

Abstract
Interferon α (IFNα) is important for antiviral and anticancer defenses. However, overproduction is associated with autoimmune disorders. Thus, the cell must precisely up- and down-regulate IFNα to achieve immune system homeostasis. The cellular FLICE-like inhibitory protein (cFLIP) is reported to inhibit IFNα production. However, the mechanism for this antagonism remained unknown. The goal here was to identify this mechanism. Here we examined the signal transduction events that occur during TLR9-induced IRF7 activation. The cFLIP long isoform (cFLIPL) inhibited the expression of IRF7-controlled natural or synthetic genes in several cell lines, including those with abundant IRF7 protein levels (e.g. dendritic cells). cFLIPL inhibited IRF7 phosphorylation; however, cFLIPL-IRF7 interactions were not detectable, implying that cFLIPL acted upstream of IRF7 dimerization. Interestingly, cFLIPL co-immunoprecipitated with IKKα, and these interactions correlated with a loss of IKKα–IRF7 interactions. Thus, cFLIP appears to bind to IKKα to prevent IKKα from phosphorylating and activating IRF7. To the best of our knowledge, this is the first report of a cellular protein that uses this approach to inhibit IRF7 activation. Perhaps this cFLIP property could be engineered to minimize the deleterious effects of IFNα expression that occur during certain autoimmune disorders.
Keywords: cFLIPL, IRF7, IKKα, IFNα, TLR9
Introduction
Type I interferons (IFNs)2 are comprised of IFNα and IFNβ, and their production is the first line of defense against virus infection (1). IFNα represents a group of cytokines (e.g. IFNα4 and IFNα6) that are predominately regulated by the interferon regulatory factor 7 (IRF7) transcription factor (2–4). In most cell types, IRF7 is expressed at low levels. However, IRF7 is expressed at high levels in hematopoietic cells like plasmacytoid dendritic cells (pDCs) (5, 6). IFNα production is increased in a variety of autoimmune diseases, including systemic lupus erythematosus, Sjögren's syndrome (7), type I diabetes (8), rheumatoid arthritis (9), and others (10, 11). This exemplifies that the precise up- and down-regulation of IFNα production is critical for proper immune system homeostasis.
IRF7 activation is required for robust IFNα expression (3). IRF7 activation occurs via the engagement of endosomal nucleic acid sensors (e.g. TLR7, TLR8, and TLR9). TLR9 homodimers are activated upon binding of viral (12) or bacterial unmethylated CpG motifs (e.g. CpG-A) (13) or DNAs involved in autoreactive immune complexes (14, 15). In all cases, the MyD88 protein is recruited to the cytoplasmic portion of these TLRs (16), acting as a critical signal adaptor molecule. Next is the assembly of a dynamic complex including at least IRAK1, IRAK4 (17), and TRAF6 (16). IKKα is subsequently recruited and activated, either by IRAK1 (18) or an unknown kinase (2, 19). Regardless, IKKα goes on to phosphorylate IRF7, whereas TRAF6 Lys-63–linked polyubiquitinates IRF7(16,17). Phospho-IRF7 then homodimerizes (20) and translocates to the nucleus, where it drives expression of IFNα genes as well as other interferon-stimulated genes (2).
Because IFNα has powerful pro-inflammatory properties, cells have mechanisms to down-regulate IFNα production in the absence of virus infection. For example, RTA-associated ubiquitin ligase (RAUL) is an E3 ligase that promotes IRF7 Lys-48–linked polyubiquitination and degradation (21). PP2A is a dephosphorylase that inactivates IRF7 (22). In contrast, 4E-BP1/2 inhibits IRF7 translation (23). The cellular aryl hydrocarbon receptor–interacting protein (AIP) inhibits IRF7 action downstream of IRF7 phosphorylation; it inhibits nuclear translocation of IRF7 homodimers (24).
The cellular FLICE-inhibitory protein (cFLIP) was originally identified as an inhibitor of extrinsic apoptosis (25). There are two major isoforms of cFLIP, the long isoform (cFLIPL) and a shorter splice variant (cFLIPS), and both are members of the FLIP family (26). Our group recently identified cFLIPL as an IRF3 antagonist; cFLIPL binds to IRF3 to prevent enhanceosome formation (27). IRF3 demonstrates considerable sequence homology to IRF7 (28), begging the question whether cFLIPL may bind to and antagonize IRF7 to control IFNα production. In support of this hypothesis is one report showing that overexpression of cFLIPS correlates with a decrease in IFNα protein expression (29). To answer this question, we examined the effect of cFLIP on different steps of the TLR9-induced IRF7 activation pathway, using CpG-A to specifically trigger IRF7 dimerization. Several lines of evidence shown here suggest that cFLIP is a bona fide inhibitor of IRF7 activation and that it disrupts IKKα–IRF7 interactions as its antagonistic function.
Results
cFLIPL inhibits IRF7-induced luciferase activity independent of IRF3 and IRF5
We showed previously that cFLIPL inhibits IRF3-driven transcription by interrupting IRF3–CBP–DNA interactions (27). Because of the sequence and structural similarities of IRF3, IRF5, and IRF7 (28, 30), it was queried whether cFLIPL could antagonize IRF5 or IRF7.
Luciferase reporter assays have been developed to specifically detect IRF5 or IRF7 activation and were used as a first step toward answering this question (31, 32). HEK293T (293T) cells were used because of their high transfection efficiency and their common use for luciferase reporter assays. Here the il12p40 promoter was fused to a luciferase gene to assess IRF5 activation (33) (Fig. 1A). Alternatively, the infa6 promoter was fused to a luciferase gene to assess IRF7 activation (34) (Fig. 1, B–E). Fig. 1A shows the specificity of the il12p40-luc plasmid for IRF5 activation; only cells transiently expressing IRF5 and TRAF6 stimulated luciferase gene expression robustly. Note that TRAF6 must be co-expressed with IRF5 for IRF5 homodimerization and subsequent IRF5 activation (35). Overexpression of a constitutively active IRF3 (IRF3CA) or IRF7 did not stimulate luciferase gene expression significantly above levels of pCI-transfected cells, as expected. Under these conditions, cFLIPL had no effect on luciferase activity, suggesting that cFLIPL did not antagonize IRF5 activation. A control for this assay was cells expressing Vpx, an HIV protein that is known to inhibit IRF5 activation (32).
Figure 1.

cFLIPL inhibits IRF7-induced gene expression independent of IRF3 and IRF5. A and B, 293T cells were co-transfected with 450 ng of pil12p40-luc (A) or 450 ng of pifna6-luc and 50 ng of pRL-TK, 1000 ng of pCI, pIRF3CA, or pIRF7 or 500 ng each of pIRF5 and pTRAF6 (B). Cells were also co-transfected with either 1000 ng of pCI, pcFLIPL, or pVpx (A) or pAIP (B). Cells were incubated for 24 h post-transfection. C, 293T cells were co-transfected with 450 ng of pifna6-luc and 50 ng of pRL-TK, 1000 ng of pCI, or 500 ng each of pIRF7 and pMyD88. Cells were also co-transfected with either 1000 ng of pCI, pcFLIPL, or pIRF7DN. Cells were incubated for 24 h post-transfection. D, 293T cells were co-transfected with 450 ng of pifna6-luc and 50 ng of pRL-TK, 1000 ng of pCI, or 250 ng each of pIRF7, pMyD88, pIKKα, and pTRAF6. Cells were also co-transfected with either 1000 ng of pCI, pcFLIPL, or pIRF7DN. Cells were incubated for 24 h post-transfection. E, HeLa cells were co-transfected with 450 ng of pifna6-luc, 50 ng of pRL-TK, 250 ng of pIFNα, and 1000 ng of pCI, pcFLIPL, or pAIP. 24 h post-transfection, cells were incubated in medium lacking or containing 3 μm CpG-A for 3 h. For all experiments, cellular lysates were examined for luciferase activities. Results are shown as -fold induction of luciferase activity relative to pCI-transfected cells. A portion of each lysate was additionally examined for protein expression by using IB to detect FLAG-tagged cFLIPL, myc-tagged Vpx, or myc-tagged AIP. Luciferase assays are representative of three technical replicates, and all luciferase assays were performed at least three times. Data are expressed as the mean ± S.D. Statistically significant differences in experimental samples versus unstimulated, pCI-transfected cells are denoted (*, p < 0.05).
Fig. 1B shows the specificity of the infa6-luc plasmid for IRF7 activation; luciferase activity was robust only in cells overexpressing IRF7 proteins. Additionally, overexpression of a IRF3CA or co-expression of IRF5 and TRAF6 did not stimulate the ifna6-controlled luciferase reporter gene significantly above levels of pCI-transfected cells, again showing specificity of ifna6-luc for IRF7. It is not fully clear how overexpressing WT IRF7 in 293T cells activates the ifna6-luc reporter, but this phenomenon has been seen in several publications (36–39). The most likely explanation is that the transfection process of plasmids mimics viral infection or CpG stimulation of TLR9 (40). cFLIPL inhibited IR7-controlled luciferase activity, suggesting that cFLIPL may act at one or more stages of the IRF7 signal transduction pathway. Note that luciferase activity is lower in cells transfected with IRF7 alone (15-fold, Fig. 1B) compared with cells co-overexpressing IRF7 and MyD88 (36-fold, Fig. 1C) or co-overexpressing IRF7, MyD88, IKKα, and TRAF6 (20-fold, Fig. 1D). The purpose of co-expressing IRF7, MyD88, TRAF6, and MyD88 (Fig. 1D) was to simulate formation of the myddosome (41). cFLIPL inhibition of luciferase activity (Fig. 1, C and D) suggested that cFLIPL antagonized one or more of these molecules or an event occurring downstream of myddosome formation. A dominant-negative mutant IRF7 (pIRF7DN) significantly inhibited ifna6-luc activity in all of these systems, as would be expected (Fig. 1, C and D). Interestingly, cFLIPL inhibited IRF7-induced infa6-luc activity to a similar extent as AIP, a cellular protein known to inhibit IRF7 activation (42) (Fig. 1B).
The experiments shown in Fig. 1B overexpressed IRF7 to stimulate IRF7 activation because 293T cells do not express sufficient levels of IRF7 to drive promoter activity (42). In contrast, HeLa cells express IRF7, and IRF7 protein levels are increased when cells are transfected with a plasmid encoding IFNα (43, 44). Using this approach, incubation of HeLa cells with CpG-A stimulates the TLR9-induced IRF7 signal transduction pathway (45). Using this system, CpG-A activated IRF7 in vector-transfected cells, similar to another published report (Fig. 1E) (45). cFLIPL significantly inhibited CpG-A-induced luciferase activity, and the extent of this inhibition was similar to the inhibition observed with AIP (Fig. 1E). Thus, cFLIPL inhibited IRF7 activity in two separate experimental systems.
cFLIPL does not associate with IRF7
We showed previously that cFLIPL binds to an IRF3–CBP complex to prevent enhanceosome formation (27). Because IRF3 and IRF7 are similar, one possibility was that cFLIPL would also interact with and inhibit IRF7.
293T cells were initially used to test this hypothesis because these cells have high rates of transfection efficiency and are used routinely to detect protein–protein interactions (27). Epitope-tagged versions of IRF7 were expressed in 293T cells because 293T cells have very low levels of endogenous IRF7 (46). As shown in Fig. 2A, despite the abundance of IRF7 in these cells, a FLAG-tagged cFLIPL was not detectable in IRF7 immunoprecipitates. It was unlikely that this lack of detection was due to suboptimal conditions for protein–protein interactions because we detected IRF7 interacting with a known binding partner (AIP) (Fig. 2A, left panel) (24). Also, we detected cFLIPL interacting with IRF3, a known cFLIPL binding partner (Fig. 2A, right panel) (27).
Figure 2.
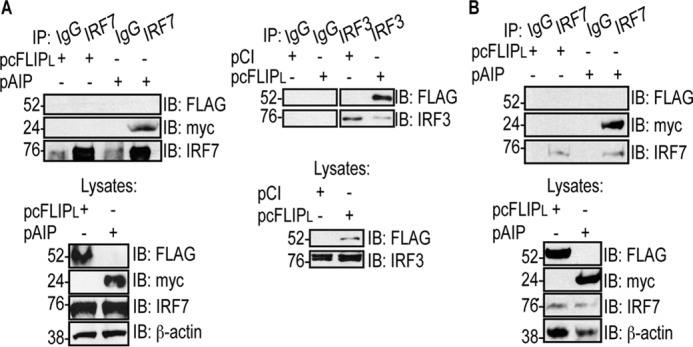
cFLIPL does not co-immunoprecipitate with IRF7. A, 293T cells were co-transfected with 500 ng of pIRF7 or pIRF3 and 1000 ng of pCI, pcFLIPL, or pAIP. 24 h post-transfection, cells were lysed. A portion of each lysate was incubated with anti-IRF7 or anti-IRF3 antibodies or nonspecific IgG. IB analysis of IP samples was performed to detect FLAG-tagged cFLIPL and myc-tagged AIP, IRF7, or IRF3. B, HeLa cells were transfected with 1000 ng of pcFLIPL or pAIP. 24 h post-transfection, cells were lysed, and a portion of each lysate was incubated with anti-IRF7 or nonspecific IgG antibodies. IB analysis of IP samples was performed to detect FLAG-tagged cFLIPL and myc-tagged AIP and IRF7. For all IPs, a portion of each whole-cell lysate was also examined by immunoblotting to confirm expression of the proteins of interest.
A similar co-immunoprecipitation was performed in HeLa cells (Fig. 2B). Endogenous IRF7 protein levels were detected in HeLa cells, allowing us to examine whether cFLIPL interacted with endogenous IRF7. Similar to Fig. 2A, cFLIPL was not detected in IRF7 immunoprecipitates. Again, IRF7–AIP interactions remained detectable, showing that conditions were optimal for detecting IRF7 binding partners. Thus, it appeared that cFLIPL did not exert its antagonistic effects via interacting with IRF7.
cFLIPL inhibits IRF7 phosphorylation
One critical step in the TLR9-induced IRF7 activation pathway is IRF7 phosphorylation at Ser-477 and Ser-479 (36). After IRF7 is phosphorylated, IRF7 changes conformation, exposing the interferon association domain to allow IRF7 homodimerization, nuclear translocation, recruitment of critical co-factors such as CBP (47), and DNA binding (20).
Because cFLIPL did not co-immunoprecipitate with IRF7 (Fig. 2), we asked whether cFLIPL prevented IRF7 phosphorylation. To test this, HeLa cells were transfected with pIFNα to increase endogenous IRF7 expression and then stimulated with CpG-A, resulting in IRF7 phosphorylation (Fig. 3A). Phospho-IRF7 was also observed when AIP was expressed in cells, and this was expected because AIP inhibits IRF7 activation downstream of IRF7 phosphorylation (24). In contrast, IRF7 phosphorylation was not detected in cFLIPL-expressing cells (Fig. 3A). This suggested that cFLIPL targeted a signaling event upstream of IRF7 phosphorylation. The data in Fig. 3B further supported this concept. In this luciferase reporter assay, IRF7CA was overexpressed. IRF7CA is sufficient to stimulate infa6-luc activity because phosphomimetic amino acid substitutions (Ser-477 and Ser-479 to Asp) yield an IRF7 protein that is constitutively active without the need for a kinase (36, 48). cFLIPL did not inhibit the activity of a constitutively active IRF7 mutant, suggesting that it works upstream of phosphorylation. AIP blocked IRF7-controlled luciferase activity, and this was expected because AIP prevents nuclear translocation of IRF7 (24) (Fig. 3B).
Figure 3.
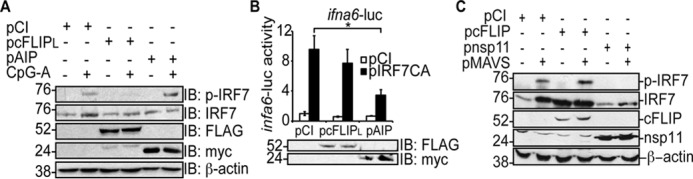
cFLIPL inhibits IRF7 phosphorylation. A, HeLa cells were transfected with 1.5 μg of pIFNα and 6 μg of pCI, pcFLIPL or pAIP. 24 h post-transfection, cells were incubated with medium lacking or containing 3 μm CpG-A for 3 h. Cells were lysed, and immunoblotting was performed to detect phospho-IRF7, IRF7, FLAG-tagged cFLIPL, myc-tagged AIP, and β-actin proteins. B, 293T cells were co-transfected with 450 ng of pifna6-luc; 50 ng of pRL-TK; 1000 ng of pCI, cFLIPL, or pAIP; and 500 ng of pCI or pIRF7CA. 24 post-transfection, cells were lysed, and luciferase activities were quantified. Luciferase assays are representative of three technical replicates, and all luciferase assays were performed at least three times. Results are shown as -fold induction of luciferase activity relative to unstimulated, pCI-transfected cells. Immunoblot analysis of whole-cell lysates also was performed to detect FLAG-tagged cFLIPL and myc-tagged AIP. C, 293T cells were transfected with 500 ng of IRF7, 500 ng of pCI or pMAVS, and1000 ng of pCI, pcFLIPL, or pnsp11. 24 h post-transfection, cells were lysed, and immunoblotting was performed to detect phospho-IRF7, IRF7, FLAG-tagged cFLIPL, FLAG-tagged nsp11, and β-actin proteins. The experiments shown here are representative of experiments performed at least three times. Data are expressed as the mean ± S.D. Statistically significant differences in experimental samples compared with cells transfected with empty vector are denoted (*, p < 0.05).
To confirm that the inhibition of phospho-IRF7 by cFLIPL was not indirectly due to CpG-A activation of TBK1–IKKϵ–mediated IRF7 phosphorylation (49), we performed an IRF7 phosphorylation assay in 293T cells expressing either empty vector, cFLIPL, or nsp11, a porcine respiratory virus protein known to inhibit IRF7 phosphorylation (50). To stimulate TBK1–IKKϵ-mediated IRF7 phosphorylation, we overexpressed the upstream signaling molecule MAVS (51). Here cFLIPL did not inhibit IRF7 phosphorylation, in contrast to nsp11 (Fig. 3C). IRF7 protein levels were greatly reduced in Nsp11-expressing cells because NSP11 is an endoribonuclease (50).These data suggest that cFLIPL does not antagonize the TBK1–IKKϵ kinase complex. This is further supported by the finding that cFLIPL does not inhibit TBK1-induced IRF3 phosphorylation (27).
The N-terminal DED-containing region of cFLIP is necessary to inhibit IRF7 phosphorylation and activation
Fig. 4A shows that cFLIPL is comprised of two death effector domains (DEDs) and a C terminus containing a caspase-like domain (CLD). In contrast, cFLIPS lacks the CLD. We showed previously that the CLD of cFLIPL is sufficient to inhibit the IRF3 activation pathway (27). Thus, the DEDs were dispensable for cFLIPL inhibition of IRF3 activity. We were curious whether the CLD also provided IRF7 inhibition. We used the same IRF7-specific luciferase reporter assay as shown in Fig. 2D to map the cFLIPL domain(s) required for inhibition. As shown in Fig. 4B, cFLIPL and cFLIPS each significantly inhibited CpG-A–induced ifna6-luc activity, suggesting that one or more DEDs possess the inhibitory function. These data also agree with the finding that cFLIPS inhibits IFNα production (29). However, the CLD did not antagonize IRF7 activation (Fig. 4B). Consistent with luciferase assay results, cFLIPL and cFLIPS, but not the CLD, inhibited IRF7 phosphorylation triggered by either CpG-A treatment of cells (Fig. 4C) or when IRF7 and IKKα were overexpressed (Fig. 4D). Thus, the DED regions of cFLIPL and cFLIPS are important for IRF7 antagonism. Equally important, Fig. 4D showed that IKKα overexpression resulted in IRF7 activation in a manner presumed to be independent of TBK1 and IKKϵ. Thus, cFLIPL inhibition of IKKα-induced IRF7 phosphorylation continues to suggest that cFLIPL does not act on the TBK1–IKKϵ complex to inhibit IRF7 activation.
Figure 4.
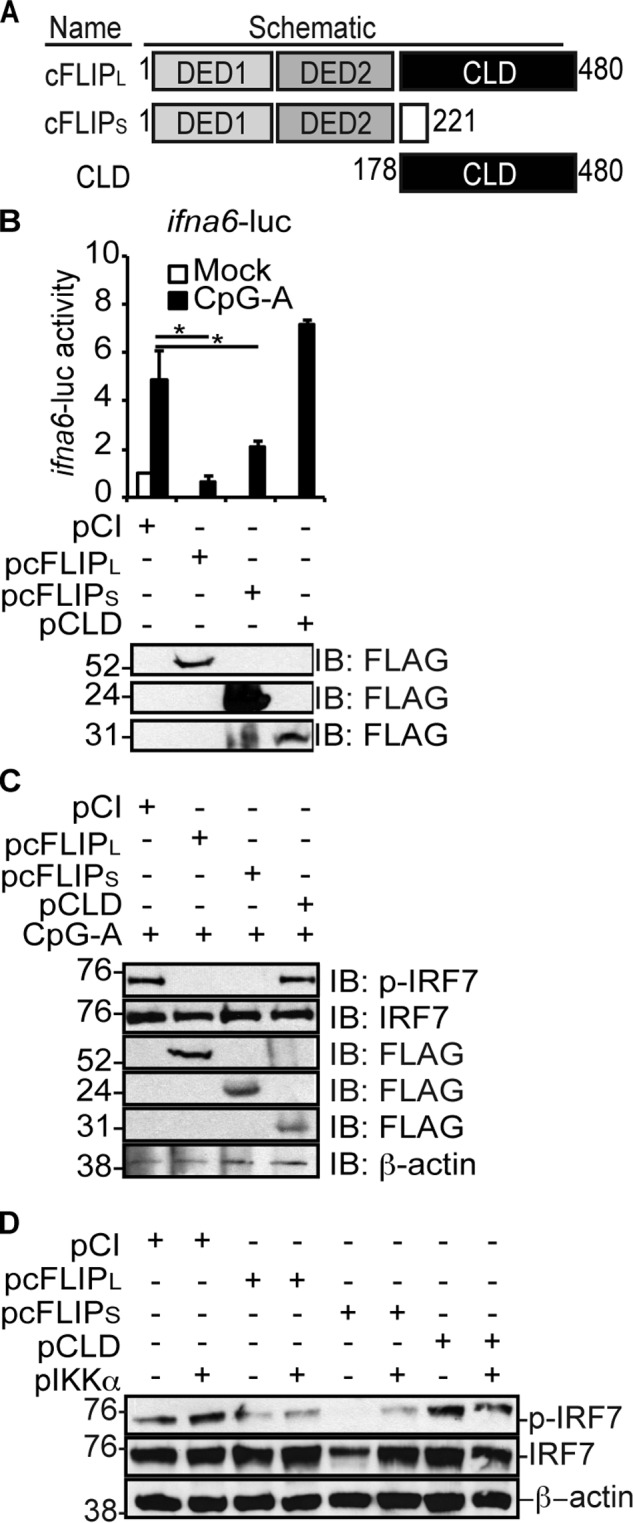
The N-terminal DEDs of cFLIP inhibit IRF7 activation. A, schematic of the wildtype and mutant (CLD) cFLIPL protein and the alternative splice variant cFLIPS. cFLIPL and cFLIPS each contain tandem DEDs, whereas only cFLIPL possesses a CLD. B, HeLa cells were co-transfected with 450 ng of pifna6-luc, 50 ng of pRL-TK, 250 ng of pIFNα, and 1000 ng of pCI, pcFLIPL, pcFLIPS, or pCLD. 24 h post-transfection, cells were incubated in medium lacking or containing 3 μM CpG-A for 3 h. Cells were lysed, and luciferase activities were quantified. Results are shown as -fold induction of luciferase activity relative to untreated pCI-transfected cells. Immunoblot analysis of lysates was also performed. Luciferase assays are representative of three technical replicates, and all luciferase assays were performed at least three times. Data are expressed as the mean ± S.D. Statistically significant differences in experimental samples compared with cells transfected with empty vector are denoted (*, p < 0.05). C, HeLa cells seeded in 10-cm dishes were transfected with 1.5 μg of pIFNα and 6 μg of pCI, pcFLIPL, pcFLIPS, or pCLD. 24 h post-transfection, cells were incubated with medium lacking or containing 3 μm CpG-A for 3 h. Cells were lysed in 100 μl to concentrate protein. D, 293T cells were transfected with 1000 ng of pCI or 500 ng of pIRF7 and pIKKα and 1000 ng of pCI, pcFLIPL, pcFLIPS, or pCLD. 24 h post-transfection, cells were lysed. Immunoblotting was performed to detect phospho-IRF7, total IRF7, each FLAG-tagged FLIP, or β-actin.
cFLIPL associates with IKKα and prevents IKKα–IRF7 interactions
The data above showed that, although cFLIPL inhibited IRF7 phosphorylation, it did not bind to IRF7. Two kinases (IRAK1 and IKKα) are reported to promote IRF7 phosphorylation during TLR9 stimulation (17, 19). The current dogma is that the IRAK1–IKKα kinase cascade leads to phosphorylation of IRF7 (19, 52). Thus, we queried whether cFLIPL disrupts members of the signaling complex that are critical for IRF7 phosphorylation. To test this, we performed IRF7 co-immunoprecipitation, where IKKα, IRF7, cFLIPL, and TRAF6 were ectopically expressed. We observed that IKKα–IRF7 interactions were greatly diminished when cFLIPL was present (Fig. 5A), implying that cFLIPL inhibited IRF7–IKKα interactions. As expected, cFLIPL–IRF7 interactions were not detected, similar to the observations shown in Fig. 2. IRF7 activation by IKKα is preceded by its ubiquitination by TRAF6 (17, 53). Interestingly, overexpression of cFLIPL did not prevent IRF7–TRAF6 interactions (Fig. 5A). This suggested that cFLIPL acted downstream of the formation of the MyD88-based complex containing TRAF6.
Figure 5.

cFLIPL associates with IKKα and prevents IKKα–IRF7 interactions. A, 293T cells were co-transfected with 500 ng of pIRF7, 1000 ng of pCI or pcFLIPL, 500 ng of pIKKα, and 500 ng of pTRAF6 as indicated. 24 h post-transfection, cells were lysed, and a portion of each lysate was immunoprecipitated with anti-IRF7 or nonspecific IgG antibodies. A separate portion of each lysate was used to examine protein expression levels. IB analysis of co-immunoprecipitated samples was performed to detect IKKα, myc-tagged TRAF6, FLAG-tagged cFLIPL, or IRF7 proteins. B, 293T cells were co-transfected with 1000 ng of pIKKα, pCI, pcFLIPL, and pIRF7 as indicated. 24 h post-transfection, cells were lysed, and a portion of each lysate was immunoprecipitated with anti-IKKα or nonspecific IgG antibodies. A separate portion of each lysate was used to examine protein expression levels. IB analysis of co-immunoprecipitated samples was performed to detect FLAG-tagged cFLIPL, IKKα, or IRF7. C, 293T cells were co-transfected with 500 ng of pIKKα and 1000 ng of pCI, pcFLIPL, pcFLIPS, or pCLD. 24 h post-transfection, cells were lysed, and a portion of each lysate was incubated with anti-IKKα or nonspecific IgG antibodies. IB analysis of IP samples was performed to detect FLAG-tagged cFLIP constructs and IKKα. For all lysates, immunoblot analysis of whole-cell lysates was also performed. The asterisk denotes the heavy chain.
We next wanted to ask whether cFLIPL disrupted IRF7–IKKα interactions by competitive inhibition. Co-immunoprecipitations were performed to examine interactions between IKKα and cFLIPL (Fig. 5B). For this experiment, epitope-tagged IKKα and cFLIPL were co-overexpressed in 293T cells. Fig. 5B shows that cFLIPL indeed co-immunoprecipitated with IKKα. This was not unexpected given that a variant of cFLIPL (p43) was reported to bind to IKKα (54). As a control, we were also able to detect IKKα–IRF7 interactions in cells ectopically expressing IKKα and IRF7 (note that the thick band representing the heavy chain has a slightly different mobility than the IRF7-containing band) (Fig. 5B). Fig. 4 suggested that the DEDs of cFLIP were critical for IRF7 inhibition, whereas the CLD is dispensable. We performed co-immunoprecipitation to identify the cFLIP region that associated with IKKα. Indeed, cFLIPL and cFLIPS co-immunoprecipitated with IRF7 whereas the CLD did not (Fig. 5C), further supporting the model that IRF7–cFLIP interactions are critical for the inhibitory mechanism of cFLIPL.
cFLIPL inhibits IRF7 in the THP-1 and CAL-1 cell lines
The above experiments showed that cFLIPL inhibited IRF7 activation in HeLa and 293T cells. IRF7 is expressed at higher levels in hematopoietic cells like macrophages and pDCs (23, 55–57). If the cFLIP function identified in HeLa and 293T cells was relevant, then cFLIPL should antagonize IRF7 activation in these professional antigen-presenting cells (APCs). There were two possible ways to test this mechanism in physiologically relevant cell lines. We could silence endogenous cFLIPL and ask whether that results in an increase in IR7 activation and IFNα gene expression, However, this approach is technically difficult because cFLIPL is required for macrophages (58) because of the anti-apoptosis properties of cFLIPL (59, 60). In our hands, attempts at silencing cFLIPL also resulted in cell death, making it difficult to collect sufficient amounts of cells for experimentation. An alternative strategy is to overexpress cFLIPL and ask whether this correlates with a decrease in IRF7 phosphorylation and IFNα expression. This approach was feasible because cFLIPL was not expressed at high levels in the THP-1 and CAL-1 cells (Figs. 6, A and C, note that cFLIP was not detected in cells transduced with the control (con) lentivirus and subsequently left untreated or treated with CpG-A). When the cFLIPL gene (cflar) was stably introduced into the THP-1 human monocyte cell line via lentivirus transduction (61), cFLIPL protein expression was detected (Fig. 6). We picked this cell line because PMA-treated THP-1 cells differentiate to macrophage-like cells (62). In this state, THP-1 cells respond to CpG-A stimulation and express high levels of IRF7-controlled IFNα and interferon-stimulated gene transcripts (63, 64). We also transduced the CAL-1 cell line with the same cFLIPL-expressing lentivirus. The CAL-1 cell line was developed for use as a surrogate for primary pDCs to study type I IFN signaling and production (65). One benefit of using this cell line as opposed to primary human cells is that it avoids donor-to-donor variation. Although CAL-1 cells produce IFNα to a lesser extent than primary pDCs (65), the IRF7 signal transduction and activation pathway is maintained (66). As a control, a separate set of THP-1 and CAL-1 cells was transduced with lentiviruses that lacked the cFLIPL gene (depicted as control in Fig. 6).
Figure 6.
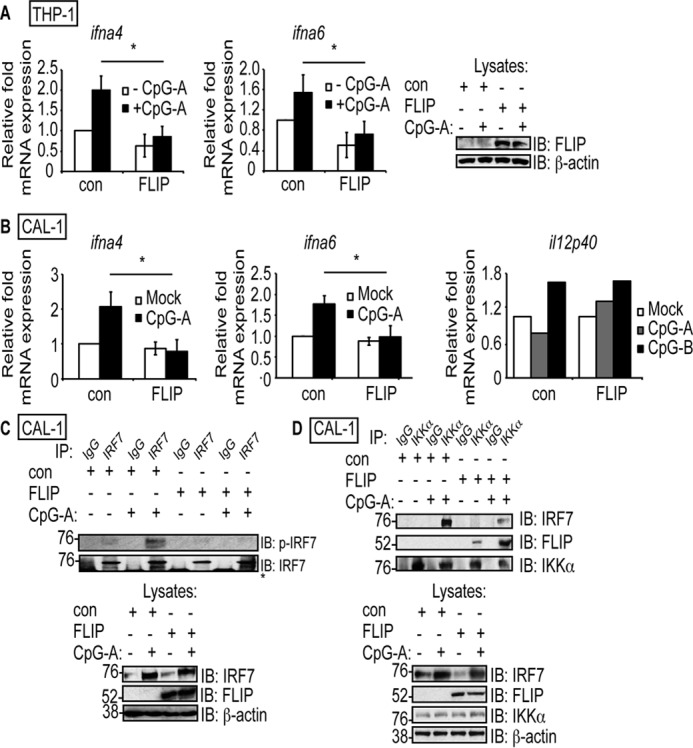
cFLIPL inhibits IRF7 activation and IKKα–IRF7 interactions in THP-1 and CAL-1 cell lines. A and B, THP-1 cells (A) and CAL-1 pDC cells (B) were transduced with a control lentivirus (con) or cFLIPL-expressing lentivirus (FLIP). THP-1 cells were differentiated into macrophages by treatment with PMA (10 ng/ml for 16 h). Transduced cells were incubated with medium lacking or containing 10 μm CpG-A or CpG-B for 5 h. Cells were lysed, and total RNA was extracted. A portion of each lysate was also used to detect cFLIPL protein expression. The levels of ifna4, ifna6, and il12p40 mRNA were quantified by using quantitative RT-PCR. C, transduced CAL-1 cells were incubated in medium lacking or containing 10 μm CpG-A for 5 h. Cells were then lysed, and a portion of lysate was immunoprecipitated with anti-IRF7 or nonspecific IgG antibodies. IB analysis of IP samples was performed to detect phospho-IRF7 or IRF7. A portion of each lysate prior to immunoprecipitation was also analyzed for expression of IRF7, cFLIPL, or β-actin. The asterisk denotes the heavy chain. D, transduced CAL-1 cells were incubated in medium lacking or containing 10 μm CpG-A for 5 h. Cells were then lysed, and a portion of lysate was immunoprecipitated with anti-IKKα or nonspecific IgG antibodies. IB analysis of IP samples was performed to detect endogenous IKKα, cFLIPL, or IRF7. A portion of each lysate prior to immunoprecipitation was also analyzed for protein expression.
Transduced THP-1 cells or CAL-1 cells were incubated with CpG-A to trigger IRF7 activation (57, 67, 68). The transcription of two genes known to be controlled by IRF7 homodimers (ifna4 and ifna6) was examined to assess the function of cFLIPL inhibition in both cell lines (69). As shown in Fig. 6, A and B, CpG-A–induced ifna4 and infa6 mRNA expression was significantly inhibited in cFLIPL-expressing THP-1 and CAL-1 cells, respectively, compared with cells transduced with a virus lacking the cFLIPL gene. As a control, the transcription of a gene not controlled by IRF7, il12p40 (2), was examined to assess the specificity of cFLIPL on TLR9-mediated, IRF7-driven transcription. CpG-B, but not CpG-A, will stimulate il12p40 expression (2). As shown in Fig. 6B, there was no significant difference in il12p40 mRNA levels in control or cFLIPL-expressing CAL-1 cells during CpG-B stimulation. There was a slight increase in il12p40 mRNA levels in cFLIPL-expressing cells versus control cells when CpG-A was used, and this may be due to the action of cFLIPL as an NF-κB activator (70). This suggests that the inhibitory role of cFLIPL is IRF7-specific, validating the luciferase results we observed in Fig. 1A.
Focusing on just CAL-1 cells, we observed that CpG-A–mediated IRF7 phosphorylation was decreased in CAL-1 cells expressing cFLIPL (Fig. 7). Fig. 7 shows cFLIPL co-immunoprecipitated with IKKα in both unstimulated and stimulated cells. Additionally, IKKα–IRF7 interactions were greatly reduced in cFLIPL-transduced cells versus cells transduced with an empty vector (Fig. 7). Thus, cFLIPL inhibits IRF7 activation by interacting with IKKα in antigen-presenting cells (Fig. 7). We attempted to examine IKKα interactions with endogenous cFLIPL but failed to reliably and consistently detect cFLIPL.
Figure 7.
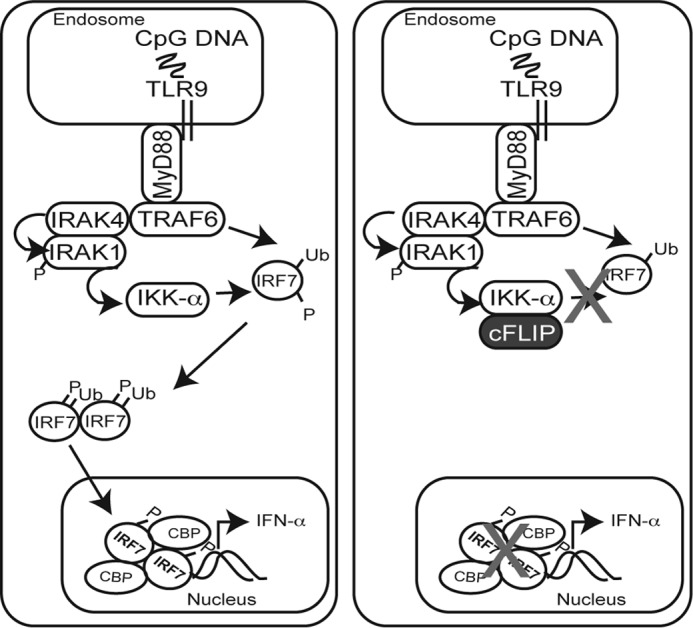
Proposed mechanism for cFLIP-mediated inhibition of IRF7-driven IFNα production. Activation of endosomal TLRs such as TLR7 by single-stranded RNAs and TLR9 by CpG motifs (e.g. CpG-A) leads to recruitment of the MyD88 protein. Next is the formation of a dynamic complex including at least IRAK4, IRAK1, and TRAF6. This complex triggers TRAF6-mediated Lys-63–linked ubiquitination of IRF7, followed by IRF7 phosphorylation. A current favored model proposes that IRAK4 phosphorylates IRAK1, leading to phosphorylation of IKKα. IKKα, in turn, activates IRF7. Phosphorylated IRF7 homodimerizes and translocates to the nucleus, where it drives expression of IFNα. The data shown here suggest that cFLIP binds to IKKα in a manner that prevents IKKα-mediated IRF7 phosphorylation and subsequent downstream IRF7 action.
Discussion
IRF7 is critical for IFNα gene expression (2–4). There is one previous report showing that cFLIP inhibits IFNα production (29). However, the antagonistic mechanism of cFLIP remained unknown. The goal here was to identify this function by examining the effect of cFLIPL on well-known signal transduction events of the TLR9-induced IRF7 activation pathway. We observed that cFLIPL prevented IRF7 phosphorylation. IKKα is one well-known IRF7 kinase (19). We performed co-immunoprecipitation assays and found that IRF7-IKKα interactions were abrogated by cFLIPL, concomitant with cFLIPL-IKKα interactions. Thus, we conclude that cFLIPL disrupts IRF7–IKKα interactions, interactions that are otherwise required for IRF7 activation (Fig. 7).
To the best of our knowledge, this is the first report of a cellular protein that disrupts IKKα–IRF7 interactions as a strategy to antagonize IRF7 activation. Most cellular IRF7 antagonists target IRF7 itself. For example, AIP binds to IRF7, and this interaction prevents IRF7 nuclear translocation (24). RAUL inhibits IRF7 (and IRF3) by targeting these IRFs for proteasomal degradation (21). Other proteins act indirectly on IRF7. Namely, transforming growth factor β1 promotes Lys-63–linked ubiquitination of TRAF6, which correlates with a decrease in IRF7 phosphorylation through unknown mechanisms (45). The myriad cellular strategies to decrease IRF7 activation are a testament to how the host cell has evolved multiple mechanisms to achieve immune system homeostasis.
Within the family of IRF proteins, IRF3 and IRF7 are most closely related (28). There are several lines of evidence showing that cFLIPL antagonizes IRF7 using a mechanism distinct from its strategy to antagonize IRF3. For example, cFLIPL inhibited CpG-A–induced IRF7 activation, a signaling pathway that does not activate IRF3 (2, 71, 72). Second, the domain of cFLIPL required for IRF7 activation (tandem DEDs) is distinct from the region required for IRF3 inhibition (CLD) (27). Third, cFLIPL co-immunoprecipitates with IRF3 but not IRF7 (27). Thus, cFLIPL has at least two separate mechanisms to antagonize type I IFN production in cells. These functions of cFLIPL may be useful considering that there is differential expression of IFNβ and IFNα by different cell types. For example, although IFNβ is produced largely by fibroblasts (73), the major expressers of IFNα are pDCs (6). Indeed, cFLIP is expressed in these cells, suggesting that cFLIP has evolved to control type I IFN production across various cell types (74). However, it appears that cFLIPL is not a pan-IRF inhibitor; cFLIPL did not inhibit IRF5-controlled il12p40-based luciferase activity in our hands.
There is one previous report that shows that cFLIPS inhibits type IFNα and IFNβ production (29). Buskiewicz et al. (29) proposed that cFLIP modulates the MAVS complex to inhibit IFNβ production, but the mechanism for inhibition of IFNα expression was not elucidated. We show here that both cFLIPS and cFLIPL inhibit IR7 activation and IFNα production. It is possible that this IKKα-binding property of cFLIP is responsible for the inhibition of IFNα production that was observed by Buskiewicz et al. (29).
There remains some controversy with respect to the roles of IRAK1 and IKKα as IRF7 kinases. Of course, each protein is critical for IFNα production (17, 19). However, it remains unknown whether IRAK1 phosphorylates IKKα, which then goes on to phosphorylate and activate IRF7, or whether IRAK1 and IKKα each phosphorylate IRF7 at different residues to activate IRF7 (52). In our hands, cFLIPL significantly reduces IRF7 phosphorylation while still allowing TRAF6–IRF7 interactions. Because TRAF6–IRF7 interactions occur downstream of IRAK1 kinase activity (17, 53), IRAK1 signaling events are probably not compromised in the presence of cFLIPL. Thus, we currently suspect that cFLIPL targets IKKα but not IRAK1.
We show here that cFLIPL co-immunoprecipitates with IKKα, resulting in a block in IRF7 activation. Neumann et al. (54) report that the p43 form of cFLIPL binds to IKKα and that this interaction activates the NF-κB pathway. It is unlikely that cFLIP-induced NF-κB activation indirectly contributed to IRF7 inhibition because NF-κB activation stimulates IRF7 expression during TNF stimulation (75). Nevertheless, it is quite interesting that cFLIPL and p43 appear to have diametrically opposed functions: cFLIPL inhibits IRF3 and IRF7, whereas p43 activates NF-κB (27, 54, 76). Thus, cFLIP may down-regulate type I IFN responses while still allowing expression of other cytokine or chemokine genes controlled by NF-κB. How this may balance an appropriate immune response remains a mystery.
Several groups target silencing of the cFLIP gene (cflar) to activate apoptosis in tumor cells that overexpress cFLIP (59, 60, 77). However, our data raise the possibility that overexpression of cFLIP may prove useful as a treatment for some types of autoimmune diseases to down-regulate IFNα production (7, 78). Thus, cFLIPL may be one protein that could be manipulated in more than one way to the benefit of human health.
Experimental procedures
Cell lines
The human embryonic kidney 293T, human cervical HeLa, and monocytic THP-1 human cell lines were obtained from the American Type Culture Collection. The CAL-1 plasmacytoid dendritic human cell line was kindly provided by Dr. Klinman (NCI, National Institutes of Health) and Dr. Maeda (Nagasaki University) (65). 293T and HeLa cells were cultured in minimum Eagle's medium supplemented with 10% FBS (Thermo Fisher Scientific) and 1% penicillin–streptomycin (Thermo Fisher Scientific). THP-1 and CAL-1 cells were cultured in RPMI medium supplemented with 10% FBS (Thermo Fisher Scientific) and 1% penicillin–streptomycin (Thermo Fisher Scientific).
Plasmids and transfections
Plasmid pCI was obtained from Promega. Plasmids encoding a FLAG-tagged human cFLIPL (pcFLIPL) or cFLIPS (pcFLIPS) were published previously (27). Plasmid pCLD encodes a FLAG-tagged caspase-like domain of cFLIPL (residues 178–480) and was a gift from Dr. Condorelli (University of Naples, Naples, Italy). Plasmid pIRF3CA, which expresses a constitutively active IRF3, pMAVS, which expresses the MAVS protein, and psnp11, which expresses the porcine respiratory virus nsp11 protein, were kind gifts from Dr. Yoo (University of Illinois). Plasmid pMyD88 was obtained from Dr. Richard Tapping (Department of Microbiology, University of Illinois). A plasmid encoding a GFP-tagged IRF3 (pIRF3) was a kind gift from Dr. Michelle Arnold (Louisiana State University Health Sciences Center, Shreveport, LA). Plasmids IRF7 (pIRF7) and pIFNα were provided by Dr. Fanxiu Zhu (Florida State University, Tallahassee, FL). Plasmid pIRF7CA, which expresses a constitutively active IRF7, and pIRF7DN, which expresses a dominant-negative IRF7, were kind gifts from Dr. Luciana Castiello (Instituto Pasteur, Rome, Italy). A plasmid encoding a myc-tagged TRAF6 protein was used in this work. Plasmid pIKKα encodes a FLAG-tagged IKKα protein and was a kind gift from Dr. Ulrich Siebenlist (National Institutes of Health, Bethesda, MD). Plasmid pAIP encodes a myc-tagged AIP protein and was a kind gift from Dr. Harhaj (Johns Hopkins University, Baltimore, MD). Plasmid pRL-TK was purchased from Promega. Plasmid pifna6-luc was kindly provided by Dr. Sun (Shanghai Institutes for Biological Sciences, Shanghai Shi, China). Plasmids pil12p40-luc, pVpx (encoding a FLAG-myc-hemagglutinin–tagged Vpx protein) and pIRF5 (encoding a GFP-tagged IRF5) were kindly provided Dr. Ratner (Washington University, St. Louis, MO). Plasmid DNA was transfected into cells using TransIT-2020 transfection reagent (Mirus Bio) following the protocol of the manufacturer.
Luciferase assays
Subconfluent 293T cellular monolayers were transfected with 50 ng of pRL-TK, 450 ng of pil12p40-luc, and either 500 ng of pIRF3CA, 500 ng of pIRF7, or 250 ng of pIRF5 and 250 ng of pTRAF6 to quantify IRF5 transcriptional activation. In this case, cells were additionally co-transfected with 1000 ng of pCI, pcFLIPL, or pVpx. To detect IRF7-specific induction of gene expression, 293T cells were transfected with 50 ng of pRL-TK, 450 ng of pinfa6-luc, and either 500 ng of pIRF3CA, 500 ng of pIRF7, or 250 ng of pIRF5 and 250 ng of pTRAF6. In this case, cells were additionally co-transfected with 1000 ng of pCI, pcFLIPL, or pAIP. To mimic myddosome-mediated, IRF7-driven gene expression, 293T cells were transfected with 50 ng of pRL-TK, 450 ng of pinfa6-luc, and either 1000 ng of pCI or 500 ng of pIRF7 and 500 ng of pMyD88, or 250 ng of pIRF7, 250 ng of pMyD88, 250 ng of pIKKα, and 250 ng of pTRAF6. In these cases, cells were additionally co-transfected with 1000 ng of pCI, pcFLIPL, or pIRF7DN. Additionally, 293T cells were co-transfected with 50 ng of pRL-TK, 450 ng of pinfa6-luc, either 500 ng of pCI or pIRF7CA, and 1000 ng of pCI, pcFLIPL, or pAIP. To detect IRF7 activation in HeLa cells, subconfluent cellular monolayers were transfected with 50 ng of pRL-TK, 450 ng of pinfa6-luc, 250 ng of pIFNα, and 1000 ng of pCI, pcFLIPL, or pAIP. 24 h post-transfection, HeLa cells were incubated in medium lacking or containing 3 μm CpG-A (ODN-2216, Invivogen) for 3 h. These same conditions were used to examine the effect of cFLIPS and CLD on IRF7 activation. In this case, HeLa cellular monolayers were transfected with 50 ng of pRL-TK, 450 ng of pinfa6-luc, 250 ng of pIFNα, and 1000 ng of pCI, pcFLIPL, pcFLIPS, or pCLD.
All cells were harvested 24–27 h post-transfection and lysed. Luciferase activities were detected using the Dual-Luciferase reporter assay system (Promega) and quantified using the Clarity luminescence microplate reader (BioTek Instruments). Analysis of firefly and sea pansy luciferase activities was performed as described previously (27). Values were normalized to those of untreated cells transfected with empty vectors. Values are shown as mean ± S.D. Student's t test was used to determine the statistical significance of inhibition of luciferase activity. A portion of each lysate was also analyzed for protein expression by immunoblotting. Luciferase assays are representative of three technical replicates, and all luciferase assays were performed at least three times.
Co-immunoprecipitations
To examine potential IRF7–cFLIPL interactions, subconfluent 293T cells were co-transfected with 500 ng of pIRF7 or 500 ng of pIRF3 and 1000 ng of pcFLIPL or pAIP. For HeLa cells, subconfluent monolayers were co-transfected with 1000 ng of pcFLIPL or pAIP. In experiments that examined IRF7–IKKα interactions, subconfluent 293T cells were co-transfected with 500 ng of pIRF7 and either 500 ng of pIKKα or pTRAF6 and 1000 ng of pCI or pcFLIPL. For co-immunoprecipitations of IKKα, 293T cells were transfected with 500 ng of pIKKα and 1000 ng of pCI, pcFLIPL, pIRF7, pcFLIPS, or pCLD. For CAL-1 cells, 108 control or cFLIPL-expressing transduced cells were treated with 10 μm CpG-A for 3 h. In all cases, cells were lysed in whole-cell lysis buffer (Abcam) 24 h post-transfection or after CpG-A treatment. Clarified supernatants were collected. A portion of each lysate was set aside for the purpose of detecting protein expression. The remaining sample was used for co-immunoprecipitations. Lysates were incubated with rabbit anti-IRF7 (Cell Signaling Technology), anti-IRF3 (Cell Signaling Technology), anti-IKKα (Cell Signaling Technology), or rabbit nonspecific IgG (Cell Signaling Technology) for 16 h at 4 °C. Protein G–Sepharose beads (Invitrogen) in a 50% slurry were added to each sample and incubated with rotation for 6 h. Beads were collected and washed three times. Pelleted beads were suspended in Laemmli buffer containing 5% 2-mercaptoethanol and boiled for 5 min. Samples were analyzed for the presence of proteins by using immunoblotting.
Immunoblotting
For all immunoblotting assays, the protein concentration of each lysate was determined by the 660-nm protein assay (Pierce). For phosphorylation assays, HeLa cells were seeded in 10-cm2 dishes, and samples were lysed in 100 μl of lysis buffer to concentrate protein levels. An equal amount of protein from each lysate was electrophoretically separated by SDS-PAGE. Proteins were transferred to polyvinylidene difluoride membranes (Millipore). Antibody–antigen reactions were detected by using chemiluminescence reagents (Amersham Biosciences and Thermo Scientific) and autoradiography. Primary antibodies included the following: monoclonal rabbit anti-IRF3 (Cell Signaling Technology), monoclonal mouse anti-FLAG (Sigma-Aldrich), monoclonal rabbit anti-FLAG (Sigma-Aldrich), monoclonal mouse β-actin (Calbiochem), monoclonal mouse anti-myc (Cell Signaling Technology), monoclonal rabbit anti-myc (Cell Signaling Technology), monoclonal rabbit anti-FLIP (Cell Signaling Technology), monoclonal mouse anti-FLIP (7F10, Enzo), mouse anti-GFP (Sigma-Aldrich), rabbit anti-IKKα (Cell Signaling Technology), mouse anti-IKKα (Santa Cruz Biotechnology), rabbit anti-IRF7 (Cell Signaling Technology), and rabbit anti-phospho-IRF7 (Cell Signaling Technology).
Transduction of cells with lentiviruses
Lentiviruses containing either cFLIPL (lenti-FLIP) or no transgene (lenti-con) were produced by co-transfecting 293T cells with the packaging plasmids pCMV-dR8.2 (Addgene, 4.5 μg) and pCMV–VSV-G (Addgene, 1.8 μg), and either an empty vector (pTRIP-IRES-GFP-control, 6 μg) or a plasmid containing the cFLIPL gene (pTRIP-cFLIPL-IRES-GFP, 6 μg) (61). 48 h post-transfection, lentiviruses were isolated from cellular supernatants. Lentiviruses were concentrated with Lent-X Concentrator (Clontech). The THP-1 or CAL-1 cell line was inoculated with lentiviruses by using spinfection. Briefly, 1 × 106 cells, 50 μl of concentrated virus, and 10 μg of Polybrene in 1 ml of virus medium (RPMI with 1% FBS) were centrifuged at 800 × g for 45 min at 37 °C. After spinfection, the medium was aspirated, and cells were resuspended in 1 ml of fresh medium (RPMI with 10% FBS) with 50 μl of concentrated virus and incubated at 37 °C. 24–72 h post-infection, GFP expression was used as a visual marker of transduction. Cellular populations with >80% GFP expression were passaged for use as stably transduced cell lines (THP-1 cells) or used immediately for experimentation (CAL-1 cells). Transduced THP-1 cells were passaged no more than four times, checking for GFP expression after each passage.
Quantitative RT-PCR
THP-1 cells were incubated in medium without or containing 10 ng/ml PMA for 16 h to differentiate cells into macrophage-like cells (62). Differentiated THP-1 or CAL-1 cells were stably transduced with a control lentivirus (lenti-con) or a lentivirus expressing cFLIPL (lenti-FLIP). Transduced cells were stimulated with 10 μm CpG-A for 5 h to stimulate the IRF7 signal transduction pathway (64). For il12p40 expression, transduced cells were stimulated with 10 μm CpG-B (ODN-2006, Invivogen) for 5 h. Total RNA was extracted from cells using the RNAeasy extraction kit (Qiagen). cDNA was generated using Moloney murine leukemia virus (M-MuLV) reverse transcriptase and poly(dT) oligonucleotides (New England Biolabs). Quantitative PCR was performed using a Mastercycler Realplex EP (Eppendorf) and SoFast EvaGreen Super Mix (Bio-Rad) according to the instructions of the manufacturer. The following primers were used to PCR-amplify cDNA: β-actin forward (5′-AGTTGCGTTACACCCTTTCT-3′), β-actinreverse (5′-ACCTTCACCGTTCCAGTTT-3′), ifna4 forward (5′-GATACTCCTGGCACAAATGG-3′), ifna4 reverse (5′-TCATGGAGGACAGAGATGG-3′), ifna6 forward (5′-CAGTTCCAGAAGGCTGAAG-3′), ifna6 reverse (5′-GAGTCCTTTGTGCTGAAGAG-3′), il12p40 forward (5′-AGAGCAGTGAGGTCTTAGG-3′), and il12p40 reverse (5′-CTTTGTGACAGGTGTACTGG-3′). Changes in gene expression levels were calculated by the 2ΔΔCt method (79). For normalization, respective β-actin mRNA quantities for each cDNA sample were measured, and then each value was normalized to that of unstimulated control cells, whose value was set to one. For all samples, data are presented as the mean ± S.D. from three independent experiments. Student's t test was used to determine statistically significant differences in mRNA expression levels compared with unstimulated cells.
Author contributions
L. T. G.-T. and J. L. S., conceptualization; L. T. G.-T., resources; L. T. G.-T., formal analysis; L. T. G.-T. and J. L. S., supervision; L. T. G.-T. and J. L. S., funding acquisition; L. T. G.-T., validation; L. T. G.-T., investigation; L. T. G.-T., visualization; L. T. G.-T., writing-original draft; J. L. S., methodology; J. L. S., project administration; J. L. S., writing-review and editing.
Acknowledgments
We thank Dr. Richard Tapping and Dr. Nick Hess for helpful discussions and Sunetra Biswas and Melissa Ryerson for critical review of the manuscript. This manuscript is part of the fulfillment of the Ph.D. requirements for L. T. G.
This work was supported by the University of Illinois and the National Institutes of Health. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- IFN
- interferon
- pDC
- plasmacytoid dendritic cell
- TLR
- Toll-like receptor
- AIP
- aryl hydrocarbon receptor–interacting protein
- cFLIP
- cellular FLICE-like inhibitory protein
- CA
- constitutively active
- HEK
- human embryonic kidney
- DN
- dominant-negative
- DED
- death effector domain
- CLD
- caspase-like domain
- PMA
- phorbol 12-myristate 13-acetate
- FBS
- fetal bovine serum
- cDNA
- complementary DNA
- IB
- immunoblot
- IP
- immunoprecipitation
- CBP
- CREB-binding protein
- CREB
- cAMP-response element-binding protein
- MAVS
- mitochondrial antiviral signaling
- IRES
- internal ribosome entry site.
References
- 1. Hoffmann H. H., Schneider W. M., and Rice C. M. (2015) Interferons and viruses: an evolutionary arms race of molecular interactions. Trends Immunol. 36, 124–138 10.1016/j.it.2015.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Honda K., Yanai H., Negishi H., Asagiri M., Sato M., Mizutani T., Shimada N., Ohba Y., Takaoka A., Yoshida N., and Taniguchi T. (2005) IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 434, 772–777 10.1038/nature03464 [DOI] [PubMed] [Google Scholar]
- 3. Honda K., Yanai H., Takaoka A., and Taniguchi T. (2005) Regulation of the type I IFN induction: a current view. Int. Immunol. 17, 1367–1378 10.1093/intimm/dxh318 [DOI] [PubMed] [Google Scholar]
- 4. Li W., Hofer M. J., Noçon A. L., Manders P., and Campbell I. L. (2013) Interferon regulatory factor 7 (IRF7) is required for the optimal initial control but not subsequent clearance of lymphocytic choriomeningitis virus infection in mice. Virology 439, 152–162 10.1016/j.virol.2013.02.015 [DOI] [PubMed] [Google Scholar]
- 5. Siegal F. P., Kadowaki N., Shodell M., Fitzgerald-Bocarsly P. A., Shah K., Ho S., Antonenko S., and Liu Y. J. (1999) The nature of the principal type 1 interferon-producing cells in human blood. Science 284, 1835–1837 10.1126/science.284.5421.1835 [DOI] [PubMed] [Google Scholar]
- 6. Ivashkiv L. B., and Donlin L. T. (2014) Regulation of type I interferon responses. Nat. Rev. Immunol. 14, 36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Crow M. K. (2014) Type I interferon in the pathogenesis of lupus. J. Immunol. 192, 5459–5468 10.4049/jimmunol.1002795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Baccala R., Kono D. H., and Theofilopoulos A. N. (2005) Interferons as pathogenic effectors in autoimmunity. Immunol. Rev. 204, 9–26 10.1111/j.0105-2896.2005.00252.x [DOI] [PubMed] [Google Scholar]
- 9. Higgs B. W., Liu Z., White B., Zhu W., White W. I., Morehouse C., Brohawn P., Kiener P. A., Richman L., Fiorentino D., Greenberg S. A., Jallal B., and Yao Y. (2011) Patients with systemic lupus erythematosus, myositis, rheumatoid arthritis and scleroderma share activation of a common type I interferon pathway. Ann. Rheum. Dis 70, 2029–2036 10.1136/ard.2011.150326 [DOI] [PubMed] [Google Scholar]
- 10. Zouali M. (2005) Molecular Autoimmunity: In Commemoration of the 100th Anniversary of the First Description of Human Autoimmune Disease, pp. 329–345, Springer, New York [Google Scholar]
- 11. Niewold T. B. (2014) Type I interferon in human autoimmunity. Front. Immunol. 5, 306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Krug A., Luker G. D., Barchet W., Leib D. A., Akira S., and Colonna M. (2004) Herpes simplex virus type 1 activates murine natural interferon-producing cells through Toll-like receptor 9. Blood 103, 1433–1437 [DOI] [PubMed] [Google Scholar]
- 13. Cortez-Gonzalez X., Pellicciotta I., Gerloni M., Wheeler M. C., Castiglioni P., Lenert P., and Zanetti M. (2006) TLR9-independent activation of B lymphocytes by bacterial DNA. DNA Cell Biol. 25, 253–261 10.1089/dna.2006.25.253 [DOI] [PubMed] [Google Scholar]
- 14. Tian J., Avalos A. M., Mao S. Y., Chen B., Senthil K., Wu H., Parroche P., Drabic S., Golenbock D., Sirois C., Hua J., An L. L., Audoly L., La Rosa G., Bierhaus A., et al. (2007) Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat. Immunol. 8, 487–496 10.1038/ni1457 [DOI] [PubMed] [Google Scholar]
- 15. Pradhan V. D., Das S., Surve P., and Ghosh K. (2012) Toll-like receptors in autoimmunity with special reference to systemic lupus erythematosus. Indian J. Hum. Genet. 18, 155–160 10.4103/0971-6866.100750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kawai T., Sato S., Ishii K. J., Coban C., Hemmi H., Yamamoto M., Terai K., Matsuda M., Inoue J., Uematsu S., Takeuchi O., and Akira S. (2004) Interferon-α induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat. Immunol. 5, 1061–1068 10.1038/ni1118 [DOI] [PubMed] [Google Scholar]
- 17. Uematsu S., Sato S., Yamamoto M., Hirotani T., Kato H., Takeshita F., Matsuda M., Coban C., Ishii K. J., Kawai T., Takeuchi O., and Akira S. (2005) Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-α induction. J. Exp. Med. 201, 915–923 10.1084/jem.20042372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferrao R., Zhou H., Shan Y., Liu Q., Li Q., Shaw D. E., Li X., and Wu H. (2014) IRAK4 dimerization and trans-autophosphorylation are induced by myddosome assembly. Mol. Cell 55, 891–903 10.1016/j.molcel.2014.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hoshino K., Sasaki I., Sugiyama T., Yano T., Yamazaki C., Yasui T., Kikutani H., and Kaisho T. (2010) Critical role of IκB kinase α in TLR7/9-induced type I IFN production by conventional dendritic cells. J. Immunol. 184, 3341–3345 10.4049/jimmunol.0901648 [DOI] [PubMed] [Google Scholar]
- 20. Marié I., Smith E., Prakash A., and Levy D. E. (2000) Phosphorylation-induced dimerization of interferon regulatory factor 7 unmasks DNA binding and a bipartite transactivation domain. Mol. Cell. Biol. 20, 8803–8814 10.1128/MCB.20.23.8803-8814.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yu Y., and Hayward G. S. (2010) The ubiquitin E3 ligase RAUL negatively regulates type I interferon through ubiquitination of the transcription factors IRF7 and IRF3. Immunity 33, 863–877 10.1016/j.immuni.2010.11.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Long L., Deng Y., Yao F., Guan D., Feng Y., Jiang H., Li X., Hu P., Lu X., Wang H., Li J., Gao X., and Xie D. (2014) Recruitment of phosphatase PP2A by RACK1 adaptor protein deactivates transcription factor IRF3 and limits type I interferon signaling. Immunity 40, 515–529 10.1016/j.immuni.2014.01.015 [DOI] [PubMed] [Google Scholar]
- 23. Colina R., Costa-Mattioli M., Dowling R. J., Jaramillo M., Tai L. H., Breitbach C. J., Martineau Y., Larsson O., Rong L., Svitkin Y. V., Makrigiannis A. P., Bell J. C., and Sonenberg N. (2008) Translational control of the innate immune response through IRF-7. Nature 452, 323–328 10.1038/nature06730 [DOI] [PubMed] [Google Scholar]
- 24. Zhou Q., Lavorgna A., Bowman M., Hiscott J., and Harhaj E. W. (2015) Aryl hydrocarbon receptor interacting protein targets IRF7 to suppress antiviral signaling and the induction of type I interferon. J. Biol. Chem. 290, 14729–14739 10.1074/jbc.M114.633065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Irmler M., Thome M., Hahne M., Schneider P., Hofmann K., Steiner V., Bodmer J. L., Schröter M., Burns K., Mattmann C., Rimoldi D., French L. E., and Tschopp J. (1997) Inhibition of death receptor signals by cellular FLIP. Nature 388, 190–195 10.1038/40657 [DOI] [PubMed] [Google Scholar]
- 26. Valmiki M. G., and Ramos J. W. (2009) Death effector domain-containing proteins. Cell Mol. Life Sci. 66, 814–830 10.1007/s00018-008-8489-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gates L. T., and Shisler J. L. (2016) cFLIPL interrupts IRF3-CBP-DNA interactions to inhibit IRF3-driven transcription. J. Immunol. 197, 923–933 10.4049/jimmunol.1502611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang L., and Pagano J. S. (1997) IRF-7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell. Biol. 17, 5748–5757 10.1128/MCB.17.10.5748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Buskiewicz I. A., Koenig A., Roberts B., Russell J., Shi C., Lee S. H., Jung J. U., Huber S. A., and Budd R. C. (2014) c-FLIP-Short reduces type I interferon production and increases viremia with Coxsackievirus B3. PLoS ONE 9, e96156 10.1371/journal.pone.0096156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Higgs R., and Jefferies C. A. (2008) Targeting IRFs by ubiquitination: regulating antiviral responses. Biochem. Soc. Trans. 36, 453–458 10.1042/BST0360453 [DOI] [PubMed] [Google Scholar]
- 31. Wang P., Zhao W., Zhao K., Zhang L., and Gao C. (2015) TRIM26 negatively regulates interferon-β production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 11, e1004726 10.1371/journal.ppat.1004726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cheng X., and Ratner L. (2014) HIV-2 Vpx protein interacts with interferon regulatory factor 5 (IRF5) and inhibits its function. J. Biol. Chem. 289, 9146–9157 10.1074/jbc.M113.534321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yang L., Zhao T., Shi X., Nakhaei P., Wang Y., Sun Q., Hiscott J., and Lin R. (2009) Functional analysis of a dominant negative mutation of interferon regulatory factor 5. PLoS ONE 4, e5500 10.1371/journal.pone.0005500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y., Yan S., Yang B., Wang Y., Zhou H., Lian Q., and Sun B. (2015) TRIM35 negatively regulates TLR7- and TLR9-mediated type I interferon production by targeting IRF7. FEBS Lett. 589, 1322–1330 10.1016/j.febslet.2015.04.019 [DOI] [PubMed] [Google Scholar]
- 35. Chang Foreman H. C., Van Scoy S., Cheng T. F., and Reich N. C. (2012) Activation of interferon regulatory factor 5 by site specific phosphorylation. PLoS ONE 7, e33098 10.1371/journal.pone.0033098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin R., Mamane Y., and Hiscott J. (2000) Multiple regulatory domains control IRF-7 activity in response to virus infection. J. Biol. Chem. 275, 34320–34327 10.1074/jbc.M002814200 [DOI] [PubMed] [Google Scholar]
- 37. Liang Q., Deng H., Sun C. W., Townes T. M., and Zhu F. (2011) Negative regulation of IRF7 activation by activating transcription factor 4 suggests a cross-regulation between the IFN responses and the cellular integrated stress responses. J. Immunol. 186, 1001–1010 10.4049/jimmunol.1002240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang J., Yang B., Hu Y., Zheng Y., Zhou H., Wang Y., Ma Y., Mao K., Yang L., Lin G., Ji Y., Wu X., and Sun B. (2013) Negative regulation of Nmi on virus-triggered type I IFN production by targeting IRF7. J. Immunol. 191, 3393–3399 10.4049/jimmunol.1300740 [DOI] [PubMed] [Google Scholar]
- 39. Sasaki I., Hoshino K., Sugiyama T., Yamazaki C., Yano T., Iizuka A., Hemmi H., Tanaka T., Saito M., Sugiyama M., Fukuda Y., Ohta T., Sato K., Ainai A., Suzuki T., et al. (2012) Spi-B is critical for plasmacytoid dendritic cell function and development. Blood 120, 4733–4743 10.1182/blood-2012-06-436527 [DOI] [PubMed] [Google Scholar]
- 40. Marié I., Durbin J. E., and Levy D. E. (1998) Differential viral induction of distinct interferon-α genes by positive feedback through interferon regulatory factor-7. EMBO J. 17, 6660–6669 10.1093/emboj/17.22.6660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kitagawa Y., Yamaguchi M., Zhou M., Nishio M., Itoh M., and Gotoh B. (2013) Human parainfluenza virus type 2 V protein inhibits TRAF6-mediated ubiquitination of IRF7 to prevent TLR7- and TLR9-dependent interferon induction. J. Virol. 87, 7966–7976 10.1128/JVI.03525-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao G. N., Jiang D. S., and Li H. (2015) Interferon regulatory factors: at the crossroads of immunity, metabolism, and disease. Biochim. Biophys. Acta 1852, 365–378 10.1016/j.bbadis.2014.04.030 [DOI] [PubMed] [Google Scholar]
- 43. Lu R., Au W. C., Yeow W. S., Hageman N., and Pitha P. M. (2000) Regulation of the promoter activity of interferon regulatory factor-7 gene: activation by interferon and silencing by hypermethylation. J. Biol. Chem. 275, 31805–31812 10.1074/jbc.M005288200 [DOI] [PubMed] [Google Scholar]
- 44. Yang S., Zhan Y., Zhou Y., Jiang Y., Zheng X., Yu L., Tong W., Gao F., Li L., Huang Q., Ma Z., and Tong G. (2016) Interferon regulatory factor 3 is a key regulation factor for inducing the expression of SAMHD1 in antiviral innate immunity. Sci. Rep. 6, 29665 10.1038/srep29665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Naiki Y., Komatsu T., Koide N., Dagvadorj J., Yoshida T., Arditi M., and Yokochi T. (2015) TGF-β1 inhibits the production of IFN in response to CpG DNA via ubiquitination of TNF receptor-associated factor (TRAF) 6. Innate Immun. 21, 770–777 10.1177/1753425915596844 [DOI] [PubMed] [Google Scholar]
- 46. Song Y. J., Izumi K. M., Shinners N. P., Gewurz B. E., and Kieff E. (2008) IRF7 activation by Epstein-Barr virus latent membrane protein 1 requires localization at activation sites and TRAF6, but not TRAF2 or TRAF3. Proc. Natl. Acad. Sci. U.S.A. 105, 18448–18453 10.1073/pnas.0809933105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang H., Lin C. H., Ma G., Baffi M. O., and Wathelet M. G. (2003) Interferon regulatory factor-7 synergizes with other transcription factors through multiple interactions with p300/CBP coactivators. J. Biol. Chem. 278, 15495–15504 10.1074/jbc.M212940200 [DOI] [PubMed] [Google Scholar]
- 48. Ning S., Campos A. D., Darnay B. G., Bentz G. L., and Pagano J. S. (2008) TRAF6 and the three C-terminal lysine sites on IRF7 are required for its ubiquitination-mediated activation by the tumor necrosis factor receptor family member latent membrane protein 1. Mol. Cell. Biol. 28, 6536–6546 10.1128/MCB.00785-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Clark K., Takeuchi O., Akira S., and Cohen P. (2011) The TRAF-associated protein TANK facilitates cross-talk within the IκB kinase family during Toll-like receptor signaling. Proc. Natl. Acad. Sci. U.S.A. 108, 17093–17098 10.1073/pnas.1114194108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Randall C. M., Biswas S., Selen C. V., and Shisler J. L. (2014) Inhibition of interferon gene activation by death-effector domain-containing proteins from the molluscum contagiosum virus. Proc. Natl. Acad. Sci. U.S.A. 111, E265–272 10.1073/pnas.1314569111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu S., Cai X., Wu J., Cong Q., Chen X., Li T., Du F., Ren J., Wu Y. T., Grishin N. V., and Chen Z. J. (2015) Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630 10.1126/science.aaa2630 [DOI] [PubMed] [Google Scholar]
- 52. Honda K., and Taniguchi T. (2006) IRFs: master regulators of signalling by Toll-like receptors and cytosolic pattern-recognition receptors. Nat. Rev. Immunol. 6, 644–658 10.1038/nri1900 [DOI] [PubMed] [Google Scholar]
- 53. Qian Y., Commane M., Ninomiya-Tsuji J., Matsumoto K., and Li X. (2001) IRAK-mediated translocation of TRAF6 and TAB2 in the interleukin-1-induced activation of NFκB. J. Biol. Chem. 276, 41661–41667 10.1074/jbc.M102262200 [DOI] [PubMed] [Google Scholar]
- 54. Neumann L., Pforr C., Beaudouin J., Pappa A., Fricker N., Krammer P. H., Lavrik I. N., and Eils R. (2010) Dynamics within the CD95 death-inducing signaling complex decide life and death of cells. Mol. Syst. Biol. 6, 352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sun L., Zhu Z., Cheng N., Yan Q., and Ye R. D. (2014) Serum amyloid A induces interleukin-33 expression through an IRF7-dependent pathway. Eur. J. Immunol. 44, 2153–2164 10.1002/eji.201344310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ning S., Pagano J. S., and Barber G. N. (2011) IRF7: activation, regulation, modification and function. Genes Immun. 12, 399–414 10.1038/gene.2011.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kui L., Chan G. C., and Lee P. P. (2017) TSG-6 Downregulates IFN-α and TNF-α expression by suppressing IRF7 phosphorylation in human plasmacytoid dendritic cells. Mediators Inflamm. 2017, 7462945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Gordy C., Liang J., Pua H., and He Y. W. (2014) c-FLIP protects eosinophils from TNF-α-mediated cell death in vivo. PLoS ONE 9, e107724 10.1371/journal.pone.0107724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wilson T. R., McLaughlin K. M., McEwan M., Sakai H., Rogers K. M., Redmond K. M., Johnston P. G., and Longley D. B. (2007) c-FLIP: a key regulator of colorectal cancer cell death. Cancer Res. 67, 5754–5762 10.1158/0008-5472.CAN-06-3585 [DOI] [PubMed] [Google Scholar]
- 60. Day T. W., Sinn A. L., Huang S., Pollok K. E., Sandusky G. E., and Safa A. R. (2009) c-FLIP gene silencing eliminates tumor cells in breast cancer xenografts without affecting stromal cells. Anticancer Res. 29, 3883–3886 [PubMed] [Google Scholar]
- 61. Wu Y. H., Kuo W. C., Wu Y. J., Yang K. T., Chen S. T., Jiang S. T., Gordy C., He Y. W., and Lai M. Z. (2014) Participation of c-FLIP in NLRP3 and AIM2 inflammasome activation. Cell Death Differ. 21, 451–461 10.1038/cdd.2013.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Park E. K., Jung H. S., Yang H. I., Yoo M. C., Kim C., and Kim K. S. (2007) Optimized THP-1 differentiation is required for the detection of responses to weak stimuli. Inflamm. Res. 56, 45–50 10.1007/s00011-007-6115-5 [DOI] [PubMed] [Google Scholar]
- 63. Chen R. F., Wang L., Cheng J. T., and Yang K. D. (2012) Induction of IFNα or IL-12 depends on differentiation of THP-1 cells in dengue infections without and with antibody enhancement. BMC Infect. Dis. 12, 340 10.1186/1471-2334-12-340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Teofilović N. K., Bihi M., Stojković M. R., Tumir L. M., Ester K., Kralj M., Majhen D., Oršolić N., Lepur A., Vrbanec D., Markotić A., Dembić Z., Weber A. N., Piantanida I., Vugrek O., et al. (2017) 1-Ethyl-3-(6-methylphenanthridine-8-il) urea modulates TLR3/9 activation and induces selective pro-inflammatory cytokine expression in vitro. Bioorg. Med. Chem. Lett. 27, 1530–1537 10.1016/j.bmcl.2017.02.048 [DOI] [PubMed] [Google Scholar]
- 65. Maeda T., Murata K., Fukushima T., Sugahara K., Tsuruda K., Anami M., Onimaru Y., Tsukasaki K., Tomonaga M., Moriuchi R., Hasegawa H., Yamada Y., and Kamihira S. (2005) A novel plasmacytoid dendritic cell line, CAL-1, established from a patient with blastic natural killer cell lymphoma. Int. J. Hematol. 81, 148–154 10.1532/IJH97.04116 [DOI] [PubMed] [Google Scholar]
- 66. Balzarolo M., Karrich J. J., Engels S., Blom B., Medema J. P., and Wolkers M. C. (2012) The transcriptional regulator NAB2 reveals a two-step induction of TRAIL in activated plasmacytoid DCs. Eur. J. Immunol. 42, 3019–3027 10.1002/eji.201242385 [DOI] [PubMed] [Google Scholar]
- 67. Sweeney S. E. (2011) Targeting interferon regulatory factors to inhibit activation of the type I IFN response: implications for treatment of autoimmune disorders. Cell. Immunol. 271, 342–349 10.1016/j.cellimm.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Tun-Kyi A., Finn G., Greenwood A., Nowak M., Lee T. H., Asara J. M., Tsokos G. C., Fitzgerald K., Israel E., Li X., Exley M., Nicholson L. K., and Lu K. P. (2011) Essential role for the prolyl isomerase Pin1 in Toll-like receptor signaling and type I interferon-mediated immunity. Nat. Immunol. 12, 733–741 10.1038/ni.2069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Julian M. W., Shao G., Bao S., Knoell D. L., Papenfuss T. L., VanGundy Z. C., and Crouser E. D. (2012) Mitochondrial transcription factor A serves as a danger signal by augmenting plasmacytoid dendritic cell responses to DNA. J. Immunol. 189, 433–443 10.4049/jimmunol.1101375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Golks A., Brenner D., Krammer P. H., and Lavrik I. N. (2006) The c-FLIP-NH2 terminus (p22-FLIP) induces NF-κB activation. J. Exp. Med. 203, 1295–1305 10.1084/jem.20051556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kerkmann M., Rothenfusser S., Hornung V., Towarowski A., Wagner M., Sarris A., Giese T., Endres S., and Hartmann G. (2003) Activation with CpG-A and CpG-B oligonucleotides reveals two distinct regulatory pathways of type I IFN synthesis in human plasmacytoid dendritic cells. J. Immunol. 170, 4465–4474 10.4049/jimmunol.170.9.4465 [DOI] [PubMed] [Google Scholar]
- 72. Pelka K., and Latz E. (2013) IRF5, IRF8, and IRF7 in human pDCs: the good, the bad, and the insignificant? Eur. J. Immunol. 43, 1693–1697 10.1002/eji.201343739 [DOI] [PubMed] [Google Scholar]
- 73. Reder A. T., and Feng X. (2013) Aberrant type I interferon regulation in autoimmunity: opposite directions in MS and SLE, Shaped by evolution and body ecology. Front. Immunol. 4, 281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Uhlén M., Fagerberg L., Hallström B. M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson Å., Kampf C., Sjöstedt E., Asplund A., Olsson I., Edlund K., Lundberg E., Navani S., Szigyarto C. A., et al. (2015) Proteomics: tissue-based map of the human proteome. Science 347, 1260419 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- 75. Chang D. W., Xing Z., Pan Y., Algeciras-Schimnich A., Barnhart B. C., Yaish-Ohad S., Peter M. E., and Yang X. (2002) c-FLIP(L) is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 21, 3704–3714 10.1093/emboj/cdf356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Baratchian M., Davis C. A., Shimizu A., Escors D., Bagnéris C., Barrett T., and Collins M. K. (2016) Distinct activation mechanisms of NF-κB regulator inhibitor of NF-κB kinase (IKK) by isoforms of the cell death regulator cellular FLICE-like inhibitory protein (cFLIP). J. Biol. Chem. 291, 7608–7620 10.1074/jbc.M116.718122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Safa A. R. (2012) c-FLIP, a master anti-apoptotic regulator. Exp. Oncol. 34, 176–184 [PMC free article] [PubMed] [Google Scholar]
- 78. Hirai M., Kadowaki N., Kitawaki T., Fujita H., Takaori-Kondo A., Fukui R., Miyake K., Maeda T., Kamihira S., Miyachi Y., and Uchiyama T. (2011) Bortezomib suppresses function and survival of plasmacytoid dendritic cells by targeting intracellular trafficking of Toll-like receptors and endoplasmic reticulum homeostasis. Blood 117, 500–509 10.1182/blood-2010-05-284737 [DOI] [PubMed] [Google Scholar]
- 79. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(−ΔΔ C(T)) Method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]