troponin I degradation or altered sarcoplasmic reticulum uptake protein expression after reversible ischemia in swine. Circ Res. 1999;85:446–456. doi: 10.1161/01.res.85.5.446. [DOI] [PubMed] [Google Scholar]
- Van der Velden J, Merkus D, Klarenbeek B, James A, Boontje N, Dekkers D, Stienen G, Lamers J, Duncker D. Alterations in myofilament function contribute to left ventricular dysfunction in pigs early after myocardial infarction. Circ Res. 2004;95:e85–e95. doi: 10.1161/01.RES.0000149531.02904.09. [DOI] [PubMed] [Google Scholar]
- Vinogradova MV, Stone DB, Malanina GG, Karatzaferi C, Cooke R, Mendelson RA, Fletterick RJ. Ca2+-regulated structural changes in troponin. Proc Natl Acad Sci U S A. 2005;102:5038–5043. doi: 10.1073/pnas.0408882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Chalovich JM, Marriott G. Structural dynamics of troponin I during Ca2+-activation of cardiac thin filaments: a multi-site Forster resonance energy transfer study. PLoS ONE. 2012a;7:e50420. doi: 10.1371/journal.pone.0050420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Grant JE, Doede CM, Sadayappan S, Robbins J, Walker JW. PKC-betaII sensitizes cardiac myofilaments to Ca2+ by phosphorylating troponin I on threonine-144. J Mol Cell Cardiol. 2006;41:823. doi: 10.1016/j.yjmcc.2006.08.016. [DOI] [PubMed] [Google Scholar]
- Wang Y, Pinto JR, Solis RS, Dweck D, Liang J, Diaz-Perez Z, Ge Y, Walker JW, Potter JD. Generation and Functional Characterization of Knock-in Mice Harboring the Cardiac Troponin I-R21C Mutation Associated with Hypertrophic Cardiomyopathy. J Biol Chem. 2012b;287:2156–2167. doi: 10.1074/jbc.M111.294306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DG, Ashton PR, Trayer HR, Trayer IP. Additional PKA phosphorylation sites in human cardiac troponin I. Eur J Biochem. 2001;268:179–185. doi: 10.1046/j.1432-1327.2001.01871.x. [DOI] [PubMed] [Google Scholar]
- Warkman AS, Atkinson BG. The slow isoform of Xenopus troponin I is expressed in developing skeletal muscle but not in the heart. Mech Dev. 2002;115:143–6. doi: 10.1016/s0925-4773(02)00096-5. [DOI] [PubMed] [Google Scholar]
- Wei B, Gao J, Huang XP, Jin JP. Mutual rescues between two dominant negative mutations in cardiac troponin I and cardiac troponin T. J Biol Chem. 2010;285:27806–16. doi: 10.1074/jbc.M110.137844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei B, Jin JP. Troponin T isoforms and posttranscriptional modifications: Evolution, regulation and function. Arch Biochem Biophys. 2011;505:144–54. doi: 10.1016/j.abb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei H, Jin JP. N-Terminal Truncated Cardiac Troponin I Enhanced the Contractility of Isolated Cardiomyocytes. Biophys J. 2013;104:154a–155a. [Google Scholar]
- Wen Y, Pinto JR, Gomes AV, Xu Y, Wang Y, Potter JD, Kerrick WG. Functional consequences of the human cardiac troponin I hypertrophic cardiomyopathy mutation R145G in transgenic mice. J Biol Chem. 2008;283:20484–94. doi: 10.1074/jbc.M801661200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Y, Xu Y, Wang Y, Pinto JR, Potter JD, Kerrick WG. Functional effects of a restrictive-cardiomyopathy-linked cardiac troponin I mutation (R145W) in transgenic mice. J Mol Biol. 2009;392:1158–67. doi: 10.1016/j.jmb.2009.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall MV, Albayya FP, Metzger JM. Functional analysis of troponin I regulatory domains in the intact myofilament of adult single cardiac myocytes. J Biol Chem. 1999;274:22508–22516. doi: 10.1074/jbc.274.32.22508. [DOI] [PubMed] [Google Scholar]
- Westfall MV, Albayya FP, Turner II, Metzger JM. Chimera analysis of troponin I domains that influence Ca2+-activated myofilament tension in adult cardiac myocytes. Circ Res. 2000;86:470–477. doi: 10.1161/01.res.86.4.470. [DOI] [PubMed] [Google Scholar]
- Westfall MV, Lee AM, Robinson DA. Differential Contribution of Troponin I Phosphorylation Sites to the Endothelin-modulated Contractile Response. J Biol Chem. 2005;280:41324–41331. doi: 10.1074/jbc.M506043200. [DOI] [PubMed] [Google Scholar]
- Westfall MV, Rust EM, Metzger JM. Slow skeletal troponin I gene transfer, expression, and myofilament incorporation enhances adult cardiac myocyte contractile function. Proc Natl Acad Sci U S A. 1997;94:5444–9. doi: 10.1073/pnas.94.10.5444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnker PJ, Li Y, Zhang P, Foster DB, Remedios CD, Van Eyk JE, Stienen GJ, Murphy AM, van der Velden J. A novel phosphorylation site, Serine 199, in the C-terminus of cardiac troponin I regulates calcium sensitivity and susceptibility to calpain-induced proteolysis. J Mol Cell Cardiol. 2015;82:93–103. doi: 10.1016/j.yjmcc.2015.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willott RH, Gomes AV, Chang AN, Parvatiyar MS, Pinto JR, Potter JD. Mutations in Troponin that cause HCM, DCM AND RCM: What can we learn about thin filament function? J Mol Cell Cardiol. 2010;48:882–892. doi: 10.1016/j.yjmcc.2009.10.031. [DOI] [PubMed] [Google Scholar]
- You B, Yan G, Zhang Z, Yan L, Li J, Ge Q, Jin JP, Sun J. Phosphorylation of cardiac troponin I by mammalian sterile 20-like kinase 1. Biochem J. 2009;418:93–101. doi: 10.1042/BJ20081340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ZB, Zhang LF, Jin JP. A proteolytic NH2-terminal truncation of cardiac troponin I that is up-regulated in simulated microgravity. J Biol Chem. 2001;276:15753–60. doi: 10.1074/jbc.M011048200. [DOI] [PubMed] [Google Scholar]
- Zakhary DR, Moravec CS, Stewart RW, Bond M. Protein kinase A (PKA)-dependent troponin-I phosphorylation and PKA regulatory subunits are decreased in human dilated cardiomyopathy. Circulation. 1999;99:505–510. doi: 10.1161/01.cir.99.4.505. [DOI] [PubMed] [Google Scholar]
- Zhang P, Kirk JA, Ji W, dos Remedios CG, Kass DA, Van Eyk JE, Murphy AM. Multiple Reaction Monitoring to Identify Site-Specific Troponin I Phosphorylated Residues in the Failing Human Heart/Clinical Perspective. Circulation. 2012;126:1828–1837. doi: 10.1161/CIRCULATIONAHA.112.096388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Zhao J, Mandveno A, Potter JD. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995a;76:1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- Zhang R, Zhao JJ, Potter JD. Phosphorylation of both serine residues in cardiac troponin I is required to decrease the Ca affinity of cardiac troponin C. J Biol Chem. 1995b;270:30773–30780. doi: 10.1074/jbc.270.51.30773. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Akhter S, Mottl S, Jin JP. Calcium-regulated conformational change in the C-terminal end segment of troponin I and its binding to tropomyosin. FEBS J. 2011;278:3348–59. doi: 10.1111/j.1742-4658.2011.08250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zile MR, Brutsaert DL. New concepts in diastolic dysfunction and diastolic heart failure: Part II: causal mechanisms and treatment. Circulation. 2002;105:1503–8. doi: 10.1161/hc1202.105290. [DOI] [PubMed] [Google Scholar]