

Abstract
Objective
To determine the ability of CSF biomarkers to predict disease progression in progressive supranuclear palsy (PSP).
Methods
We compared the ability of baseline CSF β-amyloid1–42, tau, phosphorylated tau 181 (p-tau), and neurofilament light chain (NfL) concentrations, measured by INNO-BIA AlzBio3 or ELISA, to predict 52-week changes in clinical (PSP Rating Scale [PSPRS] and Schwab and England Activities of Daily Living [SEADL]), neuropsychological, and regional brain volumes on MRI using linear mixed effects models controlled for age, sex, and baseline disease severity, and Fisher F density curves to compare effect sizes in 50 patients with PSP. Similar analyses were done using plasma NfL measured by single molecule arrays in 141 patients.
Results
Higher CSF NfL concentration predicted more rapid decline (biomarker × time interaction) over 52 weeks in PSPRS (p = 0.004, false discovery rate–corrected) and SEADL (p = 0.008), whereas lower baseline CSF p-tau predicted faster decline on PSPRS (p = 0.004). Higher CSF tau concentrations predicted faster decline by SEADL (p = 0.004). The CSF NfL/p-tau ratio was superior for predicting change in PSPRS, compared to p-tau (p = 0.003) or NfL (p = 0.001) alone. Higher NfL concentrations in CSF or blood were associated with greater superior cerebellar peduncle atrophy (fixed effect, p ≤ 0.029 and 0.008, respectively).
Conclusions
Both CSF p-tau and NfL correlate with disease severity and rate of disease progression in PSP. The inverse correlation of p-tau with disease severity suggests a potentially different mechanism of tau pathology in PSP as compared to Alzheimer disease.
Progressive supranuclear palsy (PSP) has been a focus of tau biomarker and anti-tau therapeutic development due to the high correlation between its classical phenotype, now called Richardson syndrome (PSP-RS), and PSP pathology, involving a characteristic pattern of 4 microtubule binding repeat (4R) tau deposition in various brain regions and strong genetic links to tau gene (MAPT) polymorphisms.1–4 In Alzheimer disease (AD), elevated CSF total tau and phosphorylated tau 181 (p-tau) and decreased β-amyloid1–42 (Aβ42) are useful for diagnosis and as pharmacodynamic biomarkers in clinical trials.5 In PSP, however, despite strong links to tau, CSF tau and p-tau concentrations are often lower than in age-matched controls, and do not change over the course of 1 year.6–8 An unresolved question is whether lower CSF tau and p-tau concentrations have clinical significance in PSP, similar to low Aβ42 for AD. To date, the only CSF analyte that displays clear clinical correlates in PSP is neurofilament light chain (NfL). NfL was the only fluid biomarker to show longitudinal changes correlating with clinical and regional MRI volume changes in a recent PSP clinical trial.6 Although recent studies have demonstrated the potential utility of using plasma NfL to diagnose PSP and predict disease progression,9–12 CSF analyte concentrations generally have stronger relationships to clinical features of other disorders.13 The goal of this study was to determine the clinical correlates of CSF NfL, Aβ42, tau and p-tau, and plasma NfL and their ability to predict disease progression using data from a well-characterized PSP clinical trial cohort.
Methods
Patients
Historical data were taken from the previously reported AL-108-231 international, randomized, double-blind, placebo-controlled, phase 2/3 trial of davunetide for PSP (clinicaltrials.gov, NCT 01110720).6 This trial enrolled 313 patients meeting clinical criteria for PSP-RS at 48 centers in Australia, Canada, France, Germany, the United Kingdom, and the United States. In the current study, 2 subgroups were analyzed: one with available CSF (n = 50) and another previously reported with available plasma (n = 141).11 Twenty-two patients had both CSF and plasma samples (figure e-1, links.lww.com/WNL/A66).
Standard protocol approvals, registrations, and patient consents
Patients provided informed consent at the time of recruitment and procedures were approved by local ethics committees.6
Clinical evaluation
See supplemental Methods, links.lww.com/WNL/A68.
The Mini-Mental State Examination (MMSE)14 was obtained at baseline. Neurologic assessments were based on the Progressive Supranuclear Palsy Rating Scale (PSPRS).15 The Repeatable Battery for the Assessment of Neuropsychological Disease Severity (RBANS)16 and 3 measures of executive function (color trails,17 letter-number sequencing [LNS],18 and phonemic fluency19) were used to measure neuropsychological status. The color trails test is a language-free version of the trails-making test. It consists of part 1 (CTT1) and part 2 (CTT2), which measure sustained attention and set-shifting, respectively. Assessment of overall disability relied on the Schwab and England Activities of Daily Living (SEADL) scale and a global measure of disease severity was obtained with the Clinical Global Impression of Severity (CGI-S).20 PSPRS and SEADL scores were obtained at baseline, 6-, 13-, 26-, 39-, and 52-week visits. Each of these time points is referred to as an interval. The RBANS and measures of executive function were obtained at baseline, 26-, and 52-week visits. The CGI-S was obtained at baseline and week 52.
Biomarkers
CSF and plasma collections were performed according to the Alzheimer's Disease Neuroimaging Initiative protocol at baseline and week 52.21 The INNO-BIA AlzBio3 (Fujirebio, Gent, Belgium) platform was used to measure CSF Aβ42, tau, and p-tau. CSF NfL concentrations were measured using the Uman Diagnostics (Umea, Sweden) ELISA kit (NF-Light kit). For NfL measurements, samples were diluted 4× prior to running the assay and then a correction factor of 4 was used to determine final concentration. All Alzbio3 and NfL analyses were performed in duplicate by a centralized laboratory. Plasma NfL concentrations were determined using the NF-Light kit transferred onto the Simoa platform using a homebrew kit (Quanterix, Boston, MA) as previously described.11
MRI data
Whole-brain, ventricular, midbrain, and superior cerebellar peduncle (SCP) volumetric measures obtained from 1.5T or 3T structural MRI scans at baseline and week 52 were available in 127 of 141 cases with available plasma and in 46 of 50 with available CSF (figure e-1, links.lww.com/WNL/A66). The magnet strength was determined by site availability and no patients switched scanners during the study.22 MRI scans met standards established by the Mayo Clinic's Aging and Dementia Imaging Research Laboratory and volumetric data were generated using the Boundary Shift Integral technique or label propagation in voxel-based morphometry (SPM5) as previously described.6
Statistical analyses
Baseline values were compared to those at 52 weeks using Wilcoxon signed rank test. Tests for normality were conducted with the Shapiro-Wilk test. Biomarker data were log-transformed prior to further analyses because they were not normally distributed. Initial exploratory analyses included Pearson product-moment correlations between baseline or 52-week change in CSF biomarker concentrations (Aβ42, tau, p-tau, NfL, p-tau/tau, Aβ42/tau, Aβ42/p-tau, NfL/tau, and NfL/p-tau) and baseline or 52-week change in clinical scores and brain volumes. SEADL and CGI-S were analyzed with Spearman rank correlations but considered equivalent to Pearson correlations for false discovery rate (FDR) adjustment purposes. Linear mixed effects analyses controlling for age, sex, and baseline MMSE were used to determine the ability of baseline CSF biomarkers to predict a change in clinical measures over 6, 13, 26, 39, and 52 weeks. Dependent variables included SEADL, CGI-S, PSPRS, RBANS, color trails, LNS, and phonemic fluency scores. Mixed linear models examining the ability of CSF biomarkers to predict changes in volumetric MRI data (whole-brain, ventricular, midbrain, and SCP volumes) were corrected for magnet strength and total intracranial volume, instead of MMSE. Separate models examined the effect of including baseline PSPRS score as a covariate. A compound symmetry repeated covariance matrix was used in all models and biomarker values were mean-centered. To account for the effect of baseline values, a term for biomarker by time interaction was introduced in addition to the random intercepts. In a first set of analyses, baseline CSF biomarker values were entered as continuous predictor variables. In a second set of analyses, and to test whether biomarker cutoff values could discriminate patients with a faster decline, baseline CSF biomarker values were entered as categorical variables, dichotomized at the 50th percentile value. Similar analyses were conducted with baseline plasma NfL as a predictor variable. To test the superiority of biomarkers to predict outcomes and to test for the value of combining different biomarkers in a ratio, the significance of the difference between F values from 2 given mixed linear models was computed with the Fisher distribution variance ratios. For all analyses, we accepted a p < 0.05, corrected for multiple comparisons across all dependent variables for a given biomarker, using FDR.23 Data were analyzed using SPSS (version 24; SPSS/IBM, Chicago, IL).
Results
Baseline associations of CSF analytes with clinical and imaging variables
At baseline, CSF p-tau and NfL, but not Aβ42 or tau, correlated with clinical, neuropsychological, and volumetric MRI measures (table 1 and figures e-2 to e-10, links.lww.com/WNL/A66). Biomarker ratios containing p-tau or NfL, except for p-tau/tau, correlated with clinical, executive, or volumetric variables. NfL was the only biomarker that changed median values over time (18% increase in CSF, p = 0.005, and 13% increase in plasma, p < 0.001, Wilcoxon test). CSF and plasma NfL concentrations were correlated (r = 0.64, p = 0.001). Although correlations between the annual change in NfL concentrations and the change in clinical variables have been previously reported,6 no such correlations were observed in this smaller sample for any of the biomarkers.
Table 1.
Clinical scores, brain volumes, and fluid biomarker concentrations at baseline and 1-year follow-up
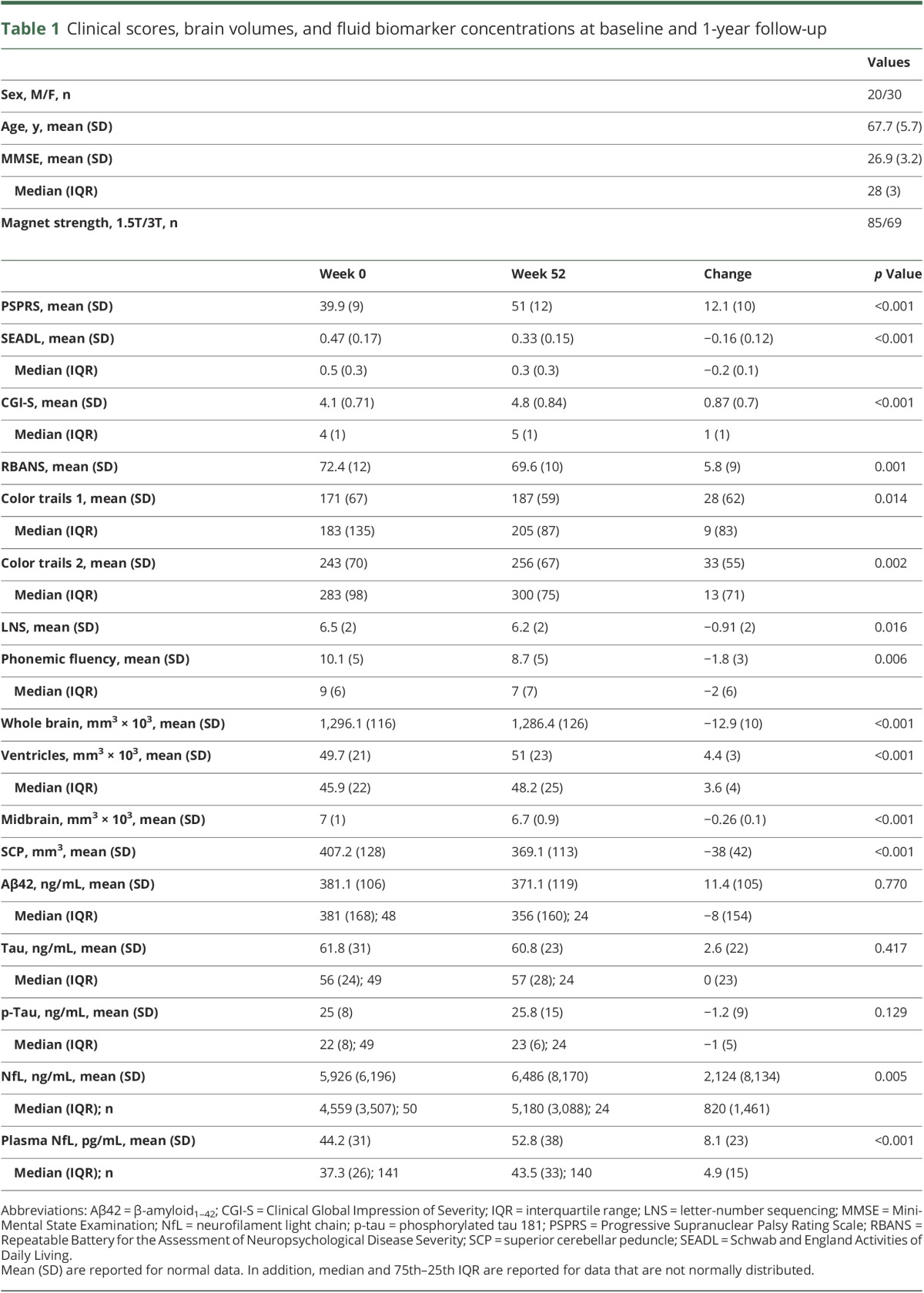
Baseline CSF values predict longitudinal change
Baseline CSF NfL, p-tau, and tau were strong predictors of the rate of change in clinical variables over 52 weeks. Biomarker concentration by time interactions were observed with higher CSF NfL concentration as a predictor of faster annual worsening in PSPRS (2.5 points per log NfL ng/mL increase per interval, 95% confidence interval [CI] 0.99–4.1, p = 0.004, FDR-corrected) (figure 1) and SEADL (−0.06 points per log NfL ng/mL increase per interval [i.e., 13-week follow-up], 95% CI −0.04 to −0.009, p = 0.008) (figure 2). Biomarker concentration by time interactions were also observed with lower baseline p-tau as a predictor of faster decline in PSPRS (−5.3 points per log p-tau ng/mL increase per interval, 95% CI −8.3 to −2.3, p = 0.004), but with higher tau as a predictor of faster worsening in SEADL (−0.04 points per log tau ng/mL increase per interval, 95% CI −0.07 to −0.01, p = 0.004, figure 1). Fixed effects of NfL without interaction with time were observed with higher NfL associated with lower SCP volumes (p = 0.029) (figure 3). No relationships were observed between Aβ42 and any variables.
Figure 1. Progressive Supranuclear Palsy Rating Scale (PSPRS) change predicted by baseline CSF biomarker concentration.

(A–C) PSPRS as a function of baseline biomarker concentration (log transformed, ng/mL) from linear mixed effects models. Each patient's longitudinal PSPRS scores appear as a vertical array of dots of increasing saturation (lightest at baseline to darkest at 52 weeks). Each individual's rate of change in PSPRS scores is depicted by the vertical distance between dots. In the examples within the purple rectangles in B and C, the rate of change is small at either high CSF phosphorylated tau 181 (p-tau) (≈log 1.8 or 63 ng/mL) or low neurofilament light chain (NfL) (≈log 3.2 or 1,600 ng/mL) concentrations (*), whereas it is relatively higher at low p-tau (≈log 1.2 or 16 ng/mL) and high NfL (≈log 4.5 or 31,600 ng/mL) (**), reflecting a time by biomarker interaction. There was no relationship between baseline CSF tau and rate of PSPRS change (A). p Values adjusted for false discovery rate (FDR) are shown for the interactions. The regression lines represent fixed effects of biomarkers on PSPRS scores and dotted lines represent 95% confidence intervals fit to the mean value of all time points for each individual. Fixed effects were present only for NfL, but did not survive FDR correction. FDR-adjusted p values are shown for (biomarker) × time interactions. Models are corrected for age, sex, and baseline Mini-Mental State Examination.
Figure 2. Schwab and England Activities of Daily Living (SEADL) scale change predicted by baseline CSF biomarker concentration.

(A–C) SEADL as a function of baseline biomarker concentration (log-transformed) from linear mixed effects models. Predicted scores are represented by arrays of dots of increasing intensity, and are associated with a particular baseline biomarker level. As revealed when comparing the purple rectangles at the extremes of the x axis, for tau (A) and neurofilament light chain (NfL) (C), there is higher variability in SEADL scores (i.e., rate of progression depicted by the vertical distance between dots) with higher values on the x axis, which reflects significant time by biomarker interactions. p Values (false discovery rate [FDR]–corrected) are shown for (biomarker) × time interactions. The regression lines represent fixed effects of biomarker concentrations on SEADL scores. Fixed effects were observed for tau and phosphorylated tau 181 (p-tau) (B), but these did not survive false discovery rate adjustment. Dotted lines represent 95% confidence intervals. Models are corrected for age, sex, and baseline Mini-Mental State Examination.
Figure 3. CSF neurofilament light chain (NfL) is correlated with superior cerebellar peduncle (SCP) volume.

(A) Tau, (B) phosphorylated tau 181 (p-tau), (C) NfL. SCP volume presented as a function of baseline biomarker concentration (log-transformed), from linear mixed effects model. A fixed effect of CSF NfL on SCP volume (p = 0.008, false discovery rate–corrected) is represented by a red regression line. Dotted lines represent 95% confidence intervals. No biomarker concentration by time interactions were observed. Models are corrected for age, sex, magnet strength, and total intracranial volume.
Baseline plasma NfL concentration interacted with time as a predictor of faster disease progression measured by PSPRS (1.8 points per log plasma NfL pg/mL increase per interval, 95% CI 0.94–2.7, p < 0.008), SEADL (−0.02 points per log plasma NfL pg/mL increase per interval, 95% CI −0.04 to −0.007, p = 0.012), and RBANS (−6.3 points per log plasma NfL pg/mL increase per interval, 95% CI −9.6 to −3.0, p < 0.004). Plasma NfL was a superior predictor of the annual rate of change of RBANS (p = 0.004) and PSPRS (p = 0.016) compared to CSF NfL (table e-1, links.lww.com/WNL/A67). For PSPRS, this superiority was not attributable to the higher number of available plasma samples, since the difference remained after a comparison was run only in cases with available CSF and plasma (n = 22, p = 0.015). In this smaller sample, no difference was observed between plasma and CSF NfL for the prediction of RBANS (n = 22, p = 0.160).
Median baseline CSF analyte values identify patients with more severe disease
To test whether biomarker threshold values could identify patients with different rates of decline, patients were grouped based on a median split in biomarker concentration. No differences in the progression rates were observed between low and high medians for any of the CSF biomarkers. Nevertheless, CSF NfL and p-tau stratified patients by disease severity at baseline. Patients with p-tau ≤22 ng/mL had SEADL values that were −0.10 points (95% CI −0.19 to −0.01, p = 0.004) lower than patients with p-tau >22 ng/mL. Patients with higher CSF NfL (>4,559 ng/mL) had values that were 11 points (95% CI 5–16, p = 0.008) worse on PSPRS, −0.13 points (95% CI −0.22 to −0.04, p = 0.012) worse on SEADL and 1 point worse (95% CI 0.68–1.3, p = 0.004) on CGI-S than patients with low NfL (≤4,559 ng/mL). For measures of executive function, patients with high NfL took 38 seconds (95% CI 0.8–76, p = 0.016) longer to complete CTT1, 45 seconds (95% CI 3–82, p = 0.013) longer to complete CTT2, and showed a −2 point (95% CI −0.79 to −4, p = 0.020) difference in LNS scores. Remarkably, high CSF NfL was also associated with more severe SCP atrophy (−84 mm3, 95% CI −0.38 to −170, p = 0.029). Higher plasma NfL (>37.3 ng/mL) predicted worse performance and more severe atrophy with significant group by time interactions for PSPRS (p = 0.025), SEADL (p = 0.029), and RBANS (p = 0.004), compared to lower plasma (≤37.3 ng/mL). When used as dichotomized categorical variables, CSF NfL had stronger associations with baseline PSPRS (p < 0.001), SEADL (p = 0.014), and CGI-S (p = 0.002), compared to plasma NfL. Controlling for baseline PSPRS did not alter the results. No relationships were observed between clinical, executive, or neuroimaging outcomes and groups defined based on CSF Aβ42 or tau.
CSF biomarker ratios predict longitudinal change better than individual values
NfL/p-tau was superior to p-tau (Fisher distribution variance ratios, p = 0.003) or CSF NfL (p = 0.001) alone in predicting annual change in PSPRS. A baseline NfL/p-tau value >198.9 was better at identifying patients with low baseline and follow-up CTT1 and CTT2 scores compared to p-tau ≤22 ng/mL (p = 0.004 and p = 0.002, respectively) and CSF NfL >4,559 ng/mL (p = 0.031 and p = 0.006, respectively). The NfL/p-tau ratio offered no advantage in predicting changes in other clinical or neuroimaging variables (table e-1, links.lww.com/WNL/A67, and figure 4). Baseline p-tau/tau was superior for predicting the rate of annual decline in SEADL to p-tau (p = 0.001), NfL (p = 0.009), NfL/p-tau (p = 0.009), and plasma NfL (p = 0.001). Other ratios including Aβ42/tau, Aβ42/p-tau, and NfL/tau did not relate to clinical or MRI measures.
Figure 4. CSF neurofilament light chain (NfL)/phosphorylated tau 181 (p-tau) predicts clinical decline and stratifies patients by disease severity.

(A–C) Clinical assessments as a function of baseline biomarker concentration, from linear mixed effects models corrected for age, sex, and baseline Mini-Mental State Examination. Significant biomarker by time interactions (i.e., differences in the rate of change in clinical scores over time as function of NfL/p-tau concentrations) are observed for Progressive Supranuclear Palsy Rating Scale (PSPRS) and Schwab and England Activities of Daily Living (SEADL) scores. False discovery rate (FDR)–adjusted p values are shown for the interactions. The regression lines represent fixed effects of biomarker concentrations on clinical scores. Fixed effects were present in PSPRS, but did not survive FDR adjustment. Dotted lines represent 95% confidence intervals. (D–F) Median values are displayed for NfL/p-tau ≤198.9 in blue and >198.9 in red. Bars represent 95% confidence intervals. Asterisks represent fixed effects of NfL/p-tau group as a categorical variable. CGI-S = Clinical Global Impression of Severity.
Discussion
We found that baseline concentrations of NfL, in CSF or plasma, and CSF p-tau were useful predictors of rates of change in clinical and neuropsychological status in PSP. These biomarkers were also associated with disease severity at baseline as reflected by clinical rating scales, neuropsychological measures of executive function scores, and SCP volume measured by MRI. Similar to studies in other diseases and a previous study of plasma NfL in PSP,11 higher CSF NfL concentrations were associated with greater disease severity and faster rates of disease progression. These associations are important because they reflect magnitudes of difference in clinical rating scales that were previously shown to be clinically meaningful.24 For example, the observed concentration range of CSF NfL spans 2 log units, which corresponds to a difference of about 5 points in PSPRS scores. This represents the difference between having normal gait vs being wheelchair-bound.
Unexpectedly, we found that lower p-tau concentrations were associated with greater disease severity and more aggressive disease progression on multiple rating scales. This was not the case for CSF tau concentration, for which higher baseline concentration predicted faster rates of SEADL change, but no other clinical or imaging association. Combining CSF NfL and p-tau in a ratio further increased the prediction value for change in PSPRS over either analyte alone. CSF was superior to plasma NfL concentration for stratification of baseline disease severity as measured by SEADL, CGI-S, and PSPRS, but plasma NfL was superior to CSF for predicting rates of change. These data suggest that both CSF NfL and p-tau are useful biomarkers to measure disease severity and predict change in PSP.
Our findings are consistent with previous studies demonstrating normal or low normal CSF tau and p-tau in PSP,7,8,25,26 and extend these results by demonstrating that low CSF p-tau has strong clinical correlates. Unlike in AD, where higher p-tau and tau concentrations are associated with increasing disease severity (asymptomatic vs mild cognitive impairment vs dementia)27 and predict the development of clinical disease,28 in PSP the opposite appears to be true. Moreover, cross-sectional correlations between CSF p-tau with clinical measures of disease severity appear to be stronger in PSP than in AD.29 CSF Aβ42 had no predictive value in PSP, but lower p-tau concentrations were associated with greater disease severity and predicted more rapid disease progression over time, which is analogous to the association of low CSF Aβ42 as an early and persistent finding in AD. The findings suggest that although AD and PSP are both tauopathies featuring insoluble deposits of hyperphosphorylated tau in neurons (and glia in PSP) at autopsy, the mechanisms by which tau protein abnormalities either cause or reflect the underlying causes of neurodegeneration in these disorders are likely to be different. One possible explanation might be that CSF tau concentrations are related to the pathogenic tau isoforms found in each disease. In AD, both 3 and 4 microtubule binding domain repeat (3R) and 4R tau are found in neurofibrillary tangles, whereas in PSP mostly 4R tau is found deposited in neurons and glia. Another possibility is that low CSF p-tau concentrations reflect a key pathogenic mechanism in PSP that is different from AD. A genetic polymorphism near the MOBP gene that increases the risk of PSP is associated with increased caspase-mediated tau cleavage via increased expression of appoptosin, which can be detected in PSP brains at autopsy.30 Therefore, low CSF p-tau might reflect increased tau cleavage in PSP. Finally, as tau can spread transcellularly in a prion-like fashion, and different tau prion strains may be associated with different diseases, low CSF p-tau in PSP might be a feature of a pathogenic tau prion strain found in PSP.31
Our results support the utility of CSF and plasma NfL as PSP biomarkers. NfL is abundant in large-caliber myelinated axons, where it contributes mechanoresistance. NfL concentrations seem to reflect active neuronal injury, based on its variations that mirror acute clinical decline, such as that seen in neurodegeneration, hypoxia, and neuroinflammation.32 Similar to AD, higher NfL concentrations were associated with worse disease and predicted faster progression.33 Nevertheless, the dynamic range and variation rate of NfL concentrations in neurodegenerative diseases are still poorly understood.
Our study has important limitations. The findings were not replicated in an independent cohort, because a cohort with comparable characterization and biospecimen availability is difficult to build. This highlights the importance of capitalizing on the unique opportunities for biomarker discovery offered by certain study designs, such as clinical trials, especially in rare diseases. Further analysis is needed to understand the relative value of CSF and plasma NfL measures. This was limited by the relatively small number of samples, which might reflect a selection bias with retention of patients with less disability and better able to undergo lumbar puncture. The comparison between the prognostic performance of CSF and plasma NfL might also have been influenced by the use of different methodologies to measure CSF and plasma NfL concentration.34 Insight into the dynamics of clinical–biomarker relationships were out of the scope of the study and will require future longitudinal biomarker analyses. We did not control for white matter disease burden, which could have affected the relationships between the fluid biomarkers and MRI measures. Only patients with the classical PSP-RS phenotype were included in this study. It is uncertain how the results relate to other variant PSP phenotypes.4 Finally, although the PSP-RS phenotype is highly specific for PSP pathology, the lack of neuropathologic data remains a relevant caveat.
This study provides strong evidence supporting the clinical relevance of NfL and p-tau for patient stratification and as predictors of neurologic, cognitive, and functional decline in clinical trials of new therapies for PSP.
Glossary
- 4R
4 microtubule binding repeat
- Aβ42
β-amyloid1–42
- AD
Alzheimer disease
- CGI-S
Clinical Global Impression of Severity
- CI
confidence interval
- CTT1
color trails test 1
- CTT2
color trails test 2
- FDR
false discovery rate
- LNS
letter-number sequencing
- MMSE
Mini-Mental State Examination
- NfL
neurofilament light chain
- p-tau
phosphorylated tau 181
- PSP
progressive supranuclear palsy
- PSP-RS
progressive supranuclear palsy–Richardson syndrome
- PSPRS
Progressive Supranuclear Palsy Rating Scale
- RBANS
Repeatable Battery for the Assessment of Neuropsychological Disease Severity
- SCP
superior cerebellar peduncle
- SEADL
Schwab and England Activities of Daily Living
Contributor Information
Collaborators: AL-108-231 Investigators, David Williams, Anne Louise Lafontaine, Connie Marras, Mandar Jog, Michael Panisset, Anthony Lang, Lesley Parker, Alistair J. Stewart, Jean-Christophe Corvol, Jean-Philippe Azulay, Philippe Couratier, Brit Mollenhauer, Stefan Lorenzl, Albert Ludolph, Reiner Benecke, Gunter Hoglinger, Axel Lipp, Heinz Reichmann, Dirk Woitalla, Dennis Chan, Adam Zermansky, David Burn, Andrew Lees, Illana Gozes, Adam Boxer, Bruce L. Miller, Iryna V. Lobach, Erik Roberson, Lawrence Honig, Edward Zamrini, Rajesh Pahwa, Yvette Bordelon, Erika Driver-Dunkley, Stephanie Lessig, Mark Lew, Kyle Womack, Brad Boeve, Joseph Ferrara, Argyle Hillis, Daniel Kaufer, Rajeev Kumar, Tao Xie, Steven Gunzler, Theresa Zesiewicz, Praveen Dayalu, Lawrence Golbe, Murray Grossman, Joseph Jankovic, Scott McGinnis, Anthony Santiago, Paul Tuite, Stuart Isaacson, Julie Leegwater-Kim, Irene Litvan, David S. Knopman, Lon S. Schneider, Rachelle S. Doody, Mary Koestler, Clifford R. Jack, Jr, Viviana Van Deerlin, Christopher Randolph, Steve Whitaker, Joe Hirman, Michael Gold, and Bruce H. Morimoto
Author contributions
Julio C. Rojas: manuscript drafting and data analysis and interpretation. Jee Bang, Iryna V. Lobach, Richard M. Tsai: data analysis and manuscript revision. Gil D. Rabinovici, Bruce L. Miller: data interpretation and manuscript revision. Adam L. Boxer: study conceptualization and design, data interpretation and manuscript revision.
Study funding
Supported by R01AG038791, U54NS092089, T32 AG23481, and the Tau Consortium.
Disclosure
J. Rojas, J. Bang, I. Lobach, and R. Tsai report no disclosures relevant to the manuscript. G. Rabinovici served on the Roche, Lundbeck, and Piramidal scientific advisory boards. He received travel or funding honoraria from GE Healthcare, Piramidal Imaging, the Alzheimer's Association, the American Academy of Neurology, Eisai, and Roche and consultant fees from Putnam. He receives research support from Avid Radiopharmaceuticals/Eli Lilly, GE Healthcare, Piramal, NIH/NIA R01-AG04561, P50-AG02350 and R01-AG048234, NIH/National Institute of Neurological Disorders and Stroke U54NS092089 and R01-AG03879, Alzheimer's Association, John Douglas French Alzheimer's Foundation, Hellman Family Foundation, Tau Consortium, American College of Radiology, Association for Frontotemporal Lobar Degeneration, and Michael J. Fox Foundation. B. Miller served on the Tau Consortium, Douglas French Foundation, Larry Hillblom Foundation, and SAB Nat'l Institute for Health Research in Dementia advisory boards. He is an external advisor for the Stanford University and Pittsburgh Alzheimer's Disease research centers. He is on the editorial boards of Neurocase, Cambridge University Press, and Guilford Publications, Inc. He receives publishing royalties for Behavioral Neurology of Dementia (Cambridge 2009), Handbook of Neurology (Elsevier 2009), and The Human Frontal Lobes (Guilford, 2008). He receives research support from NIA P50 AG23501 P01 AG19724, P50 AG16573, and CMS 1C1CMS331346-01-00. A. Boxer receives research support from NIH U54NS092089, R01AG031278, R01AG038791, R01AG032306, R01AG022983, the Tau Research Consortium, the Bluefield Project to Cure Frontotemporal Dementia, Corticobasal Degeneration Solutions, and the Alzheimer's Association. He has served as a consultant for AbbVie, Cellgener, Ionis, Janssen, and Merck, and received research support from Avid, Biogen, BMS, C2N, Cortice, Forum, Genentech, Janssen, Pfizer, Eli Lilly, Roche, and TauRx. He holds stock options in Alector and Delos. Go to Neurology.org/N for full disclosures.
References
- 1.Rojas JC, Boxer AL. Neurodegenerative disease in 2015: targeting tauopathies for therapeutic translation. Nat Rev Neurol 2016;12:74–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dickson DW, Rademakers R, Hutton ML. Progressive supranuclear palsy: pathology and genetics. Brain Pathol 2007;17:74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoglinger GU, Respondek G, Stamelou M, et al. Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord 2017;32:853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Hoglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol 2017;16:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mattsson N, Lonneborg A, Boccardi M, Blennow K, Hansson O, Geneva Task Force for the Roadmap of Alzheimer's Biomarkers. Clinical validity of cerebrospinal fluid Abeta42, tau, and phospho-tau as biomarkers for Alzheimer's disease in the context of a structured 5-phase development framework. Neurobiol Aging 2017;52:196–213. [DOI] [PubMed] [Google Scholar]
- 6.Boxer AL, Lang AE, Grossman M, et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol 2014;13:676–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wagshal D, Sankaranarayanan S, Guss V, et al. Divergent CSF tau alterations in two common tauopathies: Alzheimer's disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry 2015;86:244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansson O, Janelidze S, Hall S, et al. Blood-based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017;88:930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu CH, Macdonald-Wallis C, Gray E, et al. Neurofilament light chain: a prognostic biomarker in amyotrophic lateral sclerosis. Neurology 2015;84:2247–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Rohrer JD, Woollacott IO, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology 2016;87:1329–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rojas JC, Karydas A, Bang J, et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol 2016;3:216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinacker P, Semler E, Anderl-Straub S, et al. Neurofilament as a blood marker for diagnosis and monitoring of primary progressive aphasias. Neurology 2017;88:961–969. [DOI] [PubMed] [Google Scholar]
- 13.Bacioglu M, Maia LF, Preische O, et al. Neurofilament light chain in blood and CSF as marker of disease progression in mouse models and in neurodegenerative diseases. Neuron 2016;91:56–66. [DOI] [PubMed] [Google Scholar]
- 14.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 15.Golbe LI, Ohman-Strickland PA. A clinical rating scale for progressive supranuclear palsy. Brain 2007;130:1552–1565. [DOI] [PubMed] [Google Scholar]
- 16.Randolph C, Tierney MC, Mohr E, Chase TN. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol 1998;20:310–319. [DOI] [PubMed] [Google Scholar]
- 17.Satz P, Uchiyama CL, White T. Color Trails Test. Professional Manual. Odessa: Psychological Assessment Resources; 1996. [Google Scholar]
- 18.Kaplan RM, Saccuzzo DP. Psychological Testing: Principles, Applications and Issues. Belmont: Wadsworth; 2009. [Google Scholar]
- 19.Heaton RK, Miller SW, Taylor MJ, Grant I. Revised Comprehensive Norms for an Expanded Halstead-Reitan Battery: Demographically Adjusted Neuropsychological Norms for African American and Caucasian Adults. Lutz: PAR; 2004. [Google Scholar]
- 20.Payan CA, Viallet F, Landwehrmeyer BG, et al. Disease severity and progression in progressive supranuclear palsy and multiple system atrophy: validation of the NNIPPS—Parkinson Plus Scale. PLoS One 2011;6:e22293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Alzheimer's Disease Neuroimaging Initiative (ADNI) Protocol [online]. Available at: adni.loni.usc.edu/wp-content/uploads/2010/09/ADNI_GeneralProceduresManual.pdf. Accessed April 1, 2017. [Google Scholar]
- 22.Tsai RM, Lobach I, Bang J, et al. Clinical correlates of longitudinal brain atrophy in progressive supranuclear palsy. Parkinsonism Relat Disord 2016;28:29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Statist Soc B 1995;57:289–300. [Google Scholar]
- 24.Hewer S, Varley S, Boxer AL, Paul E, Williams DR, Investigators AL. Minimal clinically important worsening on the Progressive Supranuclear Palsy Rating Scale. Mov Disord 2016;31:1574–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hall S, Ohrfelt A, Constantinescu R, et al. Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol 2012;69:1445–1452. [DOI] [PubMed] [Google Scholar]
- 26.Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol 2014;75:116–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jack CR Jr, Holtzman DM. Biomarker modeling of Alzheimer's disease. Neuron 2013;80:1347–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mattsson N, Zetterberg H, Hansson O, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA 2009;302:385–393. [DOI] [PubMed] [Google Scholar]
- 29.Fjell AM, Walhovd KB, Fennema-Notestine C, et al. CSF biomarkers in prediction of cerebral and clinical change in mild cognitive impairment and Alzheimer's disease. J Neurosci 2010;30:2088–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao Y, Tseng IC, Heyser CJ, et al. Appoptosin-mediated caspase cleavage of tau contributes to progressive supranuclear palsy pathogenesis. Neuron 2015;87:963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaufman SK, Sanders DW, Thomas TL, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron 2016;92:796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zetterberg H. Neurofilament light: a dynamic cross-disease fluid biomarker for neurodegeneration. Neuron 2016;91:1–3. [DOI] [PubMed] [Google Scholar]
- 33.Zetterberg H, Skillback T, Mattsson N, et al. Association of cerebrospinal fluid neurofilament light concentration with Alzheimer disease progression. JAMA Neurol 2016;73:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhle J, Barro C, Andreasson U, et al. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin Chem Lab Med 2016;54:1655–1661. [DOI] [PubMed] [Google Scholar]