

Abstract
Functional cross-talk between the promoter and terminator of a gene has long been noted. Promoters and terminators are juxtaposed to form gene loops in several organisms, and gene looping is thought to be involved in transcriptional regulation. The general transcription factor IIB (TFIIB) and the C-terminal domain phosphatase Ssu72, essential factors of the transcription preinitiation complex and the mRNA processing and polyadenylation complex, respectively, are important for gene loop formation. TFIIB and Ssu72 interact both genetically and physically, but the molecular basis of this interaction is not known. Here we present a crystal structure of the core domain of TFIIB in two new conformations that differ in the relative distance and orientation of the two cyclin-like domains. The observed extraordinary conformational plasticity may underlie the binding of TFIIB to multiple transcription factors and promoter DNAs that occurs in distinct stages of transcription, including initiation, reinitiation, and gene looping. We mapped the binding interface of the TFIIB-Ssu72 complex using a series of systematic, structure-guided in vitro binding and site-specific photocross-linking assays. Our results indicate that Ssu72 competes with acidic activators for TFIIB binding and that Ssu72 disrupts an intramolecular TFIIB complex known to impede transcription initiation. We also show that the TFIIB-binding site on Ssu72 overlaps with the binding site of symplekin, a component of the mRNA processing and polyadenylation complex. We propose a hand-off model in which Ssu72 mediates a conformational transition in TFIIB, accounting for the role of Ssu72 in transcription reinitiation, gene looping, and promoter-terminator cross-talk.
Keywords: general transcription factor (GTF), RNA polymerase II, transcription initiation factor, transcription regulation, transcription termination, gene looping, promoter-terminator cross-talk, TFIIB, Ssu72
Introduction
TFIIB is an essential general transcription factor and a necessary component of the RNA polymerase II (pol II)3 preinitiation complex (PIC) along with TFIIA, TFIID, TFIIE, TFIIF, and TFIIH. TFIIB is a multidomain protein that is composed of an N-terminal zinc ribbon domain for pol II binding (1–4), a finger/reader domain for transcription start site selection and de novo transcription initiation (1, 4–6), and two C-terminal cyclin-like domains that form a ternary complex with the TATA box DNA and TATA box–binding protein (TBP) (7, 8) (Fig. 1A). TFIIB makes sequence-specific interactions with the TFIIB recognition elements (BREs) found both upstream (BREu) and downstream (BREd) of the TATA box (8–10), a structural feature that helps impose promoter directionality (11). After a ∼6–12-nucleotide nascent transcript is synthesized, TFIIB is ejected from PIC, and pol II escapes from the promoter (12–14).
Figure 1.

Structure of human TFIIBc in two new conformational states. A, schematic domain organization of human TFIIB, with individual domains and regions labeled. B, structure of the two conformations of human TFIIBc found in the asymmetric unit of the crystal. Domains are color-coded according to A.
TFIIB and TBP are the targets of the acidic activation domain of herpes simplex virus VP16 (VP16AD), which stimulates transcription by interacting with multiple transcription co-activator complexes, histone-modifying complexes, and chromatin remodeling complexes (15). One mechanism of transcriptional activation suggests that VP16AD displaces the intramolecular interaction between the N-terminal region of TFIIB (TFIIBn) and the cyclin-like domain-containing core region (TFIIBc), converting TFIIB to an active conformation for PIC assembly (16–21).
TFIIB interacts both genetically and physically with the termination factor Ssu72 (suppressor of sua7, gene 2) (22–24). Ssu72 dephosphorylates serine 5 and serine 7 in the C-terminal domain (CTD) of the pol II subunit Rpb1 during transcription termination, allowing dephosphorylated pol II to reinitiate for another round of transcription (25–27). Ssu72 binds to the N-terminal region of an RNA-processing protein, symplekin, which stimulates the phosphatase activity of Ssu72 (28). Both Ssu72 and symplekin are involved in pre-mRNA 3′-end cleavage and polyadenylation in yeast and human (24, 29).
Transcription-coupled functional cross-talk between promoters and terminators has long been noted. The general transcription factor TFIID, consisting of TBP and TBP-associated factors, was shown to recruit cleavage and polyadenylation specificity factor CPSF, to which both Ssu72 and symplekin are also bound on terminators (30, 31). In addition to Ssu72, TFIIB also interacts with the CstF64 (homolog of Rna15 in yeast) component of the cleavage stimulation factor (CstF). In particular, TFIIB phosphorylation on serine 65 is crucial for this interaction and for CstF occupancy on promoters as well (32, 33). Furthermore, both TFIIB and the general transcription factor TFIIF were found to be associated with and modulate the enzymatic activity of another pol II CTD phosphatase, Fcp1, which is involved in dephosphorylation of serine 2 on CTD (34, 35). Deletion of the transcription termination factor Pcf11 depletes TFIIB and TBP from promoters (36). Notably, a point mutation of the polyadenylation site causes defects in transcription initiation (36).
These functional connections between promoters and terminators may be understood in the framework of chromatin looping. Chromatin looping is a phenomenon in which distal genomic elements are brought within close spatial proximity of each other to influence transcription. Looping occurs between enhancer elements and promoters, intergenic regions and promoters, and promoters and terminators. Promoter-terminator gene looping was first discovered in Saccharomyces cerevisiae and was later shown to be present in other organisms, such as human and plants, as well as in mitochondria and HIV (37–44). Promoter-terminator gene looping is believed to facilitate pol II recycling during transcription reinitiation (45, 46). It is also involved in sustaining transcriptional memory and enhancing transcription directionality (47–49).
TFIIB and Ssu72 occupy both promoters and terminators and are required for gene loop formation (49, 50). Mutation of TFIIB or Ssu72 abolishes gene looping in yeast, as demonstrated by chromatin conformation capture (3C) assays (49, 50). Loss of Ssu72 leads to defects in TFIIB recruitment to promoters and elimination of TFIIB from terminators (50) (data not shown). To begin to reveal the mechanism of transcription-dependent promoter-terminator cross-talk that may be achieved at least in part through the interaction between TFIIB and Ssu72, we first conducted structural analysis of the core domain of human TFIIB captured in two new conformations. We next carried out systematic, structure-guided mapping of the binding interface of the TFIIB-Ssu72 complex using a series of in vitro binding assays and photocross-linking analysis based on site-specifically incorporated unnatural amino acids. We also generated a knowledge-based structural model for the TFIIB-Ssu72 complex. We propose a handoff mechanism to explain the role of the TFIIB-Ssu72 interaction in facilitating pol II reinitiation.
Results
The core domain of TFIIB displays a large degree of conformational plasticity
We solved the crystal structure of human apo-TFIIBc to 3.4 Å resolution by molecular replacement, with four molecules per asymmetric unit (Table 1 and Fig. S1, labeled as Molecules A–D). TFIIBc forms two distinct, previously uncharacterized conformations (Fig. 1B). Molecules A and B are found in one conformation, and C and D are in the other conformation; for simplicity, we will only discuss molecules A and D further (Fig. 1B). Notably, each conformation is characterized by prominent translational and rotational movement of the two cyclin-like domains along the flexible C-linker between them (Fig. 1, A and B). Whereas the secondary structure of each individual cyclin-like domain remains the same, their orientation and distance with respect to each other differ (Fig. S2, A and B).
Table 1.
Data collection and refinement statistics (molecular replacement)
Data were collected from a single crystal.
| Crystal | TFIIBc |
|---|---|
| Data collection | |
| Space group | P212121 |
| Cell dimensions | |
| a, b, c (Å) | 108.2, 126.5, 158.4 |
| α, β, γ (degrees) | 90, 90, 90 |
| Resolution (Å) | 50.00–3.40 (3.46–3.40)a |
| Rmerge | 0.238 (0.00)b |
| Rpim | 0.144 (0.960) |
| I/σI | 9.15 (1.20) |
| Completeness (%) | 100.0 (100.0) |
| Redundancy | 7.2 (7.2) |
| CC1/2 | (0.546) |
| Refinement | |
| Resolution (Å) | 50.00–3.40 (3.46–3.40) |
| No. of reflections | 30,767 |
| Rwork/Rfree | 0.194/0.214 |
| No. of atoms | 6640 |
| Protein | 6510 |
| Sulfate | 130 |
| Water | 0 |
| Average B-factors (Å2) | |
| Protein | 103.28 |
| Sulfate | 155.14 |
| Water | 0 |
| Root mean square deviations | |
| Bond lengths (Å) | 0.010 |
| Bond angles (degrees) | 1.24 |
| Ramachandran plot | |
| Favored | 95.4 |
| Allowed | 3.5 |
| Outliers | 1.1 |
a Values in parentheses are for highest-resolution shell.
b HKL2000 does not include values >1.
Our structure reveals an unprecedented degree of plasticity of TFIIBc, although we also note that the conformations of TFIIBc from our structure, like all crystal structures, are partially influenced by crystal packing. Although we cannot say for certain the extent to which these conformations occur in vivo, in the following, we interpret how this plasticity would influence the biological roles of TFIIB. Two separate conserved regions that would contact the TATA box (e.g. residues Arg189 and Lys193 on helix H5), the BREu (e.g. residues Ala281, Val283, Arg286, and Arg290 on helix H5′), and BREd (e.g. residues 152–154 between helices H2 and H3) are solvent-accessible in both molecules A and D (Fig. 2A). However, they are not spatially aligned and hence in an inactive conformation compared with their counterparts in TFIIBc bound to TBP and the TATA box, where the two cyclin-like domains are orientated to allow simultaneous binding of helix H5 to the TATA box, the C-linker to TBP, helix H5′ to the BREu, and the loop between H2 and H3 to the BREd (Fig. 2B and Fig. S3A) (8). Interestingly, apo-TFIIBc from Trypanosoma brucei follows essentially the same conformation as human TFIIBc in the active ternary complex, with the exception that the C-linker is replaced by a rigid bent helix (51), implying that T. brucei TFIIB may always assume an active conformation regardless of whether TBP and the TATA box are bound (Fig. 2C and Fig. S3B).
Figure 2.

Sequence alignment and structural comparison of TFIIBc. A, sequence alignment of TFIIBc from Homo sapiens (Hs), S. cerevisiae (Sc), D. melanogaster (Dm), and T. brucei (Tb). Secondary structural domain organization is labeled based on the human TFIIBc structures determined in this report. Residues mutated in experiments are labeled by blue triangles. B, alignment of TFIIBc molecule D (color-coded according to Fig. 1A) with human TFIIBc from the TFIIBc-TBPc-DNA structure (PDB code 1C9B, TFIIBc in gray schematic, DNA in multicolor schematic, and TBPc omitted). Cyclin-like domain 1 of molecule D was aligned with that of TFIIBc from 1C9B. In the subsequent panels, alignment follows the same scheme, except that the second TFIIB molecule was first aligned with TFIIBc from 1C9B to allow for modeling of the DNA. C, alignment of molecule D with TFIIBc from T. brucei (PDB code 3H4C; magenta). D, alignment of molecule D with human TFIIBc determined by NMR (PDB code 1TFB; cyan). The same TATA box DNA molecule from B is shown in C and D as well to guide the view. The purpose of the structural alignment in B, C, and D is to illustrate the conformational plasticity but not the actual new conformations of TFIIBc. E, electromobility shift assay with E4 promoter DNA, TBPc, and TFIIB variants.
Molecules A and D are also distinct from the inactive conformation of a human apo-TFIIBc determined previously by NMR (Fig. 2D and Fig. S3C) (20, 52). In the latter case, the two cyclin-like domains are stably packed against each other such that about 1900 Å2 of solvent-accessible surface area is buried. In stark contrast, the same two domains in the current structures barely contact one another, displaying an extraordinary rotational and translational plasticity that is probably subjected to cellular regulation. In line with this proposal, residues Arg286 and Arg290 from helix H5′ of the second cyclin-like domain change conformation when interacting with the archetypal acidic activator VP16 (20, 53). Thus, it appears possible that TFIIBc can assume multiple flexible, inactive conformations.
The C-linker of TFIIB is involved in ternary complex formation (7, 8). Interestingly, this region varies in length between species (Fig. 2A). To test whether the relative position of the two cyclin-like domains in TFIIBc is controlled by the C-linker, we appended a GSGS linker directly C-terminal of residue Leu208 located in the middle of the C-linker. This TFIIBc variant was found to bind TBP and the TATA box DNA to the same extent as wild-type protein (Fig. 2E, WT and GSGS-C), which agrees with the evolutionary divergence of TFIIB, in that S. cerevisiae TFIIB has an extended C-linker beyond residue Leu208 compared with human and fly TFIIB but still retains the ability to bind TBP and the TATA box (Fig. 2A). In contrast, the precise arrangement of the first part of the C-linker is critical for formation of the TFIIBc-TBP-TATA ternary complex (8), and replacing residues 200–208 with a GSG linker or adding a GSGS linker directly N-terminal of residue Lys200 significantly decreased formation of the TFIIBc-TBP-TATA box ternary complex (Fig. 2E, GSGS-C and GSGS-N). Overall, our results indicate that the semiconservative C-linker is not the only determinant of the active TFIIBc conformation.
Ssu72 binds to the first cyclin-like domain of TFIIBc
Previous studies have implicated a direct physical interaction between TFIIB and Ssu72 (23, 24). To understand the molecular basis of this interaction, we first measured the binding affinity between TFIIBc and Ssu72 by isothermal titration calorimetry (ITC), which was estimated to be around 20.2 μm (Fig. 3A). Next, we conducted pulldown assays of C-terminal FLAG-tagged human TFIIB with His6-tagged Ssu72. Human TFIIB constructs used for these assays included full-length TFIIB (TFIIBFL), TFIIBc (residues 107–316), and the isolated cyclin-like domain 1 (TFIIBc1; residues 107–206) and 2 (TFIIBc2; residues 207–316). Whereas TFIIBFL, TFIIBc, and TFIIBc1 all bound to Ssu72, TFIIBc2 could not do so (Fig. 3B), indicating that the first cyclin-like domain of TFIIBc serves as the minimal region of TFIIB for Ssu72 binding. TFIIBc2 is required for the full Ssu72-binding activity of TFIIB; in its absence, the binding of TFIIBc1 to Ssu72 became too weak to be detected by ITC (Fig. S4). Interaction of TFIIBc with Ssu72 did not appear stoichiometric in the pulldown analysis, probably due to the relatively weak interaction, as indicated by the ITC results. However, the interaction is specific, as is detailed in the studies that follow.
Figure 3.

Physical interaction of TFIIBc and Ssu72. A, ITC experiment of Ssu72 and TFIIBc. The best fit Kd value (95% confidence interval) is 20.2 (16.8–24.4) μm. B, FLAG pulldown assay with TFIIB-FLAG variants and His6-Ssu72, as visualized by Coomassie staining and Western blotting with a His6 antibody. *, IgG light chain that leeched from the resin upon elution with glycine. C, FLAG pulldown assay with TFIIBc-FLAG mutants and His6-Ssu72. D, FLAG pulldown assay with TFIIBc-FLAG WT and mutants from various species (top labels) with His6-Ssu72 from the corresponding species as visualized by Western blotting with a His6 antibody. In the figure, h represents human, d is D. melanogaster, and y is S. cerevisiae. Lanes labeled with FLAG indicate controls without TFIIBc added.
Charge reversal mutation of any pair of the positively charged residues Arg185, Lys189, Arg193, and Lys200 from helix H5 or the linker between helices H4 and H5 in TFIIBc1 decreased binding to Ssu72 compared with wild-type TFIIBc, and the K189E/R193E mutation essentially abolished the interaction (Fig. 3C). All TFIIBc variants were monomeric by gel filtration chromatography analysis, indicating that the mutations did not significantly perturb protein structure (Fig. S5). Thus, a positively charged surface of TFIIBc1 proves to be necessary for Ssu72 binding. Intriguingly, an overlapping set of residues from this region of TFIIBc1 is also implicated in promoter DNA and acidic activator binding (8, 20).
In addition, we tested whether the binding interface between TFIIBc and Ssu72 was conserved across species using purified proteins from S. cerevisiae (yTFIIBc and ySsu72) and Drosophila melanogaster (dTFIIBc and dSsu72). In both cases, TFIIB and Ssu72 were found to interact directly (Fig. 3D). Notably, whereas critical interacting residues Lys189 and Arg193 of human TFIIB are fairly conserved in yeast (as Lys201 and Lys205, respectively; Fig. 2A), residues Arg185 and Lys200 are not conserved (His197 and Asn212 in yeast; Fig. 2A). In the pulldown assays, the conserved K201E/K205E mutation of yTFIIB abolished the interaction with ySsu72 in a manner similar to the human mutation. All yTFIIBc mutants were monomeric by gel filtration analysis, indicating that mutation did not disrupt normal protein structure (Fig. S5). Surprisingly, the H197E/N212E mutation of yTFIIB also significantly reduced binding to ySsu72 (Fig. 3D), suggesting that the mode of interaction between TFIIB and Ssu72 is highly conserved from yeast to humans despite differences in protein sequence.
TFIIBc interacts with an Ssu72 surface formed by three discrete sequence patches
To probe Ssu72 residues responsible for TFIIBc binding, we used a site-specific photocross-linking approach based on incorporation of the unnatural amino acid p-benzoyl-l-phenylalanine (pBpa) via suppression of an amber stop codon (54, 55). Under UV illumination, pBpa can cross-link to residues on a binding protein within 2.4–4 Å, allowing for a clear readout of protein-protein interactions (56). Based on analysis of the human Ssu72-symplekin structure (28), several residues of Ssu72 with surface-exposed side chains were chosen for substitution. A cluster of negatively charged residues (positions 171–181) were chosen under the assumption that they may interact with positively charged residues of TFIIBc1 important to interaction (Fig. 3C). Residues around the interface with symplekin (28) were also chosen under the hypothesis that they may form a similar binding interface with TFIIB.
Ssu72 surface residues that were substituted for pBpa included Glu127, Gln128, Thr130, Cys131, Pro133, Asp169, Glu173, Leu188, Phe193, and Tyr194. Strong UV-dependent cross-linking to TFIIB was observed for all of the pBpa substitutions except E127pBpa, L188pBpa, and Y194pBpa (Fig. 4A). In agreement with the results from the pulldown assays, no cross-linking was observed with the K189E/R193E mutant of TFIIBc that loses the Ssu72-binding capacity (Fig. 4B). The photocross-linkable substitutions are clustered on an Ssu72 surface on the back of its substrate-binding site, outlining an elongated TFIIB binding surface (Fig. 4C, green dashed outline).
Figure 4.

Photocross-linking–based structural mapping of TFIIBc binding interface on Ssu72. A, cross-linking assays with TFIIBc-FLAG and Ssu72-pBpA substituted mutants, visualized by Western blotting with FLAG and His6 antibodies. B, cross-linking of TFIIBc-FLAG mutant (Mut) with Ssu72-pBpA variants, visualized by His6 antibody Western blotting. C, modeling of TFIIB-interacting residues determined by pBpA cross-linking onto the structure of human Ssu72 (from the Ssu72-symplekin structure, PDB code 3O2Q). The TFIIB-interacting surface is labeled by green outline, and residues essential for symplekin binding are labeled by blue outline. D, cross-linking assays of TFIIBc-FLAG and Ssu72-pBpA substituted mutants from the negatively charged helix region as visualized by Western blotting with FLAG and His6 antibodies. E, cross-species cross-linking of S. cerevisiae TFIIBc (y) with human Ssu72 (h) visualized by Western blotting. The topmost bands in the anti-FLAG blot are bands that are present regardless of UV illumination (Nonspecific). F, sequence alignment of Ssu72 from human (Hs), fly (Dm), and yeast (Sc). Green plus signs indicate pBpA-substituted residues in humans that cross-link to TFIIBc, whereas yellow minus signs are residues that do not cross-link to TFIIB. Orange triangles, residues critical for symplekin interaction.
Residues 128–133 of Ssu72 form a loop that is partially buried in the Ssu72-symplekin complex (28) (Fig. 4C and Fig. S5). Residue Asp169 sticks out of a helix opposite to this loop and approaches residue Pro133. Residue Glu173 is further down in the same helix and both Asp169 and Glu173 are a part of a negatively charged, surface-exposed patch that also includes residues Glu171, Glu174, and Glu181 (Fig. 4C, green sticks). When any of these residues are mutated to pBpa, cross-linking to TFIIBc was observed (Fig. 4D). Residue Phe193 was known to be critical for symplekin binding, and it now appears to contribute to the TFIIB-binding surface as well (Fig. 4C and Fig. S6) (28). Residues Glu127 and Leu188 are located close to but outside of the TFIIB-binding surface, and residue Tyr194 is partially buried during Ssu72 folding (Fig. 4C, yellow sticks). These facts not only account for the negative photocross-linking results upon UV illumination for the corresponding pBpa substitutions but also support the remarkable specificity of photocross-linking offered by pBpa. Thus, three discrete and spatially proximate patches (i.e. regions centered on residues 128–133, residues 169–181, and residue 193) of Ssu72 form the binding surface for TFIIB (Fig. 4C).
To support the conservation of Ssu72-TFIIB binding, we further showed that the pBpa substitutions of human Ssu72 within the three TFIIB binding patches cross-linked to yeast TFIIBc. The cross-linking was abolished when the K201E/K205E mutant of yeast TFIIBc was attempted (Fig. 4E), indicating that the binding mode between TFIIB and Ssu72 is highly conserved across species, although one of the three patches exhibits poor sequence conservation (Fig. 4F).
TFIIB and symplekin bind to overlapped Ssu72 surfaces
Ssu72 is known to form a stable complex with the N-terminal region (residues 30–360) of symplekin (referred to below as Symp-N) (28). In our pulldown assays, TFIIB was unable to interact with Ssu72 in the presence of Symp-N, indicating that the binding sites for TFIIB and Symp-N overlap on the Ssu72 surface (Fig. 5A). To further delineate the competitive binding of TFIIB and Symp-N to Ssu72, we utilized a competition photocross-linking assay. Ssu72 with the pBpa substitutions were prebound with an excess of either TFIIBc or Symp-N, incubated with the competitor protein, and UV-cross-linked. Symp-N strongly cross-linked to Ssu72 C131pBpa (Fig. 5B, lane 2). When cross-linked in competition with TFIIBc, only the cross-linked Ssu72–Symp-N but not Ssu72-TFIIB complex was visible (Fig. 5B, lane 6), which indicates that Symp-N outcompetes TFIIB in Ssu72 binding, probably due to a higher binding affinity (Fig. 5A). Notably, Ssu72 F193pBpa cross-linked to Symp-N in a much weaker manner compared with Ssu72 C131pBpa (Fig. 5B, compare lanes 2 and 8), possibly because the former disturbs Symp-N binding (28). The weaker interaction between Ssu72 F193pBpa and Symp-N allowed TFIIBc to cross-link to Ssu72 to a similar degree in both the presence and absence of an excess of Symp-N (Fig. 5B, compare lanes 10 and 12). In a similar experiment, the K185A mutant of Symp-N that weakens Ssu72 binding also allowed photocross-linking between Ssu72 and TFIIBc (Fig. 5C, lane 6) (28). Overall, our results indicate that Ssu72 contains an overlapping yet not identical binding surface for TFIIBc and symplekin.
Figure 5.
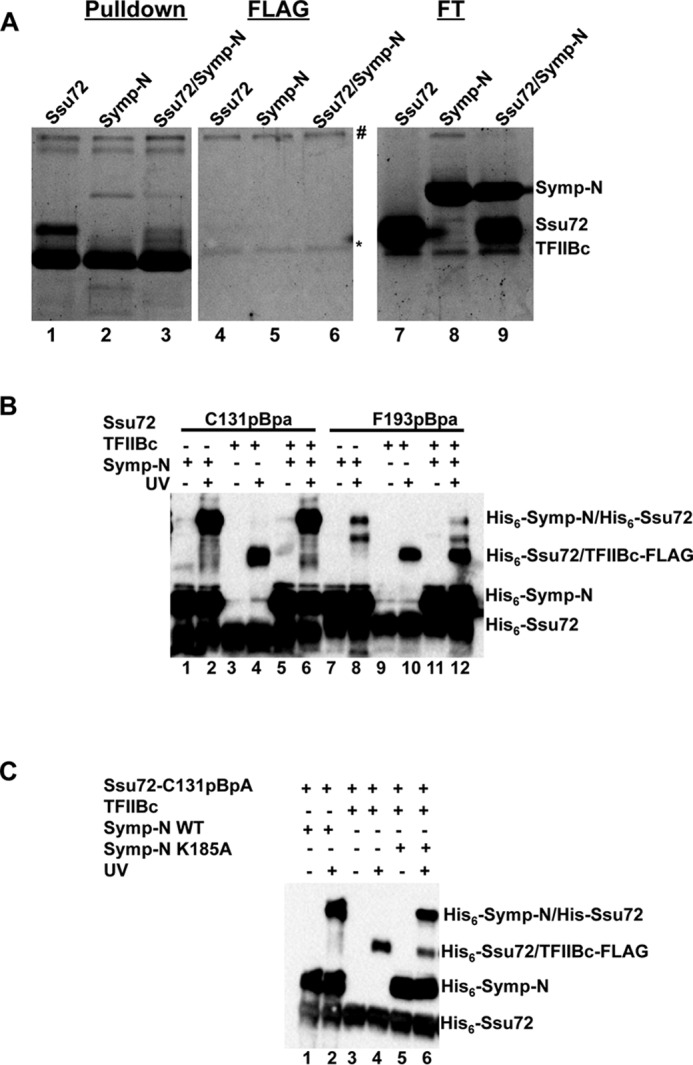
Competition of TFIIBc and symplekin for Ssu72 binding. A, competition pulldown with TFIIBc, Ssu72, and the N-terminal fragment of symplekin (Symp-N) as visualized by Coomassie staining. Lanes labeled FLAG contained only FLAG peptide as a control. Lanes labeled TFIIBc FT depict the flow-through proteins of the experimental samples that fail to bind to the resin. #, free IgG heavy chain; *, free IgG light chain that leeched from the resin during glycine elution. B, competition cross-linking between TFIIBc and symplekin for Ssu72 pBpA mutants as visualized by His6 antibody Western blotting. C, competition cross-linking assay with symplekin K185A mutant.
Although binding to an overlapped Ssu72 surface, unlike symplekin, TFIIB did not enhance the intrinsic phosphatase activity of Ssu72. In addition, TFIIB did not dramatically influence the stimulation of Ssu72 catalysis by symplekin, even in large excess (Fig. S7), confirming that TFIIB cannot outcompete symplekin for Ssu72 binding, at least at the concentration used in our assays.
Ssu72-binding site on TFIIBc overlaps with the VP16AD- and TFIIBn-binding site
Two additional interactions mediated by TFIIBc were noted to influence transcription initiation, including VP16AD acidic activator binding and TFIIBn binding, in the context of a TFIIB intramolecular complex. Because Ssu72 binds to a similar region of TFIIBc as VP16AD and TFIIBn, we further investigated such functional cross-talk with the photocross-linking competition assays.
We first established that TFIIBc could be photocross-linked to an N-terminal SUMO fusion of TFIIBn (residues 1–106) with a pBpa substitution at residue Trp52 located in the finger/reader region (Fig. 6A, compare lanes 1 and 2). In the competition assays, we next observed that unlabeled Ssu72 decreased TFIIBc-TFIIBn cross-linking compared with the GST control, probably due to competitive binding of Ssu72 and TFIIBn to TFIIBc (Fig. 6A, compare lanes 3 and 4 with lanes 6 and 7). In a reciprocal competition cross-linking experiment, GST-tagged TFIIBn could barely disrupt TFIIBc-Ssu72 cross-linking compared with the GST control (Fig. 6B, compare lanes 4 and 6 with lane 12), indicating that TFIIBn displays a comparable or a lower binding affinity toward TFIIBc when compared with Ssu72. Conversely, GST-tagged VP16AD efficiently prevented cross-linking between Ssu72 and TFIIBc (Fig. 6B, compare lanes 8 and 10 with lanes 4 and 6). Accordingly, we conclude that Ssu72 may open up the inhibitory TFIIBc-TFIIBn intramolecular complex to facilitate pol II transcription with a similar mechanism used by VP16AD (16–21).
Figure 6.
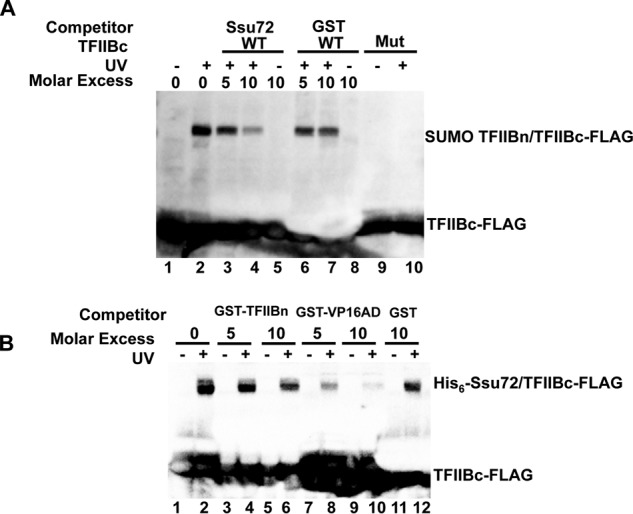
Disruption of TFIIB autoinhibition by Ssu72. A, competition cross-linking between SUMO TFIIBn W52pBpA and Ssu72 for TFIIBc WT visualized by anti-FLAG Western blotting. Lanes labeled Mut utilized the TFIIBc K189E/R193E mutant. Bottom, TFIIBc-FLAG becomes distorted in some lanes due to GST that runs in a position similar to that of TFIIBc. B, competition cross-linking assay between Ssu72 C131pBpA and either GST-TFIIBn or GST-VP16AD for TFIIBc as visualized by FLAG antibody Western blotting.
A computational model predicts the interactions of TFIIBc and Ssu72
To further elucidate how the residues identified by our experiments interact on the TFIIBc-Ssu72 interface, we devised a computational approach combining molecular dynamics (MD) simulations and knowledge-based protein-protein docking. Our approach successfully generated an interface model that agrees with all experimental observations. This model suggests a mechanism whereby Ssu72 activates TFIIB for reinitiation. Moreover, the model includes additional, hypothetical TFIIBc and Ssu72 interactions that may represent secondary binding interfaces.
We first performed MD simulations of TFIIBc and Ssu72 separately. We observed that whereas Ssu72 is a rigid protein, TFIIBc is highly flexible (Fig. S8), in line with our structural observations analyzed above (Fig. 1B). Therefore, we extracted TFIIBc structures from the MD simulations that may potentially bind to Ssu72 (example in Fig. S9) and then performed protein-protein docking to generate the binding models. Models with low HADDOCK score (less negative) and experimental violation were disregarded, resulting in a final high-quality model that best fits the experimental results.
In this model, the cross-linked residues on Ssu72 are very close to TFIIBc1. A 60-ns MD simulation of this model revealed that the average D2 distance of all cross-linked residues is well below the threshold value (Fig. 7A), indicating that this binding interface is rather stable. Consistent with the mutagenesis experiment on Lys200, Arg189, and Arg193 of TFIIBc, four salt bridges are found on the interface: Lys200-Glu121, Lys200-Asp117, Lys189-Glu167, Arg193-Glu169, with the TFIIBc residues underlined (Fig. 7B, bottom). These salt bridges are mostly mediated by TFIIBc1, consistent with its dominant role in Ssu72 binding as shown by our pulldown assay (Fig. 3B). Additional interactions include the salt bridges Lys152-Glu127, Lys188-Asp173, Arg290-Glu181, Lys272-Glu174, and Arg286-Glu178. The last three interactions involve residues of TFIIBc2. Probably, these interactions are weaker compared with those of TFIIBc1 because TFIIBc2 is not sufficient to pull down Ssu72 (Fig. 3B), and mutation of TFIIBc1 abolishes interaction with Ssu72 (Figs. 3C and 4B).
Figure 7.
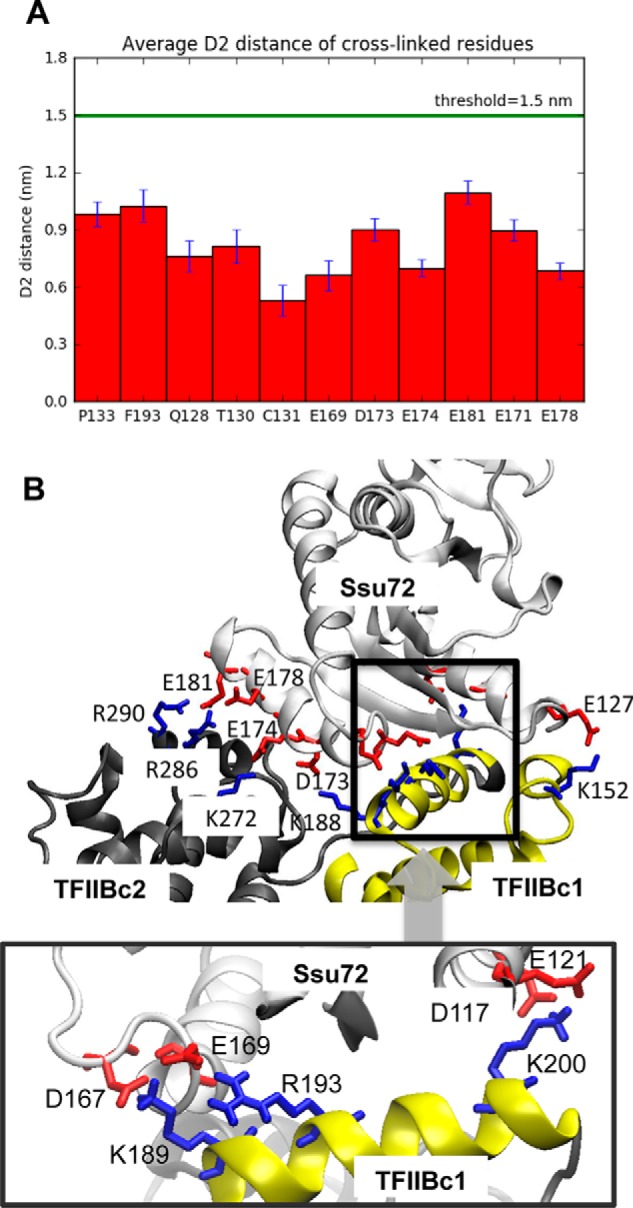
Structural modeling of a TFIIBc-Ssu72 complex. A, average D2 distance of all cross-linked residues on Ssu72 in the last 60-ns MD simulation of the TFIIBc-Ssu72 complex are shown as a bar graph. The threshold (15 Å) is shown as a green line. Error bars, S.D. B, model of the TFIIB-Ssu72 complex. Ssu72 is shown in gray, TFIIBc1 is yellow, and TFIIBc2 is black. Red or blue sticks represent negatively or positively charged residues, respectively. The inset shows a closer view of salt bridge interactions of TFIIBc1 residues Lys189, Arg193, and Lys200 with Ssu72 residues Asp167, Glu169, Asp117 and Glu121 of Ssu72.
Discussion
Whereas the role of TFIIB in transcription initiation is well established, its role in other processes such as gene looping is not clear. The current study sheds light on possible new roles of TFIIB in this process by revealing a structure of human TFIIBc that adopted two new conformations. We also examined interactions between TFIIBc and Ssu72 and other factors known to interact with these two proteins, such as VP16AD and symplekin, respectively.
The structure of human TFIIBc displays the dynamic nature of the protein and indicates that conformational plasticity of TFIIB may underlie a mechanism for functional control. Apo-TFIIB is in an autoinhibited, “closed” conformation where TFIIBn blocks the TBP and the TATA DNA-binding region of TFIIBc (16–21) (Fig. 8, “Closed” Autoinhibited state). VP16AD displaces TFIIBn by interacting with an overlapping surface on TFIIBc, promoting the structural conversion of TFIIB to an “open” conformation (20) that is then stabilized by TBP, TATA DNA, and other components of the transcription machinery during initiation. Both conformations of TFIIBc revealed by our study are essentially transcriptionally inactive because the two cyclin-like domains and C-linker are not aligned to interact with TBP and the TATA DNA due to intrinsic conformational flexibility, although structural elements for these functional interactions are all surface-exposed. The structural transition from an inactive conformation to an active conformation may be subjected to additional cellular regulation.
Figure 8.
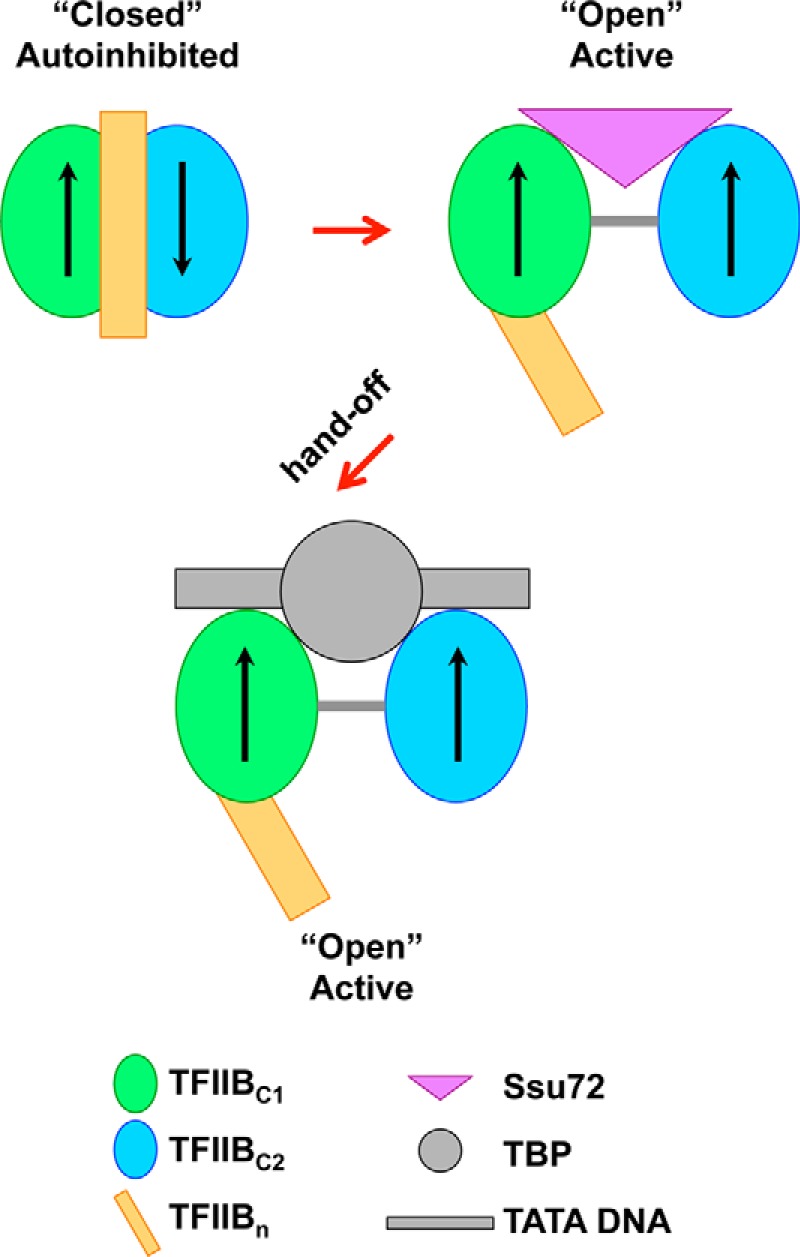
A schematic model that illustrates a possible role of the TFIIBc-Ssu72 complex in pol II transcription reinitiation. A “hand-off” model may explain the conceivable role of Ssu72 in pol II reinitiation by modulating TFIIB conformation, in a manner similar to the archetypal acidic activator VP16. Arrows, relative orientations of the cyclin domains of TFIIBc. Ssu72 may further maintain the relative orientation of the two cyclin-like domains in an active conformation.
We showed that Ssu72 predominantly interacts with the TATA-binding region of cyclin-like domain 1 of TFIIBc (TFIIBc1) and that residues Arg189 and Arg193 are critical for binding (Fig. 3C). This region is also a binding site of VP16AD, suggesting that Ssu72 could function to transition TFIIB from a closed to an open conformation in a similar manner (Fig. 8). Indeed, similar to VP16AD, Ssu72 can easily outcompete the binding of TFIIBc to TFIIBn, indicating that it can at least relieve TFIIB from autoinhibition (Fig. 8). In addition, our computational study provides a possibility that Ssu72 may further form a stable complex with the active form of TFIIBc (Fig. 8, “Open” Active state). The open active state of TFIIBc maintained by Ssu72 may facilitate formation of the TFIIB-TBP-TATA ternary complex through a “hand-off” model. This process may occur during transcription reinitiation in the context of a gene loop, through a reinitiation scaffold complex on a TATA promoter that contains TBP but not TFIIB (14). The hand-off model in transcriptional regulation has also been proposed previously for transcriptional activators (57).
Interaction between TFIIB and Ssu72 is well conserved from yeast to flies and humans (Fig. 3D), suggesting a likely functional conservation. TFIIB binds to Ssu72 over an extended surface that includes a negatively charged helix, a loop, and a region near the C terminus of the protein. The latter two TFIIB-binding regions overlap with the binding surface of symplekin, which precludes TFIIB from binding to Ssu72. The Ssu72-symplekin interaction is strong, but it can be disrupted by a point mutation of Phe193 of Ssu72 that does not impede TFIIB binding. Interestingly, residue Phe193 is close to several conserved serine, threonine, and tyrosine residues. Although we have not found a known phosphorylation motif in this area, it is possible that phosphorylation or other posttranslational modification of the C terminus of Ssu72 could be used to weaken the interaction with symplekin such that TFIIB binding is allowed.
The fact that TFIIB and Ssu72 physically interact yet are involved in disparate steps of transcription, initiation and termination, respectively, is intriguing. In the context of promoter-terminator gene looping, one can imagine a mechanism in which Ssu72 dephosphorylates the CTD of pol II during transcription termination, recruits TFIIB, and keeps it in an open conformation to allow for reassembly of PIC on the promoter for subsequent rounds of transcription. This hypothetical mechanism would explain why both TFIIB and Ssu72 are required for gene looping, and it can be further tested with separation-of-function mutants in future studies.
Materials and methods
Protein expression and purification
All proteins were produced recombinantly in Escherichia coli. DNA sequences for each protein were cloned into a pET28a vector (containing an N-terminal His6 tag), a SUMO-pET28a vector (containing a His6 SUMO tag), or pGEX4T1 (containing an N-terminal GST tag). All mutations were introduced by site-directed mutagenesis of the parental plasmid. Expression plasmids were transformed into BL21*(DE3)-Rosetta II cells and cultured in LB medium supplemented with 50 μg/ml chloramphenicol and either 50 μg/ml kanamycin or 100 μg/ml ampicillin and cultured at 37 °C until reaching an A600 of 0.6, at which point a final concentration of 0.5 mm isopropyl 1-thio-β-d-galactopyranoside was added, and cells were incubated overnight at 16 °C.
All TFIIB proteins, with the exception of GST-TFIIBn, were purified by nickel chromatography and contained a His6-SUMO tag and C-terminal FLAG tag. Cell pellets were resuspended in lysis buffer (20 mm HEPES, pH 8.0, 500 mm NaCl, 10 mm imidazole, and 2 mm β-mercaptoethanol) and lysed by sonication. Following centrifugation, the cleared lysate was applied to HisPure nickel resin (Thermo Scientific), the resin was washed with 20 column volumes of lysis buffer, and proteins were eluted with elution buffer (20 mm HEPES, pH 8.0, 500 mm NaCl, 300 mm imidazole, and 2 mm β-mercaptoethanol). Proteins that contained the N terminus of TFIIB (i.e. TFIIBFL and TFIIBn) additionally contained 10 μm zinc acetate in all buffers. The His6-SUMO tag was removed by Ulp1 cleavage overnight during dialysis into low imidazole buffer (20 mm HEPES, pH 8.0, 300 mm NaCl, 10 mm imidazole, and 2 mm β-mercaptoethanol). Proteins were then reapplied to a new nickel column to remove Ulp1 and contaminants, and the flow-through was collected. Proteins were dialyzed into a low-salt buffer (HEPES, pH 7.0 or 8.0, 100 mm NaCl, 10 mm imidazole, and 2 mm β-mercaptoethanol) and then applied to either a HiTrap heparin column (human TFIIBFL wild type and mutants) or a MonoS 5/50 column (human TFIIBc, TFIIB cyclin-like 1, and TBPc) and eluted with a gradient of NaCl (both columns were from GE Healthcare). The crystallized human TFIIBc that lacked a C-terminal FLAG tag was concentrated following the MonoS 5/50 column and then loaded onto a Superdex 200 10/300 GL column (GE Healthcare) in 20 mm HEPES, pH 8.0, 150 mm NaCl, and 1 mm DTT. Because TFIIB cyclin-like 2 and TFIIBc mutants (human K189E/K200E, R185E/R193E, R185E/K200E, K189E/R193E; yeast H197E/N212E and K201E/K205E) failed to bind to heparin, they were passed through a MonoQ 5/50 column (GE Healthcare) to remove contaminants, and the flow-through was collected.
All Ssu72 and N-terminal symplekin (residues 30–360) proteins contained an N-terminal His6 tag and were purified by nickel chromatography followed by dialysis into a low-salt buffer (HEPES, pH 8.0, 100 mm NaCl, and 2 mm β-mercaptoethanol) and a final MonoQ purification step. To assemble the Ssu72-symplekin complex, Ssu72 was mixed with symplekin in a 2:1 molar ratio, incubated on ice for 1 h, and then separated on a Superdex 200 10/300 GL column in 20 mm Tris, pH 8.5, 200 mm NaCl, and 10 mm DTT. Fractions containing the complex were pooled, concentrated, and stored at −80 °C until use.
Cells containing GST fusion proteins (TFIIBn and VP16AD) were lysed by sonication in GST lysis buffer (50 mm Tris, pH 8.0, 150 mm NaCl, and 5 mm DTT), and clarified lysate was bound to glutathione-agarose (Thermo Scientific). The resin was washed with 20 column volumes of GST lysis buffer and eluted with lysis buffer containing 20 mm reduced glutathione. The proteins were then applied to a MonoQ 5/50 column and eluted over a gradient of NaCl. Following purification, all proteins were concentrated, flash-frozen in liquid nitrogen, and stored at −80 °C until needed.
For Ssu72 proteins containing pBpA mutations, the Ssu72 plasmid containing the amber mutation of interest was transformed into BL21* (DE3) cells containing the pEVOL-pBpA plasmid that encodes for an evolved aminoacyl tRNA synthetase and tRNA that incorporate pBpA into the amber suppression site (55). Cells were cultured at 37 °C in 100 ml of 2XYT medium supplemented with 50 μg/ml chloramphenicol and 50 μg/ml kanamycin. At an A600 of 0.3, a final concentration of 0.2% arabinose was added to the culture. At an A600 of 0.6, a final concentration of 1 mm isopropyl 1-thio-β-d-galactopyranoside and 1 mm pBpA were added to the culture, and the culture was then allowed to incubate overnight at 30 °C. Proteins were purified in a manner similar to that described for Ssu72 by nickel and Q Sepharose Fast Flow (GE Healthcare) chromatography. SUMO-TFIIBn-W52pBpA, containing an N-terminal His6 tag, was expressed in the same manner as Ssu72-pBpA proteins. It was purified by nickel chromatography in a manner similar to other His-tagged proteins, with the addition of 10 μm zinc acetate in all purification buffers.
Protein crystallization, data collection, and structure solution and refinement
Human TFIIBc was crystallized at a protein concentration of 7 mg/ml in the hanging drop format, over a well solution containing 100 mm sodium acetate trihydrate, pH 4.5, and 2 m ammonium sulfate. Sucrose at a concentration of 3% (w/v) was used as an additive to improve crystal quality. Crystals were cryoprotected in mother solution supplemented with a final concentration of 30% glycerol and plunged into liquid nitrogen. Diffraction data were collected at beamline 191D of the Advanced Photon Source at Argonne National Laboratory. Data were collected over a total of 180° with oscillation of 1° and an exposure time of 2 s/degree. Diffraction data were processed with HKL2000 (58), and TFIIBc was found to be in space group P212121 (Rmeas for the lowest-resolution shell (50.00–9.21) is 6.0%). The structure of TFIIBc was solved by using the separate cyclin-like 1 and cyclin-like 2 domains from the TFIIBc-TBPc-DNA structure (8) using MOLREP (59). Manual building of connecting loops was completed with Coot (60), followed by refinement with PHENIX (61). Figures were created with PyMOL (PyMOL Molecular Graphics System, Version 1.8.6.2., Schroedinger, LLC, New York).
Isothermal titration calorimetry (ITC)
ITC was performed on a VP-ITC instrument (GE Healthcare) at 25 °C in 25 mm HEPES, pH 8.0, 150 mm NaCl, 0.5 mm tris(2-carboxyethyl)phosphine. Ssu72 (1 mm) in the syringe was titrated into TFIIBc (50 μm) in the cell over 30 injections, and data were background-corrected using a control titration where buffer was injected into Ssu72. Data were analyzed with NITPIC and Sedphat software (62). We noticed slight precipitation after the titration, which might cause an inaccuracy of the protein concentrations and the estimate of Kd.
Pulldown assays
For FLAG pulldown assays, an amount of 75 μg of TFIIBc variant with a C-terminal FLAG tag was mixed with 150 μg of N-terminal tagged Ssu72 in binding buffer (50 mm Tris, pH 8.0, 150 mm NaCl, and 0.1% Tween) and bound to 50 μl of anti-FLAG-agarose (Sigma-Aldrich) at room temperature for 1 h with rotation. For the assay conducted in Fig. 5A, TFIIBc-FLAG was prebound to the resin, followed by extensive washing before the addition of other proteins. The resin was then centrifuged for 3 min at 500 rpm at 4 °C, the supernatant was removed, and the resin was washed three times with 1 ml of binding buffer. Proteins were then eluted with 100 mm glycine, pH 3.5, neutralized with 1 μl of 1 m Tris, pH 8.0, and run on SDS-PAGE. Protein interactions were analyzed by staining with Coomassie Brilliant Blue and/or Western blotting with an anti-His6 antibody (Abcam).
Site-specific UV-cross-linking analysis
Amounts of 20 μg of TFIIBc-FLAG and 40 μg of His6-Ssu72-pBpA mutant were mixed in assay buffer (20 mm Tris, pH 8.0, 150 mm NaCl, 1 mm DTT) in a final volume of 50 μl and incubated on ice for 20 min. An amount of 25 μl was removed from each sample as a no UV control. Samples were then UV-cross-linked on ice for four cycles at energy level 9999 in a Spectrolinker cross-linker (Krackeler Scientific), with 5 min of incubation on ice between cycles. 6.25 μl of 6× SDS dye was added, samples were boiled, and 12.5 μl was run on SDS-PAGE. Cross-linking was analyzed by separate Western blots with an anti-FLAG antibody and an anti-His6 antibody (Abcam). For competition cross-linking with TFIIBn and VP16AD (Fig. 4), TFIIBc was preincubated with the molar excess of competitor, as indicated in the figure, for 15 min on ice. Samples were then processed as described above. For competition cross-linking between TFIIBc, Ssu72, and symplekin (Fig. 5), 40 μg of Ssu72 was preincubated with 30 μg of symplekin for 20 min on ice. An amount of 60 μg of TFIIBc was then added, and samples were then processed as described above.
EMSA
The template strand DNA oligonucleotide containing the E4 promoter sequence (TTTTTAGTCCTATATATACTCGCTCT) was annealed to the reverse complement strand containing a 3′-end Cy5 label (Sigma-Aldrich). TFIIBFL, TBPc, and DNA were mixed at equal molar concentrations (300 nm) in assay buffer (20 mm Tris, pH 8.0, 60 mm KCl, 5 mm MgCl2, 4% glycerol, and 10 μg bovine serum albumin in a final volume of 20 μl and incubated at 30 °C for 40 min. Samples were then subjected to electrophoresis on a 6%, 37.5:1 (acrylamide/bisacrylamide) gel at 4 °C in 25 mm Tris, pH 8.3, 190 mm glycine running buffer. Fluorescence was visualized with a Pharos imager (Bio-Rad).
Molecular dynamics simulation of TFIIBc
The two available crystal structures of TFIIBc were used as initial structures for MD simulation. The first one is TFIIBc in complex with TBP and DNA promoter (PDB code 1C9B) (8). The other one is the crystal structure of apo-TFIIBc (this report).
For TFIIBc from PDB entry 1C9B, only the TFIIBc structure was used. For apo-TFIIBc crystal structure, only molecule D was used. Both structures were solvated in a dodecahedron box with 17,779 TIP3P (63) water molecules. The system also contains 51 Na+ and 61 Cl− ions to create a neutral environment with 0.15 mol/liter salt concentration. The GROMACS version 4.5 (64) simulation package and AMBER 99SB-ILDN (65) force field were used to carry out the succeeding steps. Long-range electrostatic interactions were treated by the particle mesh–Ewald method (66). Electrostatic and van der Waals short-range interactions were calculated using a cutoff of 10 Å.
Energy minimization was performed using the steepest-descent method, followed by NVT equilibration for 100 ps with position restraints on the heavy atom of TFIIBc at 300 K using V-rescale thermostat (67), which is used in all MD simulations in succeeding steps. At this step, the positions of heavy atoms were restrained with a force constant of 1000 kJ mol−1 nm−1, and all bonds in protein molecules and water were constrained using the LINCS algorithm (68). NPT equilibration was then carried out using the Parrinello–Rahman barostat (69), to a reference pressure of 1 bar until the average pressure reached around 1 bar.
Eight production MD simulations starting from TFIIBc structure from PDB entry 1C9B were performed in the NVT ensemble with the length of each simulation ranging from 50 to 300 ns, with an accumulated time of 1600 ns for TFIIBc structure from PDB entry 1C9B. Five production MD simulations starting from apo-TFIIBc were performed in the NVT ensemble with the length of each simulation ranging from 100 to 400 ns, giving an accumulated time of 1300 ns. In the succeeding MD simulations, we used the same simulation package, force field, non-bonded interaction calculation method, barostat, thermostat, and bond constraint.
TFIIBc is an inherently flexible protein, as shown by the large root mean square fluctuation values for most of the backbone atoms (Fig. S8A). Because we obtained many TFIIBc structures other than the crystal structures, we selected the starting models based on the following criteria. In TFIIBc1, residues 189, 193, and 200 are likely to interact with Ssu72 (Fig. 3C). Given that residue Ala281 in TFIIBc2 was shown previously to bind VP16AD and that Ssu72 and VP16AD bind to an overlapping surface of TFIIBc (Fig. 6B), we assumed that residues 281–284 of TFIIBc are located on the binding interface of the TFIIBc-Ssu72 complex. Due to the flexibility of TFIIBc, these two clusters of interacting residues on TFIIBc1 or TFIIBc2 can be on the same plane (coplanar) (Fig. S9A) or not (Fig. S9B). To choose TFIIBc structures, clustering for simulations from each crystal structure was performed by K-center clustering (70) to 50 states. Eighteen structures were chosen based on 1) the coplanarity of the interacting residues that mimics the active conformation of TFIIBc when it binds to TBP and the TATA DNA and 2) the distance between the center of mass of TFIIBc1 and TFIIBc2 being less than 40 Å. The crystal structure of TFIIBc from PDB entry 1C9B was also used for protein-protein docking, which resulted in a total of 19 starting structural models of TFIIBc.
Molecular dynamics simulation of Ssu72
The starting structure for MD simulation was taken from PDB entry 3O2Q (28). It was solvated in a dodecahedron box with 11,321 TIP3P water molecules. The system also contains 44 Na+ ions and 34 Cl− ions to neutralize the system and creates a 0.15 mol/liter salt environment. Energy minimization was performed using the steepest-descent method, followed by NVT equilibration for 100 ps with position restraints (force constant = 1000 kJ mol−1 nm−1) on the heavy atom of Ssu72 at T = 300 K with coupling constant = 0.1 ps. NPT equilibration was then carried out to a reference pressure of 1 bar with coupling constant = 0.2 ps until the average pressure reached around 1 bar. Six production NVT MD simulations with different random initial velocities were performed for 50 ns each, giving an accumulated time of 300 ns. Because Ssu72 is a rigid protein (as shown by the small root mean square fluctuation value of all of the backbone atoms; Fig. S8B), we used the crystal structure to do protein-protein docking.
Protein-protein docking using HADDOCK
The 19 selected structures from MD simulation of TFIIBc were used as input for protein-protein docking, along with the crystal structure of Ssu72 (PDB code 3O2Q). The cross-linked residues on Ssu72 were used as active residues input into the HADDOCK web server (71) (i.e. Pro133, Phe193, Gln128, Thr130, Cys131, Glu169, Asp173, Glu174, Glu181. The interacting residues of TFIIBc, which were either determined by mutagenesis or prior knowledge of interaction with VP16AD, were also used as active residues (i.e. Lys189, Arg193, Lys200, Ala281, Asp282, Val283, Thr284). The intermolecular and intramolecular interactions were determined by CNS 1.3 (72, 73) along with PARALLHDG5.4 (74, 75) as force field for protein. For each docking simulation, the two input molecules were separated for 25 Å and rotated randomly around their centers of mass, followed by rigid body minimization and docking. Two hundred best structures from rigid body docking were chosen for semiflexible refinement, based on the weighted sum of several of energy terms (i.e. electrostatic, van der Waals, desolvation, ambiguous interaction restraints, and total buried surface area). Of 1000 structures generated by rigid body docking, 200 structures were used for final solvated refinement and clustering. Clustering was done based on fraction of native contacts using 0.75 similarity as the threshold. For each cluster of every docking simulation, we picked the four models with the lowest HADDOCK scores for the following analysis.
Selection of docking model
HADDOCK score and experimental results were considered to select the best model. The HADDOCK score used in the succeeding steps excluded the EAIR part of the score. For consideration of the cross-linking experiment, we defined a distance that measured the likelihood of cross-linking reaction (D2 distance, shown in Fig. S10). The D2 distance is defined as the distance between the C-α atom of cross-linked residues on Ssu72 and the C-α atom of the nearest residue on TFIIBc that can potentially form a cross-linking bond. The cross-linking reaction only occurs between pBpa and a non-quarternary carbon atom (i.e. a carbon atom that binds to at least one hydrogen atom). The threshold for the D2 distance was determined to be 15 Å. Any cross-linked residue that has a D2 distance violating the threshold is considered as 1 violation count. The contacts between residues 189, 193, 200, 281, 282, 283, and 284 on TFIIBc and Ssu72 residues were determined by using the g_contacts module in GROMACS (76), with a threshold of 3 Å. If any of these residues was not in contact, the violation count was 2 for residues 189, 193, and 200, giving priority to the mutagenesis experiment, and 1 for residues 281, 282, 283, and 284. The total experimental violation count was the sum of all of the violations. The plot of HADDOCK score versus experimental violation count is shown in Fig. S11A. All of the models with violation less than 1 were analyzed further. According to the pulldown data, the interaction between TFIIBc1 and Ssu72 is stronger than that between TFIIBc2 and Ssu72. Considering this result, we calculated the ratio of contacts between TFIIBc1 and Ssu72 and contacts between TFIIBc2 and Ssu72. The plot in Fig. S11B clearly shows one model having the largest ratio and a low HADDOCK score (represented as a star), hence fitting best to the pulldown assay experiment (Table S1).
MD simulation of TFIIB-Ssu72 complex
We solvated the TFIIBc-Ssu72 model in a dodecahedron box with a 1-nm distance between the closest point of the protein and the walls of the box. The protein was then solvated in TIP3P water molecules and an equal number of sodium and chloride ions to create a 0.15 mol/liter salt concentration.
The prepared system was then energy-minimized by the steepest-descent method, followed by NVT equilibration with T = 300 K and coupling constant = 0.1 ps as well as position restraints on heavy atoms of the protein (force constant = 1000 kJ mol−1 nm−1) for 100 ps. Next, NPT equilibration was performed to reference pressure = 1 bar and coupling constant = 0.2 ps, until the average pressure reached 1 bar. The frame from MD simulation that has a volume equal to the average volume was chosen for a 200-ps NVT MD simulation. The last frame of this short MD simulation was the starting ensemble for the production MD simulations. Finally, MD simulation in an NVT environment was performed for 60 ns, with the first 10 ns excluded in analysis.
Author contributions
X. L. conceived the study. M. B., I. C. U., X. H., and X. L. designed the experiments. M. B. carried out all of the experiments with help from M. S. I. C. U. and L. Z. performed computational structure modeling. M. B., I. C. U., X. H., and X. L. wrote the manuscript.
Supplementary Material
Acknowledgments
We thank Shih-Chia Tso, Thomas Scheuermann, and Chad Brautigam for assistance with ITC experiments and Diana Tomchick and Zhe Chen for help with X-ray data collection. We thank Haydn Ball of the UT Southwestern Protein Technology Core for peptide synthesis. We thank Qiong Wu and Jose Rizo-Rey for preliminary NMR analysis. We thank Michael Carey (Department of Biological Chemistry, UCLA, Los Angeles, CA) for generously providing the VP16AD expression plasmid. We thank Peter Schultz (Scripps Research Institute) for generously providing the pEVOL-pBpA plasmid. This research used resources of the Advanced Photon Source, a United States Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under contract DE-AC02-06CH11357. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the DOE under contract DE-AC02-05CH11231. Use of the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, is supported by the DOE, Office of Science, Office of Basic Energy Sciences under contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research and by NIGMS, National Institutes of Health, Grant P41GM103393.
This work was supported by Welch Foundation Grant I-1790, CPRIT Grant R1119, Rita Allen Foundation Grant, the UT Southwestern Medical Center Endowed Scholar fund, and National Institutes of Health Grants GM114576 and GM121662 (to X. L.). This work also received support from the Cecil H. and Ida Green Center Training Program in Reproductive Biology Sciences Research. This work was also supported by Hong Kong Research Grant Council Grant HKUST C6009–15G (to X. H.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S11.
The atomic coordinates and structure factors (code 5WH1) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- pol II
- RNA polymerase II
- PIC
- preinitiation complex
- TBP
- TATA box–binding protein
- BRE
- TFIIB recognition element
- BREu and BREd
- upstream and downstream BRE, respectively
- TFIIB
- transcription factor IIB
- TFIIBn
- N-terminal region of TFIIB
- TFIIBc
- cyclin-like domain-containing core region of TFIIB
- TFIIBFL
- full-length TFIIB
- TFIIBc1 and TFIIBc2
- TFIIB cyclin-like domains 1 and 2, respectively
- CTD
- C-terminal domain
- ITC
- isothermal titration calorimetry
- dTFIIB and yTFIIB
- D. melanogaster and S. cerevisiae TFIIB, respectively
- dSsu72 and ySsu72
- D. melanogaster and S. cerevisiae Ssu72, respectively
- pBpa
- p-benzoyl-l-phenylalanine
- TBPc
- core region of TBP
- PDB
- Protein Data Bank.
References
- 1. Bushnell D. A., Westover K. D., Davis R. E., and Kornberg R. D. (2004) Structural basis of transcription: an RNA polymerase II-TFIIB cocrystal at 4.5 Angstroms. Science 303, 983–988 10.1126/science.1090838 [DOI] [PubMed] [Google Scholar]
- 2. Kostrewa D., Zeller M. E., Armache K. J., Seizl M., Leike K., Thomm M., and Cramer P. (2009) RNA polymerase II-TFIIB structure and mechanism of transcription initiation. Nature 462, 323–330 10.1038/nature08548 [DOI] [PubMed] [Google Scholar]
- 3. Liu X., Bushnell D. A., Wang D., Calero G., and Kornberg R. D. (2010) Structure of an RNA polymerase II-TFIIB complex and the transcription initiation mechanism. Science 327, 206–209 10.1126/science.1182015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sainsbury S., Niesser J., and Cramer P. (2013) Structure and function of the initially transcribing RNA polymerase II-TFIIB complex. Nature 493, 437–440 [DOI] [PubMed] [Google Scholar]
- 5. Pinto I., Wu W. H., Na J. G., and Hampsey M. (1994) Characterization of sua7 mutations defines a domain of TFIIB involved in transcription start site selection in yeast. J. Biol. Chem. 269, 30569–30573 [PubMed] [Google Scholar]
- 6. Čabart P., Újvári A., Pal M., and Luse D. S. (2011) Transcription factor TFIIF is not required for initiation by RNA polymerase II, but it is essential to stabilize transcription factor TFIIB in early elongation complexes. Proc. Natl. Acad. Sci. U.S.A. 108, 15786–15791 10.1073/pnas.1104591108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nikolov D. B., Chen H., Halay E. D., Usheva A. A., Hisatake K., Lee D. K., Roeder R. G., and Burley S. K. (1995) Crystal structure of a TFIIB-TBP-TATA-element ternary complex. Nature 377, 119–128 10.1038/377119a0 [DOI] [PubMed] [Google Scholar]
- 8. Tsai F. T., and Sigler P. B. (2000) Structural basis of preinitiation complex assembly on human pol II promoters. EMBO J. 19, 25–36 10.1093/emboj/19.1.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lagrange T., Kapanidis A. N., Tang H., Reinberg D., and Ebright R. H. (1998) New core promoter element in RNA polymerase II-dependent transcription: sequence-specific DNA binding by transcription factor IIB. Genes Dev. 12, 34–44 10.1101/gad.12.1.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Deng W., and Roberts S. G. (2005) A core promoter element downstream of the TATA box that is recognized by TFIIB. Genes Dev. 19, 2418–2423 10.1101/gad.342405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cox J. M., Hayward M. M., Sanchez J. F., Gegnas L. D., van der Zee S., Dennis J. H., Sigler P. B., and Schepartz A. (1997) Bidirectional binding of the TATA box binding protein to the TATA box. Proc. Natl. Acad. Sci. U.S.A. 94, 13475–13480 10.1073/pnas.94.25.13475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Roberts S. G., Choy B., Walker S. S., Lin Y. S., and Green M. R. (1995) A role for activator-mediated TFIIB recruitment in diverse aspects of transcriptional regulation. Curr. Biol. 5, 508–516 10.1016/S0960-9822(95)00103-5 [DOI] [PubMed] [Google Scholar]
- 13. Zawel L., Kumar K. P., and Reinberg D. (1995) Recycling of the general transcription factors during RNA polymerase II transcription. Genes Dev. 9, 1479–1490 10.1101/gad.9.12.1479 [DOI] [PubMed] [Google Scholar]
- 14. Yudkovsky N., Ranish J. A., and Hahn S. (2000) A transcription reinitiation intermediate that is stabilized by activator. Nature 408, 225–229 10.1038/35041603 [DOI] [PubMed] [Google Scholar]
- 15. Hahn S., and Young E. T. (2011) Transcriptional regulation in Saccharomyces cerevisiae: transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 189, 705–736 10.1534/genetics.111.127019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roberts S. G., and Green M. R. (1994) Activator-induced conformational change in general transcription factor TFIIB. Nature 371, 717–720 10.1038/371717a0 [DOI] [PubMed] [Google Scholar]
- 17. Glossop J. A., Dafforn T. R., and Roberts S. G. (2004) A conformational change in TFIIB is required for activator-mediated assembly of the preinitiation complex. Nucleic Acids Res. 32, 1829–1835 10.1093/nar/gkh504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang D. Y., Dorsey M. J., Voth W. P., Carson D. J., Zeng X., Stillman D. J., and Ma J. (2000) Intramolecular interaction of yeast TFIIB in transcription control. Nucleic Acids Res. 28, 1913–1920 10.1093/nar/28.9.1913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hawkes N. A., Evans R., and Roberts S. G. (2000) The conformation of the transcription factor TFIIB modulates the response to transcriptional activators in vivo. Curr. Biol. 10, 273–276 10.1016/S0960-9822(00)00363-8 [DOI] [PubMed] [Google Scholar]
- 20. Hayashi F., Ishima R., Liu D., Tong K. I., Kim S., Reinberg D., Bagby S., and Ikura M. (1998) Human general transcription factor TFIIB: conformational variability and interaction with VP16 activation domain. Biochemistry 37, 7941–7951 10.1021/bi9801098 [DOI] [PubMed] [Google Scholar]
- 21. Roberts S. G., Ha I., Maldonado E., Reinberg D., and Green M. R. (1993) Interaction between an acidic activator and transcription factor TFIIB is required for transcriptional activation. Nature 363, 741–744 10.1038/363741a0 [DOI] [PubMed] [Google Scholar]
- 22. Sun Z. W., and Hampsey M. (1996) Synthetic enhancement of a TFIIB defect by a mutation in SSU72, an essential yeast gene encoding a novel protein that affects transcription start site selection in vivo. Mol. Cell. Biol. 16, 1557–1566 10.1128/MCB.16.4.1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu W. H., Pinto I., Chen B. S., and Hampsey M. (1999) Mutational analysis of yeast TFIIB: a functional relationship between Ssu72 and Sub1/Tsp1 defined by allele-specific interactions with TFIIB. Genetics 153, 643–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Dichtl B., Blank D., Ohnacker M., Friedlein A., Roeder D., Langen H., and Keller W. (2002) A role for SSU72 in balancing RNA polymerase II transcription elongation and termination. Mol. Cell 10, 1139–1150 10.1016/S1097-2765(02)00707-4 [DOI] [PubMed] [Google Scholar]
- 25. Krishnamurthy S., He X., Reyes-Reyes M., Moore C., and Hampsey M. (2004) Ssu72 is an RNA polymerase II CTD phosphatase. Mol. Cell 14, 387–394 10.1016/S1097-2765(04)00235-7 [DOI] [PubMed] [Google Scholar]
- 26. Zhang D. W., Mosley A. L., Ramisetty S. R., Rodríguez-Molina J. B., Washburn M. P., and Ansari A. Z. (2012) Ssu72 phosphatase-dependent erasure of phospho-Ser7 marks on the RNA polymerase II C-terminal domain is essential for viability and transcription termination. J. Biol. Chem. 287, 8541–8551 10.1074/jbc.M111.335687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. St-Pierre B., Liu X., Kha L. C., Zhu X., Ryan O., Jiang Z., and Zacksenhaus E. (2005) Conserved and specific functions of mammalian ssu72. Nucleic Acids Res. 33, 464–477 10.1093/nar/gki171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xiang K., Nagaike T., Xiang S., Kilic T., Beh M. M., Manley J. L., and Tong L. (2010) Crystal structure of the human symplekin-Ssu72-CTD phosphopeptide complex. Nature 467, 729–733 10.1038/nature09391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. He X., Khan A. U., Cheng H., Pappas D. L. Jr., Hampsey M., and Moore C. L. (2003) Functional interactions between the transcription and mRNA 3′ end processing machineries mediated by Ssu72 and Sub1. Genes Dev. 17, 1030–1042 10.1101/gad.1075203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dantonel J. C., Murthy K. G., Manley J. L., and Tora L. (1997) Transcription factor TFIID recruits factor CPSF for formation of 3′ end of mRNA. Nature 389, 399–402 10.1038/38763 [DOI] [PubMed] [Google Scholar]
- 31. Calvo O., and Manley J. L. (2003) Strange bedfellows: polyadenylation factors at the promoter. Genes Dev. 17, 1321–1327 10.1101/gad.1093603 [DOI] [PubMed] [Google Scholar]
- 32. Wang Y., Fairley J. A., and Roberts S. G. (2010) Phosphorylation of TFIIB links transcription initiation and termination. Curr. Biol. 20, 548–553 10.1016/j.cub.2010.01.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Proudfoot N., and O'Sullivan J. (2002) Polyadenylation: a tail of two complexes. Curr. Biol. 12, R855–R857 10.1016/S0960-9822(02)01353-2 [DOI] [PubMed] [Google Scholar]
- 34. Kobor M. S., Simon L. D., Omichinski J., Zhong G., Archambault J., and Greenblatt J. (2000) A motif shared by TFIIF and TFIIB mediates their interaction with the RNA polymerase II carboxy-terminal domain phosphatase Fcp1p in Saccharomyces cerevisiae. Mol. Cell. Biol. 20, 7438–7449 10.1128/MCB.20.20.7438-7449.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chambers R. S., Wang B. Q., Burton Z. F., and Dahmus M. E. (1995) The activity of COOH-terminal domain phosphatase is regulated by a docking site on RNA polymerase II and by the general transcription factors IIF and IIB. J. Biol. Chem. 270, 14962–14969 10.1074/jbc.270.25.14962 [DOI] [PubMed] [Google Scholar]
- 36. Mapendano C. K., Lykke-Andersen S., Kjems J., Bertrand E., and Jensen T. H. (2010) Crosstalk between mRNA 3′ end processing and transcription initiation. Mol. Cell 40, 410–422 10.1016/j.molcel.2010.10.012 [DOI] [PubMed] [Google Scholar]
- 37. O'Sullivan J. M., Tan-Wong S. M., Morillon A., Lee B., Coles J., Mellor J., and Proudfoot N. J. (2004) Gene loops juxtapose promoters and terminators in yeast. Nat. Genet. 36, 1014–1018 10.1038/ng1411 [DOI] [PubMed] [Google Scholar]
- 38. Ansari A., and Hampsey M. (2005) A role for the CPF 3′-end processing machinery in RNAP II-dependent gene looping. Genes Dev. 19, 2969–2978 10.1101/gad.1362305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Perkins K. J., Lusic M., Mitar I., Giacca M., and Proudfoot N. J. (2008) Transcription-dependent gene looping of the HIV-1 provirus is dictated by recognition of pre-mRNA processing signals. Mol. Cell 29, 56–68 10.1016/j.molcel.2007.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tan-Wong S. M., French J. D., Proudfoot N. J., and Brown M. A. (2008) Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proc. Natl. Acad. Sci. U.S.A. 105, 5160–5165 10.1073/pnas.0801048105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Reilly D., and Greaves D. R. (2007) Cell-type-specific expression of the human CD68 gene is associated with changes in Pol II phosphorylation and short-range intrachromosomal gene looping. Genomics 90, 407–415 10.1016/j.ygeno.2007.04.010 [DOI] [PubMed] [Google Scholar]
- 42. Le May N., Fradin D., Iltis I., Bougnères P., and Egly J. M. (2012) XPG and XPF endonucleases trigger chromatin looping and DNA demethylation for accurate expression of activated genes. Mol. Cell 47, 622–632 10.1016/j.molcel.2012.05.050 [DOI] [PubMed] [Google Scholar]
- 43. Crevillén P., Sonmez C., Wu Z., and Dean C. (2013) A gene loop containing the floral repressor FLC is disrupted in the early phase of vernalization. EMBO J. 32, 140–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Martin M., Cho J., Cesare A. J., Griffith J. D., and Attardi G. (2005) Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell 123, 1227–1240 10.1016/j.cell.2005.09.040 [DOI] [PubMed] [Google Scholar]
- 45. Yao J., Ardehali M. B., Fecko C. J., Webb W. W., and Lis J. T. (2007) Intranuclear distribution and local dynamics of RNA polymerase II during transcription activation. Mol. Cell 28, 978–990 10.1016/j.molcel.2007.10.017 [DOI] [PubMed] [Google Scholar]
- 46. Hampsey M., Singh B. N., Ansari A., Lainé J. P., and Krishnamurthy S. (2011) Control of eukaryotic gene expression: gene loops and transcriptional memory. Adv. Enzyme Regul. 51, 118–125 10.1016/j.advenzreg.2010.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Laine J. P., Singh B. N., Krishnamurthy S., and Hampsey M. (2009) A physiological role for gene loops in yeast. Genes Dev. 23, 2604–2609 10.1101/gad.1823609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tan-Wong S. M., Wijayatilake H. D., and Proudfoot N. J. (2009) Gene loops function to maintain transcriptional memory through interaction with the nuclear pore complex. Genes Dev. 23, 2610–2624 10.1101/gad.1823209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Tan-Wong S. M., Zaugg J. B., Camblong J., Xu Z., Zhang D. W., Mischo H. E., Ansari A. Z., Luscombe N. M., Steinmetz L. M., and Proudfoot N. J. (2012) Gene loops enhance transcriptional directionality. Science 338, 671–675 10.1126/science.1224350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Singh B. N., and Hampsey M. (2007) A transcription-independent role for TFIIB in gene looping. Mol. Cell 27, 806–816 10.1016/j.molcel.2007.07.013 [DOI] [PubMed] [Google Scholar]
- 51. Ibrahim B. S., Kanneganti N., Rieckhof G. E., Das A., Laurents D. V., Palenchar J. B., Bellofatto V., and Wah D. A. (2009) Structure of the C-terminal domain of transcription factor IIB from Trypanosoma brucei. Proc. Natl. Acad. Sci. U.S.A. 106, 13242–13247 10.1073/pnas.0904309106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Bagby S., Kim S., Maldonado E., Tong K. I., Reinberg D., and Ikura M. (1995) Solution structure of the C-terminal core domain of human TFIIB: similarity to cyclin A and interaction with TATA-binding protein. Cell 82, 857–867 10.1016/0092-8674(95)90483-2 [DOI] [PubMed] [Google Scholar]
- 53. Jonker H. R., Wechselberger R. W., Boelens R., Folkers G. E., and Kaptein R. (2005) Structural properties of the promiscuous VP16 activation domain. Biochemistry 44, 827–839 10.1021/bi0482912 [DOI] [PubMed] [Google Scholar]
- 54. Chin J. W., Martin A. B., King D. S., Wang L., and Schultz P. G. (2002) Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 99, 11020–11024 10.1073/pnas.172226299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Young T. S., Ahmad I., Yin J. A., and Schultz P. G. (2010) An enhanced system for unnatural amino acid mutagenesis in E. coli. J. Mol. Biol. 395, 361–374 10.1016/j.jmb.2009.10.030 [DOI] [PubMed] [Google Scholar]
- 56. Sato S., Mimasu S., Sato A., Hino N., Sakamoto K., Umehara T., and Yokoyama S. (2011) Crystallographic study of a site-specifically cross-linked protein complex with a genetically incorporated photoreactive amino acid. Biochemistry 50, 250–257 10.1021/bi1016183 [DOI] [PubMed] [Google Scholar]
- 57. Burley S. K., and Roeder R. G. (1998) TATA box mimicry by TFIID: autoinhibition of pol II transcription. Cell 94, 551–553 10.1016/S0092-8674(00)81596-2 [DOI] [PubMed] [Google Scholar]
- 58. Minor W., Cymborowski M., Otwinowski Z., and Chruszcz M. (2006) HKL-3000: the integration of data reduction and structure solution–from diffraction images to an initial model in minutes. Acta Crystallogr. D Biol. Crystallogr. 62, 859–866 10.1107/S0907444906019949 [DOI] [PubMed] [Google Scholar]
- 59. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 60. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Adams P. D., Afonine P. V., Bunkoczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Keller S., Vargas C., Zhao H., Piszczek G., Brautigam C. A., and Schuck P. (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84, 5066–5073 10.1021/ac3007522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
- 64. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 10.1093/bioinformatics/btt055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., and Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 10.1002/prot.22711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald: an N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 67. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 10.1063/1.2408420 [DOI] [PubMed] [Google Scholar]
- 68. Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M. (1997) LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 10.1002/(SICI)1096-987X(199709)18:12%3C1463::AID-JCC4%3E3.0.CO%3B2-H [DOI] [Google Scholar]
- 69. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single crystals: a new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 10.1063/1.328693 [DOI] [Google Scholar]
- 70. Gonzalez T. F. (1985) Clustering to minimize the maximum intercluster distance. Theor. Comput. Sci. 38, 293–306 10.1016/0304-3975(85)90224-5 [DOI] [Google Scholar]
- 71. de Vries S. J., van Dijk M., Bonvin A. M. J. J. (2010) The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc. 5, 883–897 10.1038/nprot.2010.32 [DOI] [PubMed] [Google Scholar]
- 72. Brunger A. T. (2007) Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2, 2728–2733 10.1038/nprot.2007.406 [DOI] [PubMed] [Google Scholar]
- 73. Brunger A. T., Adams P. D., Clore G. M., Delano W. L., Gros P., Grosse-Kunstleve R. W., Jiang J.-S., Kuszewski F., Nilges M., Pannu N. S., Read R. J., Rice L. M., Simonson T., and Warren G. L. (1998) Crystallography & NMR System: a new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 54, 905–921 10.1107/S0108767398011465 [DOI] [PubMed] [Google Scholar]
- 74. Linge J. P., and Nilges M. (1999) Influence of non-bonded parameters on the quality of NMR structures: a new force field for NMR structure calculation. J. Biomol. NMR 13, 51–59 10.1023/A:1008365802830 [DOI] [PubMed] [Google Scholar]
- 75. Linge J. P., Williams M. A., Spronk C. A. E. M., Bonvin A. M. J. J., and Nilges M. (2003) Refinement of protein structures in explicit solvent. Proteins 50, 496–506 10.1002/prot.10299 [DOI] [PubMed] [Google Scholar]
- 76. Blau C., and Grubmuller H. (2013) G-contacts: fast contact search in bio-molecular ensemble data. Comput. Phys. Commun. 184, 2856–2859 10.1016/j.cpc.2013.07.018 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.