

Abstract
Both long-term potentiation (LTP) and depression (LTD) of excitatory synapse strength require the Ca2+/calmodulin (CaM)-dependent protein kinase II (CaMKII) and its autonomous activity generated by Thr-286 autophosphorylation. Additionally, LTP and LTD are correlated with dendritic spine enlargement and shrinkage that are accompanied by the synaptic accumulation or removal, respectively, of the AMPA-receptor regulatory scaffold protein A-kinase anchoring protein (AKAP) 79/150. We show here that the spine shrinkage associated with LTD indeed requires synaptic AKAP79/150 removal, which in turn requires CaMKII activity. In contrast to normal CaMKII substrates, the substrate sites within the AKAP79/150 N-terminal polybasic membrane–cytoskeletal targeting domain were phosphorylated more efficiently by autonomous compared with Ca2+/CaM-stimulated CaMKII activity. This unusual regulation was mediated by Ca2+/CaM binding to the substrate sites resulting in protection from phosphorylation in the presence of Ca2+/CaM, a mechanism that favors phosphorylation by prolonged, weak LTD stimuli versus brief, strong LTP stimuli. Phosphorylation by CaMKII inhibited AKAP79/150 association with F-actin; it also facilitated AKAP79/150 removal from spines but was not required for it. By contrast, LTD-induced spine removal of AKAP79/150 required its depalmitoylation on two Cys residues within the N-terminal targeting domain. Notably, such LTD-induced depalmitoylation was also blocked by CaMKII inhibition. These results provide a mechanism how CaMKII can indeed mediate not only LTP but also LTD through regulated substrate selection; however, in the case of AKAP79/150, indirect CaMKII effects on palmitoylation are more important than the effects of direct phosphorylation. Additionally, our results provide the first direct evidence for a function of the well-described AKAP79/150 trafficking in regulating LTD-induced spine shrinkage.
Keywords: Ca2+/calmodulin-dependent protein kinase II (CaMKII), calmodulin (CaM), dendritic spine, synapse, synaptic plasticity, AKAP79/150, long-term depression (LTD)
Introduction
LTP7 and LTD are two opposing forms of synaptic plasticity that together are thought to underlie learning, memory, and cognition (1, 2). CaMKII and its Ca2+-independent autonomous kinase activity that is generated by Thr-286 autophosphorylation have been tightly linked to LTP for over 25 years (3–6); by contrast, an additional requirement in LTD is just emerging (7, 8). LTD also requires the phosphatase calcineurin (CaN) (9, 10), and specifically a pool of CaN co-anchored with protein kinase A (PKA) at synapses by AKAP79 (in humans) or AKAP150 (the rodent homologue) (11–13). However, somewhat paradoxically, LTD stimuli ultimately trigger the synaptic removal of AKAP79/150 (14–16), suggesting the following model: LTD stimuli quickly induce CaN-dependent dephosphorylation of the AMPA-type glutamate receptor (AMPAR) subunit GluA1 at the PKA site Ser-845 to promote AMPAR internalization; subsequent CaN-dependent AKAP79/150 removal then removes AKAP-anchored PKA from synapses to prevent GluA1 Ser-845 re-phosphorylation (12, 15). Here, we show that synaptic AKAP79/150 removal is also required for structural LTD (i.e. the shrinkage of dendritic spines) and that this removal requires CaMKII. Thus, our results provide a direct mechanistic explanation for the requirement of CaMKII in LTD as well as the first direct evidence for a requirement of the well-studied AKAP79/150 removal from spines in an LTD mechanism.
Synaptic targeting of AKAP79/150 is mediated by its N-terminal targeting domain that includes three polybasic regions (Fig. 1A) that interact with F-actin and membrane phospholipids (14, 16, 17), including via two palmitoylated Cys residues in the polybasic regions A and C (18, 19). LTD stimuli disrupt both F-actin and membrane interactions (14–16, 18), and although disruption of the F-actin interaction is required for the LTD-induced removal of AKAP79/150 from spines, it is not sufficient (14). Thus, additional mechanisms that disrupt AKAP79/150 membrane association are also likely required; however, such mechanisms remain to be elucidated.
Figure 1.
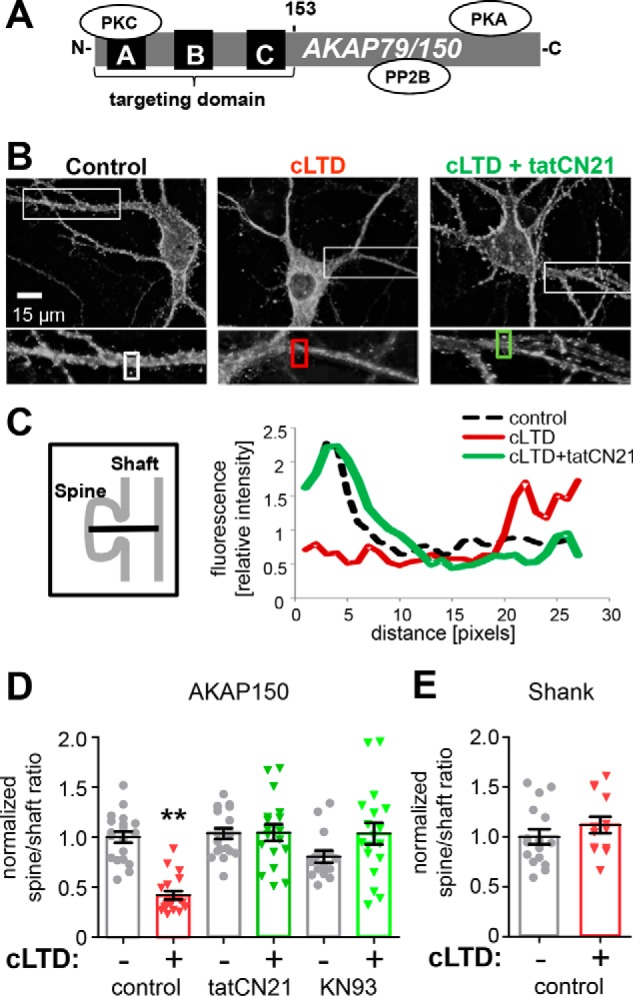
LTD-induced trafficking of AKAP is CaMKII-dependent. A, schematic of AKAP79/150 protein. B, endogenous AKAP150 antibody labeling of example neurons and enlargement of individual dendrites for control (left), cLTD (center, NMDA 30 μm), and CaMKII inhibition (right, tatCN21 5 μm) during cLTD. C, fluorescent profiles are shown for a line drawn through the spine head into the dendritic shaft of those outlined with rectangular boxes in B. AKAP150 shows a characteristic fluorescent profile with a high concentration in the spine head compared with shaft. This profile is disrupted by cLTD, indicating translocation, but is preserved in the presence of tatCN21 during cLTD. D, quantification of spine to dendritic shaft ratio (normalized to control) from experiments from three individual neuron preparations as determined by 10 masks per neuron over areas of puncta co-localization of AKAP150 and PSD95 (data not shown), and nearby dendritic shafts. Mean AKAP150 spine to shaft ratio (2.5 ± 0.14, n = 19 neurons) was significantly decreased after cLTD (1.0 ± 0.1, n = 19 neurons; p < 0.001, one-way ANOVA, Newman-Keuls post hoc analysis) compared with all other conditions: tatCN21 (2.6 ± 0.13, n = 19 neurons); tatCN21 + cLTD (2.6 ± 0.2, n = 18 neurons); KN93 (10 μm) (2.0 ± 0.15, n = 15 neurons); KN93 + cLTD (2.6 ± 0.25, n = 18 neurons). E, mean spine to shaft ratio of endogenous SHANK (2.36 ± 0.18, n = 16) revealed no translocation of SHANK with cLTD (2.64 ± 0.2, n = 13 neurons; p = 0.9885, two-tailed t test). Data shown in graphs are normalized to control.
With a role of CaMKII in LTD just emerging, only one single LTD-related CaMKII substrate is currently known: Ser-567 on the AMPAR subunit GluA1 (7). Although phosphorylation of GluA1 at Ser-831 and Ser-845 enhances channel function and synaptic localization (2), phosphorylation at Ser-567 reduces synaptic localization (20). GluA1 Ser-567 is an unusual “high-autonomy” substrate (7). Phosphorylation of regular substrates by CaMKII that has been made autonomous (i.e. Ca2+/CaM-independent) via Thr-286 autophosphorylation is still significantly further stimulated by the addition of Ca2+/CaM (21), which is also required for enhancement of synaptic strength (22). By contrast, autonomous CaMKII phosphorylates GluA1 Ser-567 equally well in presence or absence of Ca2+/CaM, resulting in ∼100% autonomy (the ratio of autonomous over maximal stimulated activity) compared with the ∼20% autonomy seen for regular substrates. In turn, this unusual regulation causes the preference of Ser-567 phosphorylation after LTD instead of LTP stimuli (7).
Here, we show that AKAP79/150 is another LTD-related substrate of CaMKII that exhibits high autonomy but through a mechanism that is distinct from high autonomy toward GluA1 Ser-567. Importantly, CaMKII inhibition prevented LTD-induced AKAP79/150 removal from dendritic spines, whereas CaMKII-mediated phosphorylation impaired AKAP79/150 interaction with F-actin and facilitated spine removal. However, AKAP79/150 spine removal additionally required CaMKII-regulated AKAP79/150 depalmitoylation. Indeed, this depalmitoylation was required for AKAP79/150 trafficking as well as for structural LTD.
Results
CaMKII inhibition blocks cLTD-induced synaptic removal of AKAP79
In agreement with previous studies (12, 15, 16), AKAP79/150 (Fig. 1A) was removed from dendritic spine synapses in cultured hippocampal neurons within minutes after cLTD stimuli by 30 μm NMDA application for 3 min (Fig. 1, B–D). As the anchored PKA is removed together with AKAP79/150, this removal is thought to aid LTD by preventing re-phosphorylation of the PKA site GluA1 Ser-845 after dephosphorylation by anchored CaN (12, 15). For quantification of this AKAP79/150 movement, we utilized the spine to shaft ratio, a measurement that was validated in our previous studies (15, 16). We decided to test whether the LTD-induced synaptic removal of AKAP79/150 is dependent on CaMKII, a kinase recently shown to be required for NMDA-receptor-dependent LTD (7). Indeed, CaMKII inhibition with the highly selective and cell-penetrating peptide inhibitor tatCN21 (23) completely blocked cLTD-induced translocation of endogenous AKAP150 in hippocampal cultures (Fig. 1, B–D), as assessed by analysis of line scans of fluorescence intensity through spines and neighboring dendrites (Fig. 1C) and spine to dendritic shaft mean fluorescence ratios (Fig. 1D) of AKAP150 immunostained neurons. CaMKII inhibition by a mechanistically different inhibitor, KN93 (24–26), had a similar effect of preventing cLTD-induced translocation (Fig. 1D). In contrast to AKAP79, the postsynaptic marker Shank1 was not significantly removed from spines in response to the cLTD stimuli (Fig. 1E). Thus, our quantification of the AKAP79 removal was not affected by the spine shrinkage caused by cLTD stimuli (see also Fig. 7). The requirement of CaMKII for cLTD-induced synaptic removal of AKAP79/150 provides a potentially new mechanism for the recently described CaMKII function in NMDA-receptor-dependent LTD (7).
Figure 7.

CaMKII activity and AKAP150 lipidation state cooperate to mediate AKAP spine removal and structural plasticity during NMDA-induced LTD. A–E, CaMKII activity is required for NMDA-elicited AKAP150 depalmitoylation. DIV 14 hippocampal cultures treated with 50 μm NMDA showed reduced AKAP150 palmitoylation as assayed by ABE and BMCC palmitoylation assays. However, this decrease was blocked by tatCN21, a small peptide inhibitor of CaMKII activity (fold change relative to vehicle (Veh) control); ABE (A and B): NMDA, 0.27 ± 0.09, p < 0.01, one-sample t test; tatCN21, 1.14 ± 0.12, p > 0.05; NMDA + tatCN21, 0.72 ± 0.09, p > 0.05; IN, input; PD, pulldown after biotin-switch assay of palmitoylation; NS, not significant; **, p < 0.01. GluA1 palmitoylation was unaffected by NMDA treatment (C). Note: this assay involves labeling all palmitoylated proteins with biotin followed by pulldown with streptavidin beads, elution, SDS-PAGE, and finally Western blotting for proteins of interest; BMCC (D and E): NMDA, 0.31 ± 0.07, p < 0.001, one-sample t test; tatCN21, 1.34 ± 0.34, p > 0.05; NMDA + tatCN21, 1.47 ± 0.31, p > 0.05; IN, input; IP, immunoprecipitation for detection of palmitoylation. Note: this assay involves immunoprecipitating the protein of interest, followed by labeling the palmitoylated fraction of the protein with biotin, processing via SDS-PAGE, and Western blotting first with streptavidin-conjugated HRP and then antibody toward the protein of interest. Irreversible lipidation of AKAP79 blocks its NMDA-induced spine-to-shaft translocation and spine shrinkage. F, 12 DIV hippocampal neurons transfected with AKAP79WT-GFP or myr-AKAP79-GFP along with an mCh cell fill were imaged before and 10 min following 30 μm NMDA application. G, NMDA treatment reduced AKAP79WT spine-to-shaft ratio but had no effect on myr-AKAP79 (AKAP79WT, 0.81 ± 0.04; myr-AKAP79, 1.02 ± 0.04, p < 0.001 by unpaired t test). ***, p < 0.001. H, NMDA application resulted in a signification reduction in mean spine cross-sectional area in AKAP79WT-expressing cells but not in those expressing myr-AKAP79 (fold change relative to pretreatment; AKAP79WT, 0.83 ± 0.03; myr-AKAP79, 1.08 ± 0.09, p < 0.01 by unpaired t test). *, p < 0.05. I, there was no significant NMDA-induced change in the spine-to-shaft ratio of the mCh cell fill in either condition. Scale bar, 5 μm. NS, not significant.
CaMKII inhibition does not significantly interfere with cLTD-induced F-actin re-organization
The AKAP79/150-targeting domain (see Fig. 1A) binds to F-actin (14) and the acidic membrane phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) (17), and AKAP79/150 removal from synapses requires cLTD-induced actin reorganization that is controlled by CaN signaling and phospholipase C (PLC)-mediated hydrolysis of PIP2 (14, 16). As the CaMKIIβ isoform binds and bundles F-actin only in its inactive state but not after Ca2+/CaM stimulation (27), CaMKII inhibition could directly or indirectly affect cLTD-induced actin dynamics. The cLTD stimuli caused a marked redistribution of F-actin from enrichment in spines to localization more in longer fibers along the dendrites (Fig. 2A), as expected from previous work (14, 16, 28). Although tatCN21 (but not KN93) increased the basal spine localization of F-actin, neither of the CaMKII inhibitors blocked the pronounced cLTD-induced F-actin redistribution (Fig. 2B). CaMKIIβ has been shown to be able to affect actin dynamics in principle (27, 29–34); however, the lack of an effect of CaMKII activity inhibition in cLTD-induced actin dynamics is consistent with our previous observation that LTD specifically requires the CaMKII α rather than the β isoform (7).
Figure 2.

F-actin re-organization during cLTD is unaltered by CaMKII inhibition. A, Texas Red phalloidin F-actin stain of DIV 12 hippocampal neurons before and 10 min after cLTD (30 μm NMDA). B, percent of F-actin synaptic localization from experiments from three individual neuron preparations, as determined by defining synaptic puncta using threshold masks as described under the “Experimental procedures.” cLTD-induced F-actin removal from spines as indicated by decreased percentage of F-actin in puncta was significant for control (before 1 ± 0.17 and after 0.5 ± 0.08, n = 19 neurons, ***, p < 0.001), tatCN21- (before 1.4 ± 0.17 and after 0.8 ± 0.07, n = 17 neurons, *, p < 0.05), and KN93 (before 1.1 ± 0.13 and after 0.6 ± 0.1, n = 19 neurons, ***, p < 0.001)-treated neurons as assessed by one-way ANOVA, Newman-Keuls post hoc analysis. Basally, tatCN21 and KN93 both stabilized F-actin in spines compared with control (##, p < 0.01; #, p < 0.05, respectively, one-way ANOVA, Newman-Keuls post hoc analysis). C, induction of the CaMKII Thr-286 autophosphorylation that generates autonomous activity after cLTP or cLTD stimuli of hippocampal cultures, assessed by Western blot analysis. The arrowhead marks 50 kDa. D, in contrast to regular substrates like MAP2, AKAP79/150 is phosphorylated more efficiently by autonomous versus Ca2+/CaM-stimulated activity. Both MAP2 and the AKAP79-targeting domain were present in the same radioactive phosphorylation assays; shown are autoradiographs after SDS-PAGE (left) and quantification of three experiments (right; mean ± S.E.). E, phosphorylation of the AKAP79-targeting domain by autonomous CaMKII reduces its interaction with F-actin in vitro, as assessed by a co-sedimentation assay. Shown is a Western blot of the supernatants (S) and pellets (P) after centrifugation at 100,000 × g that sediments F-actin together with bound proteins.
AKAP79/150-targeting domain is a high-autonomy CaMKII substrate
As CaMKII inhibition had no detectable effect on cLTD-induced F-actin re-organization that is required for synaptic removal of AKAP79/150, we next tested for direct phosphorylation of the AKAP79/150-targeting domain by CaMKII as a possible underlying mechanism for the CaMKII requirement in the cLTD-induced removal of AKAP79/150 from synapses. LTD specifically requires the autonomous (Ca2+-independent) activity of CaMKII that is induced by Thr-286 autophosphorylation (7); as expected, this Thr-286 autophosphorylation is induced by our cLTD stimuli in hippocampal neurons (Fig. 2C). Indeed, autonomous CaMKII efficiently phosphorylated the AKAP79-targeting domain (residues 1–153) in a biochemical assay with purified protein (Fig. 2D). In fact, phosphate incorporation into AKAP79 was more efficient even when compared with the classical CaMKII substrate protein MAP2 that was present within the same reaction mix (Fig. 2D). However, although further stimulation of the autonomous CaMKII by addition of Ca2+/CaM significantly enhanced MAP2 phosphorylation (as expected for traditional CaMKII substrates (21)), phosphorylation of the AKAP79-targeting domain was instead suppressed (Fig. 2D). Thus, the CaMKII autonomy (i.e. the ratio of autonomous over maximal stimulated activity) was around the traditional level of ∼20% for MAP2 phosphorylation but well over 300% for AKAP79 phosphorylation (Fig. 2D). Importantly, phosphorylation of the AKAP79-targeting domain by autonomous CaMKII decreased its binding to F-actin, as assessed by an in vitro co-sedimentation assay that showed more free AKAP79 that is not bound to F-actin in the supernatant after phosphorylation (Fig. 2E; a finding later corroborated also by imaging studies within cells, see Fig. 4).
Figure 4.

Thr-87/Ser-92 phosphorylation disrupts F-actin binding in COS7 cells but affects synaptic targeting only mildly. A, AKAP79-YFP targeting domain wild-type and Thr-87/Ser-92 phospho-deficient (AA) and phospho-mimetic (EE) mutants or YFP alone expressed in COS7 cells (left), fixed, and Texas Red phalloidin-stained (middle), merged (right). B, percentage of co-localization of expressed AKAP79 with F-actin was determined using masks, as detailed under “Experimental procedures.” The phospho-mimetic (EE) AKAP79 (19.3 ± 2.0%) was significantly less co-localized to F-actin (p < 0.001, one-way ANOVA, Newman-Keuls post hoc analysis) than both WT (26.2 ± 1.5%) and phospho-deficient (AA) (25.9 ± 2.6%), but it was not significantly more co-localized to F-actin than empty YFP (16.5 ± 1.3%). n = 11–12 cells in each condition. C, AKAP79-YFP (top left), AKAP79(AA)-YFP (bottom left), AKAP79(EE)-YFP (bottom right), and YFP alone (top right) expressed in DIV 12 hippocampal neurons. D, quantification of spine to dendritic shaft ratio (normalized to control) from experiments from four individual neuron preparations as determined by 10 masks per neuron (n = 19–20 neurons per condition). AKAP79(AA)-YFP (1.20 ± 0.05) had significantly higher localization (p < 0.001, one-way ANOVA, Newman Keuls post hoc analysis) to dendritic spines compared with AKAP79-YFP (1.00 ± 0.03), whereas AKAP79(EE)-YFP (0.86 ± 0.05) spine localization was significantly reduced relative to control. All AKAP79 constructs expressed showed greater spine localization than YFP (0.43 ± 0.02) alone. E, spine to shaft ratio of an mTurquoise2 cell fill (images not shown) was unaffected by any of the AKAP constructs. *, p < 0.05; ***, p < 0.001; NS, not significant.
Molecular mechanism for high-autonomy phosphorylation of AKAP79/150
Notably, LTP stimuli favor CaMKII-mediated phosphorylation of traditional low-autonomy substrates such as GluA1 whereas LTD stimuli favor phosphorylation of the high-autonomy substrate GluA1 Ser-567 (7). The novel high-autonomy substrate AKAP79/150 identified here exhibited a substantially further elevated level of autonomy compared with GluA1 Ser-567 (i.e. >300% versus ∼100%), further strengthening possible mechanistic links between AKAP79/150 phosphorylation by CaMKII and LTD.
Sequence analysis showed that the polybasic region B of the AKAP79-targeting domain (AKAP-B) contains three Ser/Thr residues that meet the basic criterion for CaMKII consensus sites, an Arg residue at position −3 (Fig. 3A; compare also Fig. 1A). Additionally, AKAP-B contains a predicted CaM-binding site, as determined by sequence analysis using the calmodulin target database (University of Toronto, Toronto, Ontario, Canada; http://calcium.uhnres.utoronto.ca/ctdb/ctdb/sequence.html)8 and as shown in previous studies (35, 36). Thus, addition of Ca2+/CaM may further stimulate the activity of autonomous CaMKII in principle, but may additionally specifically protect the AKAP79 phosphorylation sites from phosphorylation by binding to the overlapping binding site, thereby resulting in the apparent autonomy of over 300%. This hypothesis was tested, and it is indeed supported by the following findings.
Figure 3.
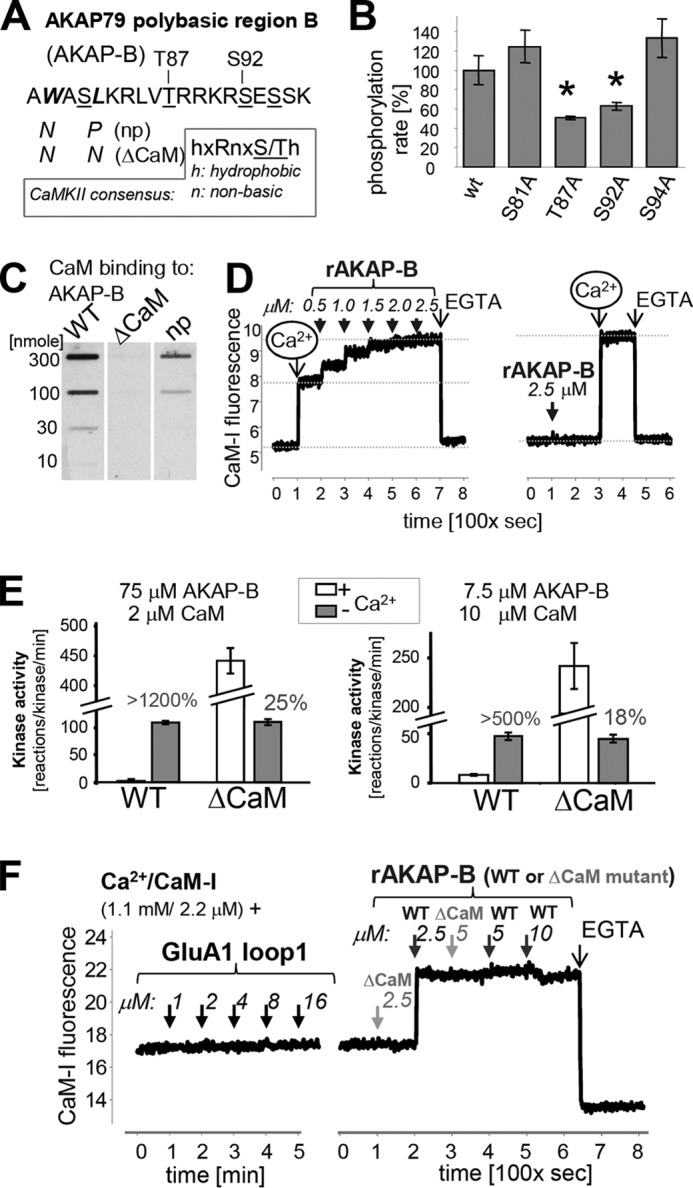
AKAP79 is protected from phosphorylation by a substrate-directed Ca2+/CaM mechanism. A, sequence of the AKAP-B peptide, which is derived from the AKAP79 polybasic region B, with mutations and potential phosphorylation sites indicated; the CaMKII phosphorylation consensus sequence is shown for comparison. B, Thr-87 and Ser-92 are substrate sites for autonomous CaMKII, as indicated by reduced in vitro phosphorylation of their respective alanine-mutant peptides. C, ΔCaM mutation (see A) prevents Ca2+/CaM binding to AKAP-B, as detected by blot-overlay with biotinylated CaM. D, rat AKAP-B region also binds CaM in a Ca2+-dependent manner, as detected using the labeled CaM-I that increases in fluorescence upon binding to Ca2+ and protein. Binding to rat AKAP-B was detected only in the presence of Ca2+. E, ΔCaM mutation abolishes the substrate protection by Ca2+/CaM and results in phosphorylation like a regular substrate, with more phosphorylation in the presence of Ca2+/CaM, resulting in ∼20% autonomy. F, high CaMKII autonomy for the phosphorylation site in the GluA1 loop1 is not due to substrate protection by Ca2+/CaM, as no binding of CaM-I was detected in the fluorescence assay. In the same assay, clear CaM-I binding to rat AKAP-B (but not its ΔCaM mutant) was detected.
Two of the predicted AKAP-B residues, Thr-87 and Ser-92, were phosphorylated by autonomous CaMKII, as determined by in vitro phosphorylation assays of the AKAP-B peptide and its various site-specific phosphorylation-incompetent mutants (Fig. 3B). Ca2+/CaM indeed bound to AKAP-B in an overlay assay with biotinylated CaM, and two mutations predicted to interfere with the CaM-binding site (np, W79N/L82P; ΔCaM, W79N/L82N) indeed disrupted or abolished the CaM binding (Fig. 3C). The hypothesized mechanism for high CaMKII autonomy by CaM-mediated substrate protection requires that CaM must bind to AKAP-B only when stimulated by Ca2+ but not in basal conditions without Ca2+. Such Ca2+-dependent binding to AKAP-B was indeed observed using an IAEDANS-labeled CaM (CaM-I) that increases in fluorescence upon binding to Ca2+ and protein or peptide (37, 38). Fluorescence of CaM-I increased after addition of Ca2+ and then increased further after addition of AKAP-B (Fig. 3D). By contrast, adding AKAP-B first did not cause any fluorescence increase, whereas subsequent addition of Ca2+ again increased fluorescence to the maximal level (Fig. 3D).
For a final test of the mechanism underlying the high autonomy of CaMKII toward AKAP79/150, the phosphorylation of AKAP-B wild-type (WT) and its CaM-binding-deficient mutant (ΔCaM) was compared in biochemical kinase assays in vitro. Both AKAP-B WT and ΔCaM were equally efficiently phosphorylated by autonomous CaMKII in the absence of Ca2+. However, although addition of Ca2+/CaM to naive CaMKII resulted in only negligible phosphorylation of AKAP-B WT, phosphorylation of AKAP-B ΔCaM was instead even 4–5-fold greater compared with the autonomous phosphorylation (Fig. 3E). Thus, abolishing the CaM-binding site transformed AKAP-B into a regular CaMKII substrate, demonstrating that the Ca2+/CaM binding is indeed the mechanism underlying suppression of phosphorylation during stimulation, thereby causing the unusually high autonomy. In the initial experiments (Fig. 3E, left panel), concentrations of substrate and CaM were used that are typical for CaMKII assays. However, these typical assay conditions contain an excess of substrate (75 μm AKAP-B) over CaM (2 μm); the observed results are likely due to sequestration of CaM by AKAP-B (which would in turn dramatically reduces stimulation of activity for the non-phosphorylated naive CaMKII), rather than to substrate protection by CaM binding. However, the same results were also observed when CaM (10 μm) was instead in excess over AKAP-B (7.5 μm), albeit at lower total phosphorylation rates (Fig. 3E, right panel), as expected at lower substrate concentration. Under these conditions, even full occupancy of AKAP-B with CaM still results in sufficient free Ca2+/CaM (2.5 μm) for maximal CaMKII stimulation. Thus, substrate-directed Ca2+/CaM binding is indeed a mechanism underlying high CaMKII autonomy and suppression of phosphorylation during Ca2+ stimulation.
AKAP79/150 and GluA1 Ser-567 belong to different LTD-related CaMKII substrate classes
As mentioned above, the other known LTD-related high-autonomy substrate of CaMKII is GluA1 Ser-567 (7), but the mechanism underlying the high autonomy is unknown in this case. Although CaMKII autonomy for the AKAP79/150-targeting domain (>300%) is significantly greater than for GluA1 Ser-567 (∼100%), we argued that this autonomy for GluA1 Ser-567 could also be caused by overlapping Ca2+/CaM binding, but perhaps with lower affinity. Indeed, the GluA1 loop1 region that contains Ser-567 contains a sequence with similarity to the “basic 1–5-10 motif” for Ca2+/CaM binding, albeit with a one amino acid insertion compared with the consensus sequence. However, in an overlay assay with biotinylated CaM, no CaM binding to the GluA1 loop1 was detected. This result could still be consistent with a lower affinity binding. However, even in the more sensitive binding assay that utilizes CaM-I fluorescence, no CaM binding to GluA1 loop1 was detectable (Fig. 3F). Thus, GluA1 Ser-567 and the AKAP79/150-targeting domain represent two distinct classes of LTD-related high-autonomy CaMKII substrates. Although the mechanism underlying high autonomy for AKAP79/150 was demonstrated here (see Fig. 3E), the mechanism for GluA1 Ser-567 remains to be elucidated.
Phosphomimetic Thr-87/Ser-92 mutations in AKAP79/150 decrease F-actin co-localization in COS7 cells but only mildly decrease synaptic localization
CaMKII activity was required for cLTD-induced synaptic removal of AKAP79/150 in neurons (see Fig. 1), and CaMKII-mediated phosphorylation of the AKAP79/150-targeting domain inhibited its binding to F-actin in vitro (see Fig. 2F). Thus, we decided to test the effect of mutating the CaMKII phosphorylation sites on the co-localization of AKAP79/150 with F-actin in heterologous cells. AKAP79 tagged with YFP on its C terminus was expressed in COS7 cells, and F-actin was stained with phalloidin-Texas Red (Fig. 4A). Mutating the CaMKII phosphorylation sites Thr-87 and Ser-92 to Ala (AA) had no effect on F-actin co-localization, as expected. By contrast, corresponding phospho-mimetic mutations to Glu (EE) still exhibited prominent localization in membrane structures but significantly reduced AKAP79 co-localization with F-actin (Fig. 4B). In fact, F-actin co-localization of the phospho-mimetic AKAP79-YFP EE mutant was statistically undistinguishable from the negative control with YFP alone (Fig. 4B). These results indicate that phosphorylation of the Thr-87/Ser-92 CaMKII sites in the targeting domain can disrupt the F-actin association of AKAP79/150 within cells. To examine the effects of the CaMKII phosphorylation sites on AKAP79/150 in neurons, AKAP79-YFP and its Thr-87/Ser-92 mutants were expressed in hippocampal cultures (Fig. 4C). Although the phosphorylation-incompetent AA mutant localized to dendritic spines significantly more than the phospho-mimetic EE mutant, this difference was rather mild (Fig. 4D). The spine localization of mTurquois2 used as a cell fill was unaffected by the co-expressed AKAP79-YFP construct (Fig. 4E). No significant differences in spine size were detected (data not shown). Thus, although the CaMKII-mediated phosphorylation of Thr-87/Ser-92 can inhibit targeting domain interaction with F-actin, it does not appear to be sufficient to induce the removal of AKAP79/150 from dendritic spines.
Combined phosphorylation and depalmitoylation removes AKAP79/150 from synapses
Next, we decided to test whether additional phosphorylation sites in the AKAP79/150 polybasic targeting domain might be responsible for the LTD-induced CaMKII-dependent removal of AKAP79/150 from spines. Mutating all five potential Ser/Thr phosphorylation sites in the B region (in the 5A mutant) reduced CaMKII-mediated phosphorylation of the AKAP79/150-targeting domain; however, mutating additional sites in the C region (in the 9A mutant) reduced phosphorylation even further (Fig. 5, A and B). More importantly, phosphorylation of both AKAP79/150 WT and 5A (but not 9A) by autonomous CaMKII was suppressed by addition of Ca2+/CaM (Fig. 5B), indicating that the additional sites in the C region are also LTD-related high autonomy sites that are protected by Ca2+/CaM, whereas the remaining phosphorylation sites in the 9A mutant, which are located outside the AKAP-targeting domain, are not protected by Ca2+/CaM. (Please also note that at least some of these remaining phosphorylation sites may actually be located in the vector-encoded linker regions flanking the C-terminal His6 epitope tag, see under “Experimental procedures”). Indeed, experiments with IAEDANS-labeled CaM showed that all of the three AKAP79/150 polybasic regions bind Ca2+/CaM, with region B showing the highest affinity, followed by regions C and A (Fig. 5C). Specificity of CaM binding was examined further by single point mutations of the predicted binding site of each region; in each case, Ca2+/CaM binding was significantly reduced, although not completely abolished (Fig. 5D). Although Ca2+/CaM binding to the A and B regions has been indicated by previous studies (35, 36, 39), this is the first report of Ca2+/CaM binding to the C region, as well as the first direct comparison of all three binding sites.
Figure 5.

AKAP79/150-targeting domain contains multiple Ca2+/CaM-protected CaMKII phosphorylation regions. A, sequence alignment of the AKAP79/150 B and C polybasic targeting regions from the indicated species showing conservation (bold if conserved across three or more species) of basic residues (blue) and putative Ser/Thr phosphorylation sites (red). Other residues are shown in black, and the conserved C palmitoylation in the C polybasic regions is indicated in green. B, autoradiogram showing the relative levels of autonomous CaMKII phosphorylation of AKAP79(1–153) WT, 9A, and 5A (top panel). Autoradiogram showing the relative phosphorylation levels of AKAP79(1–153) WT, 9SA, and 5SA with autonomous (EGTA) versus Ca2+/CaM-stimulated CaMKII (−EGTA) showing Ca2+/CaM protection for WT and B5SA but not 9A (bottom panel. Note: these samples are all from the same gel and same exposure but the lanes have been rearranged in the figure to allow easier side-by-side comparisons of the + and −EGTA conditions for each construct). C, CaM-I binds to each of the three AKAP79 polybasic regions, as indicated by increased CaM-I fluorescence upon adding increasing concentrations of peptides derived from polybasic region A, B, or C. AKAP-B bound with the highest affinity, followed by AKAP-A and -C. D, CaM-I binding is significantly reduced by the indicated individual point mutations in the respective predicted CaM-binding sites in AKAP-A, -B, or -C.
Similar to the T87E/S92E mutation, the full phospho-mimetic 9D mutation reduced basal synaptic localization of AKAP79 only mildly. However, the 9D mutation significantly facilitated the cLTD-induced synaptic removal of AKAP79. Although full removal of AKAP79 wild-type required stimulation with 30 μm NMDA, 10 μm NMDA was sufficient for full removal of the 9D mutant (Fig. 6, A and B). The corresponding phospho-incompetent 9A mutant was still fully removed from spines by 30 μm NMDA; however, in contrast to AKAP79 wild type (or 9D), it did not significantly move in response to 10 μm NMDA (Fig. 6, A and B). Thus, the AKAP79-targeting domain phosphorylation facilitates the cLTD-induced removal from spines, but is neither absolutely necessary nor sufficient.
Figure 6.

AKAP79-targeting domain phosphorylation and palmitoylation state cooperate to regulate basal spine localization and removal from spines during NMDA-induced LTD. A, time-lapse images of hippocampal neuron dendrites expressing GFP-tagged AKAP79 wild-type (WT) and targeting domain B and C region phospho-deficient (9A), phospho-mimetic (9D), and combined phospho-mimetic and palmitoylation-deficient (9DCS) mutants before (Control) and after treatment with 10 μm NMDA for 10 min followed by increasing the concentration to 30 μm NMDA for an additional 10 min. B, quantification of spine to dendritic shaft ratios for experiments as in A revealing NMDA dose-dependent 79WT translocation from spines to the dendritic shaft cytoplasm (control 1.06 ± 0.05; 10 μm NMDA 0.86 ± 0.05; 30 μm NMDA 0.61 ± 0.05; n = 11 neurons per condition). In contrast, 9A exhibited resistance (control 1.03 ± 0.09; 10 μm NMDA 0.98 ± 0.09, 30 μm NMDA 0.60 ± 0.04; n = 9 neurons per condition) and 9D exhibited sensitization (control 0.93 ± 0.05; 10 μm NMDA 0.61 ± 0.07; 30 μm NMDA 0.60 ± 0.06; n = 10 neurons per condition) to NMDA-induced translocation from spines (*, p < 0.05; **, p < 0.01 to corresponding controls, one-way ANOVA, Dunnett's post hoc). Compared with 79WT, 9D basal spine localization was slightly reduced (#, p < 0.05 by unpaired t test to 79WT control), and 9SDCS basal spine localization was strongly reduced (control 0.59 ± 0.05; ###, p < 0.0001 by unpaired t test to 79WT control). C, [3H]palmitate labeling in HEK-293 cells confirms complete loss of palmitoylation of 9DCS but lacks the resolution necessary to quantitatively compare the other AKAP mutant species. D, APEGS assay of AKAP palmitoylation mutants expressed in HEK cells reveals three bands for AKAP79WT-GFP. The highest molecular weight band corresponds to a dual palmitoylation species; the middle band represents mono-palmitoylated species, and the bottom band reveals unpalmitoylated species. Two bands are present for the C36S and C129S mutants, and a single band is present in the C36S/C129S and WT (H−) conditions. E, APEGS assay comparing palmitoylation states among the indicated AKAP79-GFP mutants. The proportion of each mutant that is palmitoylated was quantitated as a ratio of the higher molecular weight band intensities relative to the lowest molecular weight band. IN, input; A, APEGS assay of palmitoylation. F, quantification of the proportion of PEGylated (and thus palmitoylated) species in E reveals significantly reduced palmitoylation of the phosphomimetic AKAP79–9D and enhanced palmitoylation of myristoylated AKAP79 (WT, 0.44 ± 0.02; 9D, 0.25 ± 0.03; 9A, 0.51 ± 0.05; EE, 0.44 ± 0.04; myr, 0.73 ± 0.05, *, p < 0.001 by one-way ANOVA, Dunnett's post hoc).
A similarly facilitated spine removal with minimal effect on basal spine localization has been observed for the palmitoylation-incompetent AKAP79 C36S/C129S (CS) mutant (18), indicating that AKAP79 depalmitoylation also facilitates spine removal without being sufficient. Thus, the effect of the 9D mutant on facilitating synaptic AKAP79 removal could be mediated by preventing AKAP79 palmitoylation. However, AKAP79 WT, 9D, or 9A all appeared to be palmitoylated as assessed by [3H]palmitate labeling in HEK-293 cells (Fig. 6C). Nonetheless, we further investigated the effect of AKAP79 phosphorylation on its palmitoylation state using a recently developed acyl-PEGyl exchange gel shift (APEGS) assay that allows palmitoylation stoichiometry to be determined more directly (40). By labeling palmitoylated cysteines with bulky polyethylene glycol moieties and subsequent SDS-PAGE/Western blotting, palmitoylation species can be visualized as higher molecular weight (MW) bands. Consistent with the two known palmitoylation sites, APEGS analysis of AKAP79-GFP WT in HEK-293 cells revealed two bands corresponding to mono- and di-palmitoylated AKAP79 that migrated at higher MW than the unmodified/depalmitoylated species (Fig. 6C). Importantly, neither of these higher MW bands was visible for the C36S/C129S double mutant, and only one of these bands was visible for each of the C36S and C129S single mutants (Fig. 6C). Using this APEGS approach, we found a significant reduction in palmitoylation for the 9D mutant, with no changes detected for either the 9A or EE mutants (Fig. 6, E and F). Thus, although mimicking AKAP79 phosphorylation reduces palmitoylation, it does not eliminate it, indicating that phosphorylation and depalmitoylation must act in concert to mediate synaptic removal of AKAP79/150. Indeed, combining the C36S/C129S and 9D mutations was sufficient to fully mimic the cLTD-induced synaptic removal of AKAP79 such that the 9DCS combination mutant was localized predominantly in the dendritic shaft cytosol even under basal control conditions without any stimulation (Fig. 6, A and B).
CaMKII inhibition blocks cLTD-induced AKAP150 depalmitoylation
Our results show that combined AKAP79/150 phosphorylation and depalmitoylation is sufficient to remove the AKAP from dendritic spines, although individually phosphorylation or depalmitoylation are not. In agreement with our prior studies, using two complementary biotin-switch assays for the AKAP150 palmitoylation state (18, 19), we found that cLTD stimulation that removes AKAP150 from dendritic spines also caused robust AKAP150 depalmitoylation in cultured hippocampal neurons (ABE assay, Fig. 7, A and B; BMCC assay, Fig. 7, D and E). As AKAP79/150 phosphorylation facilitated its LTD-induced spine removal but was not strictly required for it, we tested whether the CaMKII dependence of this spine removal could be explained instead by CaMKII dependence of the AKAP150 depalmitoylation. Indeed, CaMKII inhibition with tatCN21 blocked the cLTD-induced AKAP150 depalmitoylation (Fig. 7, A–E). By contrast, the basal AKAP150 palmitoylation state in the absence of any cLTD stimulus was unaffected by the tatCN21 treatment. Finally, we found no effect of cLTD or CaMKII inhibition on the GluA1 palmitoylation state (Fig. 7, A and C), supporting the notion that AKAP79/150 depalmitoylation is specifically regulated and does not reflect a global depalmitoylation of synaptic proteins.
Depalmitoylation is required for both AKAP79 removal and structural LTD
To mimic persistent palmitoylation of AKAP79, we added a consensus sequence for N-terminal myristoylation, a related lipid modification that is irreversible (19). In addition, APEGS analysis of this myr-AKAP79-GFP mutant expressed in HEK-293 cells revealed significantly increased palmitoylation compared with WT (Fig. 6, E and F). Importantly, preventing de-lipidation in this myr-AKAP79 mutant completely blocked the cLTD-induced AKAP79 movement in response to 30 μm NMDA (Fig. 7, F and G). Thus, depalmitoylation is required for cLTD-induced synaptic AKAP removal, whereas phosphorylation facilitates this movement without being required. Notably, the myr-AKAP79 mutant also enabled us to directly address the question whether or not removal of AKAP79/150 from spines is also required for structural LTD, i.e. the spine shrinkage that occurs in response to LTD stimuli. Although expression of AKAP79-GFP wild-type allowed normal spine shrinkage in response to cLTD stimulation with 30 μm NMDA, expression of the myr-AKAP79 mutant completely blocked such cLTD-induced spine shrinkage (Fig. 7H). Note that spine size was determined by the area occupied by a fluorescent spine filler (mCherry), whereas AKAP79 spine localization was determined by point masks within the spines, which instead reflects its concentration. Thus, the noted movement of AKAP79/150 out of spines is not simply a direct consequence of the reduced spine size. Indeed, when mCherry localization is assessed in the same manner as the AKAP79/150 localization, no movement was detected (Fig. 7I).
Discussion
We show here that the well-studied removal of the synaptic anchoring protein AKAP79/150 from dendritic spines in response to LTD stimuli is functionally required for structural LTD, and like LTD expression, it also requires CaMKII activity. Direct phosphorylation by CaMKII facilitated the removal of AKAP79/150 from spines, but it was not required; instead, the essential function of CaMKII was additional regulation of AKAP79/150 depalmitoylation (shown schematically in Fig. 8).
Figure 8.

Model for the LTD-specific removal of AKAP79/150 by CaMKII signaling. Under basal conditions, a pool of AKAP79/150 is palmitoylated and present in dendritic spines. During brief, strong Ca2+ influx that induces LTP, any depalmitoylated AKAP is protected from phosphorylation by Ca2+/CaM-stimulated CaMKII activity due to Ca2+/CaM binding to the targeting domain. In contrast, following weak and extended Ca2+ influx, such as elicited by NMDA-LTD induction, autonomous CaMKII activity promotes AKAP79/150 depalmitoylation, removal from spines, and spine shrinkage. These effects are accomplished in part by phosphorylation of AKAP79/150's basic membrane target domains (which is no longer protected by Ca2+/CaM) but likely require additional pathways that block DHHC PAT and/or activate PPT enzymatic activities. Inhibitors of CaMKII block NMDA-induced AKAP79/150 depalmitoylation, and ectopic lipidation of AKAP79/150 blocks its NMDA-induced removal from spines and spine structural remodeling.
AKAP79/150 targeting to dendritic spines is mediated at least in part by its interaction with the F-actin cytoskeleton (14, 16), and CaMKII-mediated phosphorylation of AKAP79/150 disrupted this interaction. Although previous studies found that PKC phosphorylation of the AKAP79-targeting domain could also inhibit its interaction with F-actin in vitro (14), inhibition of PKC did not prevent LTD-induced AKAP translocation from spines (16), as we observed here for inhibition of CaMKII (Fig. 1). Thus, although both CaMKII and PKC can phosphorylate the AKAP79-targeting domain and regulate F-actin binding in vitro, only CaMKII activity appears to be important for control of AKAP79/150 targeting in neurons. A possible explanation could be that the persistent autonomous activity of CaMKII generated by LTD stimuli (Fig. 2C) (7, 41) may allow CaMKII to phosphorylate the AKAP-targeting domain when Ca2+ levels fall low enough to reverse the Ca2+/CaM protection that we observed for these substrate sites (Fig. 8). Although low Ca2+ would also allow PKC access to these previously Ca2+/CaM-protected substrate sites (14), low Ca2+ levels likely would be insufficient to maintain PKC activation. In addition, CaMKII could be promoting AKAP depalmitoylation through additional mechanisms (discussed below) as well as inhibiting AKAP interactions with MAGUK scaffold proteins to further promote spine loss (42).
Related to this Ca2+/CaM protection of targeting domain phosphorylation, we found that AKAP79/150 represents a novel class of LTD-related high-autonomy CaMKII substrates that is only appreciably phosphorylated by autonomous CaMKII activity. One other LTD-related high-autonomy CaMKII substrate identified in our previous studies is GluA1 Ser-567 (7), a site that inhibits AMPAR synaptic localization (20). However, GluA1 Ser-567 was phosphorylated equally well by autonomous or Ca2+/CaM-stimulated CaMKII activity, and it does not exhibit high autonomy due to Ca2+/CaM-mediated substrate protection as seen for AKAP79/150. A similar negative regulatory mechanism has been described previously for the CaMKII auto-phosphorylation sites Thr-305/Thr-306, which prevent subsequent stimulation of CaMKII by Ca2+/CaM (43–45) and may thereby promote LTD by suppressing phosphorylation of LTP-related regular substrates (7, 22, 46, 47). Overall, these mechanisms together would favor phosphorylation of high autonomy substrates, such as AKAP79/150 and GluA1 Ser-567, by prolonged, weak LTD stimuli, whereas phosphorylation of regular substrates such as GluA1 Ser-831 are favored by brief, strong LTP stimuli (7).
Whereas mimicking AKAP79-targeting domain phosphorylation in the 9D mutant facilitated its LTD-induced removal from spines (Fig. 6, A and B) and reduced its palmitoylation (Fig. 7, E and F), this mutation was not sufficient to disrupt AKAP spine localization. Likewise our previous studies found that mimicking AKAP79 depalmitoylation in the CS mutant also facilitated LTD-induced removal from spines but was not sufficient to disrupt basal spine localization (18). However, the 9DCS mutant that mimics both phosphorylation and depalmitoylation was able to disrupt AKAP79 basal spine targeting (Fig. 6, A and B). Yet the 9A phospho-deficient mutant did not prevent LTD-induced translocation from spines, indicating that although CaMKII phosphorylation of the targeting domain may promote AKAP depalmitoylation and LTD-induced translocation from spines, it is not strictly required. In contrast, through use of the constitutively lipidated myr-AKAP79 mutant we were able to determine that AKAP79 depalmitoylation is indeed required for LTD-induced removal from spines. Importantly, LTD-induced depalmitoylation of endogenous AKAP150 required CaMKII activity (Fig. 7, A, B, D, and E). Overall, these analyses indicate that CaMKII mediates AKAP79/150 removal from spines during LTD in part through direct phosphorylation of the targeting domain but more prominently through promoting its depalmitoylation (Fig. 8).
Protein palmitoylation is controlled by the opposing enzymatic activities of DHHC palmitoyl acyltransferases (PATs), which are integral membrane proteins that catalyze the addition of palmitate to free cysteine residues, and a subset of metabolic serine hydrolases known as protein-palmitoyl thioesterases (PPTs), which are peripheral membrane proteins that reverse this modification (40, 48, 49). Thus, CaMKII activity could either be promoting depalmitoylation by PPTs or inhibiting palmitoylation by PATs. This regulation by CaMKII may involve direct enhancement of PPT or inhibition of PAT enzymatic activities. AKAP79/150 is known to be palmitoylated by the PAT DHHC2 (19), but the PPTs that control its depalmitoylation have yet to be determined, and the signaling mechanisms regulating the enzymatic activities of DHHC2 (and other PATs) remain uncharacterized. In addition, as suggested by the reduced basal palmitoylation of the 9D phosphomimetic mutant, CaMKII phosphorylation of the AKAP-targeting domain can also indirectly regulate palmitoylation/depalmitoylation by controlling the access of PATs and/or PPTs to the embedded Cys-36/129 substrate sites. However, although the experiments with the 9A and 9D mutants suggest that phosphorylation contributes to facilitating AKAP depalmitoylation and removal from the synapse, they also indicate that phosphorylation is neither necessary nor sufficient for this regulation. Thus, future work exploring how CaMKII might suppress PAT activity and/or promote PPT activity will be highly informative.
Palmitoylation of postsynaptic scaffold proteins, including PSD-95, GRIP1, δ-catenin, and AKAP79/150, is emerging as a key signaling mechanism regulating excitatory synaptic plasticity, including both LTP/LTD and homeostatic plasticity (18, 19, 50–54). Synaptic protein palmitoylation is also affected by seizures and anticonvulsants (18, 55, 56), and several DHHC PAT mutations and substrates have been linked to neurological and neuropsychiatric disorders, including Huntington's, schizophrenia, and X-linked intellectual disability, that are characterized by altered synaptic plasticity (48, 57, 58). Our findings here reveal novel cross-talk between postsynaptic Ca2+ signaling, palmitoylation, and phosphorylation pathways that are required for synaptic plasticity and thus have implications for understanding these diseases.
Our present findings for CaMKII inhibition, along with our past findings for CaN (12, 14) and PLC inhibition (16), show that blocking the signaling pathways that mediate AKAP79/150 synaptic removal also block structural and functional aspects of LTD. Yet such correlations do not show causation, as these treatments, including inhibition of CaMKII, PLC, CaN, or stabilization of F-actin, could also have global effects that prevent structural LTD independently from their effects on AKAP79/150 movement. However, the myr-AKAP79 mutant utilized here provides the most direct evidence to date that AKAP79/150 removal from spines is indeed required for postsynaptic modifications underlying LTD. AKAP79/150 palmitoylation promotes its inclusion in lipid rafts and controls not only its localization to the plasma membrane but also to recycling endosomes that are important for surface delivery of GluA1-AMPARs that support both LTP and LTD (13, 18, 19, 59). Indeed, LTP stimuli promote AKAP palmitoylation and its recycling endosome-dependent recruitment to spines, and the palmitoylation-deficient AKAP79CS mutant interferes with both this spine recruitment and LTP-associated spine enlargement (18). In contrast, LTD stimuli decrease AKAP palmitoylation and promote its removal from spines along with spine shrinkage; as shown here, these postsynaptic changes are prevented by both the constitutively lipidated myr-AKAP79 mutant and CaMKII inhibition.
Finally, if AKAP removal from spines is a key step in LTD as our present findings indicate, then how is it mediating LTD? Many of AKAP79/150's effects on synaptic function are derived from its bound pools of PKA and CaN (11–13, 60, 61). In particular, phosphorylation/dephosphorylation of GluA1 Ser-845 is critical for AMPAR regulation in LTD (62–66), and recently, we described roles for AKAP79/150-anchored PKA and CaN in controlling GluA1 Ser-845 phosphorylation and the recruitment to and subsequent removal of GluA1-AMPARs from synapses during LTD (12, 13). CaN control of the actin-severing protein cofilin (through the phosphatase Slingshot-1L) is also thought to play a critical role in F-actin remodeling and spine shrinkage during LTD (67, 68). However, both spine F-actin depolymerization and AKAP removal from spines during LTD additionally require PLC-dependent PIP2 hydrolysis, which may both weaken AKAP membrane targeting interactions, as discussed above, and facilitate cofilin activation (16). In addition, although it is known that disruption of CaN-anchoring to AKAP79/150 inhibits its removal from spines and that inhibition of CaN phosphatase activity prevents both LTD-induced F-actin reorganization and AKAP removal, it is not known if AKAP-anchored CaN specifically controls F-actin reorganization during LTD (14, 68).
Thus, taken together, the model that emerges is that LTD induction results in activation of multiple signaling pathways involving AKAP-bound CaN, PLC, F-actin depolymerization, autonomous CaMKII, and AKAP depalmitoylation that all converge to disrupt AKAP79/150-targeting domain interactions with F-actin and lipid membranes. In turn, this inhibition of AKAP79/150 targeting interactions promotes its removal from the plasma membrane, as well as endosomes, to prevent re-phosphorylation of postsynaptic CaN substrates such as GluA1 Ser-845 that are important for LTD. Overall, our findings reveal new cross-talk between two dynamic post-translational modifications, phosphorylation and palmitoylation, and two major postsynaptic kinase/phosphatase signaling nodes, CaMKII and AKAP-PKA/CaN, that cooperatively control LTD-associated postsynaptic modifications.
Experimental procedures
Materials
CaM was from an available stock originally purified after bacterial expression as described (37). CaMKIIα was purified after baculovirus/Sf9 cell expression using a phosphocellulose column followed by CaM-Sepharose affinity chromatography as described (69, 70). MAP2 was purchased from Sigma. ATP and CaMKII inhibitor, KN93, were obtained from Calbiochem, EMD Millipore Co. (Billerica, MA). NMDA was obtained from Alexis Biochemical (San Diego). CaMKII peptide inhibitor, tatCN21, was synthesized by BioMatik Co. (Wilmington, DE). APV came from Tocris Bioscience (St. Louis, MO).
Antibodies used in this study include polyclonal AKAP150 antibody (71), monoclonal anti-PSD95 (Neuromab Davis, CA), polyclonal anti-GFP (Invitrogen), fluorescent labeled secondary antibodies, and Texas Red Phalloidin stain (Invitrogen).
Mutation of AKAP79-targeting domain regions B and C to change serine and threonine residues to phosphorylation-deficient alanine or phosphorylation-mimetic glutamate or aspartate and to change cysteine to palmitoylation-deficient serine were created using Quick-Change site-directed mutagenesis kit from Agilent Technologies, formerly Stratagene (La Jolla, CA). The T87A/S92A and T87E/S92E combination mutations in the B region are referred to as “AA” and “EE,” respectively. The S81A, T87A, S92A, S94A, and S95A mutations in the B region are referred to as “5A”. The S81A, T87A, S92A, S94A, S95A, S117A, S123A, S139A, and S142A and the S81D, T87D, S92D, S94D, S95D, S117D, S123DA, S139DA, S142D mutations in both the B and C regions are referred to as “9A” and “9D”, respectively. The palmitoylation-deficient C36S/C129S combination mutation is referred to as “CS” and was previously described by Keith et al. (18). The myristoylated (depalmitoylation-resistant) myr-AKAP79 construct is described in Woolfrey et al. (19).
Neuronal culture, cLTD stimulation, transfection, and immunostaining
Medium density hippocampal neurons were cultured from P0-1 Sprague-Dawley rats as described previously (14, 72). DIV 11–14 neurons were Lipofectamine 2000 (Life Technologies, Inc.) transfected with plasmid DNA and live-imaged or processed for immunocytochemistry 2 days later.
For experiments analyzing fixed cells, induction of cLTD was achieved by 30 μm NMDA (Tocris) treatment for 3 min followed by a 10-min recovery (unless indicated otherwise) in conditioned media and subsequent fixation. The same cLTD stimulus was used for assessing biochemical effects on CaMKII phosphorylation; the cLTP stimulation in these experiments was a 10-min incubation in Tyrode's buffer without Mg2+ and with 200 μm glycine. In experiments assessing the effects of CaMKII inhibition, neurons were treated with tatCN21 (5 μm) or KN93 (10 μm) 20 min prior to induction of cLTD. In live-imaging experiments, cLTD was induced by 10 or 30 μm NMDA for 10 min as described under the “Results” and in the figure legends.
Neurons were fixed in a 4% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA), 4% sucrose/PBS, permeabilized in 0.1% Triton/PBS at room temperature, and blocked in 10% BSA overnight at 4 °C. Fixed neurons were stained with primary antibodies in 5% BSA at room temperature, washed three times in PBS, and incubated in the indicated Alexa fluorophore or Texas Red-conjugated species-specific secondary antibodies. For additional F-actin staining, fixed neurons were stained with 0.165 μm Texas Red phalloidin.
Imaging and quantification of live and fixed hippocampal neurons
Images of live and fixed cells were taken on a Zeiss Axiovert 200M system with ×63 oil immersion objective, CoolSnap HQ charge-coupled device camera (Roper Scientific), xenon lamp LB-LS/17 (Sutter Instruments), using Slidebook software (Intelligent Imaging Innovations). Additional live imaging in Fig. 7 was conducted at 37 °C on a Zeiss Axio Observer microscope with a ×63 Plan-Apo/1.4 NA objective, using 488- and 561-nm laser excitation and a CSU-XI spinning disk confocal scan head (Yokogawa) coupled to an Evolve 512 EM-CCD camera (Photometrics) controlled by Slidebook software.
In experiments that assessed the effects of CaMKII inhibition on AKAP150 translocation following NMDA stimulation and those that assessed the synaptic localization of AKAP79 phospho- and palmitoyl-mutants, 15 images were taken at 0.5-μm intervals in the z-plane, deconvolved, and maximum-projected. Ten point masks per neuron were drawn on dendritic spines. For each spine mask, a corresponding line mask was also drawn in the dendritic shaft, and the ratio of mean fluorescence of the spine masks to the mean fluorescence of shaft masks was calculated.
For experiments evaluating the re-organization of F-actin, the co-localization of F-actin with PSD-95 (not shown in images) was assessed. Sections of dendrite were masked by hand. Dendritic spines within this mask were then defined by a PSD-95 channel threshold hold mask 1.5× mean PSD-95 fluorescence. The sum intensity of the F-actin channel within this dendritic spine mask was divided by the total dendritic F-actin channel intensity to yield the normalized synaptic localization of F-actin.
In experiments determining the sensitivity of myr-AKAP79 mutant to NMDA-induced translocation, 10 images were acquired at 1-μm intervals immediately preceding and 10 min following NMDA treatment. Maximum projection images were generated, and 10–15 spines from secondary or tertiary dendrites were randomly selected for analysis. Regions of interest for spines and dendritic shafts were manually generated. The same spines and shaft regions were created in the post-treatment neurons, and the cross-sectional area of spines and the AKAP79-GFP or mCh spine-to-shaft intensity ratio was calculated before and after treatment. These data are represented as mean fold-change ± S.E.
F-actin binding in vitro
Recombinant C-terminal His6-tagged AKAP79(1–153) fusion protein (Gomez et al. (14)) was purified from bacterial expression system using nickel-agarose as described (17) and phosphorylated by autonomous CaMKII as described below. F-actin was polymerized from G-actin in vitro for 30 min in F-Buffer (125 mm KCl, 2.5 mm MgCl2, 0.2 mm ATP, 2 mm Tris, pH 7.6). Unphosphorylated AKAP79 or CaMKII phosphorylated AKAP79(1–153) was then incubated with F-actin for 10 min and assayed for binding by co-sedimentation. Co-sedimentation was accomplished by centrifugation for 20 min at 100,000 × g. The pellets and supernatants were then analyzed by Western blotting with polyclonal anti-AKAP79 918I, 1:2000 (provided by Dr. Yvonne Lai, ICOS, Bothel, WA), as described (14).
F-actin co-localization in COS7 cells
COS7 cells were cultured on glass bottom dishes (35 mm with 14 mm glass, MatTek Co, Ashland, MA) and transfected by calcium phosphate method as described previously (73, 74) with plasmid cDNA as indicated. Cells were fixed and stained with 0.165 μm Texas Red phalloidin (Invitrogen) as done previously (27). Images were taken on a Zeiss Axiovert 200M system with ×63 oil immersion objective, CoolSnap HQ charge-coupled device camera (Roper Scientific, Trenton NJ), xenon lamp LB-LS/17 (Sutter Instrument, Novato, CA), using Slidebook software (Intelligent Imaging Innovations, Fort Collins, CO). 15 images were taken 0.5 μm apart in the z-plane per cell, deconvolved, and maximum-projected. A threshold mask (mean fluorescence plus 1 S.D.) in the CY3 channel was drawn to label F-actin. The sum fluorescence intensity of YFP within the threshold mask was determined and divided by the sum intensity of YFP within the whole cell to determine the percentage of YFP-AKAP co-localized with F-actin.
CaMKII activity assays in vitro
CaMKII autonomy was generated by pre-phosphorylating the kinase at Thr-286 by incubating 200 nm CaMKIIα subunits in 50 mm PIPES, pH 7.0–7.2, 1 nm BSA, 10 mm MgCl2, 100 μm ATP, 1 mm CaCl2, and 2 μm CaM and incubating on ice for 10 min (7, 21, 75, 76). The reaction was stopped by the addition of PIPES dilution buffer containing 5 mm EGTA. The CaMKII phosphorylation of AKAP79 region B peptides (hAKb) was then induced using a standard CaMKII assay; 2.5 nm CaMKII subunits were reacted with 7.5 μm hAKb at 30 °C for 1 min in buffer containing 50 mm PIPES, pH 7.2, 0.1 mg/ml BSA, 10 mm MgCl2, 100 μm [γ-32P]ATP (1 Ci/mmol). Reactions were spotted onto Whatman P81 phosphocellulose paper rectangles (2 × 2.5 cm). Reactions were stopped by placing the paper rectangles into 300 ml of 0.5% phosphoric acid, and then rinsed four times with water, washed for 30 min, and rinsed four more times. Radioactivity bound to peptides was quantified in a Beckman 6000TA scintillation counter by the Cherenkov method. For Fig. 5B, purified recombinant C-terminal His6-tagged AKAP79(1–153) was phosphorylated by autonomous (+EGTA) or Ca2+/CaM-stimulated (−EGTA) CaMKII in the presence [γ-32P]ATP as described above for 30 min at 30 °C followed by separation by SDS-PAGE and visualization by autoradiography. For Fig. 2C, the reaction additionally contained purified MAP2 and was carried out for 5 min. Please note that in Fig. 5B, the AKAP79(1–153) WT, 5A, and 9A proteins were expressed and purified as C-terminal His6-tagged proteins in Escherichia coli using a pET30 vector that also adds extra linker sequences that together contain three additional Ser/Thr residues. Moreover, there are 11 additional Ser/Thr residues in the AKAP79(1–153) fragment located outside of the A, B, and C polybasic membrane targeting sub-domains. Although none of these additional AKAP79 or vector-encoded Ser/Thr residues are predicted to be optimal consensus phosphorylation sites for CaMKII, they could account for the residual level of CaMKII phosphorylation observed in the 9A mutant that is not sensitive to Ca2+-CaM protection.
CaM-binding assays in vitro
CaM binding was determined by two different methods. Blot overlay with biotinylated CaM (STI Signal Transduction Products) was performed as described previously (22), with bound CaM detected by the Vectastain ABC kit (Vectastain) followed by Western Lightning (PerkinElmer Life Sciences), and chemiluminescence visualized in a ChemiImager (Alpha Innotech). For fluorescence-based detection of CaM binding, CaM was IAEDANS-labeled at a K75C mutation as described (37); the increase in CaM-I fluorescence after binding to Ca2+ and to peptide was measured in a Fluorolog2 spectrofluorometer (HORIBA Jobin Yvon) at excitation/emission wavelengths of 345/465 nm with a 16-nm bandwidth, in a stirred cuvette containing 50 mm Hepes, pH 7.4, 100 mm KCl, 10 mm MgCl2, 0.1 mg/ml BSA and CaM-I (1 μm or as indicated). Ca2+, peptide, or EGTA was added at the times indicated, with fluorescence monitored continuously.
Palmitoylation assays
[3H]Palmitate labeling in HEK-293 cells and visualization of palmitoylated AKAP79 by SDS-PAGE and fluorography were carried out as described previously (18). APEGS, ABE, and 1-biotinamido-4-[4′(maleimidomethyl)cyclohexane-carboxamido] butane (BMCC) assays were conducted as described previously (18, 19, 40). Briefly, for APEGS, neuronal cultures were rinsed in artificial cerebral spinal fluid (in mm: 126 NaCl, 5 KCl, 2 CaCl2, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 10 glucose) and then lysed in PBS buffer containing 4% SDS and 5 mm EDTA. After a 10-min spin at 1000 × g, the supernatant was tumbled with 20 mm tris(2-carboxyethyl)phosphine for 1 h at room temperature. Free thiols were then blocked by incubation with 50 mm N-ethylmaleimide (NEM) overnight at room temperature. Following a chloroform/methanol precipitation (CMP), pellets were resuspended in 4% SDS PBS buffer and incubated with 1 m hydroxylamine (HAM, Sigma) for 1 h at room temperature with end-over-end rotation. Following another CMP, free thiols were labeled with 5-kDa polyethylene glycol moieties (SUNBRIGHT maleimide PEG, NOF America) for 1 h at room temperature with rotation. After a final CMP, samples were resuspended and boiled in sample buffer with 50 mm dithiothreitol and resolved via SDS-PAGE and Western blotting with GFP antibody. For ABE, neuronal cultures were rinsed with artificial CSF and then lysed in buffer containing (in mm) 150 NaCl, 50 Tris, pH 7.4, 5 EDTA, 10 NEM, and 1.7% Triton X-100. Following a 1-h rotation at 4 °C, a CMP was performed, and the dried pellet was resuspended in 4% SDS buffer plus 50 mm Tris, pH 7.4, and 5 mm EDTA. Samples were then diluted in lysis buffer plus 50 mm NEM and tumbled overnight at 4 °C to block free thiols. Following CMP, each protein sample was resuspended in SDS buffer, halved, and diluted in labeling buffer (1 mm HPDP-biotin, EZ-link, Thermo Fisher Scientific) with or without 1 m HAM (Sigma) and tumbled for 1 h at room temperature to cleave palmitoylation thioester linkages and to biotinylate the resultant free cysteines. Following another CMP and resuspension, biotinylated proteins were affinity-purified by tumbling with streptavidin-agarose beads (Thermo Fisher Scientific) overnight at 4 °C. Proteins were eluted with dithiothreitol and resolved via SDS-PAGE and Western blotting with AKAP150 or CaMKIIα antibodies. For BMCC, neuronal cultures were rinsed with aCSF and then lysed in buffer containing (in mm) 150 NaCl, 50 Tris, pH 7.4, 10% glycerol, 1% IGEPAL CA-630 (Sigma, Nonidet P-40 analogue), and 50 NEM. AKAP150 was immunoprecipitated with AKAP150 antibody overnight at 4 °C followed by incubation with agarose beads (Thermo Fisher Scientific) for 1 h. Samples were divided into HAM-negative and -positive conditions and tumbled with or without 1 m HAM for 1 h at room temperature. Samples were then tumbled at 4 °C for 1 h in pH 6.2 buffer plus 10 μm biotin-BMCC (Thermo Fisher Scientific). Finally, beads were washed two times with pH 7.4 lysis buffer and boiled in sample buffer for 10 min at 80 °C. Samples were resolved by SDS-PAGE and Western blotting with streptavidin-HRP and AKAP150 antibody.
Experimental design and statistical analyses
Hippocampal neuron culture experiments were conducted using a minimum of three distinct culture preparations. The cultures used cells from both male and female animals. Tests for statistical significance were conducted using Prism (Graphpad). Independent sample means were compared using t-tests or one-way ANOVA as indicated in the figure legends. F values that were statistically significant were further probed by post hoc tests comparing experimental conditions to control (Dunnett's test) or among all conditions (Newman-Keuls) as indicated in the figure legends. Statistical non-significance (ns) for each condition is also reported in figure legends. Type-1 error rates for all tests were 0.05, but p values lower than 0.05 are indicated by **, p < 0.01, or ***, p < 0.001.
Author contributions
K. M. W., H. O., D. J. G., H. R. R., E. A. H., S. J. C., M. L. D., and K. U. B. designed research and designed, performed, analyzed, and interpreted experiments. K. M. W., H. O., M. L. D., and K. U. B. wrote the paper with input from all authors.
Acknowledgments
We thank Dove Keith and Nicholas Haynes for technical assistance. The Rocky Mountain Neurological Disorders Core Center was recipient of National Institutes of Health Grant P30NS04154.
This work was supported by National Institutes of Health Grants R01NS040701 (to M. L. D.), R01NS080851 and R01NS081248 (to K. U. B.), and T32GM007635 (to H. O., E. A. H., and H. R. R.). The University of Colorado holds the patent rights for tatCN21, its derivatives, and its uses (PCT/US08/077934 “Compositions and methods for improved CaMKII inhibitors and uses thereof”). K. U. B. is owner of Neurexus Therapeutics, LLC. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- LTP
- long-term potentiation
- LTD
- long-term depression
- CaM
- Ca2+/calmodulin
- CaMKII
- CaM-dependent protein kinase II
- PIP2
- phosphatidylinositol 4,5-bisphosphate
- CaN
- calcineurin
- AMPAR
- AMPA receptor
- NEM
- N-ethylmaleimide
- APEGS
- acyl-Peggy exchange gel shift
- PAT
- palmitoyl-acyltransferase
- PPT
- protein-palmitoyl thioesterase
- ABE
- acyl-biotin exchange
- BMCC
- 1-biotinamido-4-[4′(maleimidomethyl)cyclohexane-carboxamido] butane
- HAM
- hydroxylamine
- MW
- molecular weight
- CMP
- chloroform/methanol precipitation
- ANOVA
- analysis of variance
- CaM-I
- IAEDANS-labeled CaM
- DIV
- days in vitro
- cLTD
- chemical LTD
- mCh
- mCherry
- IAEDANS
- 5-({2-[(iodoacetyl)amino]ethyl}amino)naphthalene-1-sulfonic acid.
References
- 1. Collingridge G. L., Peineau S., Howland J. G., and Wang Y. T. (2010) Long-term depression in the CNS. Nat. Rev. Neurosci. 11, 459–473 10.1038/nrn2867 [DOI] [PubMed] [Google Scholar]
- 2. Huganir R. L., and Nicoll R. A. (2013) AMPARs and synaptic plasticity: the last 25 years. Neuron 80, 704–717 10.1016/j.neuron.2013.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malinow R., Schulman H., and Tsien R. W. (1989) Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 245, 862–866 10.1126/science.2549638 [DOI] [PubMed] [Google Scholar]
- 4. Silva A. J., Stevens C. F., Tonegawa S., and Wang Y. (1992) Deficient hippocampal long-term potentiation in a-calcium-calmodulin kinase II mutant mice. Science 257, 201–206 10.1126/science.1378648 [DOI] [PubMed] [Google Scholar]
- 5. Giese K. P., Fedorov N. B., Filipkowski R. K., and Silva A. J. (1998) Autophosphorylation at Thr286 of the α calcium-calmodulin kinase II in LTP and learning. Science 279, 870–873 10.1126/science.279.5352.870 [DOI] [PubMed] [Google Scholar]
- 6. Coultrap S. J., and Bayer K. U. (2012) CaMKII regulation in information processing and storage. Trends Neurosci. 35, 607–618 10.1016/j.tins.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coultrap S. J., Freund R. K., O'Leary H., Sanderson J. L., Roche K. W., Dell'Acqua M. L., and Bayer K. U. (2014) Autonomous CaMKII mediates both LTP and LTD using a mechanism for differential substrate site selection. Cell Rep. 6, 431–437 10.1016/j.celrep.2014.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Goodell D. J., Zaegel V., Coultrap S. J., Hell J. W., and Bayer K. U. (2017) DAPK1 mediates LTD by making CaMKII/GluN2B binding LTP specific. Cell Rep. 19, 2231–2243 10.1016/j.celrep.2017.05.068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mulkey R. M., Herron C. E., and Malenka R. C. (1993) An essential role for protein phosphatases in hippocampal long-term depression. Science 261, 1051–1055 10.1126/science.8394601 [DOI] [PubMed] [Google Scholar]
- 10. Zeng H., Chattarji S., Barbarosie M., Rondi-Reig L., Philpot B. D., Miyakawa T., Bear M. F., and Tonegawa S. (2001) Forebrain-specific calcineurin knockout selectively impairs bidirectional synaptic plasticity and working/episodic-like memory. Cell 107, 617–629 10.1016/S0092-8674(01)00585-2 [DOI] [PubMed] [Google Scholar]
- 11. Jurado S., Biou V., and Malenka R. C. (2010) A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat. Neurosci. 13, 1053–1055 10.1038/nn.2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sanderson J. L., Gorski J. A., Gibson E. S., Lam P., Freund R. K., Chick W. S., and Dell'Acqua M. L. (2012) AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J. Neurosci. 32, 15036–15052 10.1523/JNEUROSCI.3326-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sanderson J. L., Gorski J. A., and Dell'Acqua M. L. (2016) NMDA receptor-dependent LTD requires transient synaptic incorporation of Ca2+-permeable AMPARs mediated by AKAP150-anchored PKA and calcineurin. Neuron 89, 1000–1015 10.1016/j.neuron.2016.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gomez L. L., Alam S., Smith K. E., Horne E., and Dell'Acqua M. L. (2002) Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J. Neurosci. 22, 7027–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Smith K. E., Gibson E. S., and Dell'Acqua M. L. (2006) cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J. Neurosci. 26, 2391–2402 10.1523/JNEUROSCI.3092-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Horne E. A., and Dell'Acqua M. L. (2007) Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J. Neurosci. 27, 3523–3534 10.1523/JNEUROSCI.4340-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dell'Acqua M. L., Faux M. C., Thorburn J., Thorburn A., and Scott J. D. (1998) Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J. 17, 2246–2260 10.1093/emboj/17.8.2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Keith D. J., Sanderson J. L., Gibson E. S., Woolfrey K. M., Robertson H. R., Olszewski K., Kang R., El-Husseini A., and Dell'acqua M. L. (2012) Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J. Neurosci. 32, 7119–7136 10.1523/JNEUROSCI.0784-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Woolfrey K. M., Sanderson J. L., and Dell'Acqua M. L. (2015) The palmitoyl acyltransferase DHHC2 regulates recycling endosome exocytosis and synaptic potentiation through palmitoylation of AKAP79/150. J. Neurosci. 35, 442–456 10.1523/JNEUROSCI.2243-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lu W., Isozaki K., Roche K. W., and Nicoll R. A. (2010) Synaptic targeting of AMPA receptors is regulated by a CaMKII site in the first intracellular loop of GluA1. Proc. Natl. Acad. Sci. U.S.A. 107, 22266–22271 10.1073/pnas.1016289107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Coultrap S. J., Buard I., Kulbe J. R., Dell'Acqua M. L., and Bayer K. U. (2010) CaMKII autonomy is substrate-dependent and further stimulated by Ca2+/calmodulin. J. Biol. Chem. 285, 17930–17937 10.1074/jbc.M109.069351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Barcomb K., Buard I., Coultrap S. J., Kulbe J. R., O'Leary H., Benke T. A., and Bayer K. U. (2014) Autonomous CaMKII requires further stimulation by Ca2+/calmodulin for enhancing synaptic strength. FASEB J. 28, 3810–3819 10.1096/fj.14-250407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Vest R. S., Davies K. D., O'Leary H., Port J. D., and Bayer K. U. (2007) Dual mechanism of a natural CaMKII inhibitor. Mol. Biol. Cell 18, 5024–5033 10.1091/mbc.E07-02-0185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tokumitsu H., Chijiwa T., Hagiwara M., Mizutani A., Terasawa M., and Hidaka H. (1990) KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-l-tyrosyl]-4-phenylpiperazine, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 265, 4315–4320 [PubMed] [Google Scholar]
- 25. Sumi M., Kiuchi K., Ishikawa T., Ishii A., Hagiwara M., Nagatsu T., and Hidaka H. (1991) The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun. 181, 968–975 10.1016/0006-291X(91)92031-E [DOI] [PubMed] [Google Scholar]
- 26. Brooks I. M., and Tavalin S. J. (2011) Ca2+/calmodulin-dependent protein kinase II inhibitors disrupt AKAP79-dependent PKC signaling to GluA1 AMPA receptors. J. Biol. Chem. 286, 6697–6706 10.1074/jbc.M110.183558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. O'Leary H., Lasda E., and Bayer K. U. (2006) CaMKIIβ association with the actin cytoskeleton is regulated by alternative splicing. Mol. Biol. Cell 17, 4656–4665 10.1091/mbc.E06-03-0252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Halpain S., Hipolito A., and Saffer L. (1998) Regulation of F-actin stability in dendritic spines by glutamate receptors and calcineurin. J. Neurosci. 18, 9835–9844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fink C. C., Bayer K. U., Myers J. W., Ferrell J. E. Jr., Schulman H., and Meyer T. (2003) Selective regulation of neurite extension and synapse formation by the β but not the α isoform of CaMKII. Neuron 39, 283–297 10.1016/S0896-6273(03)00428-8 [DOI] [PubMed] [Google Scholar]
- 30. Okamoto K., Narayanan R., Lee S. H., Murata K., and Hayashi Y. (2007) The role of CaMKII as an F-actin-bundling protein crucial for maintenance of dendritic spine structure. Proc. Natl. Acad. Sci. U.S.A. 104, 6418–6423 10.1073/pnas.0701656104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin Y. C., and Redmond L. (2008) CaMKIIβ binding to stable F-actin in vivo regulates F-actin filament stability. Proc. Natl. Acad. Sci. U.S.A. 105, 15791–15796 10.1073/pnas.0804399105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sanabria H., Swulius M. T., Kolodziej S. J., Liu J., and Waxham M. N. (2009) βCaMKII regulates actin assembly and structure. J. Biol. Chem. 284, 9770–9780 10.1074/jbc.M809518200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saito A., Miyajima K., Akatsuka J., Kondo H., Mashiko T., Kiuchi T., Ohashi K., and Mizuno K. (2013) CaMKIIβ-mediated LIM-kinase activation plays a crucial role in BDNF-induced neuritogenesis. Genes Cells 18, 533–543 10.1111/gtc.12054 [DOI] [PubMed] [Google Scholar]
- 34. Kim K., Lakhanpal G., Lu H. E., Khan M., Suzuki A., Hayashi M. K., Narayanan R., Luyben T. T., Matsuda T., Nagai T., Blanpied T. A., Hayashi Y., and Okamoto K. (2015) A temporary gating of actin remodeling during synaptic plasticity consists of the interplay between the kinase and structural functions of CaMKII. Neuron 87, 813–826 10.1016/j.neuron.2015.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gorny X., Mikhaylova M., Seeger C., Reddy P. P., Reissner C., Schott B. H., Helena Danielson U., Kreutz M. R., and Seidenbecher C. (2012) AKAP79/150 interacts with the neuronal calcium-binding protein caldendrin. J. Neurochem. 122, 714–726 10.1111/j.1471-4159.2012.07828.x [DOI] [PubMed] [Google Scholar]
- 36. Seeger C., Gorny X., Reddy P. P., Seidenbecher C., and Danielson U. H. (2012) Kinetic and mechanistic differences in the interactions between caldendrin and calmodulin with AKAP79 suggest different roles in synaptic function. J. Mol. Recognit. 25, 495–503 10.1002/jmr.2215 [DOI] [PubMed] [Google Scholar]
- 37. Singla S. I., Hudmon A., Goldberg J. M., Smith J. L., and Schulman H. (2001) Molecular characterization of calmodulin trapping by calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 276, 29353–29360 10.1074/jbc.M101744200 [DOI] [PubMed] [Google Scholar]
- 38. Putkey J. A., and Waxham M. N. (1996) A peptide model for calmodulin trapping by calcium/calmodulin-dependent protein kinase II. J. Biol. Chem. 271, 29619–29623 10.1074/jbc.271.47.29619 [DOI] [PubMed] [Google Scholar]
- 39. Faux M. C., and Scott J. D. (1997) Regulation of the AKAP79-protein kinase C interaction by Ca2+/calmodulin. J. Biol. Chem. 272, 17038–17044 10.1074/jbc.272.27.17038 [DOI] [PubMed] [Google Scholar]
- 40. Yokoi N., Fukata Y., Sekiya A., Murakami T., Kobayashi K., and Fukata M. (2016) Identification of PSD-95 depalmitoylating enzymes. J. Neurosci. 36, 6431–6444 10.1523/JNEUROSCI.0419-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Marsden K. C., Shemesh A., Bayer K. U., and Carroll R. C. (2010) Selective translocation of Ca2+/calmodulin protein kinase IIα (CaMKIIα) to inhibitory synapses. Proc. Natl. Acad. Sci. U.S.A. 107, 20559–20564 10.1073/pnas.1010346107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nikandrova Y. A., Jiao Y., Baucum A. J., Tavalin S. J., and Colbran R. J. (2010) Ca2+/calmodulin-dependent protein kinase II binds to and phosphorylates a specific SAP97 splice variant to disrupt association with AKAP79/150 and modulate α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor (AMPAR) activity. J. Biol. Chem. 285, 923–934 10.1074/jbc.M109.033985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Colbran R. J. (1993) Inactivation of Ca2+/calmodulin-dependent protein kinase II by basal autophosphorylation. J. Biol. Chem. 268, 7163–7170 [PubMed] [Google Scholar]
- 44. Hanson P. I., and Schulman H. (1992) Inhibitory autophosphorylation of multifunctional Ca2+/calmodulin-dependent protein kinase analyzed by site-directed mutagenesis. J. Biol. Chem. 267, 17216–17224 [PubMed] [Google Scholar]
- 45. Colbran R. J., and Soderling T. R. (1990) Calcium/calmodulin-independent autophosphorylation sites of calcium/calmodulin-dependent protein kinase II. Studies on the effect of phosphorylation of threonine 305/306 and serine 314 on calmodulin binding using synthetic peptides. J. Biol. Chem. 265, 11213–11219 [PubMed] [Google Scholar]
- 46. Pi H. J., Otmakhov N., Lemelin D., De Koninck P., and Lisman J. (2010) Autonomous CaMKII can promote either long-term potentiation or long-term depression, depending on the state of T305/T306 phosphorylation. J. Neurosci. 30, 8704–8709 10.1523/JNEUROSCI.0133-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Elgersma Y., Fedorov N. B., Ikonen S., Choi E. S., Elgersma M., Carvalho O. M., Giese K. P., and Silva A. J. (2002) Inhibitory autophosphorylation of CaMKII controls PSD association, plasticity, and learning. Neuron 36, 493–505 10.1016/S0896-6273(02)01007-3 [DOI] [PubMed] [Google Scholar]
- 48. Fukata Y., and Fukata M. (2010) Protein palmitoylation in neuronal development and synaptic plasticity. Nat. Rev. Neurosci. 11, 161–175 10.1038/nrg2735 [DOI] [PubMed] [Google Scholar]
- 49. Lemonidis K., Werno M. W., Greaves J., Diez-Ardanuy C., Sanchez-Perez M. C., Salaun C., Thomson D. M., and Chamberlain L. H. (2015) The zDHHC family of S-acyltransferases. Biochem. Soc. Trans. 43, 217–221 10.1042/BST20140270 [DOI] [PubMed] [Google Scholar]
- 50. Brigidi G. S., Sun Y., Beccano-Kelly D., Pitman K., Mobasser M., Borgland S. L., Milnerwood A. J., and Bamji S. X. (2014) Palmitoylation of δ-catenin by DHHC5 mediates activity-induced synapse plasticity. Nat. Neurosci. 17, 522–532 10.1038/nn.3657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. El-Husseini Ael-D., Schnell E., Dakoji S., Sweeney N., Zhou Q., Prange O., Gauthier-Campbell C., Aguilera-Moreno A., Nicoll R. A., and Bredt D. S. (2002) Synaptic strength regulated by palmitate cycling on PSD-95. Cell 108, 849–863 10.1016/S0092-8674(02)00683-9 [DOI] [PubMed] [Google Scholar]
- 52. Noritake J., Fukata Y., Iwanaga T., Hosomi N., Tsutsumi R., Matsuda N., Tani H., Iwanari H., Mochizuki Y., Kodama T., Matsuura Y., Bredt D. S., Hamakubo T., and Fukata M. (2009) Mobile DHHC palmitoylating enzyme mediates activity-sensitive synaptic targeting of PSD-95. J. Cell Biol. 186, 147–160 10.1083/jcb.200903101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Thomas G. M., Hayashi T., Chiu S. L., Chen C. M., and Huganir R. L. (2012) Palmitoylation by DHHC5/8 targets GRIP1 to dendritic endosomes to regulate AMPA-R trafficking. Neuron 73, 482–496 10.1016/j.neuron.2011.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Globa A. K., and Bamji S. X. (2017) Protein palmitoylation in the development and plasticity of neuronal connections. Curr. Opin. Neurobiol. 45, 210–220 10.1016/j.conb.2017.02.016 [DOI] [PubMed] [Google Scholar]
- 55. Kang R., Wan J., Arstikaitis P., Takahashi H., Huang K., Bailey A. O., Thompson J. X., Roth A. F., Drisdel R. C., Mastro R., Green W. N., Yates J. R. 3rd., Davis N. G., and El-Husseini A. (2008) Neural palmitoyl-proteomics reveals dynamic synaptic palmitoylation. Nature 456, 904–909 10.1038/nature07605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kay H. Y., Greene D. L., Kang S., Kosenko A., and Hoshi N. (2015) M-current preservation contributes to anticonvulsant effects of valproic acid. J. Clin. Invest. 125, 3904–3914 10.1172/JCI79727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cho E., and Park M. (2016) Palmitoylation in Alzheimer's disease and other neurodegenerative diseases. Pharmacol. Res. 111, 133–151 10.1016/j.phrs.2016.06.008 [DOI] [PubMed] [Google Scholar]
- 58. Sanders S. S., and Hayden M. R. (2015) Aberrant palmitoylation in Huntington disease. Biochem. Soc. Trans. 43, 205–210 10.1042/BST20140242 [DOI] [PubMed] [Google Scholar]
- 59. Delint-Ramirez I., Willoughby D., Hammond G. V., Ayling L. J., and Cooper D. M. (2011) Palmitoylation targets AKAP79 protein to lipid rafts and promotes its regulation of calcium-sensitive adenylyl cyclase type 8. J. Biol. Chem. 286, 32962–32975 10.1074/jbc.M111.243899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lu Y., Allen M., Halt A. R., Weisenhaus M., Dallapiazza R. F., Hall D. D., Usachev Y. M., McKnight G. S., and Hell J. W. (2007) Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J. 26, 4879–4890 10.1038/sj.emboj.7601884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lu Y., Zhang M., Lim I. A., Hall D. D., Allen M., Medvedeva Y., McKnight G. S., Usachev Y. M., and Hell J. W. (2008) AKAP150-anchored PKA activity is important for LTD during its induction phase. J. Physiol. 586, 4155–4164 10.1113/jphysiol.2008.151662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fernández-Monreal M., Brown T. C., Royo M., and Esteban J. A. (2012) The balance between receptor recycling and trafficking toward lysosomes determines synaptic strength during long-term depression. J. Neurosci. 32, 13200–13205 10.1523/JNEUROSCI.0061-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Lee H. K., Barbarosie M., Kameyama K., Bear M. F., and Huganir R. L. (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959 10.1038/35016089 [DOI] [PubMed] [Google Scholar]
- 64. Lee H. K., Kameyama K., Huganir R. L., and Bear M. F. (1998) NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21, 1151–1162 10.1016/S0896-6273(00)80632-7 [DOI] [PubMed] [Google Scholar]
- 65. Lee H. K., Takamiya K., Han J. S., Man H., Kim C. H., Rumbaugh G., Yu S., Ding L., He C., Petralia R. S., Wenthold R. J., Gallagher M., and Huganir R. L. (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643 10.1016/S0092-8674(03)00122-3 [DOI] [PubMed] [Google Scholar]
- 66. Lee H. K., Takamiya K., He K., Song L., and Huganir R. L. (2010) Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J. Neurophysiol. 103, 479–489 10.1152/jn.00835.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wang Y., Shibasaki F., and Mizuno K. (2005) Calcium signal-induced cofilin dephosphorylation is mediated by Slingshot via calcineurin. J. Biol. Chem. 280, 12683–12689 10.1074/jbc.M411494200 [DOI] [PubMed] [Google Scholar]
- 68. Zhou Q., Homma K. J., and Poo M. M. (2004) Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron 44, 749–757 10.1016/j.neuron.2004.11.011 [DOI] [PubMed] [Google Scholar]
- 69. Bayer K. U., De Koninck P., Leonard A. S., Hell J. W., and Schulman H. (2001) Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 411, 801–805 10.1038/35081080 [DOI] [PubMed] [Google Scholar]
- 70. Coultrap S. J., and Bayer K. U. (2012) in Neuromethods: Protein Kinase Technologies (Mukai H., ed) pp. 49–72, Springer, New York [Google Scholar]
- 71. Brandao K. E., Dell'Acqua M. L., and Levinson S. R. (2012) A-kinase anchoring protein 150 expression in a specific subset of TRPV1- and CaV 1.2-positive nociceptive rat dorsal root ganglion neurons. J. Comp. Neurol. 520, 81–99 10.1002/cne.22692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Vest R. S., O'Leary H., Coultrap S. J., Kindy M. S., and Bayer K. U. (2010) Effective post-insult neuroprotection by a novel Ca2+/calmodulin-dependent protein kinase II (CaMKII) inhibitor. J. Biol. Chem. 285, 20675–20682 10.1074/jbc.M109.088617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bayer K. U., Harbers K., and Schulman H. (1998) αKAP is an anchoring protein for a novel CaM kinase II isoform in skeletal muscle. EMBO J. 17, 5598–5605 10.1093/emboj/17.19.5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Coultrap S. J., Barcomb K., and Bayer K. U. (2012) A significant but rather mild contribution of Thr-286 autophosphorylation to Ca2+/CaM-stimulated CaMKII activity. PLoS One 7, e37176 10.1371/journal.pone.0037176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Goodell D. J., Eliseeva T. A., Coultrap S. J., and Bayer K. U. (2014) CaMKII binding to GluN2B is differentially affected by macromolecular crowding reagents. PLoS One 9, e96522 10.1371/journal.pone.0096522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Myers J. B., Zaegel V., Coultrap S. J., Miller A. P., Bayer K. U., and Reichow S. L. (2017) The CaMKII holoenzyme structure in activation-competent conformations. Nat. Commun. 8, 15742 10.1038/ncomms15742 [DOI] [PMC free article] [PubMed] [Google Scholar]