

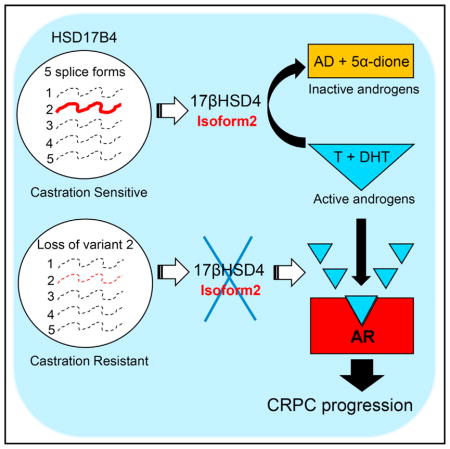
SUMMARY
Castration-resistant prostate cancer (CRPC) requires tumors to engage metabolic mechanisms that allow sustained testosterone and/or dihydrotestosterone to stimulate progression. 17β-Hydroxyste-roid dehydrogenase type 4 (17βHSD4), encoded by HSD17B4, is thought to inactivate testosterone and dihydrotestosterone by converting them to their respective inert 17-keto steroids. Counterintuitively, HSD17B4 expression increases in CRPC and predicts poor prognosis. Here, we show that, of five alternative splice forms, only isoform 2 encodes an enzyme capable of testosterone and dihydrotestosterone inactivation. In contrast with other transcripts, functional expression of isoform 2 is specifically suppressed in development of CRPC in patients. Genetically silencing isoform 2 shifts the metabolic balance toward 17β-OH androgens (testosterone and dihydrotestosterone), stimulating androgen receptor (AR) and CRPC development. Our studies specifically implicate HSD17B4 isoform 2 loss in lethal prostate cancer.
In Brief
Castration-resistant prostate cancer (CRPC) is dependent on metabolic processes that enable sustained tumor androgens, which generally require androgen-synthesizing enzymes. Ko et al. show that a single androgen inactivation enzyme splice form is lost in CRPC and encodes the only enzyme that otherwise blocks androgen action and lethal disease.
INTRODUCTION
Prostate cancer (PCa) is the most commonly diagnosed malignancy and the second leading cause of cancer-related death in men in the United States (Siegel et al., 2016). The growth and survival of PCa cells are driven by androgens through activation of the androgen receptor (AR) and its target genes. Thus, androgens, specifically testosterone (T) and dihydrotestosterone (DHT), which are the chief endogenous AR agonists, drive PCa progression (Sharifi, 2013). Androgen deprivation therapy (ADT), with medical or surgical castration, is the standard upfront treatment for advanced PCa, but patients eventually relapse with progression to castration-resistant PCa (CRPC) and ultimately die of their disease. This treatment resistance is a major challenge in the development of effective therapies (Scher and Sawyers, 2005).
Despite ADT, AR becomes reactivated in CRPC, suggesting that CRPC remains androgen signaling dependent. A major mechanism of continued AR stimulation is production of potent androgens in CRPC (Knudsen and Penning, 2010; Luu-The et al., 2008; Sharifi and Auchus, 2012). Several studies have confirmed that the concentration of androgens present in CRPC tissues is sufficient to cause persistent AR activation (Geller et al., 1978; Montgomery et al., 2008; Titus et al., 2005). CRPC generates T and/or DHT from adrenal precursor steroids and/or de novo steroidogenesis, using metabolic machinery that is composed of steroid-metabolizing enzymes (Chang et al., 2013; Knudsen and Penning, 2010; Sharifi, 2013). Finally, genetic determinants of steroidogenic enzyme activity have been shown to drive clinical behavior and predict outcomes after hormonal therapy (Agarwal et al., 2017; Chang et al., 2013; Hearn et al., 2016; Hearn et al., 2017; Almassi et al., 2017).
The 17β-OH structure is a defining feature of both potent androgens, T and DHT. 17β-OH oxidation of T and DHT to their respective 17-keto-steroids, Δ4-androstenedione (AD) and 5α-androstanedione (5α-dione), is a major mechanism of androgen inactivation that limits AR stimulation. 17β-Hydroxysteroid dehydrogenase (17βHSD) enzymes, encoded by the human HSD17B gene family, catalyze the redox reactions at C-17 (Adamski and Jakob, 2001; Mindnich et al., 2004). Of the 14 identified 17βHSDs, types 2, 4, 8, 10, 11, and 14 are thought to be oxidative enzymes, and types 1, 3, 5, and 7 are reductive enzymes (Lukacik et al., 2006).
17βHSD type 4 (17βHSD4), initially identified as an NAD+-dependent estradiolinactivating dehydrogenase (Adamski et al., 1992), is known to be a major oxidative enzyme, along with 17βHSD2 (Andersson and Moghrabi, 1997). It was originally cloned from human liver and placenta, although it is widely expressed in many tissues. The 17βHSD4 protein is 736 amino acids with a predicted molecular mass of 80 kDa and has three distinct functional domains (Adamski et al., 1995). One of the domains is an N-terminal short-chain alcohol dehydrogenase reductase (SDR; residues 1–320), known to be required for steroid metabolism.
Very little is known about the role of HSD17B4 in PCa. However, some indirect data exist that suggest that it may play a role in the development of CRPC. Exome sequencing data reveal that point mutations and copy number alterations, although uncommon, do occur in CRPC (Grasso et al., 2012). Furthermore, an intronic polymorphism in HSD17B4 is one of three germline variations that are most highly associated with progression from initiation of ADT to the development of CRPC (Ross et al., 2008). However, existing data also suggest that HSD17B4 expression is associated with poor outcomes in PCa, and that its transcript is upregulated in the transition from untreated PCa to the development of CRPC (Montgomery et al., 2008; Rasiah et al., 2009; True et al., 2006; Zha et al., 2005). The latter observations seem to run in contrast with a role for this enzyme in androgen inactivation. We set about to resolve this apparent conundrum by directly investigating the role of 17βHSD4 enzymatic activity in CRPC. Our findings suggest that only one of five HSD17B4 splice isoforms harbors enzymatic activity, which normally inactivates androgens and suppresses AR signaling, and that specific loss of this transcript-encoded enzyme enables the clinical development of CRPC.
RESULTS
17βHSD4 Expression and Androgen Inactivation in PCa
Previous studies have shown that HSD17B4 expression increases in PCa tissues when compared with paired benign tissue, and that HSD17B4 expression may increase with PCa progression. We first ascertained this relationship in our PCa cell line models, examining 17βHSD4 expression in multiple human AR-positive (LNCaP, LAPC4, 22Rv1, and VCaP) and AR-negative (PC3 and DU145) cell line models, and a nonmalignant human prostate epithelial cell line (RWPE-1). Expression was evaluated at both the mRNA and protein levels by real-time qPCR and immunoblot, respectively (Figures 1A and 1B). Among the AR-expressing models in which androgen metabolism is expected to be most relevant, LAPC4 cells had the lowest levels of both HSD17B4 transcript and 17βHSD4 protein, and the remainder of these models expressed higher levels of protein. We further investigated HSD17B4 transcript expression in clinical tissues by interrogating 26 benign prostate tissues, 40 localized PCa, and 34 CRPC tissues that we have analyzed with RNA sequencing (RNA-seq) (Figure 1C) (Beltran et al., 2016; Chakravarty et al., 2014). Neuroendocrine tumors were specifically excluded, because androgen metabolism is likely less relevant or irrelevant in this disease context. There is no significant change in the transition from benign to local PCa. In contrast, CRPC is marked by a significant loss of total HSD17B4 transcript (Figure 1C).
Figure 1. 17βHSD4 Expression and Testosterone Metabolism in Human Prostate Cancer.

(A) HSD17B4 transcript levels in human prostate cell lines. mRNA expression of the indicated cell lines was assessed by qRT-PCR, using RPLP0 as an internal control. Relative expression values were calculated by using the expression value in LAPC4 cells as a reference. Error bars represent the SD of experiments performed in triplicate.
(B) 17βHSD4 protein levels in cell lines. Equal amounts of protein (40 μg) were immunoblotted for 17βHSD4 and GAPDH (loading control).
(C) HSD17B4 transcript is lost in the transition from local untreated prostate cancer (PCa) to CRPC. Expression was assessed with RNA-seq analysis from 26 benign, 40 PCa, and 34 clinical CRPC tissues. p = 3.2e–5 for the difference between PCa and CRPC.
(D) 17βHSD and androgen metabolism pathways. All the steroids along the top (DHEA, AD, 5α-dione, and androsterone) are 17-keto-steroids, and all the steroids along the bottom (A5diol, testosterone, DHT, and 5α-diol) are 17β-OH-steroids.
(E) HPLC analysis demonstrates 17β-OH-oxidation of testosterone (T) to AD in CRPC cell lines at 24 hr. The indicated cell lines were incubated for up to 24 hr in the presence of [3H]-T (100 nM) in phenol red-free, 10% CSS medium, and androgens in the medium were separated and quantitated by HPLC. Data shown are representative from three independent experiments.
(F) Quantitation of a time course for the experiment in (E) shows that T is converted to AD over time in VCaP and 22Rv1, whereas little or no conversion of T to AD is detected in LAPC4 and LNCaP. For quantification of HPLC tracings, production of the respective steroids from [3H]-T was calculated as a percentage of all steroids.
Error bars represent the SD.
17βHSD isoenzymes are required for catalyzing the interconversion between T and DHT, which potently stimulate AR, and AD and 5α-dione, which are effectively inactive (Figure 1D). 17βHSD4 has an oxidative preference, thus converting T, DHT, and other 17β-OH androgens to their 17-keto congeners (Adamski et al., 1995; Castagnetta et al., 1997). To identify an appropriate human model for assessing the biologic consequences of oxidative 17βHSD metabolism, we incubated AR-expressing cell line models with [3H]-T and assessed for 17β-OH oxidation to AD, as well as other steroids, using high-performance liquid chromatography (HPLC). The 22Rv1 and VCaP models both converted T→AD, whereas generation of AD from T was undetectable or negligible in the LNCaP and LAPC4 models (Figures 1E and 1F), as well as the AR-negative DU145 model (Figure S1).
T Inactivation in 22Rv1 Cells Is Dependent on 17βHSD4
We next evaluated whether conversion from T to AD in VCaP and 22Rv1 cells was due to 17βHSD4 activity. Stable expression of a lentiviral short hairpin RNA (shRNA) against HSD17B4 (sh-HSD17B4) silenced transcript expression by >90% and similarly depleted protein expression (Figure 2A). 17βHSD4 ablation led to near-total inhibition of T to AD conversion in 22Rv1 cells, whereas a prominent AD signal was detected in cells expressing a control vector (sh-control), indicating the 17βHSD4 dependence of 17β-OH oxidation of T (Figures 2B and 2C). Furthermore, there was a similar loss of the generation of androsterone (AST), which like AD is also a 17-keto-steroid (Figure 1D) and which is otherwise present with endogenous 17βHSD4 expression in the control 22Rv1 cells (Figures 2B and 2C). By 48 hr, conversion to DHT was also augmented in cells with 17βHSD4 knockdown (Figure 2C). In contrast with 22Rv1 cells, the marked 17β-OH-oxidation of T in the VCaP model was not altered by 17βHSD4 silencing (Figure S2A), suggesting that another oxidative 17βHSD enzyme is operational in this model. This finding is consistent with a heterogeneity in genetic drivers of signaling that promote PCa progression (Cancer Genome Atlas Research Network, 2015; Fraser et al., 2017; Robinson et al., 2015).
Figure 2. 17βHSD4 Knockdown Suppresses T Inactivation by 17β-OH-Oxidation and Enables Sustained AR Signaling.

(A) 17βHSD4 silencing with stable lentiviral shRNA construct expression was confirmed by qPCR (in triplicate) and immunoblot in the indicated cell lines. Cells were infected with sh-control and sh-HSD17B4, and expression was assessed after puromycin selection.
(B) 17βHSD4 silencing ablates 17β-OH-oxidation of T to AD in 22Rv1 cells. The formation of metabolites in media from [3H]-T (100 nM) was assessed by HPLC at the indicated time points. Experiments were performed independently three times, in duplicate.
(C) 17βHSD4 silencing promotes sustained levels of T and DHT. Flux from [3H]-T to other steroids was calculated as a percentage of all steroids detected. Experiments were performed independently three times, in duplicate.
(D) 17βHSD4 knockdown increases nuclear accumulation of AR. 22Rv1 cells expressing sh-control (CN) and sh-HSD17B4 (sh) cells cultured in phenol red-free medium with 10% CSS for 24 hr were treated with T (1 nM) for 24–72 hr and then fractionated into cytoplasmic or nuclear portions. Each fraction was immunoblotted for AR. β-Tubulin and Lamin B protein were assessed by immunoblot as markers of the cytoplasmic or nuclear fractions, respectively.
(E) 17βHSD4 loss promotes increased PSA expression. 22Rv1 cells from the same preparation in (D) were treated with 0, 0.1, 1, or 10 nM T for 24–72 hr, and qRT-PCR for PSA expression was performed and normalized to RPLP0. Experiments were performed independently twice and in triplicate. Student’s t test was used to assess for the significance of the differences between control and sh-HSD17B4 groups. All error bars represent the SD. *p < 0.05; **p < 0.001.
To determine whether sustained active androgen levels attributable to 17βHSD4 knockdown enable augmented AR signaling, 22Rv1 cells stably expressing sh-control and sh-HSD17B4 were treated with T, and AR signaling was evaluated by assessment of AR nuclear subcellular localization and PSA expression. HSD17B4 silencing increases AR nuclear localization (Figure 2D) and PSA expression (Figure 2E). The androgen-responsive FKBP5 gene was increased with 10 nM T treatment (Figure S2B). Some increase in PSA expression was also noted in the absence of exogenous T treatment in sh-HSD17B4 cells, which might be attributable to residual steroids in charcoal-stripped serum or endogenous production.
Isoform 2 Is Predominantly Expressed in Human PCa
Transcript variants often yield distinct protein isoforms with varying biological functions that lead to differing phenotypes (Antonarakis et al., 2014; Berge et al., 2010; Dehm et al., 2008; Péterfy et al., 2005; Tazi et al., 2009). Five different human HSD17B4 transcript variants are currently known: variants 1 (GenBank: NM_001199291.2), 2 (GenBank: NM_000414.3), 3 (GenBank: NM_001199292.1), 4 (GenBank: NM_001292027.1), and 5 (Gen-Bank: NM_001292028.1), which are predicted to encode proteins of 761, 736, 718, 712, and 596 amino acids (aa), respectively (Figure 3A). We next explored expression levels of the transcripts. As a first step we examined transcript variant expression in PCa cells (Figure 3B). Variants except for variant 3 were assessed by qPCR using specific primers located in the indicated exons (Figure S3A). The products were further confirmed by sequencing to avoid possible misinterpretation. Variants 1 and 5 showed a similar expression pattern displaying the highest expression in PC3 and the second highest in RWPE1. Variant 2 was expressed predominantly in LNCaP and 22Rv1. Variant 4 was similarly abundant in all tested cell lines. We next interrogated the RNA-seq data from the clinical tissues in Figure 1C and found that HSD17B4 variant 2 is expressed at levels that generally correspond to functional expression and is specifically suppressed in the transition to CRPC (Figure 3C). All of the other four splice forms are expressed at much lower levels (Figure S3B).
Figure 3. Identification of HSD17B4 Variants and Isoforms in Human PCa.

(A) Transcript variants of HSD17B4 (top) and schematic representation of the 17βHSD4 protein encoded by isoform 2 (bottom). Human HSD17B4 has five transcript variants that differ in the 5′ coding region and encode multiple protein isoforms. Isoform 2, the major isoform, contains 736 aa and has a multidomain structure. SCAD, short-chain alcohol dehydrogenase; SCP2, sterol carrier protein 2.
(B) Differential expression of transcript variants is detected in human prostate cell lines. cDNAs from the indicated cell lines were subjected to qRT-PCR, and each variant was identified by sequencing amplicons.
(C) HSD17B4 isoform 2 is functionally expressed and specifically silenced in CRPC as assessed by RNA-seq analysis of clinical tissues.
(D) The protein encoded by HSD17B4 isoform 2 is the predominant form expressed in 22Rv1. Endogenous 17βHSD4 protein from 22Rv1 was sequenced by LC-MS/MS analysis. The coverage map of 17βHSD4 isoform 2 is shown, all of the sequences identified in the tryptic digest are shown in red, sequences identified in the chymotryptic digest are in blue, and sequences identified by both the tryptic and chymotryptic digests are in green. The yellow highlighted region indicates peptides unique to isoform 2. The protein region that is unique to isoform 2 is between aa 20 and 37 (table).
(E) The MS/MS spectra used to identify the tryptic peptide (11)VVLVTGAGAGLGR(23).
We next investigated protein isoform expression in LNCaP, 22Rv1, and DU145 cell lines with liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis. Immunoprecipitated endogenous protein complexes from each cell line were subjected to SDS-PAGE for separation and further processed for peptide analysis by LC-MS/MS. Peptide mapping followed by sequence analysis from MS/MS spectra revealed that isoform 2 predominated among the five isoforms (Figures 3D and 3E). No peptides unique to the other isoforms were detectable in any tested cell lines despite the existence of lower levels of other variant transcripts.
Expression of 17βHSD4 Isoform 2 Significantly Enhances 17β-OH-Oxidation of T, Leading to Downregulation of Androgen-Responsive Gene Expression
The N-terminal region (~300 aa) of 17βHSD4 is thought to be enzymatically responsible for steroid metabolism (Leenders et al., 1998; Möller et al., 2001). Given the isoform-dependent variability in the N-terminal structure of 17βHSD4, we suspected that the metabolic function may depend on the specific isoform expressed. We thus investigated the enzymatic activities of individual isoforms by ectopically expressing them in the LAPC4 model, which has low endogenous levels of 17βHSD4.
The cDNA for each isoform was cloned or purchased, and introduced into LAPC4 cells by transient transfection. Expression was validated by immunoblot (Figure 4A) and qPCR (Figure S4A). All protein isoforms were detectable and appeared as dominant bands corresponding to their predicted molecular weight: 83 kDa for isoform 1, 79 kDa for isoform 2, 77–78 kDa for isoforms 3 and 4, and 65 kDa for isoform 5. However, only isoform 2 expression drove T oxidation (Figure 4B). Isoform 2 robustly oxidized T to AD and its 5α-reduced metabolite, 5α-di-one, at 24 hr. In contrast, expression of other isoforms did not catalyze the conversion of T to AD, as with control cells, in which a small proportion of T was converted to DHT only, without detectable AD formation. At 48 hr, available T was nearly completely depleted in cells expressing isoform 2, whereas >70% T remained when other isoforms were expressed (Figure S4B). Over longer incubation times (24–72 hr), the generation of 5α-reduced 17β-OH steroids, DHT and 5α-diol, were replaced instead with the synthesis of 5α-dione and AST, their respective 17-keto steroids (Figure 4C). In contrast with isoform 2, other isoforms failed to convert T to AD, even at longer incubation times of T up to 72 hr (Figure S4C). Notably, in the presence of isoform 2 expression, no conversion to DHT was detectable.
Figure 4. 17βHSD4 Isoform 2 Inactivates T to 17-Keto-androgens and Suppresses AR Signaling.

(A) Immunoblot of 17βHSD4 isoforms 1–5 expressed in LAPC4 cells. Cells were transiently transfected with plasmid constructs expressing the designated 17βHSD4 isoform or empty vector, and expression was assessed by immunoblot.
(B) Only cells expressing 17βHSD4 isoform 2 are capable of converting T to AD and generating detectable 5α-dione. Cells were incubated for up to 72 hr in the presence of [3H]-T (100 nM), and steroids separated and detected by HPLC at the 24-hr time point are shown. Data shown are representative of three independent experiments.
(C) Isoform 2 expression results in increased conversion from T to AD and an increase in the generation of other 17-keto-androgens (5α-dione and AST) over time, whereas 17β-OH-androgens (DHT and 5α-diol) were predominantly produced in control cells.
(D) Isoform 2 expression decreases AR nuclear localization. Cells expressing vector or 17βHSD4 isoform 2 grown in phenol red-free medium with 10% CSS for 24 hr were treated with 0 and 1 nM T for up to 48 hr and then fractionated into cytoplasmic or nuclear portions. Each fraction was immunoblotted for AR and internal controls, β-tubulin, and Lamin B.
(E) Isoform 2 suppresses stimulation of PSA and FKBP5 expression by T in LAPC4. Gene expression was assessed in cells described in (D) in triplicate by qRT-PCR and normalized to RPLP0.
(F) Isoform 2 attenuates androgen-induced proliferation of LAPC4 cells. 1 × 105 cells were cultured in phenol red-free, 10% CSS medium and treated with or without T (1 nM) every other day, and were quantitated using the CellTiter Proliferation Assay at the indicated time points.
(G) 17βHSD4 isoform 2 suppresses CRPC tumor progression in the LAPC4 xenograft model. LAPC4 cells expressing control and 17βHSD4 (isoform2) vectors were subcutaneously injected into orchiectomized mice. Progression-free survival (time for tumors to reach 200 mm3) was assessed. The p value was determined using the log rank test.
*p < 0.05; **p < 0.001, Student’s t test.
Given the specific activity attributable to isoform 2 that inactivates T, we investigated whether androgen stimulation of AR signaling and tumor growth is blocked by isoform 2 expression in LAPC4 cells. Isoform 2 expression inhibits AR nuclear localization (Figure 4D). At early time points, isoform 2 expression had no significant inhibitory effect on expression of the androgen-responsive genes, PSA and FKBP5. However, at 48 hr, expression of both genes was markedly suppressed (Figure 4E). Furthermore, isoform 2 expression significantly suppressed cell proliferation in vitro (Figure 4F), as well as in vivo development of CRPC (Figure 4G) when compared with empty vector-expressing cells. In contrast, isoform 2 expression had no effect on cell proliferation in the AR-negative DU145 cell line model (Figure S5).
17βHSD4 Loss Promotes CRPC Xenograft Progression
To determine whether HSD17B4 variant 2 loss that occurs in patient CRPC tissues (Figure 3C) and loss of the enzymatic activity and augmented AR signaling specifically conferred by its encoded protein (Figure 4) play a causative role in the development of CRPC, we examined the effect of 17βHSD4 knockdown on tumor growth in the 22Rv1 xenograft model. Male NSG mice underwent surgical castration and dehydroepiandrosterone (DHEA) pellet implantation (5 mg 90-day sustained release) to mimic human adrenal physiology (Chang et al., 2013; Li et al., 2015, 2016) and 5 days later were subcutaneously injected with 22Rv1 cells stably expressing sh-HSD17B4 or sh-Control. Tumors were measured two to three times per week to assess for CRPC growth (Figure 5A). Progression to tumor volume ≥500 mm3 was significantly hastened in the 17βHSD4 knockdown group compared with the control group (p = 0.04), suggesting that 17βHSD4 loss enables sustained biologically active androgens required for AR signaling and CRPC development. A similar xenograft experiment performed in eugonadal mice did not show a statistically significant increase in growth for HSD17B4 knockdown xenografts, thus suggesting that the major advantage for loss of 17βHSD4 enzymatic activity likely applies to the context of the castration (Figure 5B). Transcript expression analysis was done in RNA collected from CRPC xenografts after mice were sacrificed to confirm continued HSD17B4 knockdown and sustained androgen-responsive PSA expression (Figure 5C), as was apparent with in vitro studies. Finally, we determined the total 17β-OH-androgen (i.e., T and DHT) content and their 17-keto-androgen congeners (i.e., AD and 5α-dione) in the CRPC xenografts using LC-MS/MS and found that 17βHSD4 silencing significantly shifts the equilibrium to a higher ratio of total active androgens compared with their respective inactive 17-keto-steroids (Figure 5D). Together, these results suggest that loss of HSD17B4 variant 2, which is specifically functionally expressed in prostatic tissues and silenced in the development of clinical CRPC, plays a causative role in enabling sustained levels of androgens that spur AR signaling and the development of CRPC in vivo.
Figure 5. Loss of HSD17B4 Blocks Androgen Inactivation and Promotes CRPC Tumor Progression in a Xenograft Model.

(A) Silencing HSD17B4 expression facilitates CRPC growth in orchiectomized mice. 22Rv1 tumor cells expressing sh-control and sh-HSD17B4 vectors (n = 8 mice per group) were subcutaneously injected into mice prior to castration, and time for tumors to reach 500 mm3 was assessed. The p value was determined using the log rank test.
(B) There is no statistically significant tumor growth advantage conferred by HSD17B4 loss in eugonadal mice (n = 10 mice per group).
(C) HSD17B4 silencing in CRPC xenografts from (A) is confirmed and permits sustained PSA expression. Total RNA extracted from 22Rv1 xenografts harvested on day 47 was subjected to qRT-PCR analysis. Expression was normalized to RPLP0. Error bars represent the SD of experiments performed in triplicate.
(D) HSD17B4 knockdown increases the ratio of 17β-OH-androgens that bind AR (T+DHT) to their respective and inactive 17-keto-androgens (AD+5α-dione). Intratumoral steroid levels were determined by LC-MS/MS. The p values were determined using a Wilcoxon signed rank test.
DISCUSSION
17βHSD enzymes serve as major regulators of the synthesis and loss of biologically active androgens. The absolute requirement for the reductive 17βHSD reaction is exemplified by germline genetic loss of 17βHSD3, which is normally required for gonadal conversion from AD to T synthesis in human males, leading to loss of masculinization (Moghrabi et al., 1998). In PCa and the development of CRPC tissues in particular, 17βHSD5 (also known as AKR1C3) is transcriptionally upregulated, and therefore thought to be the major reductive isoenzyme required for the synthesis of T and DHT (Bauman et al., 2006; Stanbrough et al., 2006). In contrast with certain enzymatic reactions, such as 5α-reduction, which are irreversible, C17 modification of androgens is a reversible process (Sharifi and Auchus, 2012). Therefore, along with gain of 17βHSD isoenzymes with a reductive (i.e., 17-keto → 17β-OH) preference, loss of opposing 17βHSD isoenzymes with an oxidative preference may also shift the balance in a similar fashion, favoring the generation of T and DHT, which stimulate AR signaling, thus playing a major role in the development of CRPC. 17βHSD2 and 17βHSD4 both have an oxidative preference, which defines their roles in limiting the action of androgens and estrogens (Miller and Auchus, 2011). More is known about the role of 17βHSD2 in steroid inactivation compared with 17βHSD4, in part because of evidence that suggests the latter enzyme plays a role in peroxisomal β-oxidation (van Grunsven et al., 1998) and the resultant uncertainty of the relative importance of this function versus its role in steroid metabolism. Studies implicating an increase in HSD17B4 expression as a poor prognostic marker for PCa and in the development of CRPC would seem to run in contrast with a mechanistic link to androgen inactivation (Montgomery et al., 2008; Rasiah et al., 2009). The existence of HSD17B4 splice variants that encode distinct enzymes further clouded this picture.
Of the five known variants, our data clearly point toward a single HSD17B4 variant (variant 2) that is both functionally expressed in human prostate tissues and specifically suppressed in the development of CRPC. The enzyme encoded by the same variant is the sole protein that has detectable oxidative activity that results in the inactivation of T and DHT. Although our studies with forced expression of the variants may have resulted in higher protein expression of variant 2 over the other variants, this effect appears insufficient to explain the absence of enzymatic activity attributable to the other variants. It is possible that tissue-specific regulation of variant expression contributes to determination of enzyme substrate specificity with regard to function in steroid versus peroxisomal metabolism. A limitation of our study is that we only found a single model (LAPC4) that has low endogenous expression and function of variant 2, which is amenable to genetic add-back approaches. However, our conclusions are also supported by expression data from clinical tumors and functional experiments silencing endogenous variant 2 expression in other systems.
The strong link between germline genetic variants in HSD17B4 and the development of CRPC noted by others may be driven by variations in regulatory elements that drive the type or expression levels of transcript splice forms (Ross et al., 2008). Further work is required to elucidate the possible existence of such a mechanism, which could have implications for steroid metabolism-dependent germline predictive biomarkers of hormonal therapy response (Hearn et al., 2016; Hearn et al., 2017; Almassi et al., 2017).
In summary, our data bring clarity to what has generally been a perplexing link between HSD17B4 and the development of CRPC. When viewed through the lens of the enzymatic properties of a single specific isoform encoded by HSD17B4 variant 2, which is responsible for androgen inactivation, the functional expression of this variant transcript, its loss in the clinical development of CRPC, and its effects on in vivo CRPC growth, all concordantly point toward a causal role in the development of the lethal form of PCa.
EXPERIMENTAL PROCEDURES
Detailed methods are available in the Supplemental Experimental Procedures.
Cell Culture
LNCaP, 22Rv1, DU145, and PC3 cell lines were purchased from ATCC (Manassas, VA, UA) and cultured in RPMI 1640 medium with 10% fetal bovine serum (FBS). VCaP and RWPE-1 were obtained from ATCC. VCaP was maintained in DMEM with 10% FBS, and RWPE-1 was grown in keratinocyte serum-free medium (K-SFM; Invitrogen, Carlsbad, CA, USA) supplemented with 0.05 mg/mL bovine pituitary extract and 5 ng/mL human recombinant epidermal growth factor, respectively. LAPC4 cells were generously provided by Dr. Charles Sawyers (Memorial Sloan Kettering Cancer, New York, NY, USA) and maintained in Iscove’s modified Dulbecco’s medium with 10% FBS. VCaP and all other cells were incubated in a 10% and 5% CO2 humidified incubator, respectively.
RNA Isolation and qRT-PCR
Total RNA was isolated from cells using the RNeasy Kit (Sigma-Aldrich, St. Louis, MO, USA), and 1 μg of RNA was used in a reverse-transcriptase reaction with the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). qRT-PCR was performed using the iTaq SYBR Green Supermix ROX kit (Bio-Rad) on an ABI-7500 Real-Time PCR system (Applied Biosystems, Foster City, CA, USA). Each mRNA transcript was quantitated by normalizing to the housekeeping gene RPLP0.
Western Blotting and Immunoprecipitation
For protein detection by western blotting (WB), whole cells were lysed in RIPA buffer (Sigma-Aldrich) with protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Protein concentrations were determined by a Bradford Assay kit (Bio-Rad), and 20–40 μg of protein was separated by 10% SDS-PAGE and transferred onto a nitro cellulose membrane (EMD Millipore, Billerica, MA, USA). The membrane was blocked in 5% skim milk, subsequently incubated with primary antibodies (mouse anti-17βHSD4 [Santa Cruz Biotechnology, Santa Cruz, CA, USA], mouse anti-β-actin [Sigma-Aldrich], mouse anti-GAPDH [Abcam, Cambridge, MA, USA], rabbit anti-AR [Santa Cruz], rabbit anti-tubulin [Cell Signaling, Danvers, MA, USA], and Goat anti-Lamin B [Santa Cruz]) at 4°C overnight followed by incubation with HRP-conjugated goat anti-mouse IgG or goat anti-rabbit IgG (Thermo Fisher Scientific, Rockford, IL, USA) or donkey anti-goat IgG (Santa Cruz) for 1 hr, and developed with the Pierce ECL reagent (Thermo Fisher Scientific). To purify endogenous 17βHSD4 by immunoprecipitation (IP), 0.5–1 mg of total protein lysate was precleared using control IgG, precleaned with Protein A/G Plus-Agarose (Santa Cruz Biotechnology), and incubated with mouse anti-17βHSD4 body at 4°C for 1 hr followed by addition of 20–40 μL Protein A/G Plus-Agarose and rocking overnight. After extensive washing with lysis buffer, the purified proteins were eluted with 30 μL of 2× SDS sample buffer and separated by 12% SDS-PAGE followed by Coomassie blue staining using Gel Code Blue Stain Reagent (Thermo Fisher Scientific).
Subcellular Fractionation
Cells were grown in 10-cm dishes (5–10 million cells were plated), washed twice with cold PBS, and collected in 3 mL of PBS with a cell scraper. The harvested cells were centrifuged for 5 min at 1,000 × g at 4°C and PBS was removed. After re-suspension in 1 mL of cold sucrose buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 15% sucrose) supplemented with protease inhibitor cocktail (Roche, Indianapolis, IN, USA), cells were mechanically homogenized by 12 passes through a ball-bearing homogenizer at 10-μm clearance (Isobiotec Precision Engineering, Heidelberg, Germany) on ice. Homogenates were centrifuged at 3,000 × g for 15 min at 4°C in a micro-centrifuge. The resulting supernatant fraction containing cytosolic membranes and the cytosol (C′) were transferred to new 1.5-mL Eppendorf tubes, and nuclear pellets (N′) were re-suspended with 0.8 mL of sucrose buffer. C′ and N′ samples were then spun down again at 4°C for 15 min at 3,000 × g to remove contaminants. Cleared C′ was transferred to new 1.5-mL Eppendorf tubes while washed N′ was lysed in 100 μL of RIPA buffer with a protease inhibitor cocktail. The supernatant fraction was further concentrated using a 10-kDa cutoff Amicon Ultra-2 centrifugal filter unit (EMD Millipore, Billerica, MA, USA).
Isoform Identification by LC-MS/MS Analysis
The protein digestion was performed as follows. In brief, multiple bands were cut from one Coomassie blue-stained 1D gel, and the bands were then washed/destained in 50% ethanol, 5% acetic acid, and then dehydrated in acetonitrile. The samples were then reduced with DTT and alkylated with iodoacetamide prior to the in-gel digestion. All bands were digested in-gel using trypsin, by adding 10 μL of 5 ng/μL trypsin in 50 mM ammonium bicarbonate and incubating overnight at room temperature to achieve complete digestion. The peptides were extracted from the polyacrylamide in two aliquots of 30 μL using 50% acetonitrile with 5% formic acid. These extracts were combined and evaporated to < 10 μL in a Speedvac and then resuspended in 1% acetic acid to a final volume of ~30 μL for LC-MS analysis. The LC-MS system was a Finnigan LTQ-Orbitrap Elite hybrid mass spectrometer system. The HPLC column was a C18 reversed-phase capillary chromatography column (Thermo Scientific Dionex Acclaim Pepmap C18; 15 cm × 75 μm ID; particle size, 2 μm; pore size, 100 Å). Extracts, 5 μL, were injected, and the peptides were eluted from the column using an acetonitrile/0.1% formic acid gradient at a flow rate of 0.3 μL/min. The eluted peptides were then introduced into the source of the mass spectrometer online. The nano electrospray ion source is operated at 1.9 kV. The digest was analyzed using the data-dependent multitask capability of the instrument, acquiring full scan mass spectra to determine peptide molecular weights and product ion spectra to determine aa sequence in successive instrument scans. The data were analyzed by using all CID spectra collected in each experiment to search human reference sequence databases with the search program Mascot. Sequence searches were also performed against the human 17βHSD4 protein sequence to confirm the identifications and sequence coverage.
HSD17B4 Knockdown by Lentiviral Vector
HSD17B4 shRNA (GIPZ Lentiviral Human HSD17B4 shRNA, Clone ID: V3LHS_329216) and control (GIPZ Non-silencing Lentiviral shRNA Control, RH4371) lentiviruses were purchased from Dharmacon (Lafayette, CO, USA). The sequence targeted by the shRNA was 5′-AACTGTCTGAGTCATCCGT-3′. Lentivirus expressing the shRNAs was produced in 293T cells by co-transfecting pGIPZ and packaging vectors, pMD2.G and psPAX2. Supernatants containing lentiviral particles were collected from 293T cells and filtered with a 0.45-μm nitrocellulose membrane. 1 mL of the filtered supernatants was used to infect cells in six-well plates. The infected cells were selected with 1 μg/mL puromycin for 2 weeks before evaluation of knockdown efficiency.
17βHSD4 Overexpression
For transient expression of 17βHSD4 isoforms, pcDNA3.1-HSD17B4 vector was constructed by PCR amplification followed by sub-cloning into the pcDNA3.1 vector. Plasmid DNA was transfected into cells in 6- or 12 well-plates using LipoD293 (SignaGen, Ijamsville, MD, USA) reagent according to the manufacturer’s instructions; cells were seeded in culture media 1 day before transfection. The next day, 1.0and 0.75 μg of each DNA constructwas addedperwell in6- and 12-well plates, respectively. The transfected cells were harvested 18–24 hr after transfection for WB analysis or used for the subsequent experiments.
For stable expression of HSD17B4 isoform 2 in LAPC4, the pLenti-CMV-HSD17B4-RFP-2A-Puro vector was constructed by sub-cloning PCR-amplified HSD17B4 into the pLenti-CMV-RFP-2A-Puro vector (Applied Biological Materials, BC, Canada) by ligation into filled-in Kpn1 and Xba1 sites. Virus packaging was performed in 293T cells by co-transfecting pLenti-CMV-RFP-2A-Puro and packaging vectors, pMD2.G and psPAX2. After lentiviral infection and puromycin selection for 2 weeks, 17βHSD4 expression and its oxidative activity were confirmed by western blot and steroid metabolism experiments, respectively.
Detection of Transcription Variants
cDNAs were produced using total RNA extracted from each cell line that was PCR amplified with primer sets targeting specific variants. PCR products were sequenced.
Analysis of RNA-Seq Data
We analyzed two publicly available datasets that our group reported in recent years (Beltran et al., 2016; Chakravarty et al., 2014). For this analysis, we selected 26 benign prostate tissues, 40 localized PCa, and 34 CRPC. RNA-seq and data processing were performed according to the protocol described in the respective papers (Beltran et al., 2016; Chakravarty et al., 2014). Data are available at dbGaP with accession code dbGaP: phs000909.v.p1 and in GEO with accession code GEO: GSE43988.
Cell Proliferation
LAPC4 or DU145 cells were seeded in 24-well plates (5–10 × 104 cells per well in 500 μL of culture medium) coated with poly-L-ornithine and grown in the presence of androgen (T) or vehicle control for the indicated incubation times. Cell proliferation was assessed with a kit (CellTiter 96 Aqueous One Solution Cell Proliferation Assay kit; Promega, Madison, WI, USA) according to manufacturer’s instructions with minor modifications, as follows. In brief, 50 μL of CellTiter 96 Aqueous One Solution Reagent was added to each well. After 1-hr incubation at 37°C and 5% CO2, 100 μL of culture medium from each well was transferred to a 96-well plate for absorbance measurement at 490 nm using a luminometer (Synergy 2; BioTek, Winooski, VT, USA).
Steroid Metabolism
Cells (0.5 × 106–106 cells per well) were seeded in 12-well plates coated with or without poly-L-ornithine 1 day before steroid treatment. The next day, medium was replaced with 1 mL of phenol red-free, 10% charcoal-dextran stripped FBS (charcoal-stripped serum [CSS]) medium containing [3H]-labeled steroids (100 nM, 300,000–600,000 counters per minute [cpm]) obtained from PerkinElmer (Waltham, MA, USA). Cells were incubated at 37°C; then 250-μL aliquots of medium were collected for up to 72 hr and treated with 1,000 units of β-glucuronidase (H. pomatia; Sigma-Aldrich) at 37°C for 2 hr to deconjugate glucuronidated steroids. The deconjugated steroids were extracted with 600 μL 1:1 ethylacetate:isooctane and evaporated under nitrogen. For HPLC analysis, the dried samples were dissolved in 50% methanol and injected on a Breeze 1525 system equipped with model 717+ autosampler (Waters, Milford, MA). The steroid metabolites were separated on a Luna 150 × 3 mm, 3 μM C18 reverse-phase column (Phenomenex, Torrance, CA, USA) with methanol/water gradients at 25°C. The column effluent was mixed with Liquiscint scintillation cocktail (National Diagnostics, Atlanta, GA, USA) and analyzed using a β-RAM model 3 online radioactivity flow detector (LabLogic, Brandon, FL, USA). All metabolism studies were performed in duplicate and repeated in independent experiments.
Intra-tumoral Androgen Levels by LC-MS/MS Analysis
Freshly frozen mouse xenograft tissue was homogenized with Minilys Homogenizer (Bertin, Rockville, MD, USA) and followed by liquid-liquid extraction using methyl tert-butyl ether (MTBE). In brief, homogenate in methanol/water (80/20, v/v) was rotated for 4 hr at room temperature. Then 10 μL of internal standard (50 ng/mL, AD-13C3) was added to the homogenate and vortexed. The homogenate was centrifuged (3,000 rpm, 10 min, 4°C) to separate insoluble debris, and the supernatant was transferred to a glass tube for liquid-liquid extraction. Double-distilled water, 1 mL, was added to the tubes and vortexed briefly. After addition of 2 mL of MTBE, the tubes were vortexed for 5 min and centrifuged (3,000 rpm, 5 min, 4°C), and the MTBE layer was collected. The liquid-liquid extraction was repeated and the combined MTBE layers were dried under nitrogen gas and then reconstituted in 120 μL of 50% methanol/water (v/v). The extracted steroids were quantified using LC-MS/MS. In brief, the extracted steroids were injected onto a Shimadzu UPLC system (Shimadzu Corporation, Japan), and the steroids were separated on a C18 column (Zorbax Eclipse Plus C18 column, 150 mm × 2.1 mm, 3.5 μm; Agilent, Santa Clara, CA, USA) using a gradient starting from 20% solvent B (acetonitrile/methanol [90/10, v/v] containing 0.2% formic acid) for 4 min and then to 75% solvent B for 10 min, followed by 95% solvent B for 3 min. The steroids were quantified on a Qtrap 5500 mass Spectrometer (AB Sciex; Redwood City, CA, USA) using ESI in positive ion mode and multiple reaction monitoring (MRM). Δ4-androstene-3,17-dione-2,3,4-13C3 (Cerilliant, Round Rock, TX, USA) was used as an internal standard for calibration of steroids in each sample. Data acquisition and processing were performed using MultiQuant (AB Sciex; version 3.0.1) software. The peak area ratio of the analyte over the internal standard was used for quantification. Each sample run included a calibration curve with standards for accurate quantification.
Mouse Xenografts
Male NSG mice, 6–8 weeks of age purchased from The Jackson Laboratory (Bar Harbor, ME, USA), underwent surgical orchiectomy and DHEA pellet (5 mg 90-day sustained-release; Innovative Research of America, Sarasota, FL, USA) implantation. After 5 days, 22Rv1 cells (10 × 106) stably expressing sh-HSD17B4 and sh-control were injected subcutaneously with 50% Matrigel into the right flank of mice, and tumor size was measured two to three times per week with calipers. Tumor volumes were calculated with the formula length × width × height/2. Time from tumor cell injection to tumor volume ≥500 mm3 was determined. Mice were sacrificed at day 47 and tumors were fresh frozen at −80°C for further analysis. Survival was assessed by Kaplan-Meier analysis using a log rank test to determine differences between the two groups (GraphPad Prism 5). In the second set of experiments, 22Rv1 xenografts were established in intact mice without castration to test tumor growth in the presence of gonadal T. Time from tumor cell injection to tumor volume ≥250 and 500 mm3 was assessed. For LAPC4 xenograft studies, LAPC4 cells (10 × 106) stably expressing pLenti-CMV-RFP-2A-Puro-Blank (control) or pLenti-CMV-HSD17B4-RFP-2A-Puro were subcutaneously injected into surgically orchiectomized nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mice supplemented with a DHEA (5 mg 90-day sustained-release) pellet. Tumor diameters were measured by digital calipers two or three times per week. Time from tumor injection to tumor volume >200 mm3 was assessed.
Statistical Analysis
Statistical evaluation was performed as indicated in each experiment. A p value of less than 0.05 was considered significant. Student’s t test, log rank test, and Wilcoxon signed rank test were used to determine significance differences between groups. All Student’s t tests were unpaired and two-sided.
Supplementary Material
Highlights.
Of five HSD17B4 splice forms, only isoform 2 encodes an androgen-inactivating enzyme
Isoform 2 is the only form that is expressed and lost in CRPC in patients
Isoform 2 inactivates testosterone and dihydrotestosterone
Genetic loss of isoform 2 promotes development of CRPC
Acknowledgments
This work has been supported in part by funding from a Howard Hughes Medical Institute Physician-Scientist Early Career Award (to N.S.), a Prostate Cancer Foundation Challenge Award (to N.S.), an American Cancer Society Research Scholar Award (12-038-01-CCE; to N.S.), and grants from the National Cancer Institute (R01CA168899, R01CA172382, and R01CA190289; to N.S.). The Orbitrap Elite instrument was purchased via an NIH shared instrument grant, 1S10RR031537-01.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2017.12.081.
AUTHOR CONTRIBUTIONS
H.-K.K. designed and performed the majority of experiments and wrote the paper; M.B. performed mouse experiments; Y.-M.C. performed steroid mass spectrometry; B.W. performed protein mass spectrometry; R.B., M.R., and A.S. performed RNA-seq analysis; N.S. designed experiments and wrote the paper. All authors reviewed and edited the paper.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Adamski J, Jakob FJ. A guide to 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2001;171:1–4. doi: 10.1016/s0303-7207(00)00383-x. [DOI] [PubMed] [Google Scholar]
- Adamski J, Husen B, Marks F, Jungblut PW. Purification and properties of oestradiol 17 beta-dehydrogenase extracted from cytoplasmic vesicles of porcine endometrial cells. Biochem J. 1992;288:375–381. doi: 10.1042/bj2880375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamski J, Normand T, Leenders F, Monté D, Begue A, Stéhelin D, Jungblut PW, de Launoit Y. Molecular cloning of a novel widely expressed human 80 kDa 17 beta-hydroxysteroid dehydrogenase IV. Biochem J. 1995;311:437–443. doi: 10.1042/bj3110437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal N, Hahn AW, Gill DM, Farnham JM, Poole AI, Cannon-Al-bright L. Independent validation of effect of HSD3B1 genotype on response to androgen-deprivation therapy in prostate cancer. JAMA Oncol. 2017;3:856–857. doi: 10.1001/jamaoncol.2017.0147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almassi N, Reichard CA, Li J, Russell C, Perry J, Ryan C, Friedlander T, Sharifi N. HSD3B1 and response to a non-steroidal CYP17A1 inhibitor in castration-resistant prostate cancer. JAMA Oncology. 2017 doi: 10.1001/jamaoncol.2017.3159. Published online October 12, 2017. https://doi.org/10.1001/jamaoncol.2017.3159. [DOI] [PMC free article] [PubMed]
- Andersson S, Moghrabi N. Physiology and molecular genetics of 17 β-hydroxysteroid dehydrogenases. Steroids. 1997;62:143–147. doi: 10.1016/s0039-128x(96)00173-0. [DOI] [PubMed] [Google Scholar]
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N Engl J Med. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman DR, Steckelbroeck S, Williams MV, Peehl DM, Penning TM. Identification of the major oxidative 3alpha-hydroxysteroid dehydrogenase in human prostate that converts 5alpha-androstane-3al-pha,17beta-diol to 5alpha-dihydrotestosterone: a potential therapeutic target for androgen-dependent disease. Mol Endocrinol. 2006;20:444–458. doi: 10.1210/me.2005-0287. [DOI] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BV, Varambally S, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med. 2016;22:298–305. doi: 10.1038/nm.4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berge EO, Staalesen V, Straume AH, Lillehaug JR, Lønning PE. Chk2 splice variants express a dominant-negative effect on the wild-type Chk2 kinase activity. Biochim Biophys Acta. 2010;1803:386–395. doi: 10.1016/j.bbamcr.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell. 2015;163:1011–1025. doi: 10.1016/j.cell.2015.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnetta LA, Carruba G, Traina A, Granata OM, Markus M, Pavone-Macaluso M, Blomquist CH, Adamski J. Expression of different 17beta-hydroxysteroid dehydrogenase types and their activities in human prostate cancer cells. Endocrinology. 1997;138:4876–4882. doi: 10.1210/endo.138.11.5497. [DOI] [PubMed] [Google Scholar]
- Chakravarty D, Sboner A, Nair SS, Giannopoulou E, Li R, Hennig S, Mosquera JM, Pauwels J, Park K, Kossai M, et al. The oestrogen receptor alpha-regulated lncRNA NEAT1 is a critical modulator of prostate cancer. Nat Commun. 2014;5:5383. doi: 10.1038/ncomms6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang KH, Li R, Kuri B, Lotan Y, Roehrborn CG, Liu J, Vessella R, Nelson PS, Kapur P, Guo X, et al. A gain-of-function mutation in DHT synthesis in castration-resistant prostate cancer. Cell. 2013;154:1074–1084. doi: 10.1016/j.cell.2013.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer Res. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser M, Sabelnykova VY, Yamaguchi TN, Heisler LE, Livingstone J, Huang V, Shiah YJ, Yousif F, Lin X, Masella AP, et al. Genomic hallmarks of localized, non-indolent prostate cancer. Nature. 2017;541:359–364. doi: 10.1038/nature20788. [DOI] [PubMed] [Google Scholar]
- Geller J, Albert J, Loza D, Geller S, Stoeltzing W, de la Vega D. DHT concentrations in human prostate cancer tissue. J Clin Endocrinol Metab. 1978;46:440–444. doi: 10.1210/jcem-46-3-440. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn JWD, AbuAli G, Reichard CA, Reddy CA, Magi-Galluzzi C, Chang KH, Carlson R, Rangel L, Reagan K, Davis BJ, et al. HSD3B1 and resistance to androgen-deprivation therapy in prostate cancer: a retrospective, multicohort study. Lancet Oncol. 2016;17:1435–1444. doi: 10.1016/S1470-2045(16)30227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hearn WD, Xie W, Nakabayashi M, Almassi N, Reichard CA, Pomerantz M, Kantoff PW, Sharifi N. HSD3B1 genotype and response to androgen deprivation therapy for biochemical recurrence after radiotherapy for localized prostate cancer. JAMA Oncology. 2017 doi: 10.1001/jamaoncol.2017.3164. Published online October 12, 2017. https://doi.org/10.1001/jamaoncol.2017.3164. [DOI] [PMC free article] [PubMed]
- Knudsen KE, Penning TM. Partners in crime: deregulation of AR activity and androgen synthesis in prostate cancer. Trends Endocrinol Metab. 2010;21:315–324. doi: 10.1016/j.tem.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenders F, Dolez V, Begue A, Möller G, Gloeckner JC, de Launoit Y, Adamski J. Structure of the gene for the human 17beta-hydroxys-teroid dehydrogenase type IV. Mamm Genome. 1998;9:1036–1041. doi: 10.1007/s003359900921. [DOI] [PubMed] [Google Scholar]
- Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, Liu J, Upadhyay SK, Auchus RJ, Sharifi N. Conversion of abirater-one to D4A drives anti-tumour activity in prostate cancer. Nature. 2015;523:347–351. doi: 10.1038/nature14406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Alyamani M, Li J, Rogacki K, Abazeed M, Upadhyay SK, Balk SP, Taplin ME, Auchus RJ, Sharifi N. Redirecting abiraterone metabolism to fine-tune prostate cancer anti-androgen therapy. Nature. 2016;533:547–551. doi: 10.1038/nature17954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukacik P, Kavanagh KL, Oppermann U. Structure and function of human 17beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2006;248:61–71. doi: 10.1016/j.mce.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Luu-The V, Bélanger A, Labrie F. Androgen biosynthetic pathways in the human prostate. Best Pract Res Clin Endocrinol Metab. 2008;22:207–221. doi: 10.1016/j.beem.2008.01.008. [DOI] [PubMed] [Google Scholar]
- Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindnich R, Möller G, Adamski J. The role of 17 beta-hydroxysteroid dehydrogenases. Mol Cell Endocrinol. 2004;218:7–20. doi: 10.1016/j.mce.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Moghrabi N, Hughes IA, Dunaif A, Andersson S. Deleterious missense mutations and silent polymorphism in the human 17beta-hydroxysteroid dehydrogenase 3 gene (HSD17B3) J Clin Endocrinol Metab. 1998;83:2855–2860. doi: 10.1210/jcem.83.8.5052. [DOI] [PubMed] [Google Scholar]
- Möller G, van Grunsven EG, Wanders RJ, Adamski J. Molecular basis of D-bifunctional protein deficiency. Mol Cell Endocrinol. 2001;171:61–70. doi: 10.1016/s0303-7207(00)00388-9. [DOI] [PubMed] [Google Scholar]
- Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, True LD, Nelson PS. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Péterfy M, Phan J, Reue K. Alternatively spliced lipin isoforms exhibit distinct expression pattern, subcellular localization, and role in adipogenesis. J Biol Chem. 2005;280:32883–32889. doi: 10.1074/jbc.M503885200. [DOI] [PubMed] [Google Scholar]
- Rasiah KK, Gardiner-Garden M, Padilla EJ, Möller G, Kench JG, Alles MC, Eggleton SA, Stricker PD, Adamski J, Sutherland RL, et al. HSD17B4 overexpression, an independent biomarker of poor patient outcome in prostate cancer. Mol Cell Endocrinol. 2009;301:89–96. doi: 10.1016/j.mce.2008.11.021. [DOI] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RW, Oh WK, Xie W, Pomerantz M, Nakabayashi M, Sartor O, Taplin ME, Regan MM, Kantoff PW, Freedman M. Inherited variation in the androgen pathway is associated with the efficacy of androgen-deprivation therapy in men with prostate cancer. J Clin Oncol. 2008;26:842–847. doi: 10.1200/JCO.2007.13.6804. [DOI] [PubMed] [Google Scholar]
- Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–8261. doi: 10.1200/JCO.2005.03.4777. [DOI] [PubMed] [Google Scholar]
- Sharifi N. Minireview: androgen metabolism in castration-resistant prostate cancer. Mol Endocrinol. 2013;27:708–714. doi: 10.1210/me.2013-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi N, Auchus RJ. Steroid biosynthesis and prostate cancer. Steroids. 2012;77:719–726. doi: 10.1016/j.steroids.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, Febbo PG, Balk SP. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- Tazi J, Bakkour N, Stamm S. Alternative splicing and disease. Biochim Biophys Acta. 2009;1792:14–26. doi: 10.1016/j.bbadis.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–4657. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- True L, Coleman I, Hawley S, Huang CY, Gifford D, Coleman R, Beer TM, Gelmann E, Datta M, Mostaghel E, et al. A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc Natl Acad Sci USA. 2006;103:10991–10996. doi: 10.1073/pnas.0603678103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Grunsven EG, van Berkel E, Ijlst L, Vreken P, de Klerk JB, Adamski J, Lemonde H, Clayton PT, Cuebas DA, Wanders RJ. Peroxisomal D-hydroxyacyl-CoA dehydrogenase deficiency: resolution of the enzyme defect and its molecular basis in bifunctional protein deficiency. Proc Natl Acad Sci USA. 1998;95:2128–2133. doi: 10.1073/pnas.95.5.2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zha S, Ferdinandusse S, Hicks JL, Denis S, Dunn TA, Wanders RJ, Luo J, De Marzo AM, Isaacs WB. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate. 2005;63:316–323. doi: 10.1002/pros.20177. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.