

Abstract
Objective
To evaluate the diagnostic yield and workflow of genome-scale sequencing in patients with neuromuscular disorders (NMDs).
Methods
We performed exome sequencing in 93 undiagnosed patients with various NMDs for whom a molecular diagnosis was not yet established. Variants on both targeted and broad diagnostic gene lists were identified. Prior diagnostic tests were extracted from the patient's medical record to evaluate the use of exome sequencing in the context of their prior diagnostic workup.
Results
The overall diagnostic yield of exome sequencing in our cohort was 12.9%, with one or more pathogenic or likely pathogenic variants identified in a causative gene associated with the patient's disorder. Targeted gene lists had the same diagnostic yield as a broad NMD gene list in patients with clear neuropathy or myopathy phenotypes, but evaluation of a broader set of disease genes was needed for patients with complex NMD phenotypes. Most patients with NMD had undergone prior testing, but only 10/16 (63%) of these procedures, such as muscle biopsy, were informative in pointing to a final molecular diagnosis.
Conclusions
Genome-scale sequencing or analysis of a panel of relevant genes used early in the evaluation of patients with NMDs can provide or clarify a diagnosis and minimize invasive testing in many cases.
Neuromuscular disorders (NMDs) are a heterogeneous group of disorders arising from nerve, muscle, or neuromuscular junction dysfunction and present with a variety of features including weakness, numbness, contractures, and symptoms involving other systems.1 Given the overlap of clinical features often seen in the various types of NMDs, localization of the specific lesion and diagnosis of the precise cause can be challenging. Accurate genetic diagnosis is becoming more important, as increasing numbers of experimental and clinical therapeutics are gene specific and/or mutation specific.2 Important questions remain, however, regarding the precise role of genetic testing and its position in the diagnostic algorithm for patients with suspected NMDs.3,4
As part of the North Carolina Clinical Genomic Evaluation by Next-generation Exome Sequencing (NCGENES) study, which aims to evaluate the use of exome sequencing as a diagnostic tool in a broad array of diseases, we evaluated the diagnostic yield of whole-exome sequencing (WES) in 93 patients with NMD with a previously unrevealing workup. Our study sought to answer whether genome-scale sequencing could provide or clarify a diagnosis in NMD cases in which other testing had failed to pinpoint a specific diagnosis. We specifically compared the diagnostic yield of analyzing sequence data from a subset of genes, closely related to the patient's phenotype, vs a larger set consisting of almost all known genes related to any neuromuscular disorder. Our analysis shows that patients with NMD can benefit from early application of genomic sequencing in their diagnostic workup.
Methods
Whole-exome sequencing
A cohort of 93 patients (<1–77 years of age, mean age 44 years) with suspected heritable NMDs from the NCGENES study had WES performed as part of their research evaluation. Genomic DNA was isolated from 10 mL of whole blood using PureGene chemistry in the Biospecimen Processing Facility, University of North Carolina (UNC). Library preparation, including molecular barcoding and exome capture, was done using Agilent SureSelect XT kits (Human All Exon versions 4 and 5) according to the manufacturer's guidelines. Sequencing was performed in the UNC High-Throughput Sequencing Facility on an Illumina HiSeq2500 at an average depth of 50×. Mapping (hg19), alignment, and variant calling were performed according to the Broad Institute's best practices using the Burrows-Wheeler Alignment Tool and Genome Analysis Toolkit.5,6
Variant annotation and interpretation
Exome variants were loaded into a PostgresSQL database (version 9.0.3) for annotation and facilitation of queries.6 Variants were computationally annotated with genomic position, effect on the translated protein, frequency within the Exome Aggregation Consortium database (n = 60,706 exomes),7 presence in the Human Gene Mutation Database as a “Disease Mutation,”8 and presence in ClinVar9 and predicted the effect on the resulting protein. Variants of interest were further annotated with functional domain information from the RefSeq database and with in silico pathogenicity predictions using CADD.10 Variants were then evaluated according to American College of Medical Genetics and Genomics guidelines.11
Standard protocol approvals, registrations, and patient consents
We received full approval from the UNC Institutional Review Board to perform this study. Written informed consent was obtained from all patients or guardians of patients participating in this study.
Analysis
Yield of gene lists of different size
Molecular analysis of variants identified by genome-scale sequencing is complex, particularly in singletons. Accordingly, in singletons, many laboratories evaluate variants that are present only in established disease genes related to the patient's phenotype. In this study, we evaluated the diagnostic yield when variants were filtered from a predefined list of genes associated with specific neuromuscular phenotypes matched to an individual's primary suspected diagnosis vs a list encompassing genes associated with a broad range of NMD phenotypes. Diagnostic gene lists were created from curation of the Online Mendelian Inheritance in Man database, as well as through searches of the medical literature and examination of commercial gene panels. Three diagnostic gene lists were used for analysis: a neuropathy list, containing 199 genes implicated in neuropathy phenotypes; a myopathy list focused on myopathy phenotypes consisting of 181 genes, and a third, broader list that contained all genes in the neuropathy and myopathy lists as well as over 100 additional genes, for a total of 482 genes associated with NMD phenotypes (table e-1, http://links.lww.com/NXG/A28, figure e-1, http://links.lww.com/NXG/A27). Gene-disease associations were required to have an asserted association with human disease (no “candidate genes” were included), but the strength of the evidence was not strictly evaluated before variant analysis. Postanalysis review of the clinical validity of the gene-disease association was done when qualifying variants were identified.
Patients were grouped into 3 categories based on their clinical phenotype after evaluation by a board-certified neurologist and neuromuscular disease specialist at one or more regular clinic visits before study enrollment. Evaluation consisted of a thorough physical examination and analysis of the results of any ordered relevant clinical testing, including imaging, biopsy, and genetic testing. Patients were categorized as those with a suspected primary neuropathy or suspected primary myopathy (including muscular dystrophy and neuromuscular junction disorder). Those patients with complex or nonspecific phenotypic features including the neuromuscular system that did not clearly suggest a primary neuropathy or myopathy were placed into the “complex” category. Patient sequence data were first filtered on the narrow list and then filtered on the larger NMD list. For example, a patient with a myopathy phenotype would be analyzed using the “myopathy” gene list, and then the broader “NMD” gene list, thus simulating the diagnostic yield of 2 different gene panels tested on the same patient. Case-level molecular results were classified as follows:
Positive—“definitive”: cases in which known pathogenic variants were identified with zygosity matching the inheritance pattern of a disorder matching the patient's phenotype
Positive—“probable”: cases in which likely pathogenic variants were identified with zygosity matching the inheritance pattern of a disorder matching the patient's phenotype.
Possible—“uncertain”: cases in which variants of uncertain significance (VUS) were identified with zygosity matching the inheritance pattern
Possible—“single heterozygous”: cases in which a single heterozygous known pathogenic or likely pathogenic variant was identified in a gene associated with recessive inheritance of a disorder closely matching the patient's phenotype, suggesting the possibility of a second variant in trans that was missed.
Possible—“other”: cases in which uncertainty existed for other reasons (e.g., the genetic finding might provide a partial but not full explanation for the patient's features; the clinical presentation of the patient might not match with the known phenotypic spectrum of the condition)
Yield of muscle biopsies and prior single gene testing
An author (R.J.G.B.) abstracted genetic testing data and prior diagnostic workup for each patient with NMD enrolled in the NCGENES study from the institution's electronic medical record. Three reviewers (M.C.A, K.A., and Z.F.), including a neurologist, independently evaluated pathology reports from muscle biopsies and reports from electrodiagnostic testing (defined as nerve conduction studies or electromyograms) on patients with positive genetic testing. The test was then classified as either “informative” or “noninformative” on the basis of (1) whether it assisted with the disease categorization (i.e., neuropathy or myopathy) or, in patients whose disease categorization was already known, (2) whether it further differentiated the type of neuropathy or myopathy present (e.g., distal vs proximal).
Results
The NCGENES project performed exome sequencing in a total of 643 patients; 93 of which were categorized as having primarily NMDs. Many of the patients who were enrolled in the study had already undergone genetic testing, and individuals with a known molecular diagnosis were excluded from the study.
Diagnostic yield of genome-scale sequencing
Patients judged on clinical grounds to have a predominant neuropathy phenotype (N = 21) were analyzed with the neuropathy diagnostic list as well as the comprehensive NMD list. Patients with a clinical presentation consistent with a myopathy phenotype (N = 31) were analyzed with the myopathy diagnostic list and the comprehensive NMD list. Complex patients (N = 41) with neuropathy, myopathy, and additional phenotypes (e.g., intellectual disability and retinopathy) were analyzed with all 3 diagnostic gene lists. The overall diagnostic yield of exome sequencing (“positive-definitive” or “positive-probable” case-level results) in our cohort of 93 diverse, unselected, unsolved singleton patients with NMD was 12.9%, with the yield varying depending on which gene lists were used for filtering variants (table 1). The variants identified in these 12 patients, as well as their presumptive diagnoses, are listed in table 2. In some cases, cascade testing was performed on informative family members to phase variants and support pathogenicity.
Table 1.
Positive diagnostic yield of gene lists by the patient phenotype
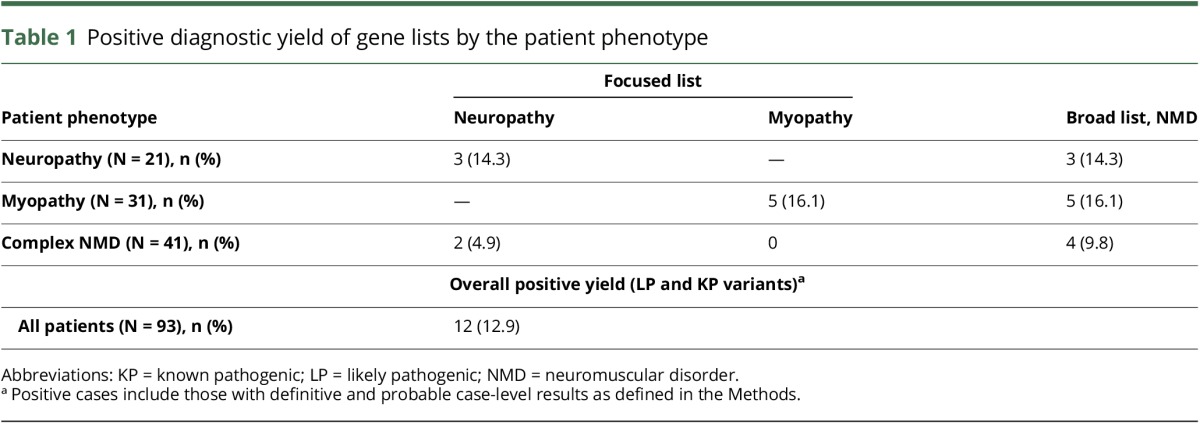
Table 2.
Pathogenic and likely pathogenic variants returned to patients with NMD as definitive or probable


In patients with a clear neuropathy or myopathy phenotype, a focused diagnostic gene list had the same diagnostic yield as a broader diagnostic gene list (14.3% and 16.1% for neuropathy and myopathy, respectively). In patients with a complex phenotype, a broader diagnostic gene list resulted in higher diagnostic yield (9.8%) compared with using neuropathy (4.9%) or myopathy (0%) diagnostic gene lists alone (table 1).
As expected, analyzing more genes increases the number of VUS identified. In our cohort, analyzing individuals in whom a diagnosis had been discovered through sequencing for variants in genes related to a phenotype they are not known to have (e.g., analyzing neuropathy patients with a myopathy diagnostic list) generated between 5 and 8 VUS. Because only patients with an existing positive diagnostic result were considered, any additional variants in other genes would most likely be irrelevant rather than possible diagnostic results, except in very rare cases of multiple genetic conditions resulting in a “blended phenotype” (no clear examples of this were seen among our cases).
Prior testing
Retrospective analysis of the NMD patient cohort revealed that most individuals had undergone an extensive prior workup, often over a period of many years. Almost half (43%) of patients had undergone a muscle biopsy before enrollment in this study, and almost 10% had undergone a nerve biopsy. A majority (86%) had prior electrodiagnostic testing. Furthermore, most patients (67.7%) had previously had negative single gene testing, with an average of roughly 2 genes tested per patient, and 20.4% of patients had prior multigene panel testing, all with negative results (since not having received a molecular diagnosis was necessary for participation in the study). All prior diagnostic workup is summarized in figure 1, illustrating the challenging nature of obtaining an accurate diagnosis in such patients.
Figure 1. Summary of prior diagnostic workup in neuromuscular disorder cases.
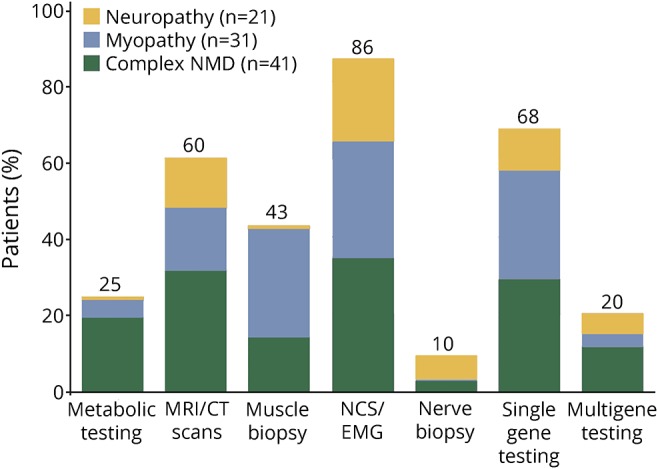
Percentage of patients in each phenotypic category with prior metabolic testing, MRI/CT scan, muscle biopsy, nerve conduction testing/electromyogram, nerve biopsy, single gene testing, and multigene testing. NCS = nerve conduction study; NMD = neuromuscular disorder.
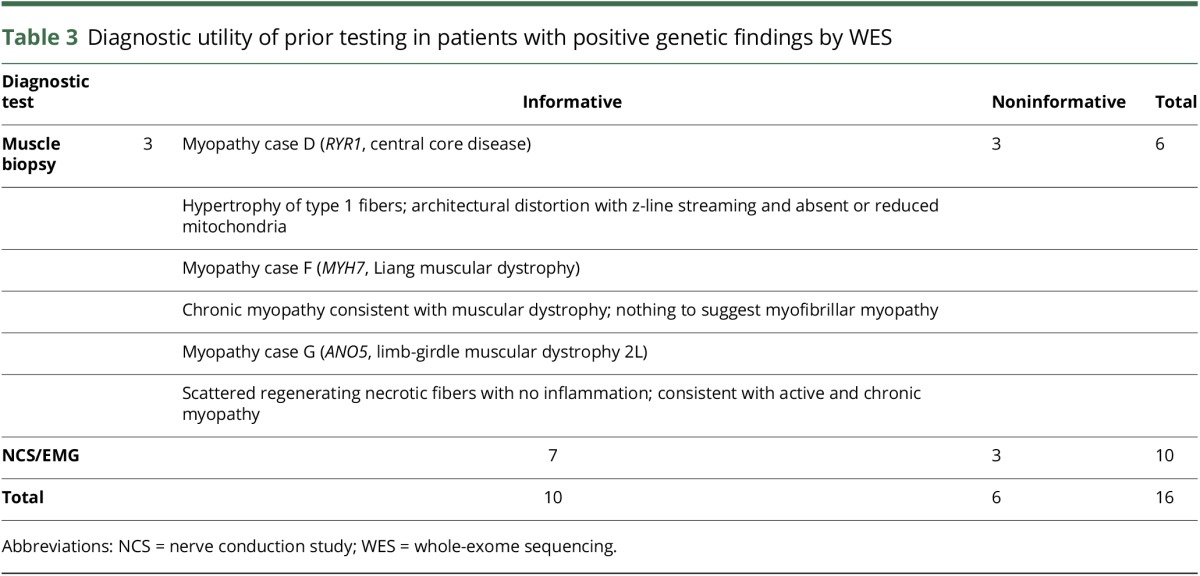
In patients with a positive result by WES, who had previously had other forms of clinical testing, it allowed us the opportunity to explore the timing and role of WES in the diagnostic workflow, including its relationship to previous invasive testing (muscle biopsy). We specifically evaluated whether the previous testing had been informative in pointing to the diagnosis before their positive molecular diagnosis by WES. Biopsies or nerve conduction studies and EMGs were considered informative or noninformative on the basis of whether it (1) assisted with the disease categorization (i.e., neuropathy or myopathy) or (2) if disease categorization was previously known, whether it further differentiated the type of neuropathy or myopathy present (e.g., distal vs proximal). For example, if a nerve conduction study or electromyogram suggested a demyelinating neuropathy, and a pathogenic variant was found in SACS (known to cause a similar phenotype) then the prior test was informative. If, however, a muscle biopsy was normal while a definitively pathogenic variant was found in COL9A3, then this was noninformative (as in case A, table 2). We found that only 10/16 (63%) prior diagnostic procedures were informative in pointing to the presumptive diagnosis in patients who had pathogenic genetic findings, including only half (3/6) muscle biopsies (table 3). However, even in cases where prior testing had been informative, a specific diagnosis was not established before exome sequencing.
Table 3.
Diagnostic utility of prior testing in patients with positive genetic findings by WES
Discussion
The diagnosis of NMDs can be challenging because patients often present with nonspecific features, symptoms, and laboratory findings. In this study, we performed exome sequencing on 93 NMD singleton patients who had defied prior diagnosis to identify a specific genetic defect that might be causative of their condition. We used 3 different diagnostic lists to evaluate optimal strategies for analysis of exome sequencing data. This large cohort includes data from both genome-scale sequencing and substantial previous testing, allowing for correlation and comparison of genetic testing with other diagnostic modalities. Most patients had significant previous workup with either negative or nonspecific laboratory findings or negative single gene testing. The overall diagnostic yield for WES in this cohort was 12.9%. Prior reports have indicated diagnostic yields of 26%–65%.12–15 The lower diagnostic rate for WES in this study likely reflects the fact that participants were singletons not trios (i.e., with inclusion of both parents), most had already had extensive workups including single gene and gene panel testing (thus removing patients who likely would also have tested “positive” on exome sequencing), and importantly, many were older than 50 years, where a monogenic etiology may be less likely. Because of this diversity, our yield may better reflect the yield for WES in a clinic seeing patients with NMD of all varieties and ages. In addition, in any study, the reported diagnostic yield depends on the thresholds for identifying variants as likely pathogenic and pathogenic. Accordingly, differences between groups in variant interpretation criteria may be reflected in reported diagnostic yield.
In specific subgroups of patients with clinically diagnosed neuropathy, clinically diagnosed myopathy, or complex phenotypes involving NMD features as well as other features such as intellectual disability or rhabdomyolysis, the diagnostic yield was 14.3%, 16.1%, and 9.8%, respectively. We also examined the use of narrow (neuropathy or myopathy associated genes) vs broad diagnostic gene lists to guide exome analysis, somewhat simulating the findings of different gene panel tests or use of an exome slice. We found that in patients with a clinically characterized neuropathy or myopathy phenotype, using a narrow diagnostic list of genes relevant only to that phenotype had a similar diagnostic yield as using the broad 481 gene list and required considerably less analytic effort. By contrast, for patients with a complex phenotype, not clearly definable as a neuropathy or a myopathy, increasing the size of the diagnostic list used to evaluate variants resulted in increased diagnostic yield. In 2 complex NMD cases (patients E and K), exome sequencing clarified their diagnosis and pointed to conditions not previously suspected for these individuals (spastic paraplegia and mitochondrial deficiency, table 2).
In at least 2 cases, WES identified molecular diagnoses that directly impacted medical treatment. In case H (table 2), this patient was thought to have a chronic inflammatory demyelinating polyneuropathy; WES identified 2 frameshifting indels in SACS, consistent with Spastic Ataxia of Charlevoix-Saguenay, and consequently, this individual was not started on immunotherapy. In 1 patient previously thought to have hereditary spastic paraplegia (case C, table 2), WES identified a nonsense variant in GCH1, indicating dopa-responsive dystonia (MIM 128230). This patient demonstrates the dramatic utility that can stem from making a correct diagnosis, as she was started on dopa therapy and regained the ability to walk without assistance.16
Molecular laboratories can establish efficient WES workflows to clarify the diagnosis in a significant fraction of NMD cases. Detailed requisition forms will help ensure that WES variants are filtered by the most appropriate gene lists. While a permissive gene filter is needed in some cases to account for ambiguity in phenotyping or phenotypic overlap, evaluating lists in a staged process can provide efficiency in the analysis. The use of WES with focused analysis of a subset of genes as a “virtual panel” can potentially overcome some of the challenges of panel testing. For example, one complexity with ordering commercial gene panels is that no 2 panels are the same, with available panels often capturing somewhat different sets of genes even if they are advertised for the same indication. Exome sequencing avoids the problem of genes missing from panels, if a systematic reanalysis of the data over time is pursued as new genes are identified to be associated with disease.
In this study, 1 patient with a positive WES result had a negative result on prior commercial gene panels, indicating that our gene list included more genes than had previously been tested for in the commercial panel. Because of the complexity in determining the optimal genetic test strategy, including the potential for secondary findings and complexities of clinical correlation of variants discovered in the course of exome sequencing, involvement of a clinician with expertise in genetics is warranted.
Only 62.5% of prior muscle biopsy or electrodiagnostic testing suggested the type of NMD in patients later found to have a pathogenic mutation in an NMD gene. Often, the prior testing could identify no specific myopathy/neuropathy or did not identify the same type of myopathy/neuropathy indicated by the genetic testing result. Uninformative muscle biopsies were often apparently normal, or consisting mainly of fat tissue, which while possibly consistent with mild myopathies, or later stage myopathy or muscular dystrophy, are not informative toward a specific diagnosis. Nerve conduction studies that were informative often delineated the affected region (proximal vs distal) and those that were uninformative often were due to being normal or limited due to the patient being uncomfortable with the procedure. One limitation of our analysis is that patients in this study were often enrolled on the basis of being undiagnosed, despite extensive prior testing, potentially biasing the present analysis in a way that makes invasive or prior testing appear to be less informative than in an unselected cohort. Nevertheless, we propose the use of sequencing early in the diagnostic workup of patients with NMD, particularly in complex cases in which additional testing may be likely to be uninformative in pointing to a specific diagnosis.
The decreasing cost of massively parallel sequencing, coupled with clear diagnostic utility, raises the question of the precise role that genomic analysis should have in a diagnostic workup for NMDs. A thorough physical examination, clinical history, and common laboratory test results should always be obtained first to identify more common, nonheritable causes of NMD. WES cannot reliably detect some major causes of several more common NMDs, including large deletions, duplications, and repeat expansions such as those causative of Duchenne muscular dystrophy, type 2 myotonic dystrophy facioscapulohumeral muscular dystrophy, or CMT1A. If a heritable condition amenable to sequencing is considered most likely, it is reasonable to consider a “sequence-early” approach, consisting of a dedicated gene panel or exome sequencing with targeted analysis.
Given the potential costs, invasiveness and incomplete yield from muscle biopsy, as well as the cost of testing for a broad array of hereditary conditions using traditional Sanger single gene testing, we suggest that genome-scale sequencing or multigene panel testing be considered early in the diagnostic process in patients likely to have a monogenic neuromuscular disorder, particularly in complex or challenging cases. Early genome-scale sequencing may shorten the diagnostic odyssey, minimize invasive testing, and provide potential opportunities for clinical and investigational therapeutics for patients with NMD.
Glossary
- NCGENES
North Carolina Clinical Genomic Evaluation by Next-generation Exome Sequencing
- NMD
Neuromuscular disorder
- UNC
University of North Carolina
- VUS
variants of uncertain significance
- WES
whole-exome sequencing
Author contributions
G.T.H. conceptualized the project, provided molecular analysis of results, and assisted with writing the manuscript. M.C.A. conceptualized the project, acquired and analyzed data, and assisted with writing the manuscript. Z.F. conceptualized the project, recruited patients, analyzed data, and assisted with writing the manuscript. K.A. assisted with data analysis, created figures, edited, and helped prepare the manuscript for publication. R.J.G.B. assisted with data acquisition and analysis. C.B. provided and maintained database annotations and infrastructure. N.C. recruited patients, analyzed data, and provided strategic direction. R.S.G. recruited patients and provided strategic direction. L.V.M. oversaw sequencing of patients, provided Sanger confirmation, and assisted in molecular analysis. Y.S.-M. recruited patients, provided clinical interpretation of results, and provided strategic direction. M.T. recruited patients and provided strategic direction. C.R.T. and A.B. prepared patient samples for sequencing and analyzed data. N.S. and K.R.C. provided molecular analysis and analyzed data. L.Z. researched and constructed new diagnostic lists, analyzed diagnostic yields, and provided edits to the manuscript. K.C.W. provided strategic direction, computational resources for sequencing, and data analysis. K.W. provided molecular analysis and strategic direction. J.P.E. and J.S.B. conceptualized the project and assisted with writing the manuscript.
Study funding
North Carolina Clinical Genomic Evaluation by NextGen Exome Sequencing (NCGENES). NIH U01HG006487.
Disclosure
G.T. Haskell, M.C. Adams, Z. Fan, K. Amin, R.J.G. Badillo, L. Zhou, C. Bizon, and N. Chahin report no disclosures. R.S. Greenwood has received research support from GW Research Ltd. L.V. Milko and Y. Sholoh-Malawsky report no disclosures. K.R. Crooks has received travel funding from Johnson & Johnson. N. Strande, M. Tennison, C.R. Tilley, and A. Brandt report no disclosures. K.C. Wilhelmsen has received research support from the NIH. K. Weck has served on the editorial boards of Genetics in Medicine, the Journal of Molecular Diagnostics, and the American Journal of Pathology; coholds a patent for Method for detecting BK virus and related compositions; has been a consultant for Laboratory Oversight Committee, Molecular Evidence Development Consortium (MED-C), and BlueCross BlueShield of North Carolina (Consultant Advisory Panel); has received research support from the NIH; and holds stock/stock options in Royal Dutch Petroleum and Wells Fargo. J.P. Evans reports no disclosures. J.S. Berg has received research support from the NIH. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/NG.
References
- 1.Jobanputra V, Naini A. New developments in molecular diagnosis of neuromuscular disorders. J Neurol Disord Stroke 2013;1:1003–1004. [Google Scholar]
- 2.Efthymiou S, Manole A, Houlden H. Next-generation sequencing in neuromuscular diseases. Curr Opin Neurol 2016;29:527–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fogel BL, Satya-murti S, Cohen BH. Clinical exome sequencing in neurologic disease. Neurol Clin Pract 2016;6:164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ankala A, da Silva C, Gualandi F, et al. A comprehensive genomic approach for neuromuscular diseases gives a high diagnostic yield. Ann Neurol 2015;77:206–214. [DOI] [PubMed] [Google Scholar]
- 5.McKenna A, Hanna M, Banks E, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reilly J, Stanley A, McGee J, Owen P, Schmitt C, Wilhelmsen K. MaPSeq, a service-oriented architecture for genomics research within an academic biomedical research institution. Informatics 2015;2:20–30. [Google Scholar]
- 7.Lek M, Karczewski K, Minikel E, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014;133:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Evilä A, Arumilli M, Udd B, Hackman P. Targeted next-generation sequencing assay for detection of mutations in primary myopathies. Neuromuscul Disord 2016;26:7–15. [DOI] [PubMed] [Google Scholar]
- 13.Chae JH, Vasta V, Cho A, et al. Utility of next generation sequencing in genetic diagnosis of early onset neuromuscular disorders. J Med Genet 2015;52:208–216. [DOI] [PubMed] [Google Scholar]
- 14.Dai Y, Wei X, Zhao Y, et al. A comprehensive genetic diagnosis of Chinese muscular dystrophy and congenital myopathy patients by targeted next-generation sequencing. Neuromuscul Disord 2015;25:617–624. [DOI] [PubMed] [Google Scholar]
- 15.Kitamura Y, Kondo E, Urano M, Aoki R, Saito K. Target resequencing of neuromuscular disease-related genes using next-generation sequencing for patients with undiagnosed early-onset neuromuscular disorders. J Hum Genet 2016;61:931–942. [DOI] [PubMed] [Google Scholar]
- 16.Fan Z, Greenwood R, Felix AC, et al. GCH1 heterozygous mutation identified by whole-exome sequencing as a treatable condition in a patient presenting with progressive spastic paraplegia. J Neurol 2014;261:622–624. [DOI] [PMC free article] [PubMed] [Google Scholar]