

Abstract
INTRODUCTION
The attenuated mycobacterium Bacille Calmette Guerin (BCG) is widely utilized as intravesical “immunotherapy” for the treatment of non-muscle invasive urothelial carcinoma. Previous studies have demonstrated that in both the laboratory and clinical setting, BCG viability is a variable that correlates with anti-tumor efficacy. This study evaluated how loss of BCG viability impacted a number of molecular and phenotypic intermediate endpoints that characterize, and/or contribute to, the direct effect of BCG on Urothelial carcinoma (UC) cells.
MATERIALS AND METHODS
Two human UC cell lines were used to study the effect of loss of BCG viability on the tumor cell response to BCG. The cellular response to BCG rendered non-viable by heat killing (hk) was compared to the response to viable BCG. The response endpoints evaluated included the induction of oxidative stress, activation of intracellular signaling pathways, gene transactivation, and phenotypic changes.
RESULTS
Loss of viability resulted in a quantitative decrease in the tumor cell response, relative to viable BCG, for all of the measured endpoints. The decrease in response varied by cell line, ranging from 15% to 100% of the response to viable BCG. While quantitatively different, non-viable BCG continued to induce responses that were qualitatively similar to BCG relative to untreated controls.
CONCLUSIONS
BCG viability is an important variable influencing the direct tumor cell response to BCG. Although the magnitude of it effects are attenuated, hkBCG remains active for the induction of BCG responsive biologic endpoints.
Keywords: Bladder cancer, BCG, viability, Heat killed BCG
Introduction
The intravesical administration of the attenuated mycobacterium BCG is currently the treatment benchmark in preventing the recurrence and progression of NMIBC.1,2,3 In both clinical and laboratory studies, BCG treatment efficacy is directly correlated with BCG viability.4,5,6 Our group has demonstrated that loss of viability reduces BCG induced cellular necrosis and High mobility group box 1 (HMGB1) release.7 In animal model systems release of HMGB1 by BCG exposed UC cells is required for the in vivo anti-tumor effect of BCG.8
Cellular and molecular mechanism of BCG therapy can be roughly separated in to interaction of BCG with tumor cells and the host immune response to the BCG exposed bladder. Direct exposure of BCG to UC cell results in complex changes in UC cell biology. BCG induced oxidative stress activates multiple stress responsive intracellular signaling pathways resulting in gene transactivation and subsequent alterations in tumor biology. In addition, this sequence triggers release of cytokines and chemokines from these cells, resulting in recruitment of various types of immune cells into the bladder.9 Through a combination of both direct and systemic effects, the interaction between UC cell and BCG plays a critical role in the ultimate response to treatment.
Presently there is limited understanding of the mechanism through which loss of viability alters the direct antitumor effect. Given the central role of the tumor cell response to BCG in treatment efficacy, this study evaluated the effect of BCG viability on molecular and phenotypic endpoints characterizing the UC cell response to BCG. Our results demonstrate that the UC cell response was compromised by loss of BCG viability. However, non-viable BCG retained activity, albeit attenuated, for inducing a direct cellular response. These qualitative similarities suggest that viability is not absolutely linked to efficacy. These observations represent an opportunity to combine non-viable BCG with “adjuvant” strategies to achieve therapeutic efficacy while eliminating the toxicity risks associated with viable BCG.
2. Materials and Methods
2.1 Cell Lines
The human UC cell line T24 was obtained from American Type Cell Culture (Rockville, MD). The 253J cell line was a kind gift of Dr. Richard Williams (University of Iowa). Cells were maintained as previously described.8
2.2 Bacillus Calmette-Guerin (BCG)
TICE BCG, were used in these experiments (Organon Inc, West Orange, NJ). Freeze dried BCG was reconstituted in complete media at an estimated concentration of 2.5 ×107 viable organisms/ml. HkBCG was prepared by incubating BCG at 80°C for 1h. Loss of viability was confirmed by plating hkBCG culture on 7H10 Middlebrooks agar medium and incubating plates at 37°C for 4 weeks. For hkBCG group, no colonies were observed, with multiple colonies observed for live BCG after 4 weeks.
2.3 Quantitative rtPCR
The effect of BCG viability on gene expression in response to BCG was measured using previously described methods and primers.8, 10 Treatment groups in the comparative analysis included control cells, and cells treated with either viable BCG or hkBCG (50:1 BCG: cells) β-actin was used to normalize all other genes tested in the same RNA sample.
2.4 Real time measurement of nitric oxide in UC cells
253J and T24 cells were plated at 1 × 104 cells/well in 96-well plates. 24 hours later, BCG and hkBCG (50:1 BCG: cells) was added to the cultures. The cells were incubated for 6 h and washed twice with sterile PBS. Intracellular NO levels were measured using fluorescence probe 4,5-Diaminofluorescein Diacetate (DAF-2DA, Calbiochem, Darmstadt, Germany) with excitation at 485 nm and emission at 535nm.
2.5 Luciferase reporter assays
The effect of BCG viability on signaling pathway activation in response to BCG was measured as described earlier.8, 10
2.6 Dye exclusion assay for cell viability
Dye exclusion assay was done as described earlier.8 Treatment groups included control cells, and cells treated with either viable BCG or hkBCG (50:1 BCG: cells).
2.7 LDH Release Assay
Lactate dehydrogenase (LDH) is a stable cytosolic enzyme, which is released upon cell lysis. Briefly, 253J and T24 cells were seeded in 24-well plate. Next day, the medium was replaced and BCG/hkBCG was added to the cultures at a ratio of 1:50 (cell:BCG). 24 hours later, LDH Released into culture supernatants is measured with a CytoTox 96® Non-Radioactive cytotoxicity assay kit (Promega, Madison, WI) according to the manufacturer’s instruction.
2.8 HMGB1 assay
HMGB1 levels in cell culture supernatant 24 hours after BCG treatment were measured by HMGB1 ELISA kit (Shino-Test, Sagamihara, Japan).
2.9 Cell Proliferation
The effect of BCG viability on tumor cell proliferation was measured using the MTT assay as previously described.11
2.10 Statistical Analysis
All experiments were performed in triplicate at three different time points. The data from individual experiments for assays employing luciferase reporter constructs was subjected to arithmetic normalization relative to average relative values from the median experiment. qRT PCR data were normalized using housekeeping gene β-actin expression. Data was analyzed using two way ANOVA for repeated measures. Results were considered significant at p < 0.05. Graphical representation of the data is shown as the mean ± S.E.
Results
3.1 Effect of BCG viability on iNOS and intracellular NO production
Exposure of UC cells to viable BCG increased the expression of iNOS mRNA 8.8- and 4.6-fold in 253J and T24 cells, respectively relative to untreated controls. This increase was reduced to 5.8- and 3.3-fold, respectively in response to hkBCG. The difference in iNOS induction between viable and hkBCG was not statistically significant (p = 0.15). These results are illustrated in Figure 1a.
Figure 1. Effect of BCG viability on iNOS mRNA Expression and NO production.
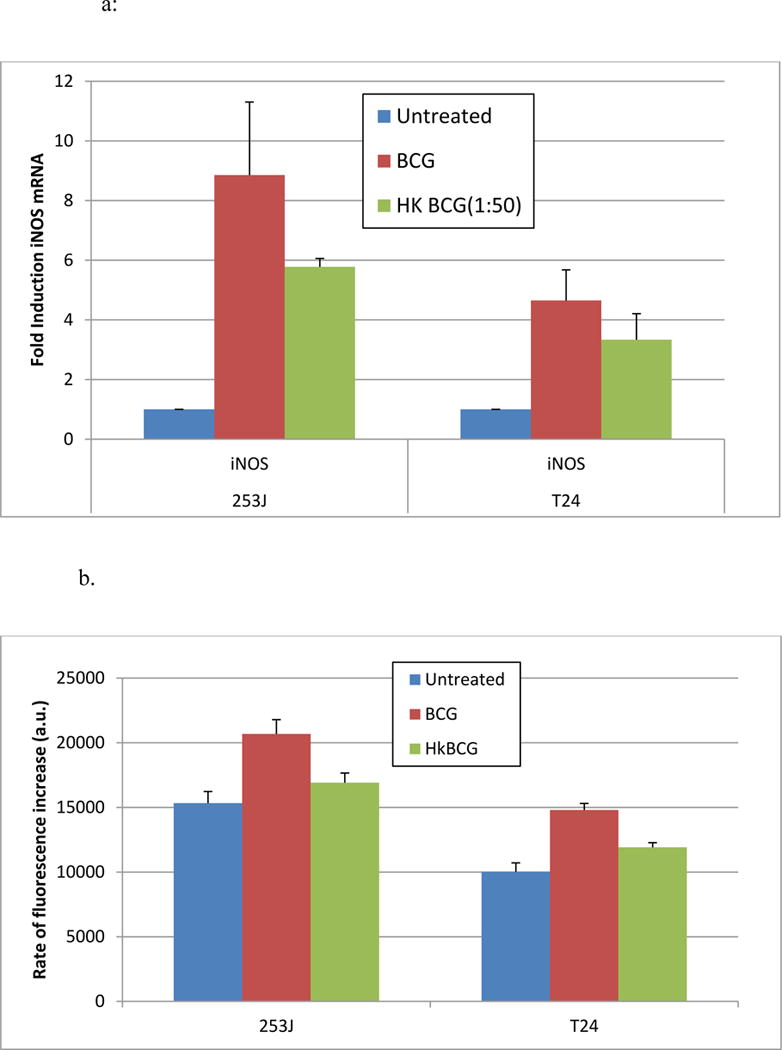
Exposure of UC cells to viable BCG increased the expression of iNOS mRNA 8.8- and 4.6-fold in 253J and T24 cells, respectively. This increase was reduced to 5.8- and 3.3-fold in response to hk BCG. The difference in iNOS induction between viable and hkBCG was not statistically significant (p = 0.15).
Exposure of UC cells to BCG significantly increased NO production in both cell lines (p < 0.05). Relative to controls, BCG treatment significantly increased NO levels in both T24 (p < 0.0001) and 253J cell lines (p < 0.05). Treatment with hk BCG significantly increased NO levels in T24 p < 0.05) but not in 253J p= 0.4). The difference in NO production between hk BCG and BCG was statistically significant (p < 0.05).
Relative to controls, BCG treatment significantly increased NO levels in both T24 (p < 0.0001) and 253J cell lines (p < 0.05). Treatment with hkBCG significantly increased NO levels in T24 (p < 0.05) but not in 253J (p= 0.4). The difference in NO production between hkBCG and BCG was statistically significant (p < 0.05, Figure 1b).
3.2 Activation of Intracellular Signaling pathways
Viable BCG significantly increased the activation of the NRF2 reporter construct 2- and 1.9-fold in 253J and T24 cells, respectively (p < 0.0001). This increase was reduced to 1.6- and 1.5-fold in response to hkBCG but remained significantly increased relative to controls (p < 0.0001). Loss of viability was associated with a statistically significant decrease in NRF2 signaling relative to viable BCG (p < 0.01, Figure 2).
Figure 2. Effect of BCG Viability on signaling pathways.
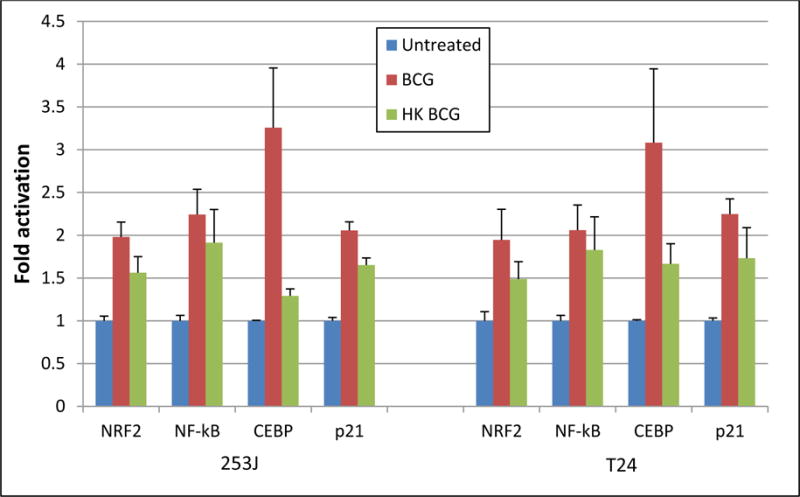
Exposure of UC cells to viable BCG significantly increased the activation of an NRF2, NF-κB, CEBP and p21 reporter constructs in both 253J and T24 cell lines. (ANOVA, p < 0.01). The difference in reporter activation between viable and hkBCG was statistically significant (p < 0.01).
Viable BCG significantly increased the activation of the NF-κB reporter construct 2.2- and 2-fold in 253J and T24 cells, respectively (p < 0.0001). This increase was reduced to 1.9- and 1.8-fold in response to hkBCG but remained significantly increased relative to controls (p < 0.001). The difference in NF-κB signaling between viable and hkBCG was not statistically significant (p = 0.3, Figure 2).
Exposure of UC cells to viable BCG significantly increased the activation of a CEBP reporter construct 3.2- and 3.1-fold in 253J and T24 cells, respectively (p < 0.01). This increase was reduced to 1.3- and 1.7-fold in response to hkBCG and remained significantly increased relative to controls (p < 0.01). Loss of viability was associated with a statistically significant decrease in CEBP signaling (p < 0.05, Figure 2).
3.3 Gene Expression
p21 expression in response to BCG has been shown to be necessary for non-apoptotic cell death and HMGB1 release.12 Exposure of UC cells to viable BCG significantly increased the activation of a p21 promoter-reporter construct 2- and 2.2-fold in 253J and T24 cells, respectively (p < 0.0001). This increase was reduced to 1.7- and 1.7-fold in response to hkBCG but remained significantly increased relative to controls (p < 0.0001). Loss of viability was associated with a statistically significant decrease in the activation of the p21 promoter-reporter construct (p < 0.01). These results are shown in Figure 2 and 3.
Figure 3. Effect of BCG Viability on UC cell Gene Expression in Response to BCG.
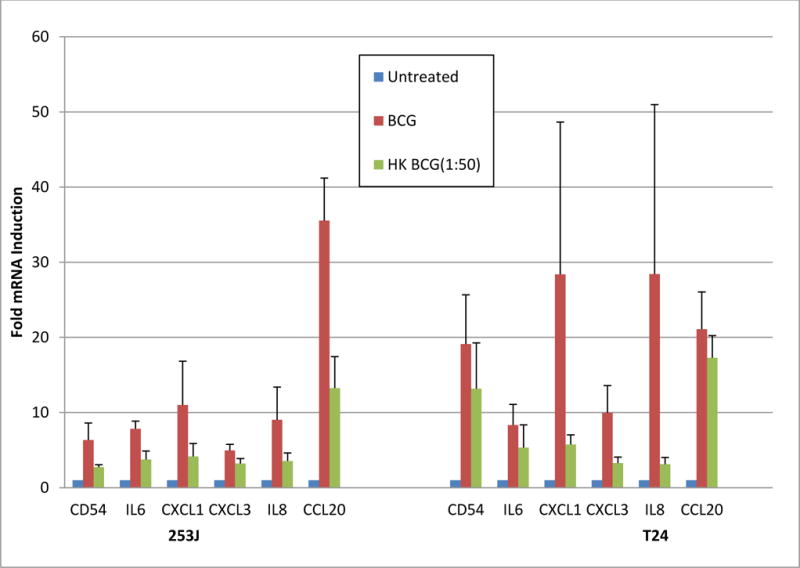
Exposure of UC cells to viable BCG significantly increased the activation of a panel of “immune response” genes in both 253J and T24 cells (ANOVA, p < 0.01). Relative to controls, hkBCG also resulted in significantly increase expression of the 6 evaluated genes (ANOVA, p < 0.01). Expression of the 6 genes in response to hkBCG, relative to viable BCG, was significantly lower in both 253J and T24 cell lines representing 52% and 62% of viable BCG values, respectively (253J p < 0.01; T24 p < 0.05).
Exposure of UC cells to viable BCG significantly increased the activation of a panel of “immune response” genes in both 253J and T24 cells (ANOVA, p < 0.01). Relative to controls hkBCG also resulted in significantly increased expression of the 6 evaluated genes (ANOVA, p < 0.01). Expression of the 6 genes in response to hkBCG, relative to viable BCG, was significantly lower in both 253J and T24 cell lines representing 52% and 62% of viable BCG values, respectively (253J p < 0.01; T24 p < 0.05). The results from this experiment are shown in Figure 3.
3.4 BCG Induced Cytotoxicity
Exposure of UC cells to viable BCG significantly increased the number of cells with loss of cytoplasmic membrane integrity as measured by vital dye exclusion. The number of cells with loss of membrane integrity was increase 14.6- and 12-fold in 253J and T24 cells, respectively relative to untreated controls (p < 0.0001, figure 4). This increase was reduced to 8- and 3.4-fold in response to hkBCG. This effect was significantly increased relative to controls (p < 0.0001) but was significantly lower relative to BCG (p < 0.0001).
Figure 4. Effect of BCG Viability on Cytoplasmic membrane integrity in UC cells.
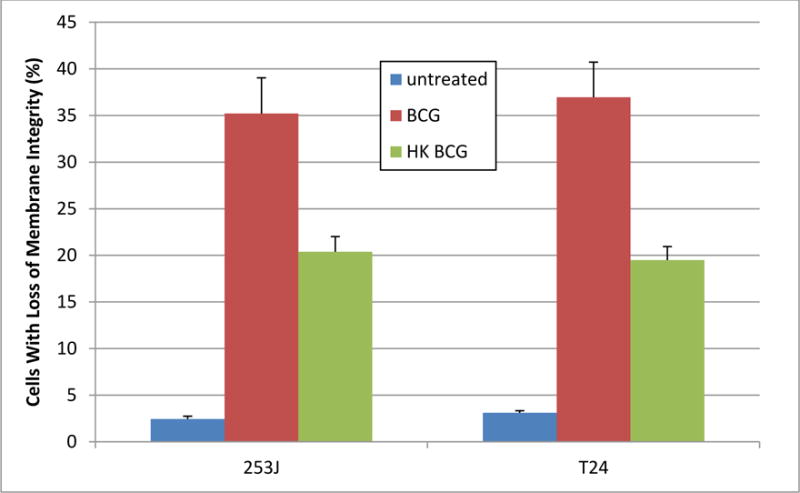
Exposure of UC cells to viable BCG significantly increased the number of cells with loss of cytoplasmic membrane integrity as measured by vital dye exclusion. The number of cells with loss of membrane integrity was increase 14.6- and 12-fold in 253J and T24 cells, respectively (p < 0.0001). This increase was reduced to 8- and 3.4-fold in response to hkBCG but remained significantly increased relative to controls (p < 0.0001). Loss of viability was associated with a statistically significant decrease in the cytotoxicity of BCG as measured by vital dye exclusion (p < 0.0001).
Figure 5 demonstrates the effect of UC cell exposure to BCG or hkBCG on LDH release, a marker of cellular injury. Exposure of UC cells to viable BCG significantly increased LDH release by UC cells. Relative to controls, the magnitude of LHD release was increased 3.5- and 4.2-fold in 253J and T24 cells, respectively (p < 0.0001). In response to hkBCG the increase relative to controls was 3- and 4.2-fold, respectively (p < 0.0001) which was not significantly different relative to viable BCG (p = 0.28).
Figure 5. Effect of BCG Viability on LDH release by UC Cell.
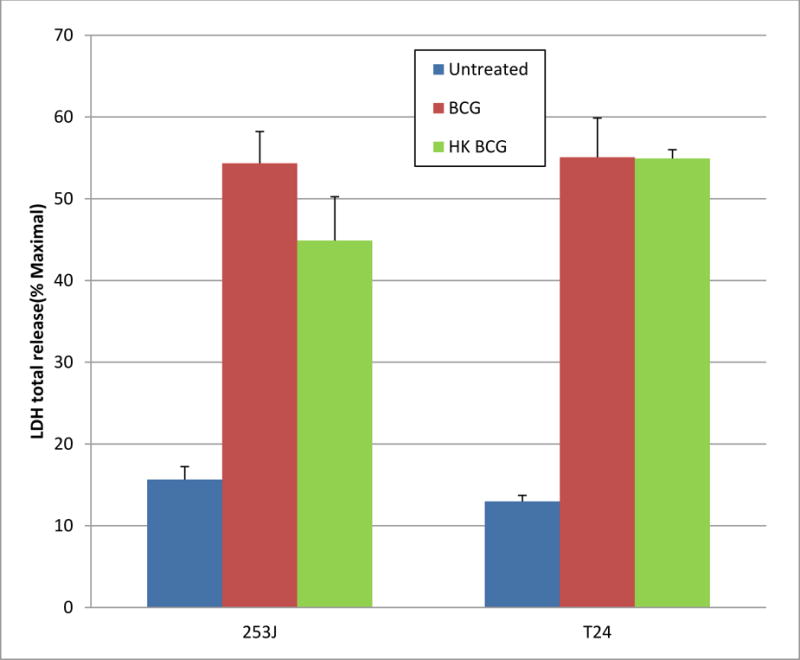
Exposure of UC cells to viable BCG significantly increased LDH release by UC cells. Relative to controls, the magnitude of LHD release was increased 3.5- and 4.2-fold in 253J and T24 cells, respectively (p < 0.0001). In response to hkBCG the increase relative to controls was 3- and 4.2-fold, respectively (p < 0.0001). There was no statistically significant difference in LDH release between viable and hkBCG (p = 0.28).
Treatment with Viable BCG showed 7- and 17-fold increase in HMGB1 release in 253J and T24 cells, respectively. HMGB1 release was 4- and 8-fold higher compare to control in response to hkBCG. The difference in HMGB1 release between viable and hk BCG was statistically significant (p < 0.001; figure 6).
Figure 6. Effect of BCG Viability on HMGB1 release by UC Cell.
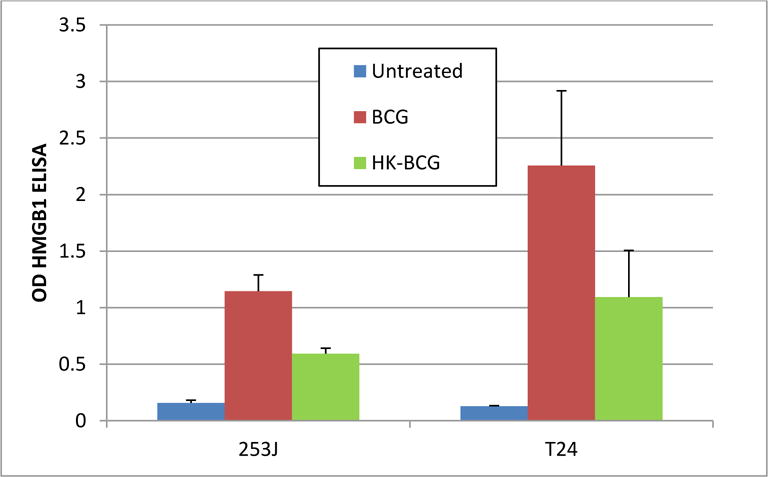
Relative to controls, the magnitude of HMGB1 release was increased 7- and 17-fold in 253J and T24 cells, respectively (p < 0.0001). In response to hkBCG the increase relative to controls was 4- and 8-fold, respectively (p < 0.0001). There was statistically significant difference in HMGB1 release between viable and hkBCG (p ˂0.001).
3.5 Dose-Response Relationship
Given the activity of hkBCG across multiple endpoints experiments were performed to determine if the biologic activity of hkBCG was dose dependent. As measured by MTT assay, BCG alone reduced the number of cells present 72 h following plating 33% and 34% of control values in 253J and T24 cells, respectively (p < 0.0001).11 For hkBCG, the values were 62% and 55% of controls. (p < 0.001). The difference in % metabolically active cells between viable and hkBCG was statistically significant (p < 0.001). When different doses of hkBCG were applied (5:1 to 250:1), there was non-significant dose dependent decrease in metabolically active cells in both 253J and T24 cell line. (p = 0.46 and p = 0.7, respectively). These results are illustrated in Figure 7.
Figure 7. Effect of Viability on the BCG induced anti-proliferative response.
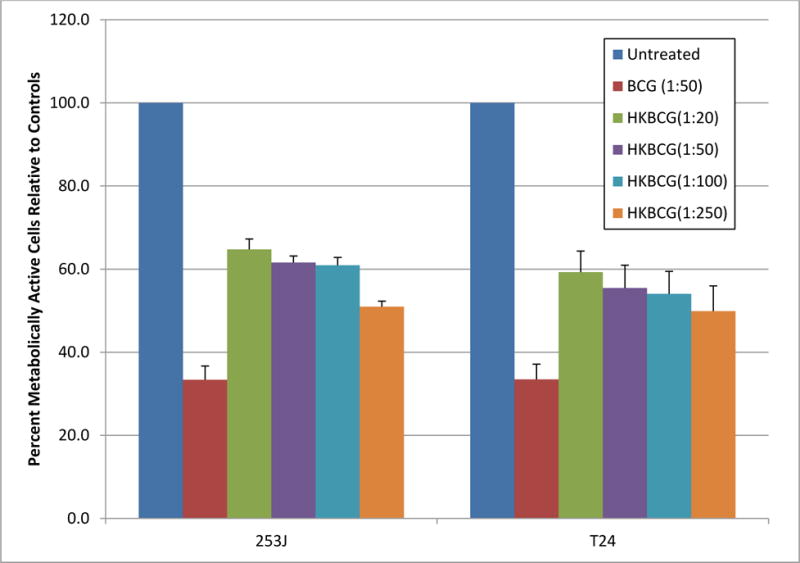
BCG treatment reduced the number of cells to 33% and 34% of control values in 253J and T24 cells, respectively (p < 0.0001). In case of hkBCG, these values were 62% and 55% of control values. (p < 0.001). The difference in % metabolically active cells between viable and hkBCG was statistically significant (p < 0.001).
Using qRTPCR we measured the relationship between hkBCG: Cell ratio on the expression of “immune response” genes. Figure 8 illustrates the relationship of gene expression to hkBCG dose across hkBCG: Cell ratios ranging from 5:1 to 1500:1. Gene expression in response to hkBCG demonstrated a positive dose-response relationship in 253J and T24 cells (p=0.11 and p<0.001, respectively). The level of gene expression in response to hkBCG doses greater than 250 organisms per cell was not significantly different than expression in response to viable BCG.
Figure 8.
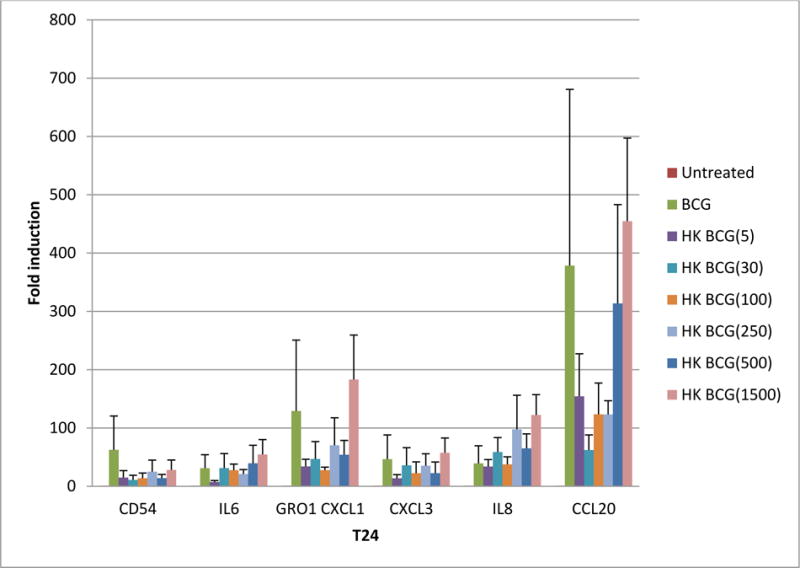
Gene expression in response to hkBCG is dose dependent. Exposure of UC cells to viable BCG increased the expression of a panel of “immune response gene relative to controls. Gene expression in response to hkBCG demonstrated a dose-response relationship in 253J and T24 cells (p = 0.11 and p < 0.001, respectively). The level of gene expression in response to hkBCG doses greater than 250 organisms per cell was not significantly different than expression in response to viable BCG. Data is shown for the T24 cell line.
Discussion
The results of prior literature reports evaluating the impact of loss of BCG viability on BCG’s anti-tumor activity have been highly variable. In the laboratory setting hkBCG retains its ability to bind tumor cells and induce a cytotoxic response.13,14 However, in contrast to viable BCG, prostaglandin inhibitors failed to increase the cytotoxic activity of hkBCG. Similar to viable BCG, hkBCG given in combination with cytokines can induce multinucleated giant cell formation as a consequence of monocyte fusion.15 In contrast, hkBCG is a poor inducer of iNOS and NO by spleen cells and the downstream expression of iNOS/NO dependent inflammatory genes.16,17 In the in vivo setting the anti-tumor activity of hkBCG appears to be dependent upon the model system. Direct intralesional injection of non-viable BCG was associated with reductions in tumor growth in sarcoma and hepatoma models.18,19,20 In orthotopic bladder cancer models autoclaved BCG had no antitumor effect and the magnitude of the antitumor/immune response correlated with BCG viability.21 In the clinical setting a direct correlation between therapeutic efficacy and BCG viability was observed.4 While highlighting the relevance of BCG viability, no prior studies have provided mechanistic insight into how bacterial viability influences the tumor response.5
The effect of BCG viability on antitumor activity is of more than just academic interest. The need to use viable BCG in patients with NMIBC subjects patients to potentially fatal, albeit infrequent, risk associated with the systemic dissemination of a living pathogen. Consequently, BCG usage is typically limited to patients with UC who are at high risk for disease progression. Understanding how loss of BCG viability influences the tumor cell response to BCG could provide a foundation for identifying alternatives to viable BCG. Recently, it was reported that HkBCG can be used to recall the response in patients who developed BCG infection after treatment with viable BCG to induce the anti-tumor immunity and BCG sensitivity. Their results showed that heat inactivated BCG did not produce grade 3 systemic toxicity, significantly reduced grade 2 local and systemic toxicity and did not increase the risk of tumor recurrence.22
Using a robust panel of intermediate endpoints that characterize and/or contribute to BCG’s direct cellular effects on UC cells, the present study sheds light on how alterations in BCG viability influence its direct effect of UC cell biology. Consistent with earlier literature, loss of BCG viability is not associated with a binary change in the effect of BCG on UC cell biology. Although attenuated relative to live BCG, hkBCG remains active in inducing direct tumor cell response. While qualitatively similar across the spectrum of studied endpoints, the magnitude of the response to hkBCG, relative to viable BCG, varies as a function of the endpoint in question. A particularly interesting observation was the positive dose-response relationship between hkBCG and gene expression and direct toxicity. Relative to viable BCG, the lower biologic activity of hkBCG could be overcome by dose increases. These findings, coupled with our prior work demonstrating a contribution of BCG viability to non-apoptotic cell death/HMGB1 release suggests that BCG viability is a variable which influences but does not abrogate BCG’s direct effect on UC cells.7
These observations raise a number of questions. What factor(s) associated with the loss of BCG viability contributes to the quantitative reduction in direct tumor effect? Could restoration of this “factor” by pharmacologic or other means restore the activity of HK BCG to that of viable BCG? How does the method of BCG inactivation influence the nature of the UC cell response? Further study is clearly warranted to better understand the mechanisms underlying the contribution of BCG viability to BCG anti-tumor activity.
Conclusions
Loss of BCG viability results in quantitative reductions in the magnitude of BCG’s direct effects on UC cell biology. However non-viable BCG continues to have activity across the range of biologic endpoints evaluated in this study. Further study will be required to identify the underlying mechanism through which BCG’s activity is attenuated as a consequence of loss of viability.
Acknowledgments
This work was supported by a grant from the Department of Veterans Affairs and the Milwaukee Veterans Affairs Medical Center.
Abbreviations
- ANOVA
analysis of variance
- BCG
Bacillus Calmette Guérin
- hkBCG
Heat killed BCG
- CD54
intercellular adhesion molecule 1
- CM
complete medium
- CXCL1
chemokine (C-X-C motif) ligand 1
- CXCL3
chemokine (C-X-C motif) ligand 3
- CCL20
chemokine (C-C motif) ligand 20
- DNA
deoxyribonucleic acid
- FBS
fetal bovine serum
- IL6
interleukin 6
- IL8
interleukin 8
- iNOS
inducible nitric oxide synthase
- LDH
lactate dehydrogenase
- mRNA
messenger ribonucleic acid
- MTT
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-κB
nuclear factor kappa B
- NO
nitric oxide
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- CEBP
CCAAT-enhancer-binding proteins
- P21
cyclin dependent kinase inhibitor p21 (cip1waf1)
- PBS
phosphate buffered saline
- RNA
ribonucleic acid
- rtPCR
reverse transcriptase polymerase chain reaction
- q rtPCR
quantitative reverse transcriptase polymerase chain reaction
- UC
urothelial carcinoma
References
- 1.Babjuk M, Oosterlinck W, Sylvester R, et al. EAU guidelines on non-muscle-invasive urothelial carcinoma of the bladder, the 2011 update. Eur Urol. 2011;59:997. doi: 10.1016/j.eururo.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 2.Kassouf W, Kamat AM, Zlotta A, et al. Canadian guidelines for treatment of non-muscle invasive bladder cancer: a focus on intravesical therapy. Can Urol Assoc J. 2010;4:168. doi: 10.5489/cuaj.10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hall MC, Cheng SS, Dalbagni G, et al. Guideline for the Management of Non-muscle Invasive Bladder Cancer: (Stages Ta, T1, and Tis): Update. Linthicum: American Urological Association; 2007. [DOI] [PubMed] [Google Scholar]
- 4.Shapiro A, Ratliff TL, Oakley DM, et al. Reduction of bladder tumor growth in mice treated with intravesical Bacillus Calmette-Guerin and its correlation with Bacillus Calmette-Guerin viability and natural killer cell activity. Cancer Res. 1983 Apr;43:1611. [PubMed] [Google Scholar]
- 5.Kelley DR, Ratliff TL, Catalona WJ, et al. Intravesical bacillus Calmette-Guerin therapy for superficial bladder cancer: effect of bacillus Calmette-Guerin viability on treatment results. J Urol. 1985;134:48. doi: 10.1016/s0022-5347(17)46976-x. [DOI] [PubMed] [Google Scholar]
- 6.Jackson A, Alexandroff A, Fleming D, et al. Bacillus-calmette-guerin (bcg) organisms directly alter the growth of bladder-tumor cells. Int J Oncol. 1994;5:697. doi: 10.3892/ijo.5.3.697. [DOI] [PubMed] [Google Scholar]
- 7.Zhang G, Chen F, Cao Y, et al. Contributors to HMGB1 Release by Urothelial Carcinoma Cells in Response to BCG. J Urol. 2013 Apr 11;:pii. doi: 10.1016/j.juro.2013.03.123. S0022-5347(13)04055-X. [DOI] [PubMed] [Google Scholar]
- 8.Zhang G, Chen F, Cao Y, et al. HMGB1 Release by Urothelial Carcinoma Cell in Responses to BCG is Required for the In Vivo Tumor Response to BCG. J Urol. 2013;189:1541. doi: 10.1016/j.juro.2012.09.123. [DOI] [PubMed] [Google Scholar]
- 9.Luo Y, Askeland EJ, Newton M, et al. Immunotherapy of Urinary Bladder Carcinoma: BCG and Beyond. Advances in Urology. 2012:181987. doi: 10.1155/2012/181987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen F, Zhang G, Cao Y, et al. MB49 murine urothelial carcinoma: molecular and phenotypic comparison to human cell lines as a model of the direct tumor response to bacillus Calmette-Guerin. J Urol. 2009;182:2932. doi: 10.1016/j.juro.2009.08.018. [DOI] [PubMed] [Google Scholar]
- 11.Mossman T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 12.See WA, Zhang G, Chen F, et al. p21 Expression by human urothelial carcinoma cells modulates the phenotypic response to BCG. Urol Oncol. 2010;28:526. doi: 10.1016/j.urolonc.2008.12.023. [DOI] [PubMed] [Google Scholar]
- 13.Klegerman ME, Zeunert PL, Lou Y, et al. Inhibition of murine sarcoma cell adherence to polystyrene substrata by bacillus Calmette-Guerin: evidence for fibronectin-mediated direct antitumor activity of BCG. Cancer Invest. 1993;11:660. doi: 10.3109/07357909309046938. [DOI] [PubMed] [Google Scholar]
- 14.Yamada H, Kuroda E, Matsumoto S, et al. Prostaglandin E2 down-regulates viable Bacille Calmette-Guerin-induced macrophage cytotoxicity against murine bladder cancer cell MBT-2 in vitro. Clin Exp Immunol. 2002;128:52. doi: 10.1046/j.1365-2249.2002.01686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gasser A, Most J. Generation of multinucleated giant cells in vitro by culture of human monocytes with Mycobacterium bovis BCG in combination with cytokine-containing supernatants. Infect Immun. 2009;67:395. doi: 10.1128/iai.67.1.395-402.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang J, Kawamura I, Zhu H, et al. Involvement of natural killer cells in nitric oxide production by spleen cells after stimulation with Mycobacterium bovis BCG. Study of the mechanism of the different abilities of viable and killed BCG. J Immunol. 1999;155:5728. [PubMed] [Google Scholar]
- 17.Yang J, Kawamura I, Mitsuyama M. Involvement of inflammatory cytokines and nitric oxide in the expression of non-specific resistance to Listeria monocytogenes in mice induced by viable but not killed Mycobacterium bovis BCG. Microb Pathog. 1997;22:79. doi: 10.1006/mpat.1996.0093. [DOI] [PubMed] [Google Scholar]
- 18.Willmott N, Pimm MV, Baldwin RW. Quantitative comparison of BCG strains and preparations in immunotherapy of a rat sarcoma. J Natl Cancer Inst. 1979 Sep;63:787. doi: 10.1093/jnci/63.3.787. [DOI] [PubMed] [Google Scholar]
- 19.Yarkoni E, Rapp HJ. Immunotherapy of guinea pigs with a transplanted hepatoma: comparison of intralesionally administered killed BCG cells and BCG cell walls. Infect Immun. 1979;25:1087. doi: 10.1128/iai.25.3.1087-1089.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreider JW, Bartlett GL, Purnell DM. Immunotherapeutic effectiveness of BCG inactivated by various modalities. Cancer. 1980;46:480. doi: 10.1002/1097-0142(19800801)46:3<480::aid-cncr2820460311>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 21.Gunther JH, Frambach M, Deinert I, et al. Effects of acetylic salicylic acid and pentoxifylline on the efficacy of intravesical BCG therapy in orthotopic murine bladder cancer (MB49) J Urol. 1999;161:1702. [PubMed] [Google Scholar]
- 22.Lamm D, Iversona T, Wanglera V. Clinical experience with heat-inactivated Bacillus calmette-guerin (bcg) immunotherapy. J Urol. 2013;189:e733. [Google Scholar]