

Abstract
Objective
To characterize, among European and Han Chinese populations, the genetic predictors of maculopapular exanthema (MPE), a cutaneous adverse drug reaction common to antiepileptic drugs.
Methods
We conducted a case-control genome-wide association study of autosomal genotypes, including Class I and II human leukocyte antigen (HLA) alleles, in 323 cases and 1,321 drug-tolerant controls from epilepsy cohorts of northern European and Han Chinese descent. Results from each cohort were meta-analyzed.
Results
We report an association between a rare variant in the complement factor H–related 4 (CFHR4) gene and phenytoin-induced MPE in Europeans (p = 4.5 × 10–11; odds ratio [95% confidence interval] 7 [3.2–16]). This variant is in complete linkage disequilibrium with a missense variant (N1050Y) in the complement factor H (CFH) gene. In addition, our results reinforce the association between HLA-A*31:01 and carbamazepine hypersensitivity. We did not identify significant genetic associations with MPE among Han Chinese patients.
Conclusions
The identification of genetic predictors of MPE in CFHR4 and CFH, members of the complement factor H–related protein family, suggest a new link between regulation of the complement system alternative pathway and phenytoin-induced hypersensitivity in European-ancestral patients.
Idiosyncratic cutaneous adverse drug reactions (cADRs) can have a genetic predisposition. The HLA-B*15:02 allele is a predictor of carbamazepine-induced Stevens-Johnson syndrome and toxic epidermal necrolysis (SJS/TEN) in individuals of Han Chinese and Southeast Asian descent, while a recent meta-analysis suggests that the allele is also a significant risk factor for oxcarbazepine-, phenytoin-, and lamotrigine-induced SJS/TEN.1,2 However, the association with HLA-B*15:02 does not extend to the milder but more common maculopapular exanthema (MPE) phenotype, and the allele is specific to individuals of Asian descent, limiting clinical utility across populations.3,4 HLA-A*31:01 has been confirmed as a transethnic risk factor for carbamazepine-induced cADRs, with the allele observed across populations of European, Japanese, and Korean descent.5–8 Recently, HLA-A*24:02 has been shown to associate with SJS in Han Chinese patients, irrespective of causal drug studied.9 Genetic variation beyond the major histocompatibility locus has also been associated with cADRs. The CYP2C9*3 allele correlates with phenytoin hypersensitivity in Han Chinese from Taiwan,10 with a similar effect reported in a Thai population.11 However, a genome-wide association study (GWAS) of lamotrigine and phenytoin-induced cADRs in Europeans did not detect significant predictors.12 A summary of the associated genetic risk variants for cADRs in various populations is provided in table e-1 (links.lww.com/WNL/A56).
The EpiPGX Consortium was established to identify genetic markers of epilepsy treatment response. The International League Against Epilepsy Complex Genetics Consortium (ILAE-CGC) facilitates the discovery of genetic variants influencing epilepsy predisposition.13 The EPIGEN consortium is a worldwide epilepsy genetics research framework and the Canadian Pharmacogenomics Network for Drug Safety (CPNDS) is an active surveillance network focused on identifying genomic markers of severe adverse drug reactions (ADRs) in children and adults.14 Collaboration among these consortia has provided detailed phenotypes and genotypes for over 15,000 epilepsy cases, for the investigation of antiepileptic drug (AED)–induced MPE.
Availing of the joint resources of EpiPGX, ILAE-CGC, EPIGEN, and CPNDS, this study aimed to characterize, among European and Han Chinese populations, the genetic predictors of MPE, a cutaneous ADR common to particular AEDs. Specifically, we set out to test the following hypotheses: (1) whether population-specific genetic variants predict MPE; (2) whether transethnic genetic variants predict MPE; (3) whether population-specific genetic variants predict AED-specific MPE; and (4) whether transethnic genetic variants predict AED-specific MPE.
Methods
Standard protocol approvals, registrations, and patient consents
All study participants provided written, informed consent for genetic analysis. Local institutional review boards approved study protocols at each contributing site.
Study design
We conducted a retrospective case-control study in individuals of European and Han Chinese ethnicity. Participants were exposed to carbamazepine, lamotrigine, phenytoin, or oxcarbazepine. Our analyses were structured to test genetic variants for association with MPE within and across both of the broad ancestral groups, through logistic regression of genotype dosage and subsequent meta-analysis of regression coefficients. We tested for association with (1) aromatic AED-induced MPE vs controls tolerant to at least 3 aromatic AEDs, (2) carbamazepine-induced MPE vs carbamazepine-tolerant controls, (3) lamotrigine-induced MPE vs lamotrigine-tolerant controls, and (4) phenytoin-induced MPE vs phenytoin-tolerant controls. Due to small sample size, oxcarbazepine-related MPE was not analyzed as an individual case cohort.
Cohorts and phenotype definition
Epilepsy cohorts from the ILAE-CGC, EpiPGX, and EPIGEN Consortia were included in the discovery GWAS meta-analysis (table 1). A European-descent replication cohort was assembled from sites in Brazil, Canada (via CPNDS), Liverpool, and other sites across the United Kingdom. Cases were defined as having MPE attributed to carbamazepine, lamotrigine, phenytoin, or oxcarbazepine as determined by their clinician, occurring within 3 months of initiation and resolving upon dose reduction or AED withdrawal. Control patients trialed carbamazepine, lamotrigine, phenytoin, or oxcarbazepine for at least 3 months without reporting a cADR. Epilepsy-specific patient demographics are presented in table e-2 (links.lww.com/WNL/A56).
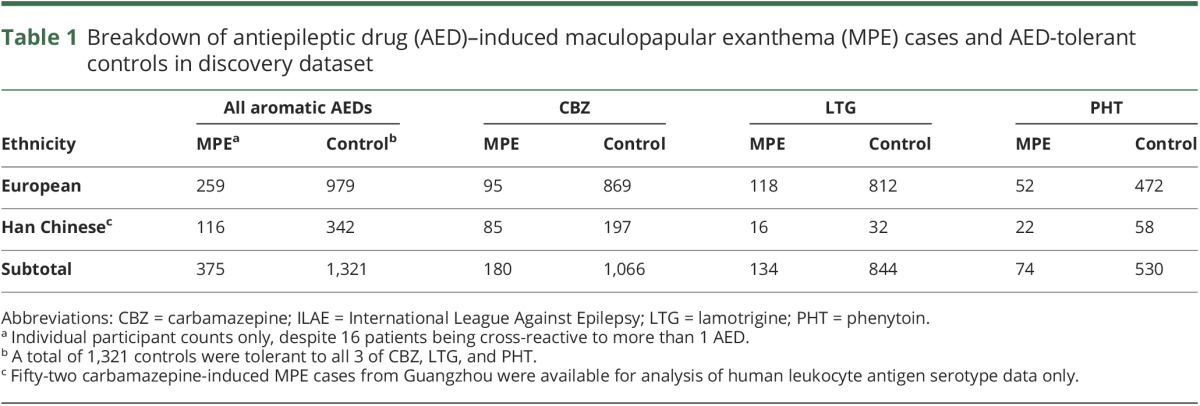
Table 1.
Breakdown of antiepileptic drug (AED)–induced maculopapular exanthema (MPE) cases and AED-tolerant controls in discovery dataset
Genotyping and imputation
Genotyping of a subset of EpiPGX samples was performed at deCODE Genetics (Reykjavik, Iceland) using Illumina (San Diego, CA) OmniExpress-12 v1.1 and OmniExpress-24 v1.1 single nucleotide polymorphism (SNP) arrays. The remainder of samples were genotyped locally using various Illumina beadchip SNP arrays, details of which are published elsewhere.13 Genotyping and imputation quality control is described in appendix e-1 (links.lww.com/WNL/A57).
Study power
We estimated that the study had 80% power to detect a genetic predictor of relative risk (approximated to odds ratio) ≥3 with an allele frequency ≥2% and an α level of 1.25 × 10−8. Power for AED-specific and population-specific analyses are detailed in appendix e-1 (links.lww.com/WNL/A57).
Statistical analyses
Association analyses were conducted within the European and Asian subgroups using an additive logistic regression model. To account for genotype uncertainty, SNPTEST was used to apply a missing data likelihood score model that included sex, clinical site, and 5 principal components as covariates to control for bias and population stratification.15 Fixed effects meta-analyses were conducted across the European and Asian subgroups using the software package METAL, applying genomic control correction within cohorts.16 The threshold for statistical significance was set at 1.25 × 10−8, reflecting an empirical Bonferroni correction for 4 tests, of the standard 5 × 10−8 genome-wide significance threshold. Conditional association analysis was performed on loci containing significant markers to establish whether other genetic variants in the region (1 Mb upstream and downstream) were independently associated with MPE. The conditional threshold for significance was set at 5 × 10−6, based on a genome-wide estimation of 10,000 imputed variants per 2 MB region.13 We applied the Stouffer z trend test to the combined results from the discovery and replication cohorts.
Confirmatory genotyping
Where an association signal satisfied the threshold for significance, additional genotyping and resequencing were performed in a subset of patients and results were compared with imputation dosage files. The variant rs78239784 was confirmed by Sanger sequencing in 100 patients from the original discovery cohort. For the purpose of replication, we genotyped the rs78239784 variant in an independent cohort of 13 phenytoin-induced MPE cases and 88 phenytoin-tolerant controls.
Results
Cohort description
In total, 375 MPE cases and 1,321 controls satisfied our criteria for inclusion in the discovery analyses (see Methods and table 1). There were 16 patients with cross-reactivity to 2 or more aromatic AEDs, 8 of whom were hypersensitive to carbamazepine and lamotrigine. Genome-wide array data for 323 cases and 1,321 controls were available for analysis. Broad European or Han Chinese ancestry was assigned to each participant according to principal components analysis (figure e-1, links.lww.com/WNL/A55).
Genome-wide association analysis of broad aromatic AED–induced MPE
After quality control (see appendix e-1, figure e-2, links.lww.com/WNL/A55, for details), 3,693,290 variants remained for analysis in the European dataset and 4,402,554 variants in the Han Chinese dataset. We only considered autosomal SNPs in our analyses. To test hypothesis (1), that population-specific genetic markers predispose to MPE, a logistic regression analysis of all MPE cases was performed separately in the European and Han Chinese ancestral subgroups. We did not observe any genome-wide significant markers for MPE due to any aromatic AED in either Europeans or Han Chinese. The study was powered to detect an effect of relative risk >3.5 in the European cohort and >5 in the Han Chinese.
To test hypothesis (2), that transethnic genetic markers predispose to MPE, a fixed-effects meta-analysis of the association results for European and Han Chinese ancestral subgroups was performed. We did not observe any genome-wide significant markers for MPE shared among European or Han Chinese subgroups (figure 1A). This analysis was powered to detect an effect size >3.
Figure 1. Meta-analysis results across European and Han Chinese cohorts.

Manhattan (a) and quantile–quantile (b) plots for the meta-analyses of maculopapular exanthema vs tolerant controls, for (A) any antiepileptic drug (genomic inflation factor [λ] = 1.01), (B) carbamazepine (λ = 1.01), (C) lamotrigine (λ = 0.99), and (D) phenytoin (λ = 0.98).
Genome-wide association analysis of specific aromatic AED–induced MPE
To test hypothesis (3), that genetic variants for MPE are AED-specific and population-specific, logistic regression analyses of AED-specific MPE was performed separately in the European and Han Chinese ancestral groups (figures e-3 and e-4, links.lww.com/WNL/A55). Within the European subgroup, HLA-A*31:01 was significantly associated with carbamazepine-induced MPE in Europeans (p = 1.47 × 10−10, odds ratio [OR] [95% confidence interval (CI)] 5.5 [3.0–10]). Conditioning on HLA-A*31:01 did not reveal additional variants within the human leukocyte antigen (HLA) region that were independently contributing to carbamazepine-induced MPE. No genome-wide significant signals for lamotrigine-induced MPE were observed in Europeans. This analysis was powered to detect an effect size >6.
For phenytoin-induced MPE, we identified a significant association with rs78239784, an intronic variant of the complement factor H–related 4 gene (CFHR4). The risk allele, G, had a minor allele frequency of 12% in our European phenytoin-induced MPE cases compared to 1.5% in European phenytoin-tolerant controls (p = 2.94 × 10−10, OR [95% CI] 8.8 [4.0–19]; figure 2). Conditioning on rs78239784 did not reveal additional variants in this locus that were independently contributing to phenytoin-induced MPE. Using 1000 Genomes Phase III European population data, rs78239784 was found to be in complete linkage disequilibrium with rs35274867 (r2 = 1 and D' = 1), a missense variant coding for an asparagine to tyrosine substitution at amino acid 1,050 of the complement factor H (CFH) gene. The missense variant was not present in our association results, as it was filtered during quality control because the imputation score was <0.95. The imputation accuracy of the top variant rs78239784 was confirmed in our cohort via Sanger sequencing and TaqMan approaches. Of 100 samples tested, a 100% concordance rate was found between imputed and resequenced genotypes. Within the Han Chinese subgroup, no significant associations were found between autosomal SNPs or HLA alleles and AED-specific MPE. Summary results for known risk loci in our dataset were scrutinized and are presented in table 2. None of these loci was even nominally significant (p > 0.05) in the Han Chinese subgroup.
Figure 2. Intronic CFHR4 variants are associated with phenytoin-induced maculopapular exanthema (MPE) in Europeans.

(A) Manhattan and (B) quantile–quantile plot of phenytoin-induced MPE in the European subgroup (λ = 1.01). The LocusZoom plot (C) highlights the most significant single nucleotide polymorphism (SNP), rs78239784 (purple dot), is an intronic variant in CFHR4.
Table 2.
Association test results for risk alleles for maculopapular exanthema (MPE) across ethnicities
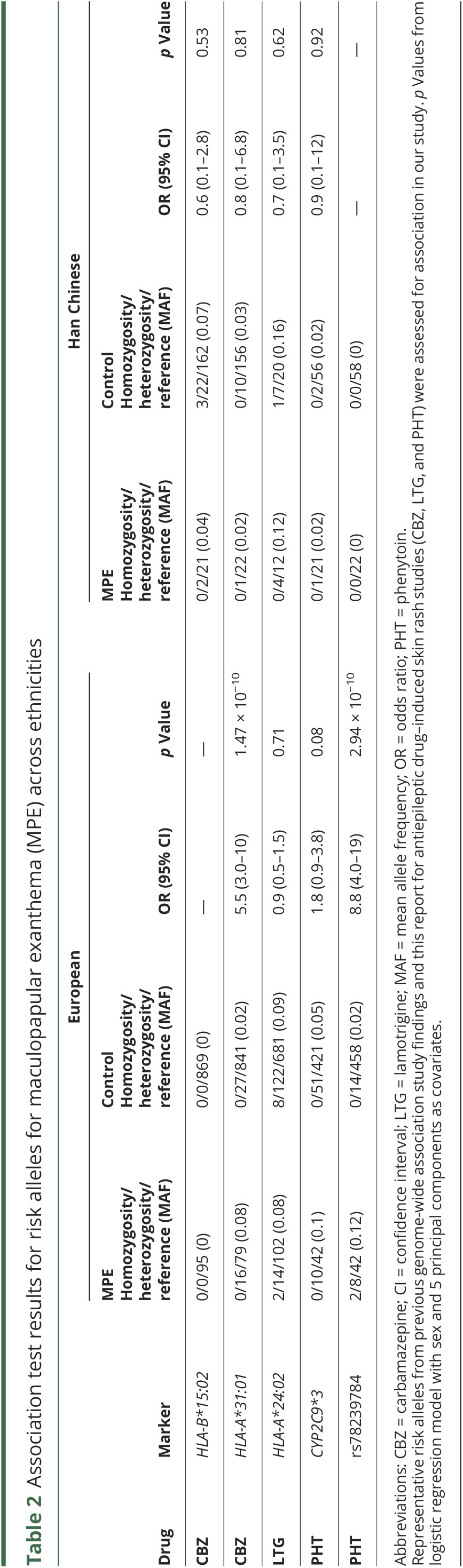
In order to test hypothesis (4), that transethnic genetic markers predispose to AED-specific MPE, we meta-analyzed p values from association results for carbamazepine, lamotrigine, and phenytoin individually, across European and Han Chinese ancestral subgroups. There were no shared genome-wide significant markers among our meta-analyses of AED-specific MPE (figure 1, B–D).
Replication of CFHR4 signal
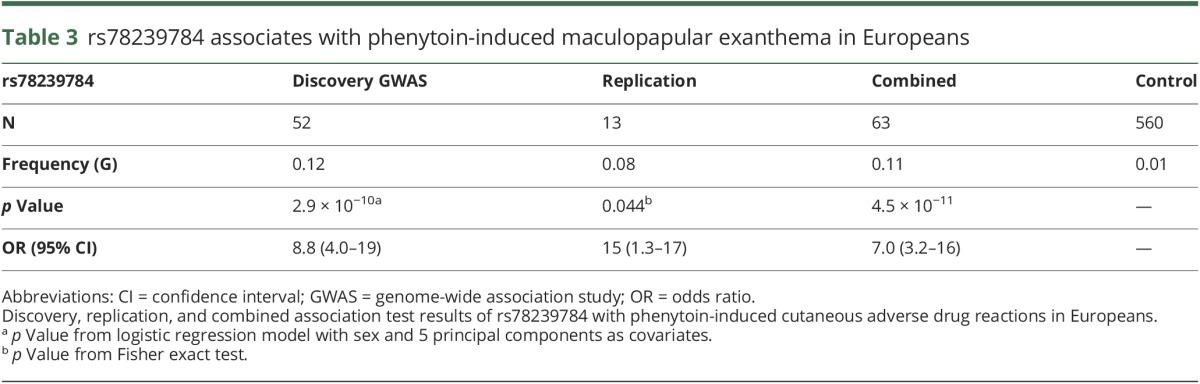
To replicate the association with phenytoin-induced MPE in an independent cohort, the variant rs78239784 was genotyped in self-reported European-descent cases and controls recruited through centers in Liverpool (United Kingdom), Sao Paolo (Brazil), and across Canada (CPNDS). Two heterozygous carriers were identified among 13 phenytoin-induced MPE cases while only a single carrier was observed among 88 phenytoin-tolerant controls yielding a 2-tailed Fisher exact p value of 0.044. Pooling all cases and controls together, we report an overall p value of 4.5 × 10−11 (with a combined OR [95% CI] 7 [3.2–16]) for the association between rs78239784 and phenytoin-induced MPE in Europeans (table 3).
Table 3.
rs78239784 associates with phenytoin-induced maculopapular exanthema in Europeans
Discussion
We detected a strong association between variants in the complement factor H regulatory pathway and phenytoin-induced MPE in a European-descent patient population. The presence of the associated genotype increases risk for MPE 6-fold. Our results indicate that risk variants for MPE tend to be drug-specific and population-specific.
These results point to the regulators of complement activation gene cluster as a genetic locus contributing to the onset of hypersensitivity to phenytoin. The most significant variant in our European subgroup analysis, rs78239784 (c.59-2448T>G), tags the missense variant rs35274867 (p.N1050Y) in CFH, suggesting aberrant complement activation as a potential causal mechanism in a subset of phenytoin-sensitive individuals. According to data from the Exome Aggregation Consortium, CFH N1050Y has an allele frequency of approximately 2% in Europeans, 3% in African subpopulations, less than 1% in South Asians, and is almost invariant in East Asians.17 Given the absence of this allele in East Asian populations, the lack of an association between the CFH locus and MPE in our Han Chinese cohort is unsurprising. We propose that population-specific, independent rare variants of large effect may explain a proportion of MPE cases, a similar paradigm to the rare variant model demonstrated in Crohn disease and ulcerative colitis.18 CFH N1050Y has previously been associated with type 2 diabetes–associated end-stage kidney disease in an African American cohort.19 Defects in CFH-related proteins are also associated with overactivation of the complement immune system and can lead to atypical hemolytic-uremic syndrome, C3 glomerulopathy, basal laminar drusen, immunoglobulin A nephropathies, and systemic lupus erythematosus.20 Further, genetic variants in CFHR4 and CFH are associated with risk for age-related macular degeneration.21 We did not detect these symptoms among N1050Y carriers. While it is unclear whether phenytoin directly interacts with circulating CFH-related proteins, it does not specifically increase serum complement levels.22 Our findings offer an expanded insight into the role of the complement alternative pathway in hypersensitivity to AEDs.
Phenytoin is still used as a first-line treatment for epilepsy in many settings and is listed on the WHO list of essential medicines.23 Epidemiologic data on prescriptions of AEDs for epilepsy in the United Kingdom show that, in 2008, phenytoin accounted for 18% of all treated person-years in epilepsy and was most frequently used in the elderly.24 Therefore, a clinically useful prognostic test for phenytoin-induced cutaneous ADRs in European-ancestral individuals would be welcome. The sensitivity of the CFHR4 variant as a prognostic marker is 16% and the specificity is 97%, which corresponds to a positive likelihood ratio of 5.93 (95% CI 2.8–12.6) and a negative likelihood ratio of 0.86 (95% CI 0.8–0.9). Assuming the pretest probability of the ADR is 5%, a positive test for this marker increases the probability of MPE to phenytoin sixfold to 30%, while a negative test reduces the probability marginally to 4.3%. There are an estimated 6 million people with epilepsy in Europe, which means approximately 90,000 people are at-risk carriers of this mutation.25 We estimate that 208 (95% CI 103–431) patients of European ancestry would need to be screened to prevent one case, based on a previously reported formula,26 which corresponds to an absolute risk reduction of 0.005 (95% CI 0.002–0.009). As a comparison, it is estimated that 442 Han Chinese patients would be needed to screened for HLA-B*15:02 in order to prevent a single carbamazepine-induced SJS/TEN case. We would suggest that the clinical utility and cost-effectiveness of implementing preemptive screening be evaluated through a prospective study.
We did not replicate the association between CYP2C9*3 (rs1057910) and MPE in our cohort, irrespective of ethnicity or AED. This is not surprising given that the original association with phenytoin in Han Chinese was largely driven by SJS/TEN cases, which were excluded from this analysis. We did, however, detect a nonsignificant enrichment of CYP2C9*3 (p = 0.08) among the European phenytoin-induced MPE cases, but the effect size we observe (OR 1.8) is smaller than previously reported for phenytoin-induced MPE in Han Chinese (OR 5.5).10 Notably, 2 CYP2C9*3 carriers among the phenytoin-induced MPE cases were also heterozygous for the CFHR4 variant while only 3 of 560 controls were jointly heterozygous. The frequency of CYP2C9*3 differs between controls from European and Han Chinese subgroups in our study, which is in accordance with background population frequency reported in the Exome Aggregation Consortium (European: 7%, East Asian: 3%). While our results do not support a significant effect of CYP2C9*3 in MPE, larger cohorts including severe cADR cases may resolve the extent of the association across populations.
HLA-A*31:01 was the most strongly associated marker with carbamazepine-induced MPE in Europeans in this study. Forty-three of the 95 cases studied here were also included in the discovery publication,8 but the effect of the allele remains significant when we restrict to new cases only (p = 4 × 10−7), thus providing an additional independent replication of the initial finding. We confirm that HLA-B*15:02 is not associated with carbamazepine-induced MPE in Han Chinese, and no novel signals emerged for carbamazepine-induced MPE in either population. No significant predictors of lamotrigine-induced MPE were observed in either population tested. HLA-A*24:02 was not significantly associated with lamotrigine-induced MPE in either of the European or Han Chinese ancestral subgroups; rather this allele was observed to be more frequent among our lamotrigine-tolerant controls.
Our meta-analyses did not reveal any significant transethnic genetic markers for MPE due to any AED. There were considerably more European-descent patients in this analysis than any other ethnicity, and we recognize this as a limitation of the study. Analysis of non-European cohorts is warranted. A second limitation of our study was the low number of Han Chinese lamotrigine-related MPE cases, relative to European-descent cases. Therefore we cannot conclusively rule out genetic predictors of modest effect size for MPE to lamotrigine in Han Chinese or other non-European descent populations. We recognize that our replication cohort for phenytoin is small and comprises self-reported ancestral Europeans. Since we did not have full genotype array data for these individuals, we relied on Fisher exact test for calculating significance rather than logistic regression with correction for principal components. Additional studies of larger sample size are required to further characterize the association, improve the estimation of the risk effect size, and determine the prognostic ability and economics of screening for this marker. Finally, as this study was not powered to investigate MPE attributed to oxcarbazepine due to low sample size, further investigation is warranted.
We have identified a genetic predictor for a common adverse reaction to phenytoin in European-descent patients, adding a new pharmacogenetic marker for potential use in the treatment of epilepsy. This finding adds to the list of genetic predictors of hypersensitivity to anticonvulsant therapy and opens up a new avenue for understanding the biology underlying cutaneous adverse reactions. This finding can advance genetic testing in the clinic as it expands the array of genetic tests available to aid clinicians in reducing overall rates of discontinuation due to adverse events and improving patient safety.
Acknowledgment
The authors thank the following coinvestigators: the members of the EpiPGX Consortium, the members of the Canadian Pharmacogenomics Network for Drug Safety (CPNDS) Consortium, the members of the International League Against Epilepsy Consortium on Complex Epilepsies (ILAE-CGC), and the members of the EPIGEN Consortium (a full list of coinvestigators is found in the e-Appendix); Ana Fulgenico-Maisch, Simona Donatello, Clare O'Kennedy, Lisa Slattery, Paula Corr, and Joanna Fay for clinical assessment of patients, technical assistance with DNA extraction, and sample management; and the patients for their participation in the study.
Glossary
- ADR
adverse drug reaction
- AED
antiepileptic drug
- cADR
cutaneous adverse drug reaction
- CFH
complement factor H
- CFHR4
complement factor H–related 4 gene
- CI
confidence interval
- CPNDS
Canadian Pharmacogenomics Network for Drug Safety
- GWAS
genome-wide association study
- HLA
human leukocyte antigen
- ILAE-CGC
International League Against Epilepsy Complex Genetics Consortium
- MPE
maculopapular exanthema
- OR
odds ratio
- SJS/TEN
Stevens-Johnson syndrome and toxic epidermal necrolysis
- SNP
single nucleotide polymorphism
Footnotes
Editorila, page 155
Contributor Information
Collaborators: International Workshop on Comorbidity in Multiple Sclerosis, Samuel F. Berkovic, Daniel H. Lowenstein, Erin L. Heinzen, Hakon Hakonarson, Russell J. Buono, Karen Oliver, Honsheng Gui, Stacey Cherny, Patrick Kwan, Larry Baum, Maxwell Kwok, Doug Speed, Rodrigo Secolin, Clarissa Yasuda, Fernando Cendes, Iscia Lopes–Cendes, Liao Wei-ping, Terence J. O’Brien, Andres Ingason, Lárus J. Gudmundsson, Kári Stefánsson, Sanjay M. Sisodiya, Andreja Avbersek, Kristin Heggeli, Costin Leu, Joseph Willis, Anna Tostevin, Krishna Chinthapalli, Chantal Depondt, Mojgansadat Borghei, Gianpiero L. Cavalleri, Norman Delanty, Mark McCormack, Sinéad Heavin, Wolfram S. Kunz, Holger Lerche, Stefan Wolking, Felicitas Becker, Sarah Rau, Christian Hengsbach, Yvonne Weber, Zvonka Rener Primec, Pasquale Striano, Antonietta Coppola, Federico Zara, Antonio Gamberdella, Amedeo Bianchi, Giuseppe Capovilla, Anna Teresa Giallonardo, Angela La Neve, Giovanni Crichiutt, Aglaia Vignoli, Roberto Michelucci, Francesca Bisulli, Carla Marini, Belcastro Vicenzo, Michael R. Johnson, Sarah R. Langley, Graeme J. Sills, Anthony G. Marson, Pauls Auce, Ben Francis, Andrea Jorgensen, Martin J. Brodie, Prashant Srivastava, Roland Krause, Martin Krenn, Fritz Zimprich, Ekaterina Pataraia, Karl Martin Klein, Felix Rosenow, Hiltrud Muhle, Dieter Dennig, Bernhard Steinhoff, Susanne Schubert-Bast, Thomas Mayer, Hartmut Baier, Rikke S. Møller, Marina Nikanorova, Sarah Weckhuysen, Peter de Jonghe, Hannah Stamberger, Josemir W. Sander, Bianca Berghuis, Bobby P.C. Koeleman, Anja C.M. Sonsma, Carolien de Kovel, David B. Goldstein, Slavé Petrovski, Rodney A. Radtke, Nicole Walley, J. Eunice Zhang, Munir Pirmohamed, Galen E.B. Wright, Colin J.D. Ross, Bruce C. Carleton, Michael Hayden, Stuart MacLeod, Reza Ghannadan, Shahrad Rod Rassekh, Henk Visscher, Folefac Aminkeng, Michelle Higginson, Nasim Massah, Fudan Miao, Adrienne Borrie, Ursula Amstutz, Claudette Hildebrand, Shevaun Hughes, Kaitlyn Shaw, Satvir Dhoot, Amit P Bhavsar, Yuling Li, Jong W Lee, Kaarina Kowalec, Jessica Stortz, Tessa Bendyshe-Walton, Mikaela Barker, Duncan Waltrip, Rachel Bader, Britt Drögemöller, Nicole McGoldrick, Cheri Nijssen-Jordan, David Johnson, Carnation Zhuwaki, Linda Verbeek, Rick Kaczowka, Patti Stevenson, Kevin Hall, Nick Honcharik, Geert 't Jong, Sara Israels, Shanna Chan, Byron Garnham, Michelle Staub, Michael Rieder, Becky Malkin, Neil Shear, Gideon Koren, Shinya Ito, Paul Nathan, Mark Greenberg, Facundo Garcia Bournissen, Miho Inoue, Sachi Sakaguchi, Toshihiro Tanaka, Hisaki Fujii, Mina Ogawa, Taro Kamiya, Smita Karande, Sholeh Ghayoori, Régis Vaillancourt, Donna Johnston, Herpreet Mankoo, Elaine Wong, Brenda Wilson, Lauren O’Connor, Caleb Hui, Cindy Yuen, Andrea Walsh, Kaviyarasan Mahalingam, Jean-François Bussières, Denis Lebel, Pierre Barret, Aurélie Clauson, Ève Courbon, Léna Cerruti, Maud Harry, Marine Aussedat, Margaret Murray, Marilyn Tiller, and Carol-anne Osborne
Affiliations
From Molecular and Cellular Therapeutics (M.M., N.D., G.L.C.) and FutureNeuro Research Centre (G.L.C.), Royal College of Surgeons in Ireland, Dublin; Centre for Genomic Sciences (H.G., S.C., L.B.), Li Ka Shing Faculty of Medicine, University of Hong Kong; deCODE Genetics/Amgen, Inc. (A.I., L.J.G., K.S.), Reykjavik, Iceland; UCL Genetics Institute (D.S.), University College London, UK; Centre for Molecular Medicine and Therapeutics, BC Children's Hospital Research Institute, Faculty of Medicine (G.E.B.W.), Departments of Pediatrics and Medical Genetics (C.J.D.R.), and Division of Translational Therapeutics, Department of Pediatrics (B.C.C.), University of British Columbia, Vancouver, Canada; Department of Molecular and Clinical Pharmacology (J.E.Z., G.J.S., A.G.M., P.A., M.P.), Institute of Translational Medicine, University of Liverpool, UK; Department of Medical Genetics (R.S., I.L.-C.), School of Medical Sciences, University of Campinas and the Brazilian Institute of Neuroscience and Neurotechnology (BRAINN); Department of Neurology (C.Y., F.C.), School of Medical Sciences, University of Campinas and the Brazilian Institute of Neuroscience and Neurotechnology (BRAINN), Campinas, SP, Brazil; Hertie Institute for Clinical Brain Research (M.K., H.L.), University of Tübingen, Germany; Department of Clinical and Experimental Epilepsy (S.W., F.B., S.R., A.A., K.H., C.L., S.M.S.) and NIHR University College London Hospitals Biomedical Research Centre (J.W.S.), UCL Institute of Neurology, London, UK; Laboratory of Experimental Neurology (C.D.), Hôpital Erasme, Université Libre de Bruxelles, Brussels, Belgium; The Walton Centre NHS Foundation Trust (A.G.M., P.A.), Liverpool; Epilepsy Unit (M.J.B.), West Glasgow ACH-Yorkhill; Department of Biostatistics (B.F.), Institute of Translational Medicine, University of Liverpool; Division of Brain Sciences (M.R.J.), Imperial College Faculty of Medicine, London, UK; Division of Neurosciences (B.P.C.K.) and Center for Molecular Medicine (M.M.), Universitair Medisch Centrum Utrecht, Netherlands; Pediatric Neurology and Muscular Diseases Unit, Department of Neurosciences, Rehabilitation, Ophthalmology, Genetics, Maternal and Child Health, University of Genoa (P.S.), and Laboratory of Neurogenetics and Neuroscience (F.Z.), Institute G. Gaslini; Department of Neuroscience (A.C.), Reproductive and Odontostomatological Sciences, Federico II University, Naples; Department of Epileptology (W.S.K.), University of Bonn, Germany; Stichting Epilepsie Instellingen Nederland (SEIN) (J.W.S.), Heemstede, Netherlands; The Chalfont Centre for Epilepsy (J.W.S., S.M.S.), Epilepsy Society, Chalfont St. Peters, Buckinghamshire, UK; Epilepsy Center Frankfurt Rhine-Main (K.M.K.), Department of Neurology, Center of Neurology and Neurosurgery, University Hospital, Goethe-University Frankfurt; Epilepsy Center Hessen (K.M.K.), Department of Neurology, University Hospitals Giessen & Marburg, and Philipps-University Marburg, Germany; Neurogenetics Group (S.W.), VIB-UAntwerp, Center for Molecular Neurology; Neurology Department (S.W.), University Hospital Antwerp, Belgium; Department of Neurology (M.K.), Medical University of Vienna, Austria; Faculty of Medicine (K.S.), University of Iceland, Reykjavik; Luxembourg Centre for Systems Biomedicine (R.K.), University of Luxembourg; University of Toronto (N.S.); Institute of Neuroscience and The Second Affiliated Hospital of Guangzhou Medical University (W.-p.L.), Key Laboratory of Neurogenetics and Channelopathies of Guangdong Province and the Ministry of Education of China, China; and Departments of Medicine and Neurology (T.J.O., P.K.), University of Melbourne, Royal Melbourne Hospital, Australia.
Author contributions
M.M.C., H.G., L.B., S.C., P.K., and G.L.C. contributed to the conception and design of the study and the acquisition, analysis, and interpretation of data. M.M.C. and H.G. performed all statistical analysis. A.I., S.M.S., R.S., S.W., F.B., S.R., K.H., C.L., A.G.M., P.A., M.J.B., B.F., M.R.J., N.S., G.E.B.W., C.J.D., B.C.C., D.S., J.E.Z., M.K., M.P., A.A., C.D., G.J.S., B.P.C.K., P.S., F.Z., A.C., W.S.K., J.W.S., H.L., K.M.K., S.W., M.K., L.J.G., R.K., N.D., C.Y., F.C., I.L.-C., T.J.O., W.-p.L., L.J.G., and K.S. contributed to the acquisition, analysis, and interpretation of data. All authors contributed to the critical revision of the final version of the manuscript for important intellectual content.
Study funding
This study was not industry-sponsored. The work was supported by a grant from the European Commission (7th Framework Programme Grant 279062, EpiPGX). M.M.C. and G.L.C. are supported by Science Foundation Ireland, grant 13/CDA/2223, and an RCSI seed funding grant GA 14-1899. This project was supported by the General Research Funds (HKU7623/08M and HKU7747/07M to S.C., CUHK4466/06M to P.K.) and Health and Medical Research Fund (HMRF 01120086 to P.K.) from Hong Kong. Some results presented in this article were prepared using the HPC facilities of the University of Luxembourg. This work was partly undertaken at UCLH/UCL, which received a proportion of funding from the Department of Health's NIHR Biomedical Research Centres funding scheme (J.W.S., S.M.S.). The work was also supported by the Epilepsy Society, UK (J.W.S., S.M.S.), by the foundation “no epilep,” the German Chapter of the ILAE (DGfE) (both to H.L.). F.C. and I.L.-C. are supported by Fundação de Amparo à Pesquisa do Estado de São Paulo, Brazil, through grant 2013/07559-3. J.E.Z. and M.P. thank the NHS Chair of Pharmacogenetics programme and MRC Centre for Drug Safety Science for support in Liverpool. B.C.C. and C.J.D.R. are supported by the Canadian Institutes of Health Research (CIHR) Drug Safety and Effectiveness Network (FRN-117588), the Canada Foundation for Innovation and the Canadian Dermatology Foundation. G.E.B.W. is supported by a CIHR Fellowship. The funders of the study had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. M.M.C., H.G., and G.L.C. had full access to all the data in the study and the corresponding authors had final responsibility for the decision to submit for publication.
Disclosure
M. McCormack and H. Gui report no disclosures relevant to the manuscript. A. Ingason is an employee of deCODE Genetics/Amgen. D. Speed, G. Wright, E. Zhang, R. Secolin, C. Yasuda, M. Kwok, S. Wolking, F. Becker, and S. Rau report no disclosures relevant to the manuscript. A. Avbersek is employed by UCB Pharma SPRL, Belgium, as Associate Director. K. Heggeli, C. Leu, C. Depondt, and G. Sills report no disclosures relevant to the manuscript. A. Marson was awarded grants from GSK, Eisai, and UCB Pharma, which funded the National Audit of Seizure Management in Hospitals. P. Auce, M. Brodie, B. Francis, M. Johnson, B. Koeleman, P. Striano, A. Coppola, F. Zara, and W. Kunz report no disclosures relevant to the manuscript. J. Sander has served on scientific advisory boards for UCB Pharma and Eisai; has served on speaker's bureaus for UCB Pharma, Eisai, Teva, and Lundbeck; has received research support from UCB Pharma, GSK, Eisai, The Marvin Weil Epilepsy Research Fund, and NL Nationaal Epilepsie Fonds; and his current position is endowed by the UK Epilepsy Society. H. Lerche has received speaker or consultancy fees or travel support from Bial, Desitin, Eisai, GlaxoSmithKline, Pfizer, UCB Pharma, or Valeant. K. Klein reports personal fees from UCB Pharma, Novartis Pharma AG, and Eisai, outside of the submitted work. AC was awarded a grant from EISAI and personal fees for speaking from Eisai, outside of the submitted work. S. Weckhuysen and M. Krenn report no disclosures relevant to the manuscript. L. Gudmundsson is an employee of deCODE Genetics/Amgen. K. Stefánsson is an employee of deCODE Genetics/Amgen. R. Krause, N. Shear, C. Ross, and N. Delanty report no disclosures relevant to the manuscript. M. Pirmohamed reports grants from MRC and grants from UK Department of Health during the conduct of the study. B. Carleton, through the Pharmaceutical Outcomes Programme (POPi), has received financial support for its pharmacogenomics research from the Canada Foundation for Innovation (CFI), Canadian Institutes of Health Research, Genome Canada, Genome British Columbia, and the Provincial Health Services Authority. Prof. Carelton has also received support by the University of British Columbia, Child & Family Research Institute, Vancouver, and Pfizer. Prof. Carleton has a patent and applications pending for biomarkers of anthracycline-induced cardiotoxicity and cisplatin-induced ototoxicity but no relevant financial disclosures relevant to this study. F. Cendes reports speaking fees from UCB Pharma, outside of the current work. I. Cendes-Lopes and W. Liao report no disclosures relevant to the manuscript. T. O'Brien reports grants from The Royal Melbourne Foundation during the conduct of the study. S. Sisodiya and S. Cherny report no disclosures relevant to the manuscript. P. Kwan has received speaker or consultancy fees and/or research grants from Eisai, GlaxoSmithKline, Johnson & Johnson, Pfizer, and UCB Pharma. L. Baum and G. Cavalleri report no disclosures relevant to the manuscript. Go to Neurology.org/N for full disclosures.
References
- 1.Li X, Yu K, Mei S, et al. HLA-B*1502 increases the risk of phenytoin or lamotrigine induced Stevens-Johnson syndrome/toxic epidermal necrolysis: evidence from a meta-analysis of nine case-control studies. Drug Res 2015;65:107–111. [DOI] [PubMed] [Google Scholar]
- 2.Chung WH, Hung SI, Hong HS, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature 2004;428:486. [DOI] [PubMed] [Google Scholar]
- 3.Man CB, Kwan P, Baum L, et al. Association between HLA-B*1502 allele and antiepileptic drug-induced cutaneous reactions in Han Chinese. Epilepsia 2007;48:1015–1018. [DOI] [PubMed] [Google Scholar]
- 4.Lonjou C, Thomas L, Borot N, et al. A marker for Stevens-Johnson syndrome: ethnicity matters. Pharmacogenomics J 2006;6:265–268. [DOI] [PubMed] [Google Scholar]
- 5.Amstutz U, Ross CJ, Castro-Pastrana LI, et al. HLA-A 31:01 and HLA-B 15:02 as genetic markers for carbamazepine hypersensitivity in children. Clin Pharmacol Ther 2013;94:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim SH, Lee KW, Song WJ, et al. Carbamazepine-induced severe cutaneous adverse reactions and HLA genotypes in Koreans. Epilepsy Res 2011;97:190–197. [DOI] [PubMed] [Google Scholar]
- 7.Ozeki T, Mushiroda T, Yowang A, et al. Genome-wide association study identifies HLA-A*3101 allele as a genetic risk factor for carbamazepine-induced cutaneous adverse drug reactions in Japanese population. Hum Mol Genet 2011;20:1034–1041. [DOI] [PubMed] [Google Scholar]
- 8.McCormack M, Alfirevic A, Bourgeois S, et al. HLA-A*3101 and carbamazepine-induced hypersensitivity reactions in Europeans. N Engl J Med 2011;364:1134–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi YW, Min FL, Zhou D, et al. HLA-A*24:02 as a common risk factor for antiepileptic drug-induced cutaneous adverse reactions. Neurology 2017;88:2183–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chung WH, Chang WC, Lee YS, et al. Genetic variants associated with phenytoin-related severe cutaneous adverse reactions. JAMA 2014;312:525–534. [DOI] [PubMed] [Google Scholar]
- 11.Tassaneeyakul W, Prabmeechai N, Sukasem C, et al. Associations between HLA class I and cytochrome P450 2C9 genetic polymorphisms and phenytoin-related severe cutaneous adverse reactions in a Thai population. Pharmacogenet Genomics 2016;26:225–234. [DOI] [PubMed] [Google Scholar]
- 12.McCormack M, Urban TJ, Shianna KV, et al. Genome-wide mapping for clinically relevant predictors of lamotrigine- and phenytoin-induced hypersensitivity reactions. Pharmacogenomics 2012;13:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.International League Against Epilepsy Consortium on Complex Epilepsies. Genetic determinants of common epilepsies: a meta-analysis of genome-wide association studies. Lancet Neurol 2014;13:893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Carleton B, Poole R, Smith M, et al. Adverse drug reaction active surveillance: developing a national network in Canada's children's hospitals. Pharmacoepidemiol Drug Saf 2009;18:713–721. [DOI] [PubMed] [Google Scholar]
- 15.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007;39:906–913. [DOI] [PubMed] [Google Scholar]
- 16.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beaudoin M, Goyette P, Boucher G, et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet 2013;9:e1003723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonomo JA, Palmer ND, Hicks PJ, et al. Complement factor H gene associations with end-stage kidney disease in African Americans. Nephrol Dial Transpl 2014;29:1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Skerka C, Chen Q, Fremeaux-Bacchi V, Roumenina LT. Complement factor H related proteins (CFHRs). Mol Immunol 2013;56:170–180. [DOI] [PubMed] [Google Scholar]
- 21.Fritsche LG, Igl W, Bailey JN, et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat Genet 2016;48:134–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Basaran N, Kansu E, Hincal F. Serum immunoglobulins, complement levels and lymphocyte subpopulations in phenytoin-treated epileptic patients. Immunopharmacol Immunotoxicol 1989;11:335–346. [DOI] [PubMed] [Google Scholar]
- 23.The selection and use of essential medicines. World Health Organ Tech Rep Ser 2015: vii–xv:1–546. [PubMed] [Google Scholar]
- 24.Nicholas JM, Ridsdale L, Richardson MP, Ashworth M, Gulliford MC. Trends in antiepileptic drug utilisation in UK primary care 1993-2008: cohort study using the General Practice Research Database. Seizure 2012;21:466–470. [DOI] [PubMed] [Google Scholar]
- 25.Baulac M, de Boer H, Elger C, et al. Epilepsy priorities in Europe: a report of the ILAE-IBE Epilepsy Advocacy Europe Task Force. Epilepsia 2015;56:1687–1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen Z, Liew D, Kwan P. Real-world efficiency of pharmacogenetic screening for carbamazepine-induced severe cutaneous adverse reactions. PLoS One 2014;9:e96990. [DOI] [PMC free article] [PubMed] [Google Scholar]