

Abstract
Activation of the tumor necrosis factor receptor 1 (TNFR1) death receptor by TNF induces either cell survival or cell death. However, the mechanisms mediating these distinct outcomes remain poorly understood. In this study, we report that the ST6Gal-I sialyltransferase, an enzyme up-regulated in numerous cancers, sialylates TNFR1 and thereby protects tumor cells from TNF-induced apoptosis. Using pancreatic and ovarian cancer cells with ST6Gal-I knockdown or overexpression, we determined that α2-6 sialylation of TNFR1 had no effect on early TNF-induced signaling events, including the rapid activation of NF-κB, c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and Akt (occurring within 15 min). However, upon extended TNF treatment (6–24 h), cells with high ST6Gal-I levels exhibited resistance to TNF-induced apoptosis, as indicated by morphological evidence of cell death and decreased activation of caspases 8 and 3. Correspondingly, at these later time points, high ST6Gal-I expressers displayed sustained activation of the survival molecules Akt and NF-κB. Additionally, extended TNF treatment resulted in the selective enrichment of clonal variants with high ST6Gal-I expression, further substantiating a role for ST6Gal-I in cell survival. Given that TNFR1 internalization is known to be essential for apoptosis induction, whereas survival signaling is initiated by TNFR1 at the plasma membrane, we examined TNFR1 localization. The α2-6 sialylation of TNFR1 was found to inhibit TNF-induced TNFR1 internalization. Thus, by restraining TNFR1 at the cell surface via sialylation, ST6Gal-I acts as a functional switch to divert signaling toward survival. These collective findings point to a novel glycosylation-dependent mechanism that regulates the cellular response to TNF and may promote cancer cell survival within TNF-rich tumor microenvironments.
Keywords: apoptosis, β-galactoside α2-6 sialyltransferase 1 (ST6Gal-I), cell signaling, glycosylation, tumor cell biology, tumor necrosis factor (TNF), TNFR1, death receptor
Introduction
Tumor necrosis factor (TNF)2 is the prototypical member of the TNF superfamily of cytokines. Although it was first recognized for its anti-tumor activity, TNF has since been identified as a highly pleiotropic cytokine that mediates multiple cellular processes, including inflammation, cell differentiation, cell survival and proliferation, and apoptosis. TNF can bind and activate two receptors: tumor necrosis factor receptor 1 (TNFR1) and tumor necrosis factor receptor 2 (TNFR2), the latter of which is chiefly expressed in hematopoietic cells (1). TNFR1, which is responsible for the majority of TNF-induced events (2, 3), is a ubiquitously expressed member of the death receptor subgroup of the TNF receptor superfamily. Death receptors, which also include Fas and the TRAIL receptors DR4 and DR5, are characterized by a cytoplasmic “death domain” (DD), a conserved sequence vital for apoptosis induction (4). TNFR2 lacks a DD and therefore cannot induce cell death.
When initially isolated in 1975, TNF was described as an “endotoxin” capable of inducing necrosis of tumors (5). However, over the next two decades, the promise that TNF could serve as an antitumor therapy faded, as research revealed a contradictory role for TNF as an inducer of cell survival (1). In recent years, intensive investigation has centered on defining the mechanisms by which the TNF/TNFR1 signaling axis pivots between cell survival and cell death. The TNFR1 receptor has a complex mechanism of regulation mediated by diverse processes, including receptor oligomerization, lipid raft recruitment, endocytosis, and TNFR1 ubiquitination and shedding (6–10). The events downstream of TNFR1 activation are directed by two distinct signaling complexes, one associated with survival (complex I) and the other with apoptosis (complex II). Complex I, composed of TRADD, TRAF2, RIP-1, and cIAP1/2, initiates and propagates survival signaling via activation of the NF-κB and MAPK pathways (11, 12). Conversely, apoptotic signaling is initiated by TNFR1 internalization into endosomes, followed by formation of complex II, the death-inducing signaling complex (DISC) (11, 12). The DISC consists of the DD adaptor proteins TRADD and FADD and the apoptosis initiator caspase-8, which is autoproteolytically cleaved into its active form upon recruitment to the DD. Activated caspase-8 then cleaves caspase-3, resulting in the activation of the effector arm of apoptosis.
Adding to the complexity of TNF/TNFR1 signaling, we previously reported that the glycosylation profile on the TNFR1 receptor significantly affects TNFR1 activity. TNFR1 was identified as a substrate for the ST6Gal-I sialyltransferase (13), a Golgi enzyme that adds α2-6–linked sialic acids to N-glycans on select receptors. The addition of α2-6 sialic acid to TNFR1 was shown to block TNF-induced apoptosis in human monocytic cells as well as monocytes from ST6Gal-I–overexpressing mice (13). However, in this prior investigation, neither the mechanism by which sialylation regulates TNFR1 nor the specific signaling cascades downstream of TNFR1 sialylation were examined, thus limiting a fundamental understanding of the role of glycosylation in regulating TNFR1 function. Furthermore, the contribution of TNFR1 sialylation to cancer cell behavior has not been investigated previously, which is significant in that TNF was originally defined as an anti-tumor effector. Importantly, ST6Gal-I is up-regulated in many diverse cancer types (14–18), implicating α2-6 sialylation of TNFR1 as a common feature of cancer cells.
In this study, we define a novel sialylation-dependent mechanism that inhibits TNF-induced TNFR1 internalization and shifts the balance of TNFR1 signaling to favor survival. Using ovarian and pancreatic cancer cell models with ST6Gal-I overexpression or knockdown, we find that ST6Gal-I–mediated TNFR1 sialylation blocks the apoptotic arm of TNFR1 signaling while leaving NF-κB- and Akt-mediated survival signaling intact. These findings highlight the importance of receptor glycosylation in the regulation of a key cell survival pathway critical to numerous physiologic and pathophysiologic processes.
Results
ST6Gal-I–mediated sialylation of TNFR1 confers resistance to TNF-induced cell death
To investigate the effect of ST6Gal-I–mediated sialylation on TNFR1 activity, the MiaPaCa-2 pancreatic cancer cell line, which has substantial ST6Gal-I expression, was stably transduced with a lentivirus encoding shRNA for ST6Gal-I to knock down levels of the enzyme (Fig. 1A). Parallel to ST6Gal-I protein expression, knockdown (KD) of ST6Gal-I resulted in a decrease in surface α2-6 sialylation compared with empty vector (EV) control cells (Fig. 1B), as measured by binding of the SNA lectin, which specifically recognizes α2-6 sialic acids. The level of α2-6 sialylation on TNFR1 was subsequently examined. α2-6–sialylated proteins were precipitated by SNA–agarose beads, and then precipitates were immunoblotted for TNFR1. As shown in Fig. 1C, TNFR1 expressed by KD cells had reduced α2-6 sialylation compared with EV cells.
Figure 1.

Manipulating ST6Gal-I expression leads to altered α2-6 sialylation of TNFR1. A, using a lentivirus, MiaPaCa-2 cells were stably transduced with shRNA for ST6Gal-I, and ST6Gal-I KD was confirmed via immunoblotting. Control cells were generated by stable transduction of an EV lentiviral construct. B, MiaPaCa-2 EV and KD cells were stained with SNA-FITC and analyzed by flow cytometry. Solid black line, EV; dashed gray line, KD. C, MiaPaCa-2 cell lysates were precipitated (Precip) with agarose-conjugated SNA and blotted for TNFR1. WB, Western blot. D, OV4 cells were stably transduced with a lentivirus encoding ST6Gal-I, and ST6Gal-I OE was confirmed by immunoblotting. E, OV4 EV and OE cells were stained with SNA–FITC and analyzed by flow cytometry. Solid black line, EV; dashed gray line, OE. F, OV4 cell lysates were precipitated with agarose-conjugated SNA and blotted for TNFR1.
To complement the MiaPaCa-2 cell model, the effect of ST6Gal-I overexpression was evaluated. The OV4 ovarian cancer cell line, one of the few cancer lines lacking detectable ST6Gal-I, was engineered to stably overexpress ST6Gal-I (Fig. 1D). SNA labeling experiments revealed that cells with ST6Gal-I OE displayed greatly increased surface expression of α2-6 sialic acids (Fig. 1E). Furthermore, TNFR1 was found to be α2-6–sialylated in OE cells, as measured by SNA precipitation assays, whereas no α2-6–sialylated TNFR1 was detected in EV cells (Fig. 1F).
Based on previous findings that ST6Gal-I protects monocytes against TNF-induced cell death (13), we examined the effect of TNF on MiaPaCa-2 and OV4 cell viability. MiaPaCa-2 and OV4 cells were treated with TNF for 24 h and then monitored for changes in cell density and morphology. As shown in Fig. 2A, MiaPaCa-2 KD cells were sensitized to TNF-induced cell death, indicated by their spindle-shaped morphology and detachment from the culture plate. In contrast, MiaPaCa-2 EV cells showed limited evidence of TNF-induced cytotoxicity, suggesting that ST6Gal-I has a protective effect. These data are further substantiated in Fig. 2B, in which OV4 OE cells appear highly resistant to TNF treatment, whereas marked cell death is observed in OV4 EV cells, which lack ST6Gal-I.
Figure 2.
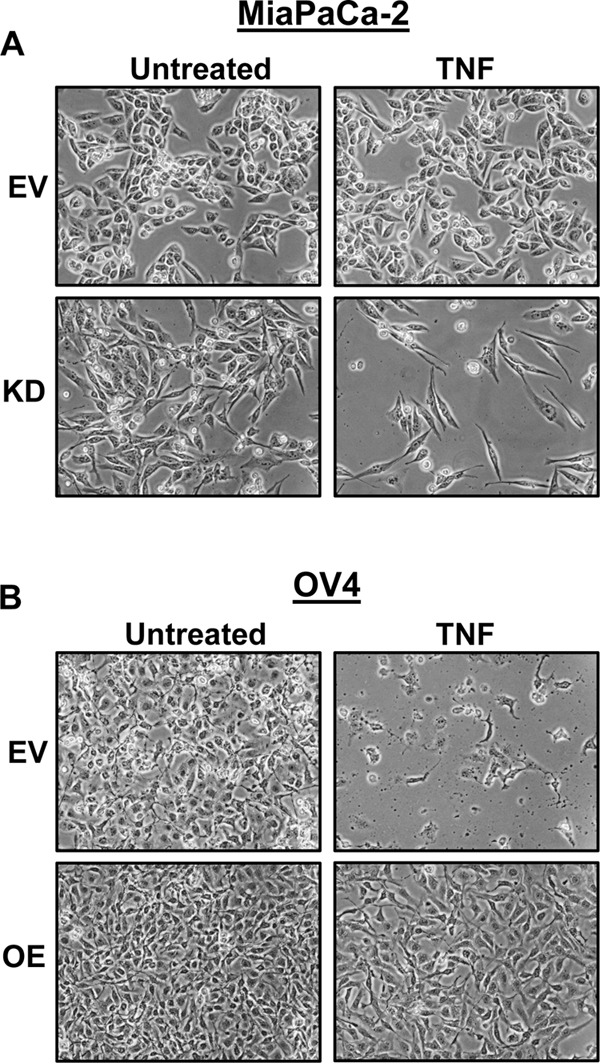
Cells with high ST6Gal-I expression are protected against TNF-induced cell death. A, MiaPaCa-2 EV and KD cells were treated with TNF for 24 h or left untreated and imaged to observe morphological changes. B, OV4 EV and OE cells were treated with TNF plus CHX for 24 h or left untreated and imaged to observe morphological changes.
To further interrogate the protective effect of ST6Gal-I, MiaPaCa-2 EV and KD cells were prelabeled individually with their own distinct CellTracker fluorescence dyes, seeded at equivalent densities into the same tissue culture dish, and then treated with TNF. Following a 24-h TNF treatment, fluorescence microscopy revealed that the red-labeled EV cells preferentially survived relative to their green-labeled KD counterparts, with EV cells comprising 73% of the total cell population following TNF treatment compared with the approximate 50:50 EV:KD ratio of the initial population prior to treatment (Fig. 3A, Untreated). Similar results were noted with OV4 cells; the green-labeled OE cells exhibited significantly better survival than EV cells following TNF treatment, with OE cells encompassing 87% of the TNF-treated population (Fig. 3B).
Figure 3.

ST6Gal-I levels are enriched in TNF-treated cells. A, MiaPaCa-2 EV and KD cells were prelabeled with their own distinct CellTracker fluorescence dye (EV, red; KD, green), seeded at equal densities into tissue culture wells, and then treated with TNF. After 24 h, a fluorescence microscope was used to image the cells. Four distinct fields of the representative experiment were quantified via ImageJ and plotted as the percentage mean of total cell population per field ± S.D. *, p < 0.05. B, OV4 EV and OE cells were prelabeled with distinct fluorescence dyes (EV, red; OE, green), seeded at equal densities, and then treated with TNF plus CHX. After 24 h, cells were imaged, and the number of red versus green cells was enumerated. Four distinct fields of the representative experiment were quantified via ImageJ and plotted as the percentage mean of total cell population per field ± S.D. *, p < 0.05. C, MiaPaCa-2 EV cells were treated with TNF, and after 24 h, lysates were prepared from the cells still adhered to the plate and immunoblotted for ST6Gal-I. D, OV4 OE cells were treated with TNF plus CHX, and after 24 h, lysates derived from the remaining cells were immunoblotted for ST6Gal-I. The immunoblots in A and B are duplicates of the blots in Fig. 1, A and D, respectively. These images were duplicated to clarify the experimental design. UT, untreated.
Prior studies from our group have suggested that cytotoxic stimuli or cell stressors such as serum deprivation, anchorage-independent spheroid growth, or chemotherapy drugs exert selection for clonal variants with high ST6Gal-I expression (19–22). Accordingly, we investigated whether sustained exposure to TNF would select for cells with high ST6Gal-I levels. MiaPaCa-2 EV cells were treated with TNF for 24 h, and viable cells present after treatment were harvested. Lysates from these remaining cells were immunoblotted for ST6Gal-I. Interestingly, the EV cells that selectively survived TNF treatment (TNF-resistant) displayed an enrichment in ST6Gal-I compared with the EV population prior to treatment (Fig. 3C, UT). Similar results were observed for OV4 OE cells, in which ST6Gal-I levels were greater in TNF-treated OE cells relative to the starting population (Fig. 3D). The ST6Gal-I OE cell line is a polyclonal population created by lentiviral transduction, and the ST6Gal-I construct is driven by a constitutive promoter. Thus, the increased levels of ST6Gal-I in TNF-treated OE cells are likely a consequence of clonal selection rather than gene induction of ST6Gal-I.
α2-6 sialylation of TNFR1 does not alter initial receptor activation
TNF activation of TNFR1 induces the rapid recruitment of complex I proteins to the TNFR1 DD, followed immediately by activation of IκB kinase (IKK) and IKK-mediated phosphorylation of IκBα, a negative regulator of NF-κB. The phosphorylation of IκBα leads to its release from NF-κB and subsequent proteasome-mediated degradation, ultimately facilitating activation and nuclear translocation of NF-κB. To investigate whether ST6Gal-I affects early TNFR1-directed NF-κB signaling, MiaPaCa-2 EV and KD cells were incubated with TNF for 10 min, and lysates were immunoblotted for IκBα. Robust IκBα degradation was observed in both the MiaPaCa-2 EV and KD cell lines (Fig. 4A). Furthermore, fluorescence microscopy revealed that nuclear translocation of NF-κB occurs in both EV and KD cells upon a 15-min incubation with TNF (Fig. 4B). Consistent with NF-κB activation, a similar degree of NF-κB phosphorylation was elicited by TNF in EV and KD cells (Fig. 4C). Comparable results were noted in the OV4 cell model. For both EV and OE cells, TNF stimulated IκBα degradation, NF-κB nuclear translocation, as well as NF-κB phosphorylation (Fig. 4, D–F). This lack of effect of α2-6 sialylation on early NF-κB–related signaling indicates that TNFR1 sialylation does not hinder binding of TNF to TNFR1 or subsequent receptor activation. Moreover, these data also suggest that TNFR1 sialylation does not alter the initial recruitment of complex I proteins to the receptor or the ensuing activation of IKK.
Figure 4.

α2-6 sialylation of TNFR1 does not alter initial activation of NFκB. A, MiaPaCa-2 EV and KD cells were treated with TNF for 10 min and immunoblotted for IκBα. B, MiaPaCa-2 cells were treated with TNF for 15 min and immediately washed with ice-cold PBS and fixed. Cells were then permeabilized and stained with anti-NF-κBp65 overnight, and anti-rabbit IgG Alexa Fluor 488 was used for fluorescence visualization. UT, untreated. C, MiaPaCa-2 cells were treated with TNF for 15, 30, or 120 min and immunoblotted for pNF-κB–p65. D, OV4 EV and OE cells were treated with TNF for 10 min and immunoblotted for IκBα. E, OV4 cells were treated with TNF for 15 min and then immunostained for NF-κBp65 as described in B. F, OV4 cells were treated with TNF for 15, 30, or 120 min, and immunoblotted for pNF-κB–p65.
Early TNFR1-dependent survival signaling is unaffected by α2-6 sialylation
Considering that ST6Gal-I did not inhibit early activation of NF-κB, we next examined the effect of α2-6 sialylation on TNFR1-mediated activation of other pro-survival signaling molecules, including members of the MAPK family and Akt. To evaluate the acute signaling response to TNF, MiaPaCa-2 EV and KD cells were incubated with TNF for 15, 30, and 120 min. As shown in Fig. 5A, JNK, ERK, and Akt all exhibited similar activation and overall expression patterns in EV versus KD lines. In line with these results, OV4 EV and OE cells displayed a similar temporal pattern of TNF-induced activation of ERK, JNK, and Akt (Fig. 5B).
Figure 5.
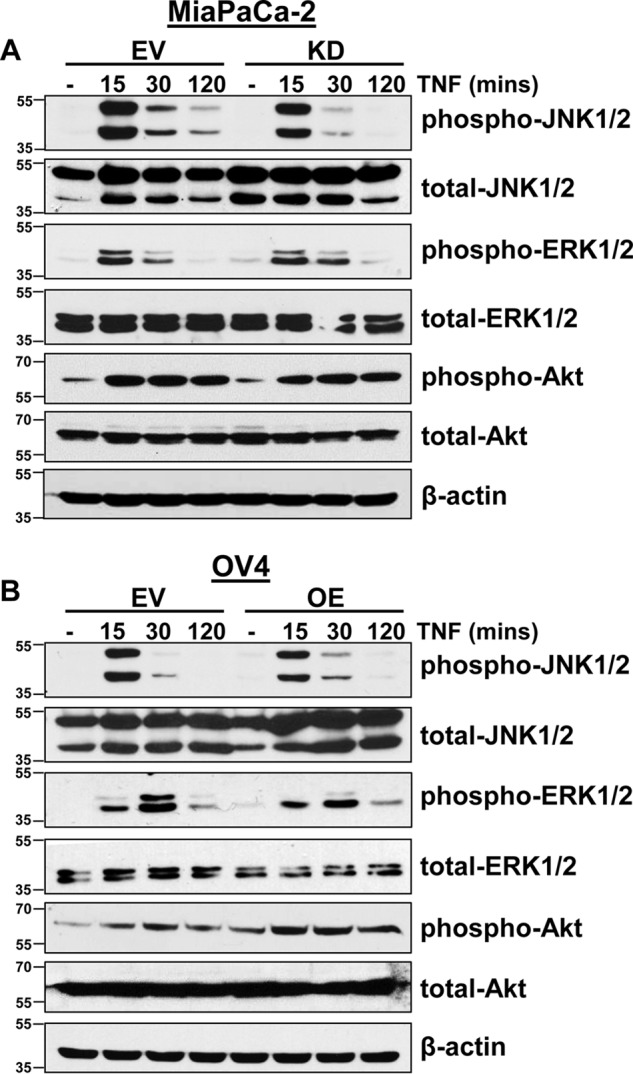
ST6Gal-I does not affect the early activation of MAPKs and Akt. A and B, MiaPaCa-2 EV and KD cells (A) and OV4 EV and OE cells (B) were treated with TNF for 15, 30, or 120 min and immunoblotted for p-JNK, p-ERK, and p-Akt.
ST6Gal-I promotes TNFR1-mediated survival signaling under extended TNF stimulation
Although the data in Figs. 4 and 5 suggest that α2-6 sialylation of TNFR1 does not affect early TNF-induced signaling, Figs. 2 and 3 clearly show that ST6Gal-I provides a protective effect against TNFR1-mediated cell death. We thus examined later time points in TNF signaling that may be more indicative of the overall balance between TNF-induced survival and apoptosis. To this end, MiaPaCa-2 EV and KD cells were incubated with TNF for 6 and 24 h and then immunoblotted for multiple pro-survival molecules (Fig. 6A). MiaPaCa-2 EV cells demonstrated robust activation of Akt and NF-κB upon a 6-h treatment with TNF, and enhanced activation of these molecules was maintained at 24 h relative to untreated cells. However, ST6Gal-I KD dramatically decreased the TNF-induced activation of Akt and NF-κB at both 6 and 24 h. In addition, expression of cIAP2, an apoptosis inhibitor and transcriptional target of NF-κB, is elevated in MiaPaCa-2 EV cells upon treatment with TNF, whereas no increase in cIAP2 was noted in the KD line.
Figure 6.

ST6Gal-I promotes TNFR1-mediated survival signaling. A and B, MiaPaCa-2 EV and KD (A) and OV4 EV and OE cells (B) were treated with TNF for 6 and 24 h and immunoblotted for p-Akt, pNF-κB–p65, and cIAP2. C, OV4 EV or OE cells were cultured with or without an anti-TNFR1 neutralizing antibody for 24 h, and lysates were immunoblotted for p-Akt, pNF-κB–p65, and cIAP2.
As with MiaPaCa-2 cells, OV4 cells with high ST6Gal-I expression displayed enhanced long-term activation of survival molecules, although there were some differences in the signaling pattern between the two cell lines. In particular, OV4 cells with ST6Gal-I OE exhibited increased basal levels of pAkt, pNF-κB, and cIAP2 compared with EV cells (Fig. 6B). TNF had no apparent effect on Akt activation at 6 and 24 h, although clearly pAkt levels were higher in OE versus EV cells at all time points. On the other hand, a 6-h TNF treatment induced strong activation of NF-κB in both EV and OE cells. However, at 24 h, EV cells had no detectable pNF-κB, whereas OE cells maintained substantial NF-κB activation. cIAP2 expression was comparably increased by TNF in EV and OE cells; however, as noted above, OE cells exhibited greater basal cIAP2 expression.
Because pAkt, pNF-κB, and cIAP2 levels were basally higher in OV4 OE cells, we next explored the potential contribution of TNFR1 to this enrichment. We hypothesized that α2-6 sialylation may have influenced inherent TNFR1 signaling irrespective of TNF or alternately affected autocrine TNF signaling. To address this hypothesis, OV4 cells were incubated with a TNFR1-neutralizing antibody for 24 h, and then lysates were immunoblotted for signaling molecules. As shown in Fig. 6C, treatment with the TNFR1-blocking antibody ablated basal differences in pAkt, pNF-κB, and cIAP2 between EV and OE cells. These data indicate that sialylation-mediated differences in basal signaling are dependent upon TNFR1.
ST6Gal-I inhibits TNF-induced apoptosis
Considering that ST6Gal-I potentiated TNFR1-mediated survival signaling, we next evaluated the effects of α2-6 sialylation on the apoptotic arm of TNFR1. MiaPaCa-2 cells were treated with TNF for 6 and 24 h and evaluated for caspase activation. As shown in Fig. 7A, activation of the initiator caspase, caspase-8, was substantially higher in KD versus EV cells upon 6-h TNF treatment. Likewise, KD cells exhibited enhanced cleavage of the effector caspase, caspase-3, upon 8- and 24-h TNF treatment (Fig. 7B). Using a luminescence assay that measures the activity of caspases 3 and 7, greater caspase activation was noted in MiaPaCa-2 KD cells (Fig. 7C).
Figure 7.

ST6Gal-I inhibits TNF-induced apoptosis. A, MiaPaCa-2 EV and KD cells were treated with TNF for 6 and 24 h and immunoblotted for cleaved caspase-8 (cl. caspase 8). The β-tubulin blot is a duplicate of the β-tubulin blot shown in Fig. 6A because the data from Figs. 6A and A are from the same experiment. B, MiaPaCa-2 cells were treated with TNF for 8 and 24 h and immunoblotted for cleaved caspase-3 (cl. caspase-3). C, MiaPaCa-2 cells were treated with TNF for 4 and 24 h and analyzed via a luminescence assay that detects caspase-7 and -3 activity. A representative experiment performed in quadruplicate is shown; data are plotted as mean ± S.D. D, OV4 EV or OE cells were treated with TNF plus CHX for 3, 6, and 24 h and immunoblotted for cleaved caspase-8. E, OV4 cells were treated with TNF plus CHX for 24 h and immunoblotted for cleaved caspase-3. F, OV4 cells were treated with TNF plus CHX for 4 and 24 h and analyzed via a luminescence assay for caspase-7 and -3 activity. A representative experiment in quadruplicate is shown; data are plotted as mean ± S.D.
In the OV4 cell line, TNF induced strong activation of caspases 8 and 3 in EV cells, as measured by immunoblotting (Fig. 7, D and E, respectively). However, cleavage of caspases 8 and 3 was markedly inhibited by ST6Gal-I OE. These data were mirrored by caspase luminescence assays (Fig. 7F), which revealed heightened TNF-induced caspase-3/7 activation in EV versus OE cells. Taken together, these data suggest that ST6Gal-I-mediated TNFR1 sialylation hinders caspase-mediated apoptosis, which may then foster sustained activation of survival-associated molecules.
α2-6 sialylation of TNFR1 blocks apoptosis by preventing TNFR1 internalization
We next addressed potential mechanisms by which α2-6 sialylation may affect TNFR1 function. It was noted in some of our OV4 immunoblots (Fig. 8A) that the TNFR1 protein can be resolved into two distinct bands, a phenomenon not always apparent because of the small difference in molecular weight. Treatment of OV4 parental (Par) and EV cells with TNF for 10 min reduced the upper TNFR1 band, whereas the upper band was retained in TNF-treated OE cells. Notably, the upper band was still present in OE cells even after 1 h of TNF treatment. To test whether this pattern of TNFR1 processing was specific to OV4 cells, U937 leukemia cells with ST6Gal-I overexpression were also utilized. We reported previously that ST6Gal-I OE in U937 cells protects against TNF-induced apoptosis (13). As shown in Fig. 8B, the response of U937 cells to TNF was comparable with that of OV4 cells. TNF treatment of parental and EV U937 cells induced a loss in the upper TNFR1 band, whereas this band was retained in TNF-treated U937 OE cells. These data suggest that α2-6 sialylation of TNFR1 modulates some aspect of TNF-induced TNFR1 processing. We considered two mechanisms as possible explanations for differential receptor processing: TNFR1 shedding and TNFR1 internalization. We hypothesized that sialylation-dependent changes in TNFR1 shedding were unlikely because the size of the known proteolytic product following shedding is inconsistent with the molecular weight differences in the two bands shown in Fig. 8. Following cleavage by ADAM17/TACE, the released extracellular TNFR1 domain is ∼25 kDa, and the retained membrane-bound form is ∼35 kDa (7). Nonetheless, ELISA assays were conducted to quantify shed TNFR1 in the culture medium. U937 cells are a well-known model for evaluating TNFR1 shedding, and treatment of these cells with PMA is a strong inducer of shedding (23), thus offering a positive control. As shown in Fig. 8C, TNF-induced TNFR1 shedding was minimal over a 24-h interval (in contrast to the PMA positive control), and no differences in the degree of shedding were noted between EV and OE cells.
Figure 8.

α2-6 sialylation of TNFR1 prevents TNF-induced TNFR1 internalization. A and B, OV4 parental (Par), EV, and OE cells (A) or U937 Par, EV, and OE cells (B) were treated with TNF for 10 min or 1 h and immunoblotted for TNFR1 using an antibody that recognizes the intracellular domain (Cell Signaling Technology, 3736). C, U937 EV and OE cells were treated with TNF or PMA for 24 hr, and then the culture supernatant was analyzed for shed TNFR1 using a soluble TNFR1 ELISA kit. D, OV4 EV and OE cells were incubated with TNF-biotin at 4 °C to allow TNF binding to surface TNFR1. Cells were either maintained in 4 °C (control), or the temperature was switched to 37 °C for 10 min to permit TNFR1 internalization. Cells incubated at 4 °C or 37 °C were then stained with streptavidin–FITC and analyzed by flow cytometry to measure levels of surface TNF-biotin–TNFR1complexes.
Because sialylation had no effect on shedding, we next examined TNFR1 internalization. In Fig. 8, A and B, the TNFR1 molecular weight changes following TNF treatment are consistent with observations reported by Schütze and co-workers (10). They have developed a method to isolate endosomes, and they have found that full-length TNFR1 enters the endosomal compartment within ∼3 min following TNF treatment and is then processed within 10–30 min into a lower-molecular-weight form that is comparable in size to the changes shown in Fig. 8, A and B (10). Also, it is well known that a substantial amount of TNFR1 is retained intracellularly within the endosomes and Golgi in the absence of TNF treatment (10, 24–26), which provides a possible explanation for the double TNFR1 bands in untreated OV4 and U937 cells. To examine whether sialylation prevented TNFR1 internalization, we performed flow cytometry to quantify levels of surface TNFR1 following TNF-induced receptor activation. OV4 Par or OE cells were incubated with TNF-biotin at 4 ºC, which allows TNF to bind TNFR1 but not induce internalization. Temperature was then switched to 37 ºC for 10 min to induce internalization, and the remaining surface-bound TNF-biotin–TNFR1 complexes were quantified by labeling cells with streptavidin–FITC. As shown in Fig. 8D, a temperature shift to 37 ºC induced TNFR1 internalization in parental cells but not in OE cells, suggesting that α2-6 sialylation of TNFR1 impedes TNF-induced TNFR1 internalization.
To further address the hypothesis that a sialylation-dependent block in TNFR1 internalization is responsible for abrogating TNF-induced apoptosis, OV4 cells were co-incubated for 24 h with TNF and an inhibitor of internalization, Dyngo-4a (a dynamin inhibitor). As shown in Fig. 9, TNF-induced apoptosis in OV4 EV cells was inhibited by Dyngo-4a, recapitulating the protective effects observed in TNF-treated OE cells. Of note, Dyngo-4a had no apparent effect on OE cells. We also incubated cells with an inhibitor, TAPI-1, of the ADAM17/TACE protease responsible for TNFR1 shedding. As shown in Fig. 9, the TAPI-1 inhibitor did not protect against TNF-induced apoptosis in either EV or OE cells, consistent with the concept that the anti-apoptotic function of ST6Gal-I is not mediated through a shedding mechanism. In the aggregate, studies in this report suggest that α2-6 sialylation of TNFR1 inhibits TNF-induced apoptosis by preventing internalization, which, in turn, potentiates survival signaling.
Figure 9.
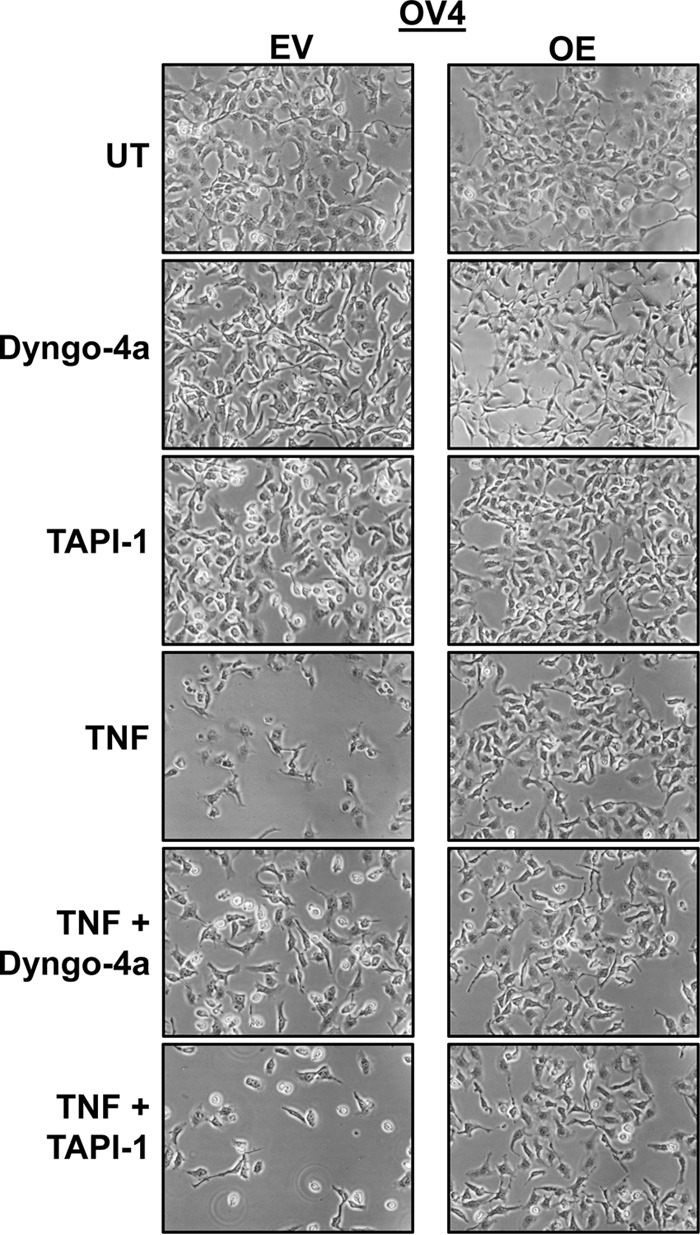
Pharmacological inhibition of endocytosis rescues OV4 EV cells from TNF-induced cell death. OV4 EV and OE cells were co-cultured with TNF and either Dyngo-4a (a dynamin inhibitor) or TAPI-1 (an ADAM17/TACE inhibitor). Images were obtained following 24 h of treatment. UT, untreated.
Discussion
Dysregulation of the TNF/TNFR1 signaling axis contributes to a diversity of pathologies, including autoimmune diseases (4, 27) and cancer (1, 28, 29). Although TNF was originally described as a tumoricidal molecule, it has since emerged that cancer cells are often resistant to TNF-induced cell death, and, in fact, TNF has potent tumor-promoting functions (1, 30). The TNF/TNFR1 pathway fuels tumor growth via multiple conduits, including direct stimulation of tumor cell proliferation (1) and induction of epithelial–mesenchymal transition (31, 32). In addition, impaired apoptotic signaling by TNFR1 likely contributes to tumor evasion from immune surveillance, given that immune cells are the major source of TNF (4, 33). In view of these factors, a better understanding of the molecular mechanisms that subvert TNFR1 signaling to drive tumorigenesis is greatly needed.
In this investigation, we show that ST6Gal-I–mediated α2-6 sialylation of TNFR1 selectively blocks the apoptotic effects of TNF. Importantly, TNFR1 sialylation does not influence initial TNF-stimulated events such as IκBα destruction, NF-κB nuclear translocation, or rapid activation of Akt, JNK, and ERK. However, striking phenotypic and signaling differences in cells with variant ST6Gal-I expression become apparent upon prolonged TNF treatment (≥6 h). Under chronic TNF stimulation, cells with high ST6Gal-I levels exhibit an increase in survival characteristics, indicated by morphological evidence as well as sustained activation of the survival-associated molecules NF-κB and Akt. Correspondingly, ST6Gal-I–mediated α2-6 sialylation inhibits TNFR1-induced apoptosis, evidenced by diminished activation of caspases 8 and 3. Hence, ST6Gal-I activity shifts the overall balance of TNFR1 signaling toward survival.
We further investigated potential mechanisms by which α2-6 sialylation of TNFR1 might regulate intracellular signaling. Our combined studies point to a sialylation-dependent block in TNFR1 internalization as the principal driver of a signaling switch. This finding is in line with our prior studies of the Fas death receptor, which revealed that α2-6 sialylation of Fas prevents apoptosis by blocking Fas internalization (34). Significantly, TRAIL-induced apoptosis is unaffected by ST6Gal-I activity, and the TRAIL receptor DR5 has no consensus sequences for N-glycosylation (34). The lack of effect on TRAIL-mediated apoptosis indicates that ST6Gal-I activity modulates the function of specific surface receptors and does not fundamentally alter the apoptotic machinery. It is well established that the localization of TNFR1 is critical for downstream signaling (12). Many studies have shown that the activation of TNFR1 by TNF at the cell surface initiates NF-κB-mediated survival signaling, whereas TNFR1 internalization into endosomes is essential for robust formation of the DISC and apoptosis induction (10, 35, 36). As examples, preventing TNFR1 internalization through pharmacologic intervention or by deleting the TNFR1 internalization domain obstructs caspase activation while simultaneously preserving NF-κB survival signaling (10, 35, 36). However, the biological mechanisms that regulate the partitioning of TNFR1 between these compartments remain elusive. As ST6Gal-I expression is dynamically regulated in many cell types, fluctuating levels of TNFR1 α2-6 sialylation are physiologically relevant and offer a novel mechanism for dictating translocation of TNFR1 between the cell surface and endosomes.
Elevated receptor α2-6 sialylation is prevalent in tumor cells because of the up-regulation of ST6Gal-I in diverse human malignancies (16, 18, 37). Immunohistochemical studies show that ST6Gal-I is highly expressed in pancreatic, ovarian, and colon cancer, whereas ST6Gal-I levels are low in the normal epithelium of these organs (21, 22). High ST6Gal-I expression is correlated with poor patient prognosis in ovarian, colon, breast, and pancreatic cancer (22, 38–40), and animal models support a tumor-promoting function for ST6Gal-I (22, 40). In the azoxymethane/dextran sulfate sodium (AOM/DSS) inflammation-associated colon tumorigenesis model, for which the TNF/TNFR1 axis plays a major role (41), mice with forced ST6Gal-I overexpression have an increased incidence of tumor development and overall greater tumor burden (22). As another example, ST6Gal-I overexpression propels pancreatic cancer metastasis in an orthotopic transplant model (40). ST6Gal-I activity likely contributes to malignant progression through imparting metastatic cellular behaviors such as invasiveness (40, 42–45), epithelial–mesenchymal transition (46), and hallmark cancer stem cell characteristics, including anchorage-independent tumor spheroid growth and tumor-initiating potential (22).
Accumulating evidence suggests that one of the main functions of ST6Gal-I may be to protect cancer cells against a variety of microenvironmental assaults. In addition to inhibiting Fas (34) and TNFR1-dependent cell death, α2-6 sialylation of select receptors inhibits apoptosis induced by extracellular galectins (47–49), a family of galactose-binding lectins. ST6Gal-I also protects cells from cytotoxicity induced by serum deprivation (19), radiation (50), and chemotherapy drugs (20, 22, 51). Our results add to this body of literature by elucidating a novel pathway by which ST6Gal-I may promote tumor cell survival within TNF-rich inflammatory tumor microenvironments. Furthermore, this study addresses one of the major knowledge gaps in TNF signaling; namely, the mechanisms that dictate whether TNF induces cell survival or cell death.
Experimental procedures
Cell culture
MiaPaCa-2 cells were purchased from the ATCC, and OV4 cells were obtained from Dr. Timothy Eberlein at Harvard University (Boston, MA). Cells were grown in Dulbecco's modified Eagle's medium/F12 (OV4) or Dulbecco's modified Eagle's medium (MiaPaCa-2) containing 10% fetal bovine serum and 1% antibiotic/antimycotic supplements (GE Healthcare Hyclone). Stable polyclonal cell lines were created by transducing cells with an empty vector lentivirus (Sigma) or a lentivirus encoding either the ST6Gal-I gene (Genecopoeia) or shRNA against ST6Gal-I (Sigma, TN00000035432, sequence CCGGCGTGTGCTACTACTACCAGAACTCGAGTTCTGGTAGTAGTAGCACACGTTTTTG), followed by selection with 10 μg/ml of puromycin (Sigma). Puromycin was removed from the medium at least 2 days prior to all experiments.
ST6Gal-I expression and activity confirmation assays
ST6Gal-I overexpression or knockdown was verified by immunoblotting. Briefly, cells were lysed in radioimmune precipitation assay buffer supplemented with 1× protease and phosphatase inhibitors (Pierce). Total protein concentration was measured by BCA (Pierce). Samples were resolved by SDS-PAGE and transferred to polyvinylidene difluoride membranes. Membranes were incubated with 5% nonfat dry milk in TBS buffer containing 0.1% Tween 20 (TBS-T) and then probed with a goat polyclonal antibody against ST6Gal-I (R&D Systems, AF5924, lot CDSF0114101). Membranes were incubated with anti-goat secondary antibody and developed using ECL detection methods. In addition to immunoblotting, ST6Gal-I activity was monitored by using the SNA lectin to detect surface α2-6 sialic acids. MiaPaCa-2 or OV4 cells were stained with a 1:100 or 1:500 dilution, respectively, of FITC-conjugated SNA lectin (Vector, B-1305) for 30 min at 4 °C. SNA lectin binding was quantified via flow cytometry. To confirm that TNFR1 is a substrate for ST6Gal-I and that α2-6 sialylation of TNFR1 is altered by ST6Gal-I manipulation, cell lysates (500 μg for MiPaCa-2 and 200 μg for OV4) were incubated with 50 μl of SNA-conjugated agarose beads (Vector, AL-1303) overnight at 4 °C. α2-6–sialylated proteins bound to the beads were then precipitated by centrifugation, washed twice with PBS, and resuspended in 1× sample buffer (Bio-Rad) plus 10% 2-mercaptoethanol (Sigma). Proteins were resolved by SDS-PAGE and immunoblotted against TNFR1 (Cell Signaling Technology, 3736, lot 2).
TNF treatments
All studies involving TNF treatments were done in medium containing 1% serum (2 h preincubation in 1% serum-containing medium, followed by administration of TNF in 1% serum). For studies evaluating cellular morphological changes and ST6Gal-I enrichment, MiaPaCa-2 cells, which are highly sensitive to TNF-induced cell death, were cultured with 100 ng/ml recombinant human TNF (R&D Systems, 210-TA) for 24 h, whereas OV4 cells were cultured with a combination of 100 ng/ml TNF plus 5 μm cycloheximide (CHX, Sigma) to amplify the cytotoxicity of TNF. To examine TNF-induced signaling, cells were cultured with 10 ng/ml TNF for the indicated times, and the only exception to this method was the evaluation of caspase-3/8 activation in OV4 cells, in which 5 μm CHX was used in addition to 10 ng/ml TNF. To examine the contribution of TNFR1 to basal signaling differences in OV4 cells, OV4 EV or OE cells were incubated with anti-TNFR1 neutralizing antibody (R&D Systems, MAB225, lot IP0914031) in 1% serum-containing medium, and lysates were collected 24 h later and immunoblotted.
Immunoblotting
Cells were treated with TNF for the indicated times and then lysed in radioimmune precipitation assay buffer as described previously. Following SDS-PAGE and transfer, membranes were blocked in 5% nonfat dry milk in TBS-T. Immunoblots were probed with antibodies against ST6Gal-I (R&D Systems, AF5924, lot CDSF0114101), cIAP2 (Cell Signaling Technology, 3130, lot 6), pAkt (Ser-473, Cell Signaling Technology, 4060, lot 19), total Akt (Cell Signaling Technology, 4691, lot 20), pNF-κB–p65 (Ser-536, Cell Signaling Technology, 3033, lot 14), total NF-κB–p65 (Cell Signaling Technology, 8242, lot 4), pJNK (Thr-183/Tyr-185, Cell Signaling Technology, 9255, lot 32), total JNK (Cell Signaling Technology, 9258, lot 17), IκBα (Cell Signaling Technology, 4812, lot 9), pErk1/2 (Thr-202/Tyr-204, Cell Signaling Technology, 4370, lot 15), total Erk1/2 (Cell Signaling Technology, 9102, lot 26), cleaved caspase-3 (Asp-175, Cell Signaling Technology, 9661, lot 43), cleaved caspase-8 (Asp-391, Cell Signaling Technology, 9496, lot 7), and TNFR1 (Cell Signaling Technology, 3736, lot 2). Protein loading was verified using horseradish peroxidase (HRP)–conjugated anti-actin (Abcam, ab20272, lot GR201277) or HRP-conjugated anti-tubulin (Abcam, ab21058, lot GR284232). Membranes were incubated with appropriate HRP-coupled secondary antibodies (anti-rabbit and anti-mouse IgG, Cell Signaling Technology; anti-goat IgG, Santa Cruz Biotechnology), and protein was detected by ECL (Pierce), Clarity (Bio-Rad), or SuperSignal West Femto substrate (Pierce).
Immunofluorescence assays
Cells of the same background were prelabeled with 25 μm of their own distinct red or green CellTracker dyes (Thermo Scientific) (Thermo Scientific) and incubated at 37 °C for 30 min. Cells were then washed three times with 1× PBS and seeded at equal densities into the same tissue culture–treated plate. After allowing the cells to adhere for 4 h, the cells were then treated with 100 ng/ml TNF (OV4, TNF plus 5 μm CHX). After 24-h treatment, the cells were imaged with an EVOS fluorescence microscope. To visualize NF-κB subcellular localization, cells were seeded onto lysine-coated chamber slides and treated with 10 ng/ml TNF for 15 min. The cells were immediately washed in ice-cold PBS to block TNF signaling and fixed with 4% paraformaldehyde for 10 min at room temperature. Cells were then permeabilized using 0.1% Triton X-100 (Fisher) for 10 min. The cells were blocked using 10% newborn calf serum (Atlanta Biologicals) and then incubated with total NF-κB-p65 antibody overnight at 4 °C. The primary antibody was washed off, followed by incubation with a donkey anti-rabbit IgG secondary antibody conjugated with Alexa Fluor 488 (Life Technologies). Coverslips were then mounted, and the slides were imaged with a fluorescence microscope (Nikon) fitted with a Nikon CoolSNAP camera.
Caspase-3/7 luminescence assays
Cells were seeded at equal densities into a 96-well tissue culture plate and allowed to adhere overnight. Prior to TNF treatment, cells were incubated in 1% serum-containing medium for 2 h and then treated with 100 ng/ml TNF (OV4, TNF plus 5 μm CHX) for the indicated times. Reconstituted Caspase-Glo 3/7 assay reagent (Promega) was then added to each well, mixed via an orbital shaker, and incubated at room temperature for 45 min. Luminescence was quantified with a BioTek Synergy H1 instrument. In the same experiment, with a separate 96-well plate of the same treatment conditions, the CellTiter-Glo (ATP) assay system (Promega) was utilized as a representation of overall cell number. The values represented are the ratios of values for Caspase-Glo relative to CellTiter-Glo.
TNFR1 shedding and internalization assays
To evaluate TNFR1 shedding, U937 EV or OE cells were treated with either TNF (100 ng/ml) or PMA (200 ng/ml) for 24 h. The supernatant was then collected and analyzed via an ELISA kit specific for soluble TNFR1 (R&D Systems). To examine surface TNFR1 levels, OV4 EV or OE cells were incubated in serum-free medium with TNF-biotin (R&D Systems) at 4 °C for 1 h. Unbound TNF-biotin was washed off with ice-cold PBS. Cells were then incubated with prewarmed 37 °C serum-free medium for 10 min. Control cells were maintained at 4 °C. The cells were fixed with 2% paraformaldehyde for 20 min at room temperature. Following fixation, the cells were incubated with 5 μg/ml streptavidin–FITC (Invitrogen) for 30 min on ice. Cells were then washed twice with PBS, and residual surface-bound TNF-biotin–TNFR1 complexes were quantified via flow cytometry. To analyze the effect of internalization and shedding inhibition, OV4 EV or OE cells were preincubated for 1 h with 5 μm Dyngo-4a or 50 μm TAPI-1 (both from Selleckchem) and then treated with TNF (100 ng/ml) plus CHX (1 μm). Images were obtained following 24 h of treatment.
Author contributions
A. T. H. was responsible for the acquisition and analysis of the data. C. M. B. aided with the collection and analysis of results. A. T. H. and S. L. B. were responsible for the concept and design of this study and together wrote the manuscript.
Acknowledgments
We gratefully acknowledge technical assistance from Dr. Zhongyu Liu (University of Alabama at Birmingham). The University of Alabama at Birmingham Flow Cytometry Core Facility was funded by National Institutes of Health Grants P30AR048311 and P30AI027767.
This work was supported by National Institutes of Health Grants R01 GM111093 and R21 CA192629 (to S. L. B.), a T32 HL007918 cardiovascular pathophysiology training grant fellowship (to A. T. H.), and an American Heart Association predoctoral fellowship (to A. T. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- TNF
- tumor necrosis factor
- DD
- death domain
- MAPK
- mitogen-activated protein kinase
- DISC
- death-inducing signaling complex
- shRNA
- short hairpin RNA
- KD
- knockdown
- EV
- empty vector
- OE
- overexpression
- IKK
- IκB kinase
- JNK
- c-Jun N-terminal kinase
- ERK
- extracellular signal-regulated kinase
- Par
- parental
- PMA
- phorbol 12-myristate 13-acetate
- TBS
- Tris-buffered saline
- CHX
- cycloheximide
- HRP
- horseradish peroxidase
- SNA
- Sambucus nigra agglutinin
- TACE
- tumor necrosis α–converting enzyme
- TRAIL
- TNF-related apoptosis-inducing ligand
- FADD
- Fas-associated death domain protein
- TRADD
- tumor necrosis factor receptor type 1-associated death domain protein.
References
- 1. Ham B., Fernandez M. C., D'Costa Z., and Brodt P. (2016) The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 11, 1–27 [PMC free article] [PubMed] [Google Scholar]
- 2. Barbara J. A., Smith W. B., Gamble J. R., Van Ostade X., Vandenabeele P., Tavernier J., Fiers W., Vadas M. A., and Lopez A. F. (1994) Dissociation of TNF-α cytotoxic and proinflammatory activities by p55 receptor- and p75 receptor-selective TNF-α mutants. EMBO J. 13, 843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Puimège L., Libert C., and Van Hauwermeiren F. (2014) Regulation and dysregulation of tumor necrosis factor receptor-1. Cytokine Growth Factor Rev. 25, 285–300 10.1016/j.cytogfr.2014.03.004 [DOI] [PubMed] [Google Scholar]
- 4. Locksley R. M., Killeen N., and Lenardo M. J. (2001) The TNF and TNF receptor superfamilies: integrating mammalian biology. Cell Death Differ. 104, 487–501 [DOI] [PubMed] [Google Scholar]
- 5. Carswell E. A., Old L. J., Kassel R. L., Green S., Fiore N., and Williamson B. (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. U.S.A. 72, 3666–3670 10.1073/pnas.72.9.3666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ali M., Fritsch J., Zigdon H., Pewzner-Jung Y., Schütze S., and Futerman A. H. (2013) Altering the sphingolipid acyl chain composition prevents LPS/GLN-mediated hepatic failure in mice by disrupting TNFR1 internalization. Cell Death Dis. 4, e929 10.1038/cddis.2013.451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chhibber-Goel J., Coleman-Vaughan C., Agrawal V., Sawhney N., Hickey E., Powell J. C., and McCarthy J. V. (2016) γ-Secretase activity is required for regulated intramembrane proteolysis of tumor necrosis factor (TNF) receptor 1 and TNF-mediated pro-apoptotic signaling. J. Biol. Chem. 291, 5971–5985 10.1074/jbc.M115.679076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fritsch J., Stephan M., Tchikov V., Winoto-Morbach S., Gubkina S., Kabelitz D., and Schütze S. (2014) Cell fate decisions regulated by K63 ubiquitination of tumor necrosis factor receptor 1. Mol. Cell. Biol. 34, 3214–3228 10.1128/MCB.00048-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Legler D. F., Micheau O., Doucey M. A., Tschopp J., and Bron C. (2003) Recruitment of TNF receptor 1 to lipid rafts is essential for TNFα-mediated NF-κB activation. Immunity 18, 655–664 10.1016/S1074-7613(03)00092-X [DOI] [PubMed] [Google Scholar]
- 10. Schneider-Brachert W., Tchikov V., Neumeyer J., Jakob M., Winoto-Morbach S., Held-Feindt J., Heinrich M., Merkel O., Ehrenschwender M., Adam D., Mentlein R., Kabelitz D., and Schütze S. (2004) Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity 21, 415–428 10.1016/j.immuni.2004.08.017 [DOI] [PubMed] [Google Scholar]
- 11. Micheau O., and Tschopp J. (2003) Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 114, 181–190 10.1016/S0092-8674(03)00521-X [DOI] [PubMed] [Google Scholar]
- 12. Schütze S., Tchikov V., and Schneider-Brachert W. (2008) Regulation of TNFR1 and CD95 signalling by receptor compartmentalization. Nat. Rev. Mol. Cell Biol. 9, 655–662 10.1038/nrm2430 [DOI] [PubMed] [Google Scholar]
- 13. Liu Z., Swindall A. F., Kesterson R. A., Schoeb T. R., Bullard D. C., and Bellis S. L. (2011) ST6Gal-I regulates macrophage apoptosis via α2-6 sialylation of the TNFR1 death receptor. J. Biol. Chem. 286, 39654–39662 10.1074/jbc.M111.276063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Büll C., Stoel M. A., den Brok M. H., and Adema G. J. (2014) Sialic acids sweeten a tumor's life. Cancer Res. 74, 3199–3204 10.1158/0008-5472.CAN-14-0728 [DOI] [PubMed] [Google Scholar]
- 15. Dall'Olio F., Malagolini N., Trinchera M., and Chiricolo M. (2014) Sialosignaling: sialyltransferases as engines of self-fueling loops in cancer progression. Biochim. Biophys. Acta 1840, 2752–2764 10.1016/j.bbagen.2014.06.006 [DOI] [PubMed] [Google Scholar]
- 16. Lu J., and Gu J. (2015) Significance of β-galactoside α2,6 sialyltransferase 1 in cancers. Molecules 20, 7509–7527 10.3390/molecules20057509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Harduin-Lepers A., Krzewinski-Recchi M. A., Colomb F., Foulquier F., Groux-Degroote S., and Delannoy P. (2012) Sialyltransferases functions in cancers. Front Biosci. 4, 499–515 [DOI] [PubMed] [Google Scholar]
- 18. Schultz M. J., Swindall A. F., and Bellis S. L. (2012) Regulation of the metastatic cell phenotype by sialylated glycans. Cancer Metastasis Rev. 31, 501–518 10.1007/s10555-012-9359-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Britain C. M., Dorsett K. A., and Bellis S. L. (2017) The glycosyltransferase ST6Gal-I protects tumor cells against serum growth factor withdrawal by enhancing survival signaling and proliferative potential. J. Biol. Chem. 292, 4663–4673 10.1074/jbc.M116.763862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schultz M. J., Swindall A. F., Wright J. W., Sztul E. S., Landen C. N., and Bellis S. L. (2013) ST6Gal-I sialyltransferase confers cisplatin resistance in ovarian tumor cells. J. Ovarian Res. 6, 25 10.1186/1757-2215-6-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Swindall A. F., Londoño-Joshi A. I., Schultz M. J., Fineberg N., Buchsbaum D. J., and Bellis S. L. (2013) ST6Gal-I protein expression is upregulated in human epithelial tumors and correlates with stem cell markers in normal tissues and colon cancer cell lines. Cancer Res. 73, 2368–2378 10.1158/0008-5472.CAN-12-3424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schultz M. J., Holdbrooks A. T., Chakraborty A., Grizzle W. E., Landen C. N., Buchsbaum D. J., Conner M. G., Arend R. C., Yoon K. J., Klug C. A., Bullard D. C., Kesterson R. A., Oliver P. G., O'Connor A. K., Yoder B. K., and Bellis S. L. (2016) The tumor-associated glycosyltransferase ST6Gal-I regulates stem cell transcription factors and confers a cancer stem cell phenotype. Cancer Res. 76, 3978–3988 10.1158/0008-5472.CAN-15-2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arribas J., and Borroto A. (2002) Protein ectodomain shedding. Chem. Rev. 102, 4627–4638 10.1021/cr010202t [DOI] [PubMed] [Google Scholar]
- 24. Bradley J. R., Thiru S., and Pober J. S. (1995) Disparate localization of 55-kd and 75-kd tumor necrosis factor receptors in human endothelial cells. Am. J. Pathol. 146, 27–32 [PMC free article] [PubMed] [Google Scholar]
- 25. Chambaut-Guérin A. M., Rouher C., and Gauthereau X. (1997) p55 tumour necrosis factor receptors distribution in neuroblastoma cells. Neuroreport 8, 1451–1456 10.1097/00001756-199704140-00025 [DOI] [PubMed] [Google Scholar]
- 26. Jones S. J., Ledgerwood E. C., Prins J. B., Galbraith J., Johnson D. R., Pober J. S., and Bradley J. R. (1999) TNF recruits TRADD to the plasma membrane but not the trans-Golgi network, the principal subcellular location of TNF-R1. J. Immunol. 162, 1042–1048 [PubMed] [Google Scholar]
- 27. Bradley J. R. (2008) TNF-mediated inflammatory disease. J. Pathol. 214, 149–160 10.1002/path.2287 [DOI] [PubMed] [Google Scholar]
- 28. Oshima H., Ishikawa T., Yoshida G. J., Naoi K., Maeda Y., Naka K., Ju X., Yamada Y., Minamoto T., Mukaida N., Saya H., and Oshima M. (2014) TNF-α/TNFR1 signaling promotes gastric tumorigenesis through induction of Noxo1 and Gna14 in tumor cells. Oncogene 33, 3820–3829 10.1038/onc.2013.356 [DOI] [PubMed] [Google Scholar]
- 29. Charles K. A., Kulbe H., Soper R., Escorcio-Correia M., Lawrence T., Schultheis A., Chakravarty P., Thompson R. G., Kollias G., Smyth J. F., Balkwill F. R., and Hagemann T. (2009) The tumor-promoting actions of TNF-α involve TNFR1 and IL-17 in ovarian cancer in mice and humans. J. Clin. Invest. 119, 3011–3023 10.1172/JCI39065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang X., and Lin Y. (2008) Tumor necrosis factor and cancer, buddies or foes? Acta Pharmacol. Sin. 29, 1275–1288 10.1111/j.1745-7254.2008.00889.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bates R. C., and Mercurio A. M. (2003) Tumor necrosis factor-α stimulates the epithelial-to-mesenchymal transition of human colonic organoids. Mol. Biol. Cell 14, 1790–1800 10.1091/mbc.E02-09-0583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang H., Wang H. S., Zhou B. H., Li C. L., Zhang F., Wang X. F., Zhang G., Bu X. Z., Cai S. H., and Du J. (2013) Epithelial-mesenchymal transition (EMT) induced by TNF-α requires AKT/GSK-3β-mediated stabilization of snail in colorectal cancer. PLoS ONE 8, e56664 10.1371/journal.pone.0056664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Guicciardi M. E., and Gores G. J. (2009) Life and death by death receptors. FASEB J. 23, 1625–1637 10.1096/fj.08-111005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Swindall A. F., and Bellis S. L. (2011) Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J. Biol. Chem. 286, 22982–22990 10.1074/jbc.M110.211375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schütze S., Machleidt T., Adam D., Schwandner R., Wiegmann K., Kruse M. L., Heinrich M., Wickel M., and Krönke M. (1999) Inhibition of receptor internalization by monodansylcadaverine selectively blocks p55 tumor necrosis factor receptor death domain signaling. J. Biol. Chem. 274, 10203–10212 10.1074/jbc.274.15.10203 [DOI] [PubMed] [Google Scholar]
- 36. Schneider-Brachert W., Tchikov V., Merkel O., Jakob M., Hallas C., Kruse M. L., Groitl P., Lehn A., Hildt E., Held-Feindt J., Dobner T., Kabelitz D., Krönke M., and Schütze S. (2006) Inhibition of TNF receptor 1 internalization by adenovirus 14.7K as a novel immune escape mechanism. J. Clin. Invest. 116, 2901–2913 10.1172/JCI23771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Dall'Olio F., and Chiricolo M. (2001) Sialyltransferases in cancer. Glycoconj. J. 18, 841–850 10.1023/A:1022288022969 [DOI] [PubMed] [Google Scholar]
- 38. Lise M., Belluco C., Perera S. P., Patel R., Thomas P., and Ganguly A. (2000) Clinical correlations of α2,6-sialyltransferase expression in colorectal cancer patients. Hybridoma 19, 281–286 10.1089/027245700429828 [DOI] [PubMed] [Google Scholar]
- 39. Recchi M. A., Hebbar M., Hornez L., Harduin-Lepers A., Peyrat J. P., and Delannoy P. (1998) Multiplex reverse transcription polymerase chain reaction assessment of sialyltransferase expression in human breast cancer. Cancer Res. 58, 4066–4070 [PubMed] [Google Scholar]
- 40. Hsieh C. C., Shyr Y. M., Liao W. Y., Chen T. H., Wang S. E., Lu P. C., Lin P. Y., Chen Y. B., Mao W. Y., Han H. Y., Hsiao M., Yang W. B., Li W. S., Sher Y. P., and Shen C. N. (2017) Elevation of β-galactoside α2,6-sialyltransferase 1 in a fructose-responsive manner promotes pancreatic cancer metastasis. Oncotarget 8, 7691–7709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Popivanova B. K., Kitamura K., Wu Y., Kondo T., Kagaya T., Kaneko S., Oshima M., Fujii C., and Mukaida N. (2008) Blocking TNF-α in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 118, 560–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lin S., Kemmner W., Grigull S., and Schlag P. M. (2002) Cell surface α2,6 sialylation affects adhesion of breast carcinoma cells. Exp. Cell Res. 276, 101–110 10.1006/excr.2002.5521 [DOI] [PubMed] [Google Scholar]
- 43. Christie D. R., Shaikh F. M., Lucas J. A. 4th, Lucas J. A. 3rd, Bellis S. L. (2008) ST6Gal-I expression in ovarian cancer cells promotes an invasive phenotype by altering integrin glycosylation and function. J. Ovarian Res. 1, 3 10.1186/1757-2215-1-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhu Y., Srivatana U., Ullah A., Gagneja H., Berenson C. S., and Lance P. (2001) Suppression of a sialyltransferase by antisense DNA reduces invasiveness of human colon cancer cells in vitro. Biochim. Biophys. Acta 1536, 148–160 10.1016/S0925-4439(01)00044-8 [DOI] [PubMed] [Google Scholar]
- 45. Shaikh F. M., Seales E. C., Clem W. C., Hennessy K. M., Zhuo Y., and Bellis S. L. (2008) Tumor cell migration and invasion are regulated by expression of variant integrin glycoforms. Exp. Cell Res. 314, 2941–2950 10.1016/j.yexcr.2008.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lu J., Isaji T., Im S., Fukuda T., Hashii N., Takakura D., Kawasaki N., and Gu J. (2014) β-Galactoside α2,6-sialyltranferase 1 promotes transforming growth factor-β-mediated epithelial-mesenchymal transition. J. Biol. Chem. 289, 34627–34641 10.1074/jbc.M114.593392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhuo Y., Chammas R., and Bellis S. L. (2008) Sialylation of β1 integrins blocks cell adhesion to galectin-3 and protects cells against galectin-3-induced apoptosis. J. Biol. Chem. 283, 22177–22185 10.1074/jbc.M800015200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Fukumori T., Takenaka Y., Yoshii T., Kim H. R., Hogan V., Inohara H., Kagawa S., and Raz A. (2003) CD29 and CD7 mediate galectin-3-induced type II T-cell apoptosis. Cancer Res. 63, 8302–8311 [PubMed] [Google Scholar]
- 49. Toscano M. A., Bianco G. A., Ilarregui J. M., Croci D. O., Correale J., Hernandez J. D., Zwirner N. W., Poirier F., Riley E. M., Baum L. G., and Rabinovich G. A. (2007) Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 8, 825–834 10.1038/ni1482 [DOI] [PubMed] [Google Scholar]
- 50. Lee M., Park J. J., and Lee Y. S. (2010) Adhesion of ST6Gal I-mediated human colon cancer cells to fibronectin contributes to cell survival by integrin β1-mediated paxillin and AKT activation. Oncol. Rep. 23, 757–761 [PubMed] [Google Scholar]
- 51. Chen X., Wang L., Zhao Y., Yuan S., Wu Q., Zhu X., Niang B., Wang S., and Zhang J. (2016) ST6Gal-I modulates docetaxel sensitivity in human hepatocarcinoma cells via the p38 MAPK/caspase pathway. Oncotarget 7, 51955–51964 10.18632/oncotarget.10192 [DOI] [PMC free article] [PubMed] [Google Scholar]