

Abstract
Indole is a universal signal that regulates various bacterial behaviors, such as biofilm formation and antibiotic resistance. To generate mechanistic probes of indole signaling and control indole-mediated pathogenic phenotypes in both Gram-positive and Gram-negative bacteria, we have investigated the use of desformylflustrabromine (dFBr) derivatives to generate highly active indole mimetics. We have developed non-microbicidal dFBr derivatives that are 27–2000 times more active than indole in modulating biofilm formation, motility, acid resistance, and antibiotic resistance. The activity of these analogues parallels indole, because they are dependent on temperature, the enzyme tryptophanase TnaA, and the transcriptional regulator SdiA. This investigation demonstrates that molecules based on the dFBr scaffold can alter pathogenic behaviors by mimicking indole-signaling pathways.
Keywords: desformylflustrabromine, mechanistic probes, natural products, phenotype control, structure, activity relationships
Introduction
Bacteria coordinate collective behaviors by using different cell–cell communication systems to protect the community when subjected to adverse conditions. Quorum sensing (QS) describes one such behavior, in which secreted chemical signals, called autoinducers (AI), are used to coordinate gene expression based on population density.[1,2] QS signals include several classes of molecules encompassing acylhomoserine lactones (AHLs) in Gram-negative bacteria,[3] autoinducing peptides (AIPs) in Gram-positive bacteria[4] and autoinducer-2 (AI-2) shared by both Gram-negative and Gram-positive species (Figure 1A).[5]
Figure 1.
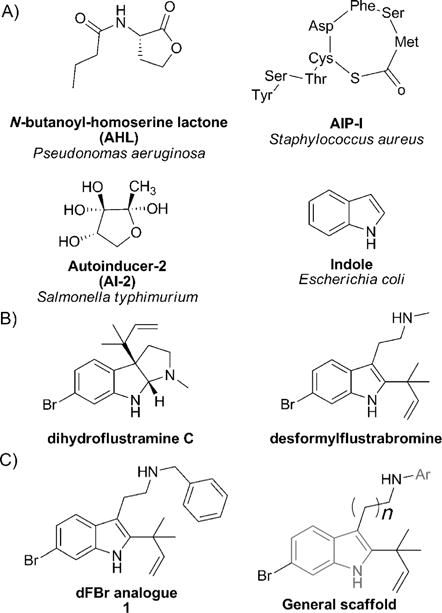
A) General structures of major classes of autoinducers. B) Flustramine family inspiration for the design of a synthetic dFBr analogue. C) General scaffold of the desformylflustrabromine library.
Outside of AI-2 communication pathways, indole signaling has recently received attention as a putative universal signaling network.[6] First to be identified in the indole-positive bacterial species Escherichia coli, indole is synthesized in high concentration by the action of tryptophanase (Tna), which converts l-tryptophan into indole, pyruvate, and ammonia during the stationary phase of growth.[7] To date, over 85 bacterial species, both Gram negative and Gram positive, have been reported to produce indole, whereas both indole-producing and non-indole-producing strains of bacteria will adapt their behavior in response to extracellular indole.[8,9,10] It has been shown that indole controls a variety of key pathogenic phenotypes. In E. coli, indole decreases acid resistance,[11] induces the expression of multidrug exporter genes and increases antibiotic resistance.[12,13] In addition, indole has been shown to decrease E. coli biofilm formation in a non-toxic manner by repressing motility, chemotaxis, and cell adherence,[14,15] whereas it promotes biofilm formation in the non-indole-producing bacterial strain Pseudomonas aeruginosa.[16] In E. coli, phenotypical changes, such as biofilm formation and antibiotic resistance, have been found to be more significant at 25°C.[13] At 37°C, indole is believed to control biofilm formation through the transcriptional regulator SdiA although the exact mechanism has not yet been determined.[14]
Many natural and synthetic indole derivatives have been shown to control bacterial phenotypes in a variety of Gram-positive and Gram-negative bacteria and have been investigated as potential candidates for antivirulence therapies.[17,18,19,20] Because indole derivatives are believed to modulate a large panel of bacterial behaviors by competing with indole or promoting indole biding to its signal receptor, indole-based natural products have been employed in our group as structural inspiration for the design of small molecules to potentially mimic indole signaling and modulate pathogenic phenotypes. In previous work, we investigated the potential of flustramine metabolites, isolated from the marine invertebrate Flustra foliacea, containing a pyrroloindoline or indolic core as a template to construct small molecules that inhibit bacterial biofilm formation (Figure 1B).[21] We observed the inhibition of E. coli and Staphylococcus aureus biofilm formation by the natural products flustramine C and desformylflustrabromine, analogous to the effect of indole itself. Tuning the flustramine scaffold by developing a desformylflustrabromine (dFBr) library generated the most potent analogue 1, which inhibited biofilm formation by S. aureus and E. coli 10–1000 times more potently than indole itself through a non-microbicidal mechanism, with IC50 values of 5.9 μm and 53 μm, respectively. Mechanistic studies in wild-type and knockout E. coli strains have shown that, parallel to indole itself, the activity of compound 1 is dependent on temperature, TnaA, and the transcriptional regulator SdiA, demonstrating the capacity of 1 to modulate indole-based signaling pathways. This indicated that we may be mimicking indole signaling with our dFBr derivatives.
Encouraged by the biological activity of compound 1, we elected to further explore the structure–activity relationship (SAR) of 1 and synthesize a second generation of analogues by introducing different aromatic substituents on the aliphatic nitrogen and increasing the tryptamine chain length (Figure 1C). Considering the fact that indole and compound 1 control biofilm formation of wild-type and knockout E. coli strains similarly, we hypothesized that compound 1 and our new generation of analogues may mimic other indole-dependent bacterial behaviors. Our ultimate goal is to develop highly active modulators of indole signaling that control pathogenic behavior through indole-signaling pathways in both Gram-negative and Gram-positive bacteria, to employ these molecules to address fundamental questions about indole signaling in disparate bacterial species, and to explore the impact of disrupting indole signaling both in vitro and in vivo.
Results and Discussion
The introduction of alternative aromatic substituents off the aliphatic nitrogen to access analogues of compound 1 was achieved by alkylation of the tryptamine derivative 3 (Scheme 1). All second-generation dFBr analogues of 1 were accessed from tryptamine 2 in five steps. The alkylation of nosyl-protected tryptamine 3 was followed by installation of the reverse prenyl group at the C2 position by applying Danishefsky’s tert-prenylation methodology to give intermediates 5a–f in 34–54% yield.[22] Treatment of 5a–f with N-bromosuccinimide (NBS) in AcOH/HCO2H (3:1)[23] gave selective bromination at the C6 position and gave intermediates 6a–f in 11–61% yield. Deprotection of the aliphatic nitrogen was carried out under Fukayama conditions by using thiophenol with potassium carbonate to give final compounds 7a–f in 80–90% yield.[24]
Scheme 1.
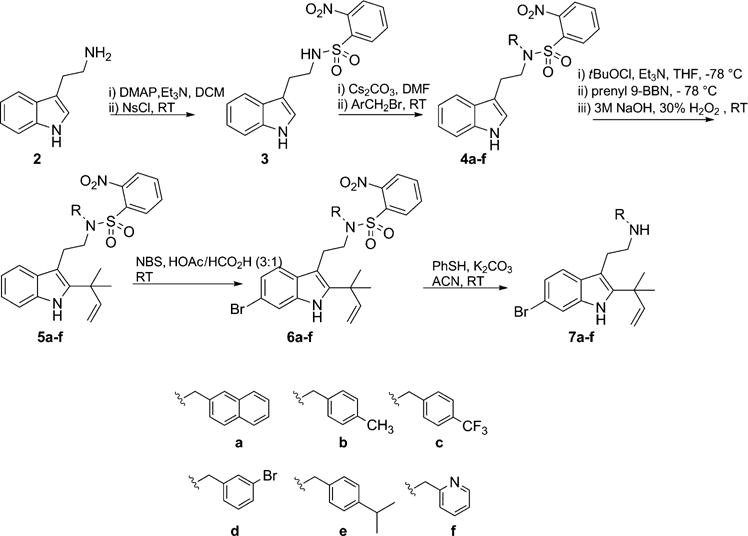
Synthetic route introducing structural diversity on the aliphatic nitrogen.
dFBr analogues inhibit biofilm formation of E. coli, Acinetobacter baumannii, S. aureus and methicillin-resistant Staphyloccocus aureus (MRSA): by synthesizing this second generation of analogues, our goal was to delineate additional SAR parameters on the dFBr scaffold and to develop highly active modulators of indole signaling with antibiofilm activity that surpass compound 1, while still acting through a non-toxic mechanism. Compounds were screened to investigate their effects on biofilm development by using A. baumannii (ATCC 19606), E. coli (K12 ER2718), S. aureus (ATCC 29213), and MRSA (ATCC BAA44) as our clinically relevant model bacteria, and effects were quantified by determining IC50 values. Herein, we define the compound concentration required to inhibit 50% of biofilm formation relative to an untreated control as the IC50 value. Growth-curve analysis was performed for each active compound to determine their toxicity towards planktonic bacteria. This analysis revealed if a compound inhibited biofilm formation through a non-toxic mechanism similar to indole, or acted through a bactericidal mechanism. Because bacteria tolerate and adapt to microbicidal therapies, we aimed to develop non-toxic compounds to avoid evolutionary pressure and thereby impart a reduced risk of resistance development. The results of these studies are summarized in Table 1.
Table 1.
Inhibition activity of dFBr analogues. All IC50 values are reported at μm concentrations.
| Compound | E. coli | A. baumannii | S. aureus | MRSA |
|---|---|---|---|---|
| 1 | 53.0 | 70.3[a] | 5.9 | 4.3[b] |
| 7a | 15.6 | 26.3[b] | 7.7[b] | 14.6[a] |
| 7b | 18.5 | 34.9[b] | 11.7[b] | 10.3[a] |
| 7c | 19.2 | 200 | 11.9[b] | 14.3[a] |
| 7d | 19.7 | 49.0[b] | 14.7[b] | 14.1[a] |
| 7e | 31.4 | 103.5[b] | 2.3 | 6.3 |
| 7 f | 90.8 | >200 | 18.4 | 37.0[a] |
| 11a | 16.3 | 56.7[b] | 13.4 | 12.0[b] |
| 11b | 32.1 | 39.4[b] | 9.8[b] | 9.0[b] |
| 11c | 52.5 | 73.2[b] | 10.8 | 15.7[a] |
Indicates inhibition of biofilm formation through a toxic mechanism as was determined by growth-curve analysis.
Indicates not complete toxicity, bacterial-growth delay was noted in the first 8 h, bacterial density was identical at 24 h.
All the synthesized compounds (except 7 f) equaled or surpassed the antibiofilm activity of compound 1 against E. coli. Although many analogues affected the planktonic cell growth of A. baumannii, S. aureus, and MRSA, none of these analogues presented a bactericidal effect on E. coli. Compounds 7a and b inhibited biofilm formation of E. coli with 3 to 3.5-fold greater efficacy than compound 1 with IC50 values of 15.6 and 18.5 μm, respectively. Because indole is an active signal that inhibits E. coli and S. aureus biofilms at 250–1000 μm,[6,14] our most potent compounds 7a and b are 27–130 times more active than indole. In the same manner, analogues 7a and b inhibited biofilm formation of A. baumannii with 2 to 2.5-fold greater efficacy compared to compound 1, with IC50 values of 26.3 and 34.9 μm, respectively. In addition to these two hits, compound 7d presented an increased activity against A. baumannii biofilm formation in comparison to 1. Unfortunately, this new generation of analogues did not exhibit increased antibiofilm activity towards Gram-positive bacteria except for 7e, which presented an IC50 value of 2.3 μm against S. aureus.
The impact of chain length on biological activity was also investigated while preserving the structural features proven to be fundamental for biological activity (the bromine atom at the C6 position and the reverse prenyl group at the C2 position).[21] For these new analogues, the aromatic substituent appended to the aliphatic nitrogen was selected by considering the three most potent compounds of the dFBr library in terms of antibiofilm activity against E. coli. Commercially available carboxylic acid 8 was treated with ethyl chloroformate and triethylamine in dry tetrahydrofuran leading to the corresponding amide 9. This intermediate was reduced by using lithium aluminum hydride to give the corresponding 3-indoylalkylamine 10 in 92% yield,[25] which was then nosyl-protected, alkylated, prenylated, and brominated by using the same conditions, as was previously described. This synthetic route gave final products 11a–c (Scheme 2).
Scheme 2.
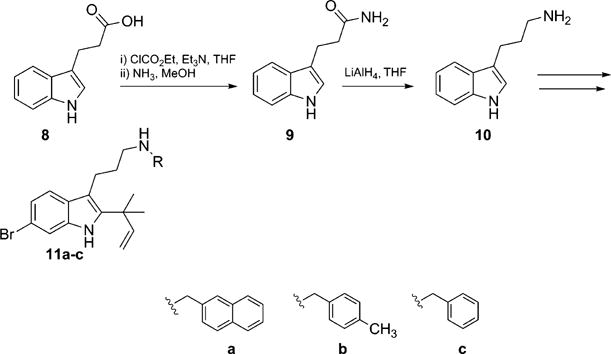
Synthesis of dFBr series 11a–c.
These new compounds were screened for antibiofilm activity against E. coli, A. baumannii, S. aureus and MRSA. We found that extending the tryptamine chain with one additional carbon led to either a conservation of the antibiofilm activity or a decrease up to fourfold for all four bacterial strains. Because compounds 1, 7a and b presented the best antibiofilm activity, these analogues were selected to further mechanistic studies.
Indole and dFBr analogues reduce the biofilm formation of E. coli to a greater extent at 25 °C
It has been established that temperature can have an important effect on bacterial signaling.[26,27] Indole signaling has been primarily observed at temperatures lower than 37°C. It has been shown that indole-mediated reduction of E. coli biofilm formation is greater at 25 than 37°C, and indole controls biofilm formation through the transcriptional regulator SdiA at 37°C.[13] In our previous work, mechanistic studies based on biofilm inhibition of E. coli at different temperatures demonstrated that the activity of analogue 1 paralleled indole. because its activity was dependent on temperature, SdiA, and TnaA.[21] Based on this activity, we first established temperature and gene-dependent biofilm-inhibition studies with lead compounds 7a and b and compared their activity to indole and dFBr analogue 1 by using the wild-type E. coli strain BW25113 and the isogenic knockout mutants ΔtnaA and ΔsdiA (Figure 2). Similar to indole, we found that these second generation of dFBr analogues inhibited biofilm formation in a dose-dependent manner that was more pronounced at 25 than at 37°C for all three strains of bacteria without affecting planktonic bacterial growth. At 25°C, wild-type E. coli biofilm was reduced by 82% in the presence of 100 μm analogue 7a compared to 33% at 37°C. Because the ΔtnaA mutant does not produce indole, this strain exhibited biofilm inhibition to a greater extent than the wild-type upon addition of exogenous indole, which was amplified at 25°C. The same result was observed upon addition of compounds 1 and 7b, which inhibited biofilm formation of the ΔtnaA mutant by 79 and 95%, respectively at 25°C, compared to 69 and 59% for the wild-type at 100 μm. At 37°C, the inhibition of wild-type bacteria by our analogues, especially 1, was more pronounced than their effects on the ΔsdiA knockout mutant strain. At a concentration of 100 μm, analogue 1 inhibited biofilm formation of the wild-type strain by 81 versus 52% for the ΔsdiA knockout mutant strain. These results indicate that the activity of these compounds depend on both TnaA and SdiA at 37°C.
Figure 2.
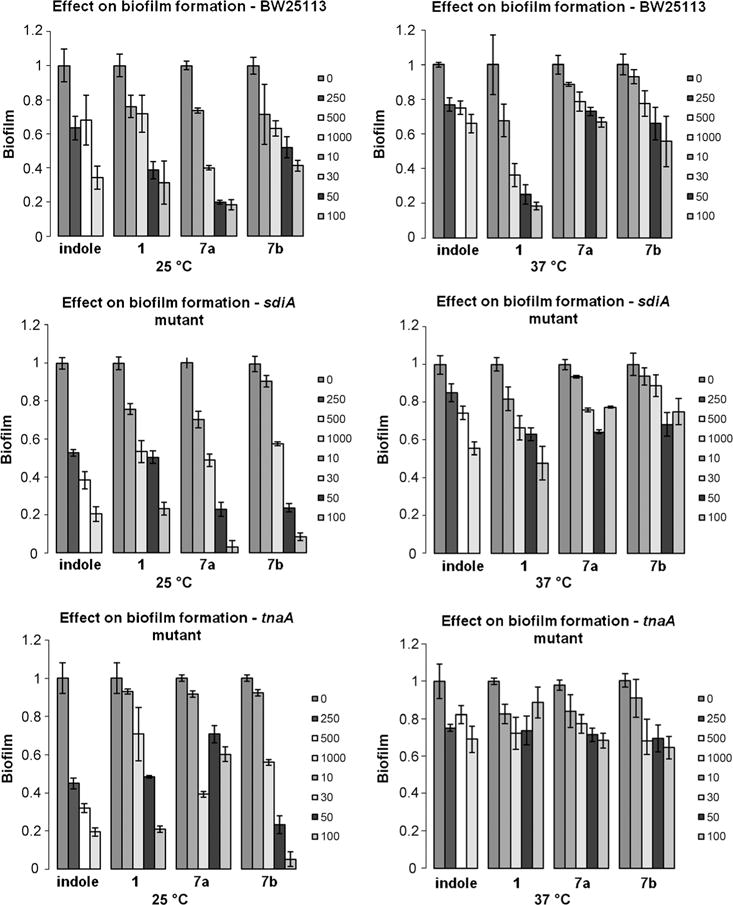
Biofilm inhibition activity of indole and dFBr analogues against wild-type and knockout E. coli strains at 25 and 37°C. Concentrations are reported in μm.
Indole and dFBr analogues promote P. aeruginosa PAO1 biofilm formation
Although P. aeruginosa does not produce indole, it has been shown that indole is a signal that stimulates biofilm formation of this strain at 30 and 37°C.[6,19] To evaluate the effect of indole and our dFBr analogues on P. aeruginosa biofilm formation, we tested different concentrations of indole and synthetic compounds for their ability to modulate PAO1 biofilm formation after 24 h at 25°C, because at this temperature, the biofilms formed by this bacterial species have been found to be more robust than biofilms formed at 37°C. Given that P. aeruginosa may encounter E. coli in different environments,[19] we decided to test indole at concentrations comparable to those produced by E. coli in a rich medium.[28] Indole promoted P. aeruginosa biofilm formation in a dose-dependent manner. At 500 μm it increased biofilm formation by 1.3-fold compared to the negative control (Figure 3), and did not affect planktonic cell growth at this concentration.
Figure 3.
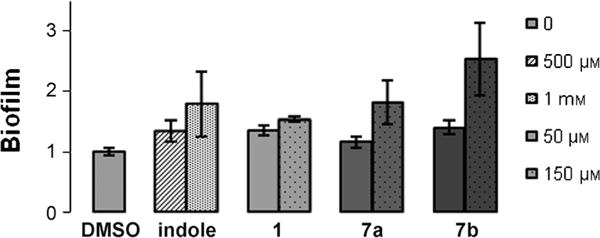
Biofilm formation of PAO1 at 25°C; effect of indole and dFBr analogues on PAO1 biofilms.
Paralleling this activity, the three dFBr analogues increased PAO1 biofilm formation to a greater extent than indole at lower concentrations. Compound 1 increased biofilm formation by 35% at 50 μM compared to the 500 μM indole required to achieve the same increase. Similarly at 150 μM, analogues 7a and b elicited a 1.8-fold and 2.5-fold increase in P. aeruginosa biofilm formation compared to the negative control. In comparison, it requires a minimum of 1.0 mM indole to promote P aeruginosa biofilm formation to a similar degree.
Indole and dFBr analogues repress E. coli and P. aeruginosa motility
Motility has been shown to be dependent on indole signaling and has an essential role in the colonization of new environments. In both E. coli and P. aeruginosa, indole has been shown to decrease bacterial motility.[19,29] To investigate the effect of our dFBr analogues on motility, compounds 1, 7a and b were added to motility agar and bacterial movement was quantified. In addition to the wild-type bacterial strain and the ΔsdiA knockout mutant, the isogenic mutant strains ΔtnaA, ΔtrpE, and ΔtnaC were used, because these genes are also involved in E. coli indole synthesis. The experiments were performed at 37°C and motility was measured at 8 h (see the Supporting Information) and 24 h (Figure 4A). Bacteria that do not express the tnaA gene are unable to produce indole, whereas the ΔtrpE or ΔtnaC knockout mutants produce less indole than the wild-type E. coli. Therefore, the motility of the wild-type strain is repressed in comparison with its knockout mutants.[14] As expected, we observed that the ΔtnaA isogenic knockout mutant is more motile than the wild-type strain of E. coli, which produces indole. The presence of 500 μM indole in the motility agar reduced the motility of all strains, whereas an equivalent or greater effect was observed in the presence of 50 μM of dFBr analogues. For example, with the ΔtnaA mutant the swimming halo diameters at 24 h for this bacterial strain were 1.63±0.31 (negative control), 0.99±0.19 (indole), 0.78±0.24 (analogue 1), 0.50±0.28, and 0.53±0.04 cm (analogues 7a and b, respectively).
Figure 4.
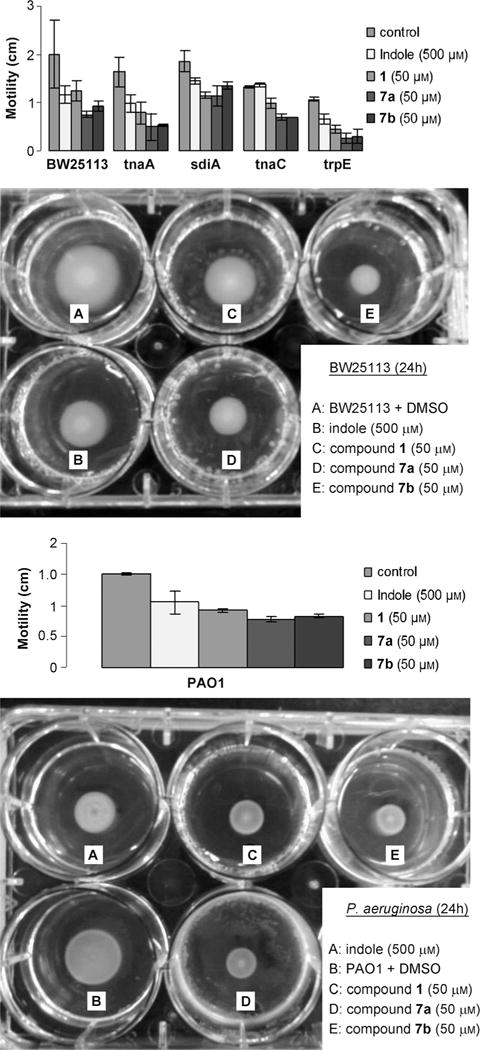
A) Motility of wild-type E. coli and knockout mutants after 24 h. B) Motility of P. aeruginosa PAO1 after 24 h.
As expected, the effect of indole and our synthetic analogues on motility was considerably muted for the ΔsdiA mutant in comparison to the other E. coli strains. Indole, analogue 1, and analogues 7a and b were 1.3 to 1.74-fold more effective at suppressing motility in the wild-type E. coli strain in comparison to the ΔsdiA mutant (33 and 47% inhibition for the wild-type compared to 22 and 27% for the ΔsdiA mutant in the presence of 500 μM of indole and 50 μM of 7b, respectively). Therefore, similar to indole, the activity of our dFBr analogues is partially dependent on SdiA at 37°C. Indole and our dFBr analogues both decreased the swimming motility of P. aeruginosa (Figure 4B). The motility halo diameter was decreased by 1.4-fold in the presence of 500 μM of indole, whereas a decrease of 1.7-fold, 2-fold, and 1.8-fold was observed for compounds 1 and 7a and b, respectively at a concentration ten times lower than indole.
Indole and dFBr analogues decrease acid resistance of E. coli
Indole has been shown to repress several E. coli acid-resistance genes and in the process, significantly reduce the bacterium acid tolerance. We evaluated the effect of our dFBr analogues on acid resistance by submitting wild-type E. coli BW25113 to an acid challenge at pH 2.5 (Figure 5).
Figure 5.
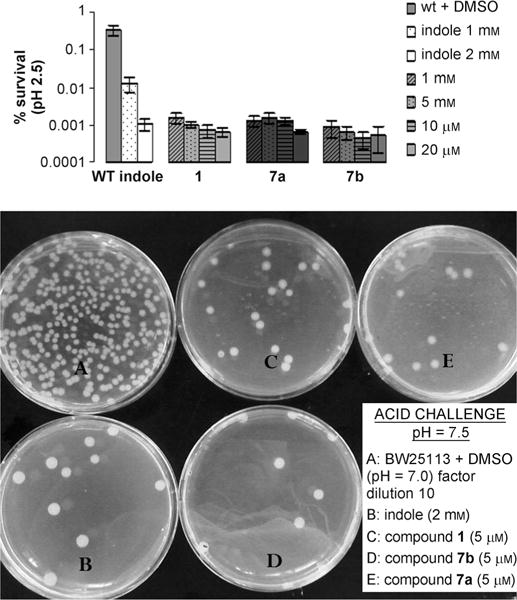
Acid resistance of wild-type E. coli at pH 2.5 at 37°C.
Because indole is a signal produced during the stationary phase of growth, we performed our experiments in log phase to directly observe the phenotypical changes attributed to growth in exogenous indole or synthetic compound. Growth in indole or compounds 1 and 7a and b decreased acid resistance in a dose-response manner. Growth in the presence of 2 mM indole produced a decrease of 319-fold in acid resistance, whereas 10 μM of analogue 7a was required to achieve a similar effect. Only 1 or 5 μM of synthetic compounds 7b and 1 were necessary to produce a decrease of 529-fold and 650-fold, respectively, making our dFBr analogues 200–2000 times more active than indole itself in mediating acid-resistance of E. coli.
Indole and dFBr analogues increase antibiotic resistance of E. coli
Because indole confers drug resistance to E. coli through the upregulation of multidrug exporter genes,[12] we assayed the antibiotic resistance of wild-type BW25113 grown to log phase in presence of indole (1 and 2 mM) or different concentrations of dFBr analogues. Norfloxacin and ampicillin belong to two different classes of broad spectrum antibiotics that kill growing cells with distinct modes of action; the fluoroquinolone norfloxacin inhibits DNA replication, whereas the β-lactam ampicillin inhibits cell-wall synthesis. We performed our experiments in log phase to directly compare the phenotypical changes controlled by growth in exogenous indole to our synthetic compounds. The survival of E. coli was evaluated after three hours at 37°C (Figure 6A and B). As was expected, we observed that indole and our dFBr compounds induced antibiotic resistance of E. coli against the two antibiotics.
Figure 6.
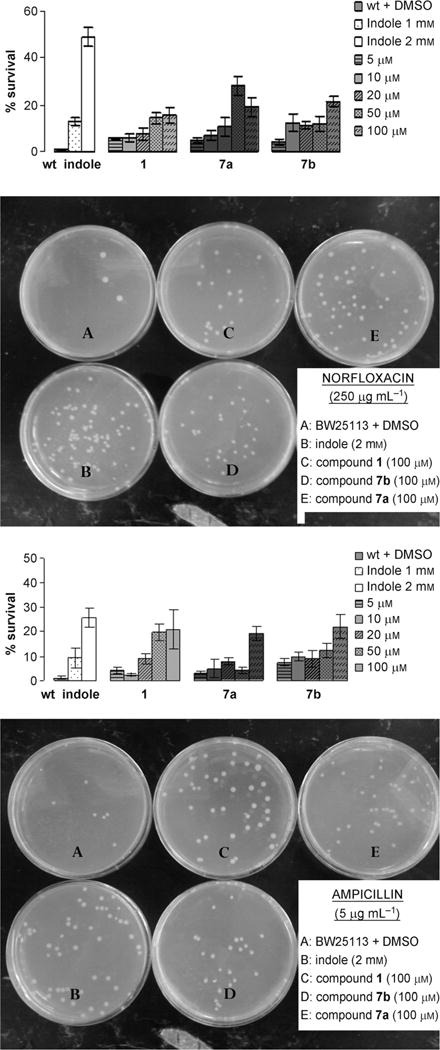
A) Effect of indole and dFBr analogues on norfloxacin resistance (0.250 ngmL−1) after 3 h. B) Effect of indole and dFBr analogues on ampicillin resistance (5 μgmL−1) after 3 h.
In comparison to the negative control, growth in 2 mM indole increased survival rate by 27% when the bacteria were subjected to 5 μgmL−1 ampicillin (Figure 6A) and 50% for 0.25 μgmL−1 norfloxacin (Figure 6B). Analogues 1 and 7a–b increased antibiotic resistance of BW25113 in a dose-response manner. Growth in 1 mM indole increased the survival rate by 13% in the presence of norfloxacin and 9% in the presence of ampicillin, whereas an equivalent or greater effect was observed upon growth in 10 μM of compound 7b in presence of both antibiotics.
Conclusions
The desformylflustrabromine scaffold was used to develop analogues that are able to control multiple bacterial behaviors. We have shown that our dFBr analogues are able to 1) inhibit biofilm formation of E. coli in a manner dependent on temperature, SdiA, and TnaA; 2) promote P aeruginosa biofilm formation; 3) repress E. coli and P. aeruginosa motility; 4) decrease acid resistance of E. coli; and 5) increase the antibiotic resistance of E. coli, all in a manner that parallels indole itself (Figure 7). Because our analogues are upwards of 2000 times more potent than indole itself at altering pathogenic behaviors, the dFBr scaffold presents potential for the design of small molecules to probe antivirulence therapies in vivo. In addition to modulating indole-controlled bacterial behaviors, compounds 1 and 7a and b can be used as mechanistic probes to potentially deconvolute indole signaling in Gram-positive and Gram-negative bacteria to further delineate fundamental questions about indole signaling itself.
Figure 7.
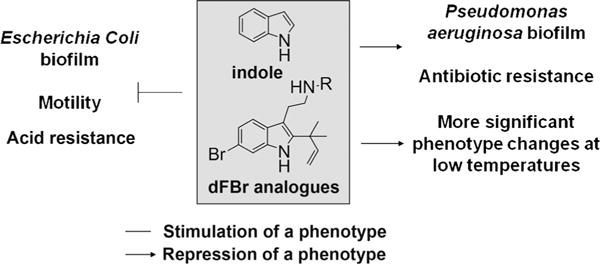
Phenotypes regulated by indole and dFBr analogues.
Experimental Section
Bacterial strains and culture conditions
A. baumannii (ATCC 19606), E. coli (K12 ER2718), S. aureus (ATCC 29213), MRSA (ATCC BAA44) and P aeruginosa (PAO1) strains were used for biofilm inhibition assay. Wild-type E. coli (BW25113) and the isogenic deletion mutations ΔtnaA (JW3686–7), ΔtrpE (JW1256–1), ΔtnaC (JW3685–1), and ΔsdiA (JW1901–5) from the Keio collection were used for mechanistic studies. Luria–Bertani (LB) broth and agar were used as the growth media for the wild-type E. coli strains and were supplemented with kanamycin (32 μgmL−1) for the use of the knockout mutants. Tryptic soy (TSB) broth supplemented with 5% glucose and tryptic soy agar were used for growth of the S. aureus and MRSA strains. No salt LB (1% tryptone+0.5% yeast extract) broth (LBNS) was used as the growth media for P. aeruginosa. Bacteria were streaked from a −80°C glycerol stock on the appropriate agar plate, and a single colony was inoculated in 3 mL of media and cultured at 37°C overnight with agitation at 200 rpm. Prior to the antibiotic- and acid-resistance assays, the overnight cultures were diluted in LB in the presence of indole or dFBr analogue and were regrown to mid-log phase (A600=1.0).
Biofilm-inhibition procedure
The inhibitory effects of compounds against biofilm formation was determined under static conditions by using a crystal-violet (CV) reporter assay.[30] Inhibition assays were performed by taking an overnight culture and sub-culturing it to an OD600 of 0.01 into the appropriate media; LB was used for A. baumannii and E. coli and TSB supplemented with 5% Glucose (TSBG) was used for S. aureus and MRSA and LBNS for P. aeruginosa. Stock solutions of predetermined concentrations of the test compounds were then made in DMSO (biology grade) and added to the inoculum to give the desired concentrations, untreated inoculum served as the control. This was aliquoted (100 μL) into the wells of a 96-well PVC microtiter plate. Sample plates were wrapped in GLAD Press n’ Seal® followed by incubation under stationary conditions for 24 h at 25°C for P. aeruginosa and 37°C for all the other strains of bacteria. After incubation, the media was discarded and the plates were washed with water. The sample plates were then stained with 110 μL of 0.1% solution of CV and incubated at RT for 30 min. The CV stain was then discarded and the plates were washed with water. The remaining stain was solubilized with 200 μL of 95% ethanol for 10 min and a sample of 125 μL of solubilized CV-stained ethanol was transferred from each well into the corresponding wells of a polystyrene microtiter dish. Biofilm inhibition was quantified by measuring the OD540 of each well. A negative control lane with no biofilm was measured to determine background staining and was subtracted out. The percent inhibition was calculated by the comparison of the OD540 for control wells versus treated wells under identical conditions.
Motility-test procedure
Motility was determined by using plates containing agar (0.3%) with tryptone (1%) and NaCl (0.25%).[31] Bacteria were grown from diluted overnight cultures to a turbidity of 1.0 at 600 nm, and 1.5 μL of the bacterial culture was placed into the center of the agar plate. The motility halos were measured at 8 h and 24 h. The effect of indole and synthetic compounds was tested by adding indole (500 μM) or compounds (50 μM) dissolved in DMSO (biological grade) to the agar. An equivalent volume of DMSO was added as the negative control. Each experiment was performed in duplicate by using two independent cultures.
Acid-resistance assay
LB overnight cultures were diluted 1:100 and grown with indole (1 mM or 2 mM) or synthetic compound dissolved in DMSO (biological grade) at different concentrations. When the turbidity reached 1.0 at 600 nm, the culture was then diluted 40-fold into a phosphate-buffered saline solution (pH 7.2) or in acidified LB (pH 2.5). Bacteria in acidic media were incubated at 37°C during 1 h without shaking. An equivalent volume of DMSO was added as the negative control. The number of colony-forming units (CFU) per milliliter was determined by plating serial dilutions on agar plates. The percentage of cells surviving the acid challenge was calculated as the number of CFU after acid treatment divided by the CFU before treatment. Each experiment was performed in triplicate by using two independent cultures.
Antibiotic resistance
LB overnight cultures were diluted 1:100 and grown to a OD600 of 1.0 in the presence of indole (2 mM), different concentrations of synthetic compound or a corresponding volume of DMSO (biological grade) as a negative control. Antibiotics (norfloxacin 0.25 μgmL−1 and ampicillin 5 μgmL−1) were mixed with cells and incubated for three hours at 37°C with shaking. The percentage of cells surviving the antibiotic treatment was calculated as the number of CFU after antibiotic treatment divided by the CFU before treatment. Each experiment was performed in triplicate by using two independent cultures.
Supplementary Material
Acknowledgments
The authors would like to thank the National Institutes of Health (GM055769) for their support.
Footnotes
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/chem.201303510.
References
- 1.Camilli A, Bassler B. Science. 2006;311:1113–1116. doi: 10.1126/science.1121357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rutherford S, Bassler B. Cold Spring Harbor Perspect Med. 2012;2:a012427. doi: 10.1101/cshperspect.a012427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Geske GD, Wezeman RJ, Siegel AP, Blackwell HE. J Am Chem Soc. 2005;127:12763. doi: 10.1021/ja0530321. [DOI] [PubMed] [Google Scholar]
- 4.Mayville P, Ji G, Beavis R, Yang H, Goger M, Novick R, Muir TW. Proc Natl Acad Sci USA. 1999;96:1218–1223. doi: 10.1073/pnas.96.4.1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller M, Bassler B. Annu Rev Microbiol. 2001;55:165. doi: 10.1146/annurev.micro.55.1.165. [DOI] [PubMed] [Google Scholar]
- 6.Lee J, Bansal T, Jayaraman A, Bentley W, Wood T. Appl Environ Microbiol. 2007;73:4100. doi: 10.1128/AEM.00360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vega N, Allison K, Samuels A, Klempner M, Collins J. Proc Natl Acad Sci USA. 2013;110:1218–1223. doi: 10.1073/pnas.1318744110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee JH, Lee J. FEMS Microbiol Rev. 2010;34:426. doi: 10.1111/j.1574-6976.2009.00204.x. [DOI] [PubMed] [Google Scholar]
- 9.Nikaido E, Giraud E, Baucheron S, Yamasaki S, Wiedemann A, Okamoto K, Takagi T, Yamaguchi A, Cloeckaert A, Nishino K. Gut Pathog. 2012;4:5. doi: 10.1186/1757-4749-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kim D, Sitepu I, Hashidoko Y. Appl Environ Microbiol. 2013;79:4845–4852. doi: 10.1128/AEM.01209-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee J, Page R, Garcia-Contreras R, Palermino JM, Zhang XS, Doshi O, Wood TK, Peti W. J Mol Biol. 2007;373:11–26. doi: 10.1016/j.jmb.2007.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirakawa H, Inazumi Y, Masaki T, Hirata T, Yamaguchi A. Mol Microbiol. 2005;55:1113–1126. doi: 10.1111/j.1365-2958.2004.04449.x. [DOI] [PubMed] [Google Scholar]
- 13.Lee J, Zhang X, Hedge M, Bentley W, Jayaraman A, Wood T. ISME J. 2008;2:1007. doi: 10.1038/ismej.2008.54. [DOI] [PubMed] [Google Scholar]
- 14.Lee J, Jayaraman A, Wood T. BMC Microbiol. 2007;7:42. doi: 10.1186/1471-2180-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bansal T, Englert D, Lee J, Hegde M, Wood T, Jayaraman A. Infect Immun. 2007;75:4597. doi: 10.1128/IAI.00630-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hentzer M, Wu H, Riedel K, Rasmussen TB, Bagge N, Kumar N, Schembri MA, Song Z, Kristoffersen P, Manefield M, Costerton J, Molin S, Eberl L, Steinberg P, Kjelleberg S, Hoiby N, Givskov M. EMBO J. 2003;22:3805–3815. doi: 10.1093/emboj/cdg366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Worthington R, Richards J, Melander C. Org Biomol Chem. 2012;35:310–315. doi: 10.1039/c2ob25835h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JH, Cho MH, Lee J. Environ Microbiol. 2011;13:62–73. doi: 10.1111/j.1462-2920.2010.02308.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Attila C, Cirillo SLG, Cirillo JD, Wood TK. J Microb Biotechnol. 2009;2:75. doi: 10.1111/j.1751-7915.2008.00061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee JH, Kim YG, Cho MH, Kim JA, Lee J. FEMS Microbiol Lett. 2012;329:36. doi: 10.1111/j.1574-6968.2012.02500.x. [DOI] [PubMed] [Google Scholar]
- 21.Bunders C, Minvielle M, Worthington R, Ortiz M, Cavanagh J, Melander C. J Am Chem Soc. 2011;133:20160. doi: 10.1021/ja209836z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schkeryantz JM, Woo JC, Danishefsky S. J Am Chem Soc. 1995;117:7025. [Google Scholar]
- 23.Lindel T, Brauchle L, Golz G, Bohrer P. Org Lett. 2007;9:283. doi: 10.1021/ol0627348. [DOI] [PubMed] [Google Scholar]
- 24.Fukuyama T, Jow C, Cheung M. Tetrahedron Lett. 1995;36:6373. [Google Scholar]
- 25.Ang SH, Kratsel P, Leong SY, Tan LJ, Wong WLJ, Yeug B, Zou B. WO 2009132921A1 20091105. PCT Int Appl. 2009
- 26.Hasegawa H, Chatterjee A, Cui Y, Chatterjee AK. Appl Environ Microbiol. 2005;71:4655–4663. doi: 10.1128/AEM.71.8.4655-4663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han TH, Lee JH, Han MH, Wood TK, Lee J. Res Microbiol. 2011;162:108–116. doi: 10.1016/j.resmic.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li G, Young K. Microbiology. 2013;159:402–410. doi: 10.1099/mic.0.064139-0. [DOI] [PubMed] [Google Scholar]
- 29.Ma Z, Gong S, Richard H, Tucker DL, Conway T, Foster JW. Mol Microbiol. 2003;49:1309–1320. doi: 10.1046/j.1365-2958.2003.03633.x. [DOI] [PubMed] [Google Scholar]
- 30.O’Toole GA, Kolter R. Mol Microbiol. 1998;30:295. doi: 10.1046/j.1365-2958.1998.01062.x. [DOI] [PubMed] [Google Scholar]
- 31.Sperandio V, Torres AG, Kaper JB. Mol Microbiol. 2002;43:809–821. doi: 10.1046/j.1365-2958.2002.02803.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.