

Abstract
Store-operated Ca2+ entry (SOCE) is critical for salivary gland fluid secretion. We report that radiation treatment caused persistent salivary gland dysfunction by activating a TRPM2-dependent mitochondrial pathway, leading to caspase-3–mediated cleavage of stromal interaction molecule 1 (STIM1) and loss of SOCE. After irradiation, acinar cells from the submandibular glands of TRPM2+/+, but not those from TRPM2−/− mice, displayed an increase in the concentrations of mitochondrial Ca2+ and reactive oxygen species, a decrease in mitochondrial membrane potential, and activation of caspase-3, which was associated with a sustained decrease in STIM1 abundance and attenuation of SOCE. In a salivary gland cell line, silencing the mitochondrial Ca2+ uniporter or caspase-3 or treatment with inhibitors of TRPM2 or caspase-3 prevented irradiation-induced loss of STIM1 and SOCE. Expression of exogenous STIM1 in the salivary glands of irradiated mice increased SOCE and fluid secretion. We suggest that targeting the mechanisms underlying the loss of STIM1 would be a potentially useful approach for preserving salivary gland function after radiation therapy.
INTRODUCTION
Salivary gland fluid secretion is regulated by neurotransmitter-stimulated increases in the cytosolic concentration of Ca2+ ([Ca2+]i) in acinar cells, which activates the function of key ion channels and transporters that concertedly generate the osmotic gradient required to drive fluid secretion (1, 2). Physiologically, [Ca2+]i increase in salivary gland acinar cells is initiated in response to inositol 1,4,5-trisphosphate–mediated release of Ca2+ from the endoplasmic reticulum (ER) Ca2+ store(s) and influx of Ca2+ into the cells through plasma membrane channels. Fluid secretion depends on [Ca2+]i increase resulting from store-operated Ca2+ entry (SOCE), involving the channels TRPC1 (transient receptor potential canonical 1) and Orai1, both of which are regulated by the ER-Ca2+ sensor protein STIM1 (stromal interaction molecule 1) (1–6). A serious consequence of head and neck radiation treatment in patients is irreversible loss of salivary gland fluid secretion, leading to xerostomia (7–9). The exact mechanism(s) underlying irradiation-induced salivary gland dysfunction is not yet understood. The loss of salivary gland fluid secretion that occurs after relatively low doses of radiation treatment cannot be correlated with disruption of cellular morphology, apoptosis, or loss of acinar cells (10–16). We have reported that agonist-induced cell volume reduction is attenuated in acinar cells from mice after irradiation (14), which suggests that radiation might compromise acinar cell function. However, the exact consequences of irradiation on acinar cells and on mechanisms involved in the regulation of fluid secretion are not yet known.
We have previously reported that Ca2+ entry through TRPM2 (transient receptor potential melastatin 2), a reactive oxygen species (ROS)–sensitive Ca2+-permeable nonselective cation channel (17, 18), is activated by irradiation and critically contributes to radiation-induced salivary gland dysfunction (14). TRPM2 has also been reported to be activated by ionizing radiation treatment in cell lines and by electromagnetic radiation in rat brain (19–21). Our previous study showed that in contrast to TRPM2+/+ mice, TRPM2−/− mice do not display persistent loss of fluid secretion after radiation treatment. Rather, salivary gland function in these mice transiently decreases soon after the treatment but recovers to >75% of preradiation amounts by about 30 days. Thus, when TRPM2 is present in salivary gland acinar cells and is activated by irradiation, it converts an inherently reversible response to the treatment into a persistent loss of function. Here, we investigated the mechanism(s) that links the early activation of TRPM2 by radiation to the persistent loss of salivary fluid secretion. We found that irreversible loss of fluid secretion was caused by sustained reduction of SOCE due to loss of STIM1 that occurred as a result of TRPM2 activation by irradiation. Our data showed that the critical early consequences of radiation treatment in salivary gland acinar cells were TRPM2-mediated increase in [Ca2+]i, which triggered an increase in mitochondrial [Ca2+] ([Ca2+]mt) and ROS, and loss of mitochondrial membrane potential. There was also a relatively slower activation of caspase-3, although the enzyme remained active for a longer time. In cells from TRPM2−/− mice, the effects of radiation on mitochondria, capsase-3, STIM1 abundance, SOCE, and salivary fluid secretion were attenuated and transient when compared to the changes seen in acini from TRPM2+/+ mice. We also showed that radiation treatment of a human salivary gland (HSG) cell line induced decreases in STIM1 and that SOCE was prevented by blocking TRPM2 activation and knockdown of mitochondrial Ca2+ uniporter (MCU) or caspase-3 or by treatment with a caspase inhibitor, suggesting that caspase-3 is responsible for the loss of STIM1 and SOCE. Finally, we showed that adenovirus-mediated expression of STIM1 in the glands of irradiated TRPM2+/+ mice led to recovery of SOCE and fluid secretion. Together, these data indicated that the dysfunction in mouse salivary glands after irradiation was primarily caused by activation of a TRPM2-dependent mitochondrial pathway that resulted in caspase-3 cleavage of STIM1 and loss of SOCE. We suggest that targeting the mechanisms that underlie the loss of STIM1 would be a potentially useful approach for preserving salivary gland function after radiation treatment.
RESULTS
Radiation-induced activation of TRPM2 leads to loss of SOCE, STIM1, and fluid secretion in mouse submandibular gland
We measured resting [Ca2+]i (basal, in the absence of external Ca2+) and Ca2+ entry (after readdition of external Ca2+ medium) in acinar cells from submandibular glands (SMGs) of irradiated TRPM2+/+ and TRPM2−/− and control nonirradiated mice (noted as IR and CTL in figures and figure legends). Addition of external Ca2+ to acinar cells from irradiated TRPM2+/+ mice elicited an increase in [Ca2+]i, which was highest 1 day after radiation (about 40-fold greater than control acinar cells from nonirradiated mice), and decreased by 3 and 10 days after treatment to amounts 20- and 8-fold higher than that in control acinar cells, respectively (Fig. 1, A and B). Basal [Ca2+]i in TRPM2+/+ cells was higher on days 1 and 3 after irradiation compared to control cells and recovered to pretreatment values by 10 days (Fig. 1, A and C). Ca2+ addition to acini from irradiated TRPM2−/− mice induced a slow and small increase in [Ca2+]i that was detected only on day 1 (Fig. 1, D and E; 8-fold in TRPM2−/−compared to 40-fold in TRPM2+/+ cells 1 day after irradiation), whereas there was no significant increase in basal [Ca2+]i (Fig. 1, D and F). Thus, TRPM2-independent enhancement of plasma membrane Ca2+ permeability induced by irradiation was substantially less and more transient compared to that seen in response to activation of TRPM2 by irradiation.
Fig. 1. Radiation treatment causes transient activation of TRPM2 but persistent loss of SOCE, STIM1, and fluid secretion in submandibular acinar cells.

(A to F) SMG acinar cells were isolated from nonirradiated mice (CTL) and on 1, 3, and 10 days after 15-Gy radiation treatment (IR-1, IR-3, and IR-10). Fura-2 fluorescence ratio (340/380, which reflects [Ca2+]i) was monitored in cells from TRPM2+/+ (A) and TRPM2−/− (D) mice in Ca2+-free medium and after readdition of 1 mM external Ca2+. Quantitation of data from traces in (A) and (D) for peak increase in [Ca2+]i after Ca2+ add-back (B and E) and basal [Ca2+]i in Ca2+-free medium (C and F). Data were obtained from four CTL-TRPM2+/+ and four CTL-TRPM2−/− mice (210 acini in each case), 200 acini from three TRPM2+/+ mice and 212 acini from three TRPM2−/− mice (IR-1), 190 acini from three TRPM2+/+ mice and 188 acini from three TRPM2−/− mice (IR-3), and 210 acini from three TRPM2+/+ mice and 206 acini from three TRPM2−/− mice (IR-10). *P < 0.05 and **P < 0.01, comparing the indicated values on different days after irradiation with the respective control value in each set using unpaired t test (B, C, E, and F). (G) Pilocarpine-stimulated salivary fluid secretion was monitored in TRPM2+/+ and TRPM2−/− mice on 10 (28 TRPM2+/+ mice and 28 TRPM2−/− mice), 30 (26 TRPM2+/+ mice and 26 TRPM2−/−mice), and 60 (22 TRPM2+/+ mice and 22 TRPM2−/−mice) days after irradiation (**P < 0.01, comparing secretion in TRPM2+/+ mice to TRPM2−/− mice at each time point, unpaired t test). (H) Thapsigargin (Tg)–stimulated intra-cellular Ca2+ release and Ca2+ entry were measured in acini at 60 days after irradiation (data were obtained from 220 acini each from four nonirradiated TRPM2+/+ mice and four irradiated TRPM2+/+ mice and 215 acini from four IR-TRPM2−/− mice). (I) SOCE was measured at 10 (216 acini from four TRPM2+/+ acini and 210 acini from four TRPM2−/− mice), 30 (206 acini from four TRPM2+/+ mice and 202 acini from four TRPM2−/− mice), and 60 (186 acini from four TRPM2+/+ mice and 192 acini from four TRPM2−/− mice) days after irradiation (**P < 0.01, comparing TRPM2+/+ to TRPM2−/− cells at each time point, unpaired t test). (J) Western blot showing the abundance of STIM1, TRPC1, and NKCC1 in salivary gland samples from TRPM2+/+ and TRPM2−/− mice at 0, 10, 30, and 60 days after irradiation (as indicated). Data are representative of results with three salivary gland samples each from TRPM2+/+and TRPM2−/− mice at all time points shown. (K) Ca2+ influx was measured in HSG cells that were not irradiated (CTL; 224 cells from three independent experiments) or 1 hour after irradiation (IR, 214 cells from three independent experiments; IR + 3AB, 212 cells from three independent experiments; and IR + siTRPM2, 220 cells from three independent experiments). Inset shows Ca2+ influx in nonirradiated cells and in cells 4 hours after irradiation (200 to 240 cells from three independent experiments). (L) Thapsigargin-stimulated SOCE in control and irradiated cells. Inset shows the quantitation (**P < 0.01, comparing the indicated value with the respective control in each set, unpaired t test; 200 to 220 cells from three independent experiments). (M) Western blot of STIM1 in nonirradiated and irradiated HSG cells with or without 3AB treatment (four independent experiments).
Unlike radiation-induced TRPM2 activity in acinar cells from TRPM2+/+ mice, which almost recovered to pretreatment values by 10 days, the decrease in salivary fluid secretion was persistent with about 75% reduction by 10 days after treatment and no further changes at 30 and 60 days (Fig. 1G). Consistent with our previous study, saliva secretion was transiently decreased in irradiated TRPM2−/− mice, with about 65% decrease at 10 days, 35% at 30 days, and <25% at 60 days. Together, these data show that, although irradiation induces sustained loss of salivary gland fluid secretion in TRPM2+/+ mice, TRPM2−/− mice show initial loss with >75% recovery of function 2 months after treatment.
To elucidate the mechanism underlying radiation-induced salivary gland dysfunction, we measured SOCE in acinar cells, a process critically required for fluid secretion (2, 3). Acinar cells obtained from TRPM2+/+ mice 60 days after radiation treatment displayed a 70% reduction in SOCE, whereas those from irradiated TRPM2−/− mice showed <10% loss (Fig. 1H) and no change in intracellular Ca2+ release (fig. S1A). Carbachol (CCh)–stimulated Ca2+ influx was similarly decreased by radiation in TRPM2+/+ acinar cells but not in TRPM2−/− acinar cells (fig. S1, B and C). Further, SOCE was reduced by about 75% at 10 days after radiation in TRPM2+/+ cells with no further change until 60 days (Fig. 1I). In contrast, cells from irradiated TRPM2−/− mice showed 55% decrease in SOCE on 10 days, which was transient and recovered to 75% of preradiation amounts by 30 and 60 days (Fig. 1I). At all the time points, SOCE in cells from irradiated TRPM2−/− mice was significantly greater than that in cells from irradiated TRPM2+/+ mice (Fig. 1I). Western blots demonstrated that irradiation caused a reduction in STIM1 abundance in acinar cells from TRPM2+/+ mice 10 days after treatment with further decreases by 30 and 60 days, whereas samples from control and irradiated TRPM2−/− mice (days 10 and 30) showed similar STIM1 abundance (Fig. 1J). The Ca2+ channel TRPC1, the sodium potassium chloride cotransporter NKCC1, and Orai1, all of which are required for SOCE and fluid secretion in this gland (2, 5), were not changed by irradiation in TRPM2+/+ or TRPM2−/− mice (Fig. 1J and fig. S1D). Together, these data demonstrate that radiation induced an early transient increase in TRPM2 function in salivary gland acinar cells, which was associated with a persistent loss of STIM1 abundance and SOCE, which could account for the impairment of fluid secretion. In the absence of TRPM2, the effects of irradiation were reduced and transient with about 80% recovery in STIM1, SOCE, and salivary gland function within 60 days after treatment.
TRPM2 activity was also enhanced by irradiation in HSG cells with an increase detected at 1 hour after treatment, which decreased to preirradiation values in 4 hours (Fig. 1K). Further, increase in TRPM2 activity in response to irradiation was attenuated by treating cells with siTRPM2 or 3-aminobenzamide (3AB), which inhibits poly[adenosine diphosphate (ADP)–ribose] polymerase to suppress generation of the TRPM2 activator ADP ribose (ADPR) (Fig. 1K) (17, 18). Thapsigargin (Fig. 1L) and CCh-induced Ca2+ influx were significantly reduced 3 days after irradiation but not at 24 hours after treatment (fig. S1, E and F). Irradiation or H2O2-induced TRPM2 activity was not affected by siSTIM1 or siOrai1, although SOCE was eliminated (fig. S1, G to I). Consistent with the data from mice, radiation induced a decrease in STIM1 abundance in HSG cells (Fig. 1M), which was prevented by 3AB treatment of cells and thus was associated with activation of TRPM2.
Activation of TRPM2 by radiation causes an increase in [Ca2+]mt
Irradiation is suggested to trigger the intrinsic mitochondrial apoptotic pathway in many different cell types (22–25). Additionally, mitochondria generate ROS and ADPR, either of which could contribute to TRPM2 activation (18). We assessed the impact of radiation and TRPM2 activation on [Ca2+]mt concentration. Resting [Ca2+]mt (basal) was measured in Rhod2-loaded salivary gland acini that were perfused with Ca2+-free medium, after which external Ca2+ was readded to measure changes in [Ca2+]mt due to Ca2+ influx. Addition of external Ca2+ entry induced significant increases in [Ca2+]mt in acini from TRPM2+/+ mice on days 1, 3, and 10 after radiation (Fig. 2, A and B). Basal [Ca2+]mt in these cells was significantly increased 1 and 3 days after treatment, whereas at 10 and 30 days, it was similar to that in nonirradiated cells (Fig. 2, A and C). In contrast, Ca2+ influx into TRPM2−/− cells induced a small increase +/+ in [Ca2+]mt (about 80% less than in TRPM2 cells) that was higher than that in control cells only immediately after irradiation (Fig. 2, D and E). However, basal [Ca2+]mt was not significantly increased (Fig. 2, D and F). This pattern was similar to the effect of radiation on TRPM2 activity (Fig. 1, D to F). Consistent with these findings, H2O2 induced a greater increase in [Ca2+]mt in TRPM2+/+ than in TRPM2−/− acini (fig. S2, A and B).
Fig. 2. Irradiation causes a TRPM2- and MCU-dependent increase in [Ca2+]mt.

(A to F) [Ca2+]mt was monitored in acinar cells from nonirradiated mice (control) and from TRPM2+/+ (A to C) and TRPM2−/− mice (D to F) on 1, 3, 10, and 30 days after 15-Gy radiation treatment (IR-1, IR-3, IR-10, and IR-30) by measuring Rhod2 fluorescence in cells maintained in Ca2+-free medium and after Ca2+ was added to the external medium (A and D). Quantitation of data in (A) and (D) is shown for the peak increase in [Ca2+]i after readdition of Ca2+ (B and E) and basal [Ca2+]i in Ca2+-free medium (C and F) (CTL, 188 acini from four TRPM2+/+ mice and 224 acini from four TRPM2−/− mice; IR-1, 214 acini from three TRPM2+/+ mice and 212 acini from three TRPM2−/− mice; IR-3, 210 acini from three TRPM2+/+ mice and 200 acini from three TRPM2−/− mice; IR-10, 204 acini from three TRPM2+/+ mice and 210 acini from three TRPM2−/− mice; and IR-30, 204 acini from three TRPM2+/+ mice). *P < 0.05 and **P < 0.01, comparing the indicated value to its respective control value using unpaired t test. (G and H) A similar protocol was used to measure [Ca2+]mt in control and irradiated HSG cells with or without siMCU treatment 1 hour (G) and 4 hours (H) after irradiation (180 to 210 cells from three independent experiments). Inset: Western blot in (G) shows MCU abundance in control and siMCU-treated cells (representative of results in three independent experiments). (I to K) Similar measurements were made in HSG cells treated with 3AB before irradiation (180 to 220 cells from three independent experiments) or maintained under Ca2+-free conditions during and 1 hour after irradiation (200 to 212 cells from three independent experiments). **P < 0.01, the indicated value compared to its respective control using unpaired t test.
Irradiation of HSG cells also increased [Ca2+]mt, which was detected 1 hour (Fig. 2G) but not 4 hours after treatment (Fig. 2H). Knockdown of the MCU attenuated radiation-induced changes in [Ca2+]mt, both basal and after Ca2+ readdition (Fig. 2G). Further, the increase in [Ca2+]mt was attenuated in cells pretreated with 3AB or maintained in Ca2+-free medium during irradiation (Fig. 2I and J; quantitation of the data in Fig. 2, G, I, and J, is shown in Fig. 2K). Ca2+ uptake into mitochondria lowered [Ca2+]i as seen by the greater and more sustained [Ca2+]i increase induced by irradiation of cells treated with siMCU (fig. S2, C and D) or H2O2 (fig. S2, E and F). Together, these findings suggest that TRPM2-mediated Ca2+ entry after radiation treatment triggered MCU-dependent increase in [Ca2+]mt.
Radiation-induced activation of TRPM2 leads to increase in mitochondrial ROS
Mitochondrial ROS production can be directly increased by radiation or by an increase in [Ca2+]mt (22, 24, 26). Measurement of MitoSOX fluorescence, an indicator of mitochondrial ROS, showed that addition of external Ca2+ increased mitochondrial ROS in acini from irradiated TRPM2+/+ mice. The greatest increase was seen on day 1 after radiation, which decreased by 3 days and returned to control amounts by 10 days. Similarly, basal mitochondrial ROS was significantly higher in irradiated TRPM2+/+ acini on days 1 and 3 but was similar to that in nonirradiated cells by day 10 (Fig. 3, A to C). In contrast, acinar cells from TRPM2−/− mice displayed an attenuated increase in mitochondrial ROS in response to addition of external Ca2+, which was detected only 1 day after radiation treatment, but not at later time points (Fig. 3, D and E). Basal mitochondrial ROS in TRPM2−/−acini was not affected by irradiation (Fig. 3F). Thapsigargin or CCh stimulation of HSG cells induced very small and transient increases in [Ca2+]mt, as well as mitochondrial ROS (fig. S2, G and H). H2O2-induced mitochondrial ROS was also much greater in TRPM2+/+ acini compared to TRPM2−/− acini (fig. S3, A and B). Thus, like [Ca2+]mt, the radiation-induced increase in mitochondrial ROS was also associated with TRPM2, whereas the TRPM2-independent increase in mitochondrial ROS was relatively small.
Fig. 3. Radiation treatment causes a TRPM2 and MCU-dependent increase in mitochondrial ROS.

(A to F) Mitochondrial ROS as assessed by measuring MitoSOX fluorescence in acini from control and irradiated TRPM2+/+ (A to C) and TRPM2−/− (D to F) on days 1, 3, 10, and 30 after irradiation, as indicated (IR-1, IR-3, IR-10, and IR-30). Fluorescence changes were measured in cells maintained in Ca2+-free medium and after Ca2+ was added to the external medium (A and D). Quantitation of data from (A) and (D) is shown for peak increase after readdition of Ca2+ (B and E) and basal in Ca2+-free medium (C and F) (CTL, 188 to 256 acini from five TRPM2+/+ mice and TRPM2−/− mice each; IR-1, 200 acini from three TRPM2+/+ mice and 212 acini from three TRPM2−/− mice; IR-3, 210 acini from three TRPM2+/+ mice and 200 acini from three TRPM2−/− mice; IR-10, 214 acini from three TRPM2+/+ mice and 218 acini from three TRPM2−/− mice; and IR-30, 210 acini from three TRPM2+/+ mice). *P < 0.05 and **P < 0.01, comparing the value with its respective control, unpaired t test. (G and H) A similar protocol was used to measure mitochondrial ROS in control HSG cells (G and H), in irradiated HSG cells maintained under Ca2+-free conditions during and 1 hour after irradiation (G), and in irradiated HSG cells with or without siMCU (H) [200 to 260 cells from four independent experiments in (G) and 210 to 240 cells from four independent experiments in (H)]. (I) Quantitation of basal and peak changes in fluorescence after Ca2+ addition. (J and K) HSG cells were treated with MitoTEMPO before irradiation. Data from control, irradiated, and irradiated MitoTEMPO cells are shown (220 cells in each group in three independent experiments). *P < 0.05 and **P < 0.01, comparing each indicated value with its respective control, unpaired t test (I to K).
Irradiation also caused an increase in basal mitochondrial ROS in HSG cells, which was further increased when Ca2+ was added to the external medium (Fig. 3G). Basal mitochondrial ROS was significantly lower when cells were irradiated in a Ca2+-free medium as compared to cells irradiated in normal medium. However, the addition of Ca2+ to cells irradiated in a Ca2+-free medium induced greater increase in mitochondrial ROS as compared to cells irradiated in normal medium (Fig. 3G). Both basal amounts of mitochondrial ROS and the increase upon readdition of external Ca2+ were attenuated by knockdown of MCU before radiation (Fig. 3H; quantitation of data in Fig. 2, G and H, is shown in Fig. 2I). In addition, increased mitochondrial ROS was not detected in cells 4 hours after radiation (fig. S3C). Thus, these results suggest that radiation-induced increases in [Ca2+]mt and mitochondrial ROS depended on MCU and plasma membrane Ca2+ entry mediated by TRPM2. The role of mitochondrial ROS was further examined by treating cells with MitoTEMPO, which preferentially scavenges free radicals in mitochondria (27). Enhancement of mitochondrial ROS by irradiation was significantly dampened in cells treated with MitoTEMPO, which abolished the increase in basal mitochondrial ROS and reduced the increase after plasma membrane Ca2+ entry by about 80% (Fig. 3J). MitoTEMPO treatment before irradiation resulted in a small but significant loss of radiation-induced basal [Ca2+]i increase, as well as plasma membrane Ca2+ entry (Fig. 3K). We have previously shown that scavenging cytosolic ROS with TEMPOL prevents TRPM2 activation in irradiated HSG cells (14). Here, we showed that MitoTEMPO treatment attenuated the radiation-induced increase in basal [Ca2+]mt and that caused by plasma membrane Ca2+ entry (fig. S3, D and E). Together, these findings implicate an amplification loop whereby an increase in plasma membrane Ca2+ permeability through TRPM2 triggers mitochondrial Ca2+ accumulation and enhancement of mitochondrial ROS. ROS in mitochondria can generate ADPR locally within the organelle, which is then released into the cytosol, leading to amplification of TRPM2 activity. A similar role for mitochondrial ROS in increasing TRPM2 function in neutrophils has been previously suggested (17, 18). Contribution due to possible Ca2+-dependent regulation of TRPM2, which is reported to occur in the presence of ADPR, cannot be excluded (28)
Activation of TRPM2 by irradiation leads to a decrease in mitochondrial membrane potential
Increases in [Ca2+]mt and mitochondrial ROS can reduce mitochondrial membrane potential, ψm (22, 24). Rhodamine 123 (Rh-123) fluorescence was monitored using a similar experimental protocol as described above to assess changes in ψm. There was a significant decrease in resting ψm in acinar cells from irradiated TRPM2+/+ mice on 1 and 3 days after treatment compared to cells from nonirradiated mice (Fig. 4, A and B). Upon addition of Ca2+ to the external medium, there was a further decrease in ψm in irradiated cells, which again was significant 1 and 3 days, but not 10 days, after treatment (Fig. 4, A and C). Consistent with the data shown above, changes in ψm were attenuated in cells from irradiated TRPM2−/− mice (Fig. 4, D to F). Although resting ψm was not affected in these cells, the decrease in ψm in response to addition of external Ca2+ was reduced and transient and was detected only on the first day after treatment. Knockdown of MCU or inhibition of plasma membrane Ca2+ entry with Gd3+ in irradiated HSG cells inhibited changes in ψm due to Ca2+ influx but only partly suppressed the reduction of basal ψm (Fig. 4, G to I). It is possible that part of the de-polarization in HSG cells, but not in salivary gland acinar cells, might be caused by direct effects of radiation on mitochondria.
Fig. 4. Irradiation causes a TRPM2- and MCU-dependent decrease in mitochondrial membrane potential.

(A to F) ψm was measured using Rh-123 fluorescence in acini from control and irradiated TRPM2+/+ (A to C) and TRPM2−/− (D to F) on days 1, 3, and 10 after irradiation, as indicated (IR-1, IR-3, and IR-10). Fluorescence changes in cells maintained in Ca2+-free medium and after Ca2+ was added to the external medium (A and D). Quantitation of data from (A) and (D) is shown for basal in Ca2+-free medium (B and E) and peak increase after readdition of Ca2+ (C and F) (CTL, 200 to 220 acini from four TRPM2+/+ mice and TRPM2−/− mice; IR-1, 200 acini from three TRPM2+/+ mice and 210 acini from three TRPM2−/− mice; IR-3, 206 acini from three TRPM2+/+ mice and 204 acini from three TRPM2−/−mice; IR-10, 210 acini from three TRPM2+/+ mice and 204 acini from three TRPM2−/− mice; and IR-30, 190 acini from three TRPM2+/+ mice). *P < 0.05 and **P < 0.01 were compared with their respective control using unpaired t test (B, C, and F). FCCP, carbonyl cyanide p-trifluoromethoxyphenylhydrazone. (G to I) A similar protocol was used to measure ψm in control and irradiated HSG and HSG cells irradiated in the presence of extracellular Gd3+ or after treatment with siMCU (180 to 240 cells from three independent experiments). Values marked **P < 0.01 were compared to their respective controls using unpaired t test (H and I).
Activation of TRPM2 after irradiation promotes caspase-3 activation and loss of STIM1 and SOCE
Irradiation activates caspase-3 in acinar cells (29, 30), although <20-gray (Gy) radiation results in minimal apoptosis but substantial loss of salivary fluid secretion that is detected soon after the treatment (13, 15). Acinar cells from TRPM2+/+ and TRPM2−/− mice had similar amounts of activated caspase-3 before irradiation (control), whereas after irradiation, cells from TRPM2+/+ mice showed 10-fold higher caspase-3 at 3 and 10 days and about 5-fold at 30 days (Fig. 5A). In comparison, significantly less cleaved caspase-3 was detected in SMG acini from irradiated TRPM2−/− mice, and the change in caspase-3 was transient. Three days after irradiation, activated caspase-3 was threefold greater than in respective control cells, whereas 10 days after treatment, caspase-3 activity in cells from radiated TRPM2−/− mice was similar to that in cells from mice before irradiation (Fig. 5, B and C). Thus, the reduction of STIM1 abundance in the gland after irradiation correlated with the activation of caspase-3.
Fig. 5. TRPM2 channel contributes to activation of caspase-3 after irradiation.

(A and B) Immunofluorescence staining of active caspase-3 in submandibular acinar cells. Anti–active caspase-3 antibody staining in acinar cells from TRPM2+/+ mice (A) (CTL, four TRPM2−/−mice and four TRPM2+/+ mice; IR-3 and IR-10, six TRPM2−/− mice and six TRPM2+/+ mice; and IR-30, four TRPM2−/− mice and four TRPM2+/+ mice). (B) Anti–active caspase-3 antibody staining in acinar cells from TRPM2−/− mice (CTL, three TRPM2−/− mice and three TRPM2+/+ mice; IR-3 and IR-10, six TRPM2−/− and six TRPM2+/+ mice). Scale bars, 30 μm (A and B). The area of caspase-3 signal/total surface acinar area is shown in (C). **P < 0.01 compares TRPM2+/+ mice with TRPM2−/−mice group at the same time point after radiation using χ2 test; #P < 0.05, ##P < 0.01 compares the values to their respective control, χ2 test; the number of mice for each is shown in parentheses above.
HSG cells also displayed activated caspase-3 6 hours after radiation treatment, which further increased at 96 hours. Treatment of cells with zVAD, an inhibitor of procaspase-3 cleavage, before irradiation prevented the increase in cleaved caspase-3 (Fig. 6, A and B). In irradiated HSG cells, the abundance of procaspase-3 and STIM1 decreased progressively starting at 24 hours after treatment (Fig. 6C). The decreases in both these proteins were prevented by treatment of cells with zVAD before irradiation. This finding suggests that STIM1 could be a target for caspase-3, although we could not detect low–molecular weight fragments of STIM1 in HSG cells after irradiation. It is possible that, because the decrease in STIM1 under these conditions is a relatively slow process, the fragments were further degraded before they could be detected. However, as proof of principle, we used two additional methods of activating caspase-3: treatment with staurosporine or H2O2 (fig. S4, A and B), either of which decreased STIM1 protein abundance after radiation treatment. For staurosporine, this decrease was prevented by zVAD. Lower–molecular weight fragments of STIM1 were detected when blots were exposed for longer time periods, and these were reduced in abundance in zVAD-treated cells. Treatment with siMCU or 3AB prevented irradiation-induced loss of STIM1 and procaspase-3 (Fig. 6D). In contrast, TRPC1 or Orai1 abundance was not affected up to 4 days after irradiation (Fig. 6E). Finally, we showed that SOCE did not decrease after irradiation in cells treated with zVAD, siMCU, or 3AB (Fig. 6, F and G) or siCaspase-3 (Fig. 6, H and I, and fig. S5, A to C). These data reveal a critical link between activation of TRPM2 by irradiation and MCU-dependent accumulation of Ca2+ into mitochondria with activation of caspase-3 and reduction in STIM1.
Fig. 6. The coordinated effects of TRPM2, MCU, and caspase-3 govern loss of STIM1 and SOCE in irradiated HSG cells.

(A and B) Detection of caspase-3 activation in HSG cells at different times after irradiation with or without pretreatment with caspase-3 inhibitor zVAD. (A) Cells with activated caspase-3 (indicated by green fluorescence signal) in control or irradiated cells and with zVAD treatment. (B) Percentage of caspase-3–positive cells in each group. *P < 0.05, **P < 0.01, comparing marked values to control. ##P < 0.01 compares irradiated cells with zVAD treatment to those without zVAD treatment at the same time point after irradiation using χ2 test. Scale bar, 20 μm. Data were obtained from >200 cells in each group from three independent experiments. (C) Western blots showing STIM1 and procaspase-3 in irradiated cells and those treated with zVAD at the indicated time points after irradiation (three independent experiments). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (D) Western blots for STIM1 and procaspase-3 in control and irradiated cells with siMCU or 3AB treatment (three independent experiments). (E) Western blots showing TRPC1 and Orai1 in control and irradiated cells at the indicated time points after irradiation (three independent experiments). (F) Thapsigargin-induced Ca2+ release and influx were monitored in control and irradiated cells with or without siMCU, zVAD, and 3AB treatments. (G) Quantitation of Ca2+ influx from traces shown in (F) (220 to 240 cells from three independent experiments). (H) Thapsigargin-activated Ca2+ release and influx measured in control, IR–siCaspase-3, and IR-siControl HSG cells. (I) Quantitation of Ca2+ influx from traces shown in (H) (190 to 200 cells from three independent experiments). **P < 0.01 compares marked value with the control using unpaired t test (G and I).
Adenovirus-mediated expression of STIM1 in salivary glands of irradiated mice leads to recovery of SOCE and fluid secretion
To further support our suggestion that loss of STIM1 could be the primary underlying cause of radiation-induced reduction in salivary gland function, we used adenovirus cytomegalovirus (AdCMV)–STIM1 to induce expression of STIM1 in salivary glands of irradiated TRPM2+/+ mice. Virus was delivered to mouse salivary glands 15 days after irradiation, and saliva was measured 15 days after that (that is, on day 30 after radiation treatment). The mice that received AdCMV-STIM1 secreted twofold more fluid than irradiated mice receiving control virus, which was about 60% of the secretion measured in non-irradiated mice (Fig. 7A; also see Fig. 1G). SOCE was also increased about twofold in acinar cells from irradiated mice that received AdCMV-STIM1 compared to those receiving control virus (Fig. 7, B and C). Similar to fluid secretion, SOCE in these cells also recovered to about 60% of preradiation amounts. Together, these data demonstrate that STIM1 depletion in salivary glands after radiation treatment is a key cause for loss of fluid secretion and restoration of STIM1 in acinar cells induces recovery of fluid secretion.
Fig. 7. Adenovirus-mediated expression of STIM1 in irradiated mice leads to recovery of SOCE and fluid secretion.
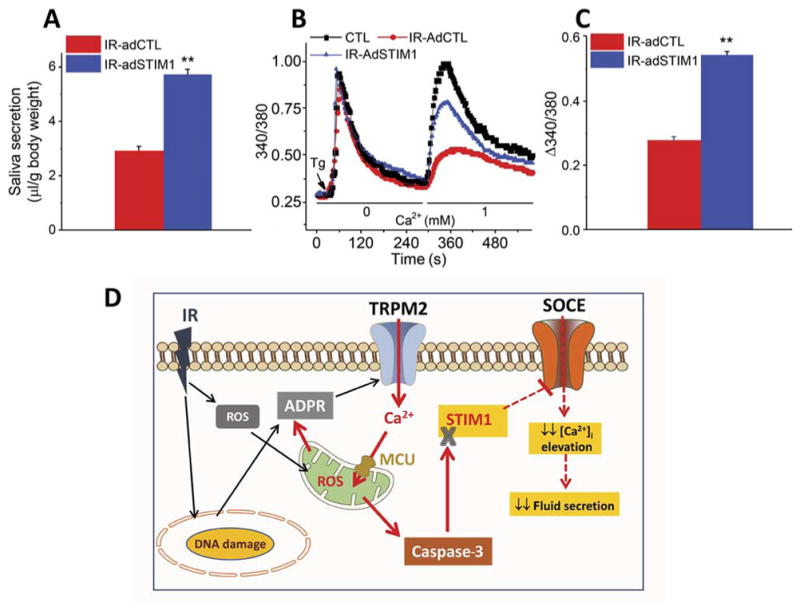
(A) Salivary gland secretion was assessed in control nonirradiated mice and irradiated mice that received AdCMV–green fluorescent protein (GFP) or AdCMV-STIM1GFP in salivary glands 15 days after irradiation with saliva collection done at 30 days after irradiation (12 mice each with AdCMV-GFP or AdCMV-STIM1GFP). (B) Thapsigargin-mediated Ca2+ release and influx were measured in acini from control (220 acini from three mice), IR-AdCTL (210 acini from three mice), and IR-AdSTIM1 (200 acini from three mice) mice. (C) Quantitation of data from (B). **P < 0.01, comparing indicated value with IR-AdCTL (A and C). (D) Model showing the early consequences of radiation treatment and mechanisms underlying persistent loss of salivary secretion. Effects of irradiation (black arrows) lead to TRPM2 activation. TRPM2-mediated Ca2+ entry causes an increase in [Ca2+]mt and ROS, depolarization of ψm, and caspase-3 activation, which lead to loss of STIM1 (solid red arrows). Consequently, SOCE is reduced, which attenuates agonist-stimulated sustained increases in [Ca2+]i that are required to activated Ca2+-dependent ion channels that drive fluid secretion (red dashed arrows).
DISCUSSION
Our data elucidate a critical TRPM2-dependent mechanism that underlies salivary gland dysfunction caused by radiation treatment. Our findings demonstrated that radiation-induced loss of salivary gland function was caused by a persistent reduction in STIM1 protein and SOCE. We showed that activation of TRPM2 by irradiation led to the loss of STIM1, SOCE, and salivary fluid secretion. Mice lacking TRPM2 had transient loss of salivary fluid secretion with >70% recovery of function by 30 days that was associated with recovery of STIM1. A major finding of this study is that TRPM2-dependent decrease in STIM1 and SOCE after radiation treatment was mediated through effects on mitochondria (Fig. 7D). We showed that an early event after irradiation was a TRPM2-mediated [Ca2+]i increase, which triggered MCU-dependent uptake of Ca2+ into mitochondria. Consequently, [Ca2+]mt and mitochondrial ROS were increased, whereas mitochondrial membrane potential was decreased. This was accompanied by a relatively slower appearance of activated caspase-3, which caused loss of STIM1 and, consequently, attenuation of SOCE. A reduction in the SOCE-mediated [Ca2+]i increase in acinar cells after irradiation resulted in decreased activation of Ca2+-dependent ion channels required for fluid secretion. The final outcome of the radiation-induced disruption of Ca2+ signaling in salivary glands was persistent loss of fluid secretion.
The early effects of 15-Gy radiation on TRPM2, [Ca2+]mt, and ROS were transient with the greatest increase in cells 1 day after treatment and a gradual decrease thereafter. By 30 days after irradiation, these functions were similar to those in nonirradiated cells. Enhancement of caspase-3 by radiation treatment was more sustained and persisted at 30 days after irradiation. In contrast, acini from irradiated TRPM2−/− mice displayed substantial attenuation of irradiation-induced increases in [Ca2+]i, [Ca2+]mt, and caspase-3. We suggest that these abrogated effects of radiation on TRPM2 and mitochondrial function, as well as caspase-3 in acinar cells of TRPM2−/− mice, likely reflect TRPM2-independent mechanisms, which might include direct consequences of radiation on cellular membranes and other components. These small and transient changes in function did not cause persistent loss of STIM1, SOCE, or fluid secretion. Instead, they were associated with maintenance of STIM1 and recovery of SOCE and salivary gland fluid secretion in TRPM2−/− mice. Although further studies are required to identify the mechanism involved in TRPM2-independent effects on acinar cell function, our findings provide evidence that a TRPM2-dependent mechanism mediated through mitochondria leads to the irreversible loss of salivary gland fluid secretion after radiation treatment. Our studies involved a single 15-Gy dose of radiation treatment. Although low-dose fractionated radiation also results in similar loss of salivary gland fluid secretion, further studies are required to determine possible involvement of TRPM2 and mitochondria in the process.
We have previously shown that, despite >60% loss of saliva secretion, salivary gland acinar cells retain normal morphology and abundance of key proteins related to fluid secretion such as NKCC1 and AQP5 (14). Our present findings revealed that reduction of SOCE in acinar cells after irradiation could fully account for the impairment of salivary fluid secretion and that of agonist-stimulated decrease in acinar cell volume (31). We showed that this impairment of SOCE was due to loss of STIM1, which occurred downstream of caspase-3 activation. The increase in [Ca2+]mt after radiation was due to TRPM2-mediated Ca2+ influx and contributed to caspase-3 activation and loss of STIM1. Although caspase is activated in salivary glands after irradiation, there is minimal apoptosis after a single dose of radiation (2.5 to 17.5 Gy) (13, 15), which cannot account for the early onset and persistent loss of acinar cell function. Our data suggest a role for caspase-3 in depletion of STIM1, thus linking the activated protease to loss of salivary gland function after irradiation rather than apoptosis. Consistent with this notion, caspase-3 can target and cleave various proteins not involved in apoptosis and thus contribute to regulation of critical cellular functions such as proliferation and differentiation (32–35). The mechanism that determines whether caspase-3 activation triggers apoptosis has not yet been established. On the basis of these previous and present data, we suggest that caspase-3 might have a noncanonical role in acinar cells after low-dose radiation. Our model suggests that as a sequelae to radiation-induced enhancement of TRPM2 function and increased [Ca2+]mt and ROS, caspase-3 was activated and cleaved STIM1, causing reduction in SOCE and loss of fluid secretion. We showed that replenishment of STIM1 in salivary gland acinar cells of irradiated mice induced recovery of SOCE and salivary fluid secretion. Regulation of STIM1 abundance or loss of protein can affect other cellular processes. Cleavage of STIM1 by calpain and γ-secretase is associated with stress and Alzheimer’s disease, respectively (36, 37). Further, proteasome inhibition reduces SOCE and promotes autophagy-mediated degradation of STIM1/2 (38). Loss of STIM1 is also associated with the autoimmune exocrinopathy, Sjögren’s syndrome, in which salivary gland function is affected (39, 40).
A role for TRPM2 in ROS-associated cell death has been noted in various cell types including cerebral cortical neurons and myocytes (17, 41). TRPM2 has been linked to inflammasome assembly (42) and ischemia-reperfusion–induced cellular injury (43). However, downstream targets and signaling pathways of ROS-induced TRPM2 activity that lead to loss of cell viability compared to modulation of cell function are poorly understood. Here, we described a critical link between TRPM2 and mitochondria that was triggered by radiation treatment and that resulted in persistent salivary gland dysfunction. Our findings show that activation of TRPM2 by radiation treatment triggered an increase in [Ca2+]mt and mitochondrial ROS, decrease in mitochondrial membrane potential, activation of caspase-3, and loss of STIM1 and SOCE. Loss of salivary gland fluid secretion was due to decreased STIM1 and SOCE because a gene therapy strategy to express STIM1 in vivo in salivary glands of irradiated mice rescued function. Insulin-like growth factor 1 and interleukin-6 treatments, which protect salivary gland function after irradiation, promote the antiapoptotic factor BCL2 (9, 15). Thus, treatment with these factors would lead to suppression of cytochrome C release from mitochondria, as well as the subsequent activation of caspase-3. We have previously shown that inhibitors of TRPM2, as well as free radical scavengers such as TEMPOL, promote recovery of salivary gland function after irradiation (14). Our present findings suggest that targeting the mechanisms underlying loss of STIM1 would be a potentially useful approach for preserving salivary gland function after radiation treatment.
MATERIALS AND METHODS
Animals and cell culture
HSG cells were cultured on glass coverslips with Earle’s minimum essential medium (EMEM). EMEM was supplemented with 10% fetal calf serum, 2 mM glutamine, and 1% penicillin/streptomycin at 37°C in 5% CO2. TRPM2+/+ and TRPM2−/− mice were bred from TRPM2+/−animals (C57BL/6 background) obtained from Kyoto University, Japan and genotyped using the protocol described earlier (14) with the primers (ROSC1-13F, 5'-cttgggttgcagtcatatgcaggc-3'; ROSC1-10R, 5'-gccctcac-catccgcttcacgatg-3'; and Pneo5'a, 5'-gccacacgcgtcaccttaatatgcg-3'). For obtaining samples of salivary glands, mice were euthanized by CO2 asphyxiation and salivary glands were immediately excised. Breeding and all other procedures were carried out using a protocol approved by the National Institute of Craniofacial Research–Animal Care and Use Committee in compliance with the Guide for the Care and Use of Laboratory Animal Resources National Research Council.
Construction of recombinant adenoviral vector and in vivo animal experiments
First-generation, E1-deleted, replication-defective serotype 5 adenoviral vector encoding human STIM1 (AdCMV-hSTIM1) was used in this study. The 4000 base pairs of hSTIM1 complementary DNA were excised from pCMV6-XL5C3 (OriGene) with Eco RI/Sma I and ligated into the Eco RI/Sma I sites of pACCMV-pLpA to create pACCMV-hSTIM1. Adenoviral vector was generated by homologous recombination of pACCMV-hSTIM1 with pJM17 (Microbix Biosystems Inc.), subsequently amplified in 293 cells (Microbix Biosystems Inc.), and purified by CsCl gradient centrifugation as described previously (44). Purified vector was titered by quantitative polymerase chain reaction with primers from the E2 region of adenovirus, E2q1 (5'-GCAGAAC-CACCAGCACAGTGT-3') and E2q2 (5'-TCCACGCATTTCCTTC-TAAGCTA-3'). The titer was expressed as viral genome/ml. Mice were anesthetized with ketamine (60 mg/kg) and xylazine (8 mg/kg) intramuscularly. AdCMV-hSTIM1 was administered to both SMGs by local ductal cannulation at 5 × 109, 1 × 109, or 1 × 1010 particles per gland. Glands were infused with AdCMV-STIM1 or control virus 15 days after irradiation by retrograde perfusion through the salivary ducts, and salivary gland fluid secretion was measured 2 weeks later (at 30 days after irradiation).
Irradiation of HSG cells and mice
HSG cells were cultured in glass-bottom dishes and irradiated in a Gammacell 1000 irradiator with a single dose of radiation or using x-ray as described previously (14). Salivary gland irradiation was accomplished by placing each animal into a specially built Lucite jig such that the animal could be immobilized without the use of anesthetics. In addition, the jig was fitted with a Lucite cone that surrounded the head and prevented head movement during the radiation exposure (14, 31). Lead shields were designed to cover the jigs with the mice with a small aperture in the lead shield that allowed radiation to the salivary gland area of the immobilized animal. A single radiation dose of 15 Gy was delivered to the animal by a Therapax DXT300 X-ray irradiator (Pantak Inc.) using 2.0-mm Al filtration (300-kV peak) at a dose rate of 1.9 Gy/min. Immediately after irradiation, the animals were removed from the Lucite jig and housed (five animals per cage) in a climate- and a light/dark-controlled environment and allowed free access to dough diet (soft food).
Saliva secretion measurements
The salivary secretion was measured in control and irradiated mice 10, 30, or 60 days after irradiation. For whole saliva collection, anesthetized mice were treated with pilocarpine solution (0.5 mg/kg) subcutaneously, after which the saliva was collected as described earlier (14). For whole saliva collection, anesthetized mice were treated with pilocarpine solution (0.5 mg/kg) subcutaneously, after which the saliva was collected as described earlier (14).
Acinar cell preparation
SMG acinar cells were isolated by enzyme digestion using Liberase TL (Roche Diagnostics). The dissected glands were finely minced and digested in Eagle’s minimum essential medium containing 0.015% trypsin for 10 min at 37°C and then centrifuged. The cell pellet was rinsed with Eagle’s minimum essential medium containing 0.2% trypsin inhibitor and then digested for 25 min in the presence of Liberase TL (0.5 mg/ml). After centrifugation, the cell pellet was further digested with Liberase TL for another 25 min and finally resuspended in Eagle’s minimum essential medium. Cells were continuously gassed with a mixture of 95% O2 and 5% CO2.
[Ca2+]i measurements
[Ca2+]i were measured in Fura-2–loaded cells using an Olympus × 51 microscope (Olympus Centre Valley), with an ORCA-ER camera (Hamamatsu) attached to a Polychrome V (Till Photonics LLC) for HSG cells or acini from mouse SMG. Cells were excited at 340/380 nm with an emission of 510 nm. MetaFluor (Molecular Devices) was used to acquire images and process data.
[Ca2+]mt measurement
Cells were loaded with 1 μM Rhod2/acetoxymethyl for 30 min or acini from mouse SMG and washed three times with SES (standard external solution) solution for 15 min at 37°C. Cells were excited at 550 nm wavelengths and the emission intensities were excited at 580 nm. Mitochondrial Ca2+ concentrations were expressed as normalized fluorescence ratio (F/F0, where F0 is the baseline fluorescence).
Mitochondrial ROS measurement
HSG cells seeded on glass coverslips or acini from mouse SMGs were loaded with the mitochondrial O2-specific fluorescent probe MitoSOX (1 μM) for 10 min at 37°C. MitoSOX fluorescence was analyzed using Polychrome V using 510 nm excitation wavelength. The relative fluorescence intensity was used to indicate mitochondrial ROS as F/F0 (MitoSOX).
Mitochondrial membrane potential measurement
Mitochondrial membrane potential (ψm) was assessed using Rh-123 (45). At low concentrations, the fluorescence intensity depends on dye accumulation in mitochondria, which, in turn, is directly related to mitochondrial potential. Cells were cultured on glass coverslips and loaded for 15 min at 37°C with Rh-123 (100 nM) in the culture medium. Rh-123 fluorescence was measured with a Polychrome V fluorescence microscope using 505 nm excitation wavelength. The relative fluorescence intensity was used to indicate ψm as F/F0 (Rh-123).
Detection of cleaved caspase-3 in HSG and acinar cells
Active caspase-3 was detected in HSG cells using the CellEvent Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific) with an absorption/emission maximum of 502/530 nm. HSG cells were plated on glass coverslips and incubated with the reagent at 1 μM concentration for 30 min. The reagent was detected using an Olympus × 51 microscope (Olympus), with an ORCA-ER camera (Hamamatsu) attached to a Polychrome V (Till Photonics LLC). MetaFluor (Molecular Devices) was used to acquire images and process data as described above. Apoptotic cells with activated caspase-3/7 have bright green nuclei, whereas cells without activated caspase-3 have minimal fluorescence signal.
Isolated submandibular acini were plated on glass coverslips for 3 min at room temperature before fixation with cold methanol. After fixation, the acini were rinsed in phosphate-buffered saline (PBS), quenched with 500 mM glycine in PBS for 10 min at 4°C, and washed with solution A (0.37% saponin and 0.21% Triton X-100 in PBS). The samples were blocked for 1 hour in 5% goat serum (Jackson Immuno-Research) diluted in solution A and stained with anti–caspase-3 active form antibody (5 μg/ml; EMD Millipore) diluted in solution A containing 1% bovine serum albumin (BSA) overnight at 4°C. Subsequently, the samples were washed with solution A and incubated for 30 min at room temperature with anti-rabbit Alexa 488 (Thermo Fisher Scientific) diluted in solution A containing 1% BSA. Finally, the acini were washed in solution A, and coverslips were mounted with Fluormount-G (Electron Microscopy Sciences). Confocal images were acquired using a FluoView 1000 (Olympus). Area of caspase-3 staining and total surface of acinar area were quantified using Volocity software (PerkinElmer).
Western blotting
HSG cell protein was mixed with tris-glycine sodium dodecyl sulfate sample buffer (Invitrogen) and loaded onto gels for Western blot analysis. Rabbit polyclonal anti-TRPM2 antibody (1:200 dilution, Abcam), anti–caspase-3 antibody (1:3000 dilution, Abcam), and rabbit polyclo-nal anti–β-actin antibody (1:500 dilution, Abcam) were used for immu-noblotting. After incubation with secondary antibodies, signals were detected by chemiluminescence using SuperSignal West Femto Maximum Sensitivity Substrate (Thermo Scientific).
Statistics
Data analyses were performed using Origin 9.0 (OriginLab) and Graph-Pad Prism (GraphPad Software). Statistical significance was determined using unpaired t test between two groups and the χ2 test for responses in cell populations. Data are expressed as means ± SE. Differences in the mean values were considered to be significant at *P < 0.05 or #P < 0.05 and **P < 0.01 or ##P < 0.01.
Supplementary Material
Acknowledgments
Funding: I.S.A. is funded by National Institute of Dental and Craniofacial Research–Division of Intramural Research, NIH (Z01-DE00438-31).
Footnotes
Author contributions: X.L. designed and carried out the experiments, analyzed the data, prepared the figures, and wrote the manuscript. B.G. designed and performed the experiments in HSG cells and analyzed the data. L.B.d.S. designed and carried out the caspase measurements in acinar cells, analyzed the data, and prepared the figures. K.P.S. assisted in the Western blot analysis and siCaspase-3 experiments. H.L.O. analyzed the data, prepared the reagents, carried out the experiments in HSG cells, and was involved in the manuscript preparation. K.T.C. carried out the Western blots with salivary glands and cells. W.S. assisted in the immunofluorescence measurements and analysis. C.Z. irradiated the mice, carried out the in vivo delivery of adenovirus in the gland, performed the saliva secretion measurements, analyzed the data, and prepared the figures. Y.M. provided the TRPM2−/− mice, helped in planning the design of study, and contributed to the manuscript preparation. I.S.A. designed and planned the study, provided all the reagents and materials except the TRPM2−/− mice, analyzed the data, and wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
REFERENCES AND NOTES
- 1.Melvin JE, Yule D, Shuttleworth T, Begenisich T. Regulation of fluid and electrolyte secretion in salivary gland acinar cells. Annu Rev Physiol. 2005;67:445–469. doi: 10.1146/annurev.physiol.67.041703.084745. [DOI] [PubMed] [Google Scholar]
- 2.Ambudkar IS. Calcium signalling in salivary gland physiology and dysfunction. J Physiol. 2016;594:2813–2824. doi: 10.1113/JP271143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ambudkar IS. Ca2+ signaling and regulation of fluid secretion in salivary gland acinar cells. Cell Calcium. 2014;55:297–305. doi: 10.1016/j.ceca.2014.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hong JH, Li Q, Kim MS, Shin DM, Feske S, Birnbaumer L, Cheng KT, Ambudkar IS, Muallem S. Polarized but differential localization and recruitment of STIM1, Orai1 and TRPC channels in secretory cells. Traffic. 2011;12:232–245. doi: 10.1111/j.1600-0854.2010.01138.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X, Cheng KT, Bandyopadhyay BC, Pani B, Dietrich A, Paria BC, Swaim WD, Beech D, Yildrim E, Singh BB, Birnbaumer L, Ambudkar IS. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1−/− mice. Proc Natl Acad Sci USA. 2007;104:17542–17547. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng KT, Liu X, Ong HL, Swaim W, Ambudkar IS. Local Ca2+ entry via Orai1 regulates plasma membrane recruitment of TRPC1 and controls cytosolic Ca2+ signals required for specific cell functions. PLOS Biol. 2011;9:e1001025. doi: 10.1371/journal.pbio.1001025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen SB, Pedersen AML, Vissink A, Andersen E, Brown CG, Davies AN, Dutilh J, Fulton JS, Jankovic L, Lopes NNF, Mello ALS, Muniz LV, Murdoch-Kinch CA, Nair RG, Napeñas JJ, Nogueira-Rodrigues A, Saunders D, Stirling B, von Bültzingslöwen I, Weikel DS, Elting LS, Spijkervet FKL, Brennan MT Salivary Gland Hypofunction/Xerostomia Section, Oral Care Study Group; Multinational Association of Supportive Care in Cancer (MASCC)/International Society of Oral Oncology (ISOO) A systematic review of salivary gland hypofunction and xerostomia induced by cancer therapies: Prevalence, severity and impact on quality of life. Support Care Cancer. 2010;18:1039–1060. doi: 10.1007/s00520-010-0827-8. [DOI] [PubMed] [Google Scholar]
- 8.Baum BJ, Alevizos I, Chiorini JA, Cotrim AP, Zheng C. Advances in salivary gland gene therapy—Oral and systemic implications. Expert Opin Biol Ther. 2015;15:1443–1454. doi: 10.1517/14712598.2015.1064894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grundmann O, Mitchell GC, Limesand KH. Sensitivity of salivary glands to radiation: From animal models to therapies. J Dent Res. 2009;88:894–903. doi: 10.1177/0022034509343143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coppes RP, Roffel AF, Zeilstra LJ, Vissink A, Konings AW. Early radiation effects on muscarinic receptor-induced secretory responsiveness of the parotid gland in the freely moving rat. Radiat Res. 2000;153:339–346. doi: 10.1667/0033-7587(2000)153[0339:ereomr]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 11.Paardekooper GM, Cammelli S, Zeilstra LJ, Coppes RP, Konings AW. Radiation-induced apoptosis in relation to acute impairment of rat salivary gland function. Int J Radiat Biol. 1998;73:641–648. doi: 10.1080/095530098141898. [DOI] [PubMed] [Google Scholar]
- 12.Peter B, Van Waarde MAWH, Vissink A, ’s-Gravenmade EJ, Konings AWT. Radiation-induced cell proliferation in the parotid and submandibular glands of the rat. Radiat Res. 1994;140:257–265. [PubMed] [Google Scholar]
- 13.Coppes RP, Meter A, Latumalea SP, Roffel AF, Kampinga HH. Defects in muscarinic receptor-coupled signal transduction in isolated parotid gland cells after in vivo irradiation: Evidence for a non-DNA target of radiation. Br J Cancer. 2005;92:539–546. doi: 10.1038/sj.bjc.6602365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu X, Cotrim A, Teos L, Zheng C, Swaim W, Mitchell J, Mori Y, Ambudkar I. Loss of TRPM2 function protects against irradiation-induced salivary gland dysfunction. Nat Commun. 2013;4:1515. doi: 10.1038/ncomms2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marmary Y, Adar R, Gaska S, Wygoda A, Maly A, Cohen J, Eliashar R, Mizrachi L, Orfaig-Geva C, Baum BJ, Rose-John S, Galun E, Axelrod JH. Radiation-induced loss of salivary gland function is driven by cellular senescence and prevented by IL6 modulation. Cancer Res. 2016;76:1170–1180. doi: 10.1158/0008-5472.CAN-15-1671. [DOI] [PubMed] [Google Scholar]
- 16.Vissink A, Down JD, Konings AW. Contrasting dose-rate effects of γ-irradiation on rat salivary gland function. Int J Radiat Biol. 1992;61:275–282. doi: 10.1080/09553009214550911. [DOI] [PubMed] [Google Scholar]
- 17.Ogawa N, Kurokawa T, Mori Y. Sensing of redox status by TRP channels. Cell Calcium. 2016;60:115–122. doi: 10.1016/j.ceca.2016.02.009. [DOI] [PubMed] [Google Scholar]
- 18.Sumoza-Toledo A, Penner R. TRPM2: A multifunctional ion channel for calcium signalling. J Physiol. 2011;589:1515–1525. doi: 10.1113/jphysiol.2010.201855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klumpp D, Misovic M, Szteyn K, Shumilina E, Rudner J, Huber SM. Targeting TRPM2 channels impairs radiation-induced cell cycle arrest and fosters cell death of T cell leukemia cells in a Bcl-2-dependent manner. Oxid Med Cell Longev. 2016;2016:8026702. doi: 10.1155/2016/8026702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masumoto K, Tsukimoto M, Kojima S. Role of TRPM2 and TRPV1 cation channels in cellular responses to radiation-induced DNA damage. Biochim Biophys Acta. 2013;1830:3382–3390. doi: 10.1016/j.bbagen.2013.02.020. [DOI] [PubMed] [Google Scholar]
- 21.Naziroğlu M, Celik Ö, Özgül C, Çiğ B, Doğan S, Bal R, Gümral N, BRodríguez A, Pariente JA. Melatonin modulates wireless (2.45 GHz)-induced oxidative injury through TRPM2 and voltage gated Ca2+ channels in brain and dorsal root ganglion in rat. Physiol Behav. 2012;105:683–692. doi: 10.1016/j.physbeh.2011.10.005. [DOI] [PubMed] [Google Scholar]
- 22.Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ROS: A mutual interplay. Redox Biol. 2015;6:260–271. doi: 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wu CC, Bratton SB. Regulation of the intrinsic apoptosis pathway by reactive oxygen species. Antioxid Redox Signal. 2013;19:546–558. doi: 10.1089/ars.2012.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhivotovsky B, Orrenius S. Calcium and cell death mechanisms: A perspective from the cell death community. Cell Calcium. 2011;50:211–221. doi: 10.1016/j.ceca.2011.03.003. [DOI] [PubMed] [Google Scholar]
- 25.O-Uchi J, Ryu SY, Jhun BS, Hurst S, Sheu S-S. Mitochondrial ion channels/transporters as sensors and regulators of cellular redox signaling. Antioxid Redox Signal. 2014;21:987–1006. doi: 10.1089/ars.2013.5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kobashigawa S, Kashino G, Suzuki K, Yamashita S, Mori H. Ionizing radiation-induced cell death is partly caused by increase of mitochondrial reactive oxygen species in normal human fibroblast cells. Radiat Res. 2015;183:455–464. doi: 10.1667/RR13772.1. [DOI] [PubMed] [Google Scholar]
- 27.Liang HL, Sedlic F, Bosnjak Z, Nilakantan V. SOD1 and MitoTEMPO partially prevent mitochondrial permeability transition pore opening, necrosis, and mitochondrial apoptosis after ATP depletion recovery. Free Radic Biol Med. 2010;49:1550–1560. doi: 10.1016/j.freeradbiomed.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ. Critical intracellular Ca2+ dependence of transient receptor potential melastatin. 2 (TRPM2) cation channel activation. J Biol Chem. 2003;278:11002–11006. doi: 10.1074/jbc.M210810200. [DOI] [PubMed] [Google Scholar]
- 29.Bralic M, Muhvic-Urek M, Stemberga V, Golemac M, Jurkovic S, Borcic J, Braut A, Tomac J. Cell death and cell proliferation in mouse submandibular gland during early post-irradiation phase. Acta Med Okayama. 2005;59:153–159. doi: 10.18926/AMO/31948. [DOI] [PubMed] [Google Scholar]
- 30.Avila JL, Grundmann O, Burd R, Limesand KH. Radiation-induced salivary gland dysfunction results from p53-dependent apoptosis. Int J Radiat Oncol Biol Phys. 2009;73:523–529. doi: 10.1016/j.ijrobp.2008.09.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Teos LY, Zheng CY, Liu X, Swaim WD, Goldsmith CM, Cotrim AP, Baum BJ, Ambudkar IS. Adenovirus-mediated hAQP1 expression in irradiated mouse salivary glands causes recovery of saliva secretion by enhancing acinar cell volume decrease. Gene Ther. 2016;23:572–579. doi: 10.1038/gt.2016.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han MH, Jiao S, Jia JM, Chen Y, Chen CY, Gucek M, Markey SP, Li Z. The novel caspase-3 substrate Gap43 is involved in AMPA receptor endocytosis and long-term depression. Mol Cell Proteomics. 2013;12:3719–3731. doi: 10.1074/mcp.M113.030676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hüttemann M, Pecina P, Rainbolt M, Sanderson TH, Kagan VE, Samavati L, Doan JW, Lee I. The multiple functions of cytochrome c and their regulation in life and death decisions of the mammalian cell: From respiration to apoptosis. Mitochondrion. 2011;11:369–381. doi: 10.1016/j.mito.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jana K, Banerjee B, Parida PK. Caspases: A potential therapeutic targets in the treatment of Alzheimer’s disease. Transl Med. 2013;S2:006. [Google Scholar]
- 35.Lamkanfi M, Festjens N, Declercq W, Vanden Berghe T, Vandenabeele P. Caspases in cell survival, proliferation and differentiation. Cell Death Differ. 2007;14:44–55. doi: 10.1038/sj.cdd.4402047. [DOI] [PubMed] [Google Scholar]
- 36.Tong BCK, Lee CSK, Cheng WH, Lai KO, Foskett JK, Cheung KH. Familial Alzheimer’s disease–associated presenilin 1 mutants promote γ-secretase cleavage of STIM1 to impair store-operated Ca2+ entry. Sci Signal. 2016;9:ra89. doi: 10.1126/scisignal.aaf1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Prins D, Michalak M. STIM1 is cleaved by calpain. FEBS Lett. 2015;589:3294–3301. doi: 10.1016/j.febslet.2015.09.015. [DOI] [PubMed] [Google Scholar]
- 38.Keil JM, Shen Z, Briggs SP, Patrick GN. Regulation of STIM1 and SOCE by the ubiquitin-proteasome system (UPS) PLOS ONE. 2010;5:e13465. doi: 10.1371/journal.pone.0013465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng KT, Alevizos I, Liu X, Swaim WD, Yin H, Feske S, Oh-hora M, Ambudkar IS. STIM1 and STIM2 protein deficiency in T lymphocytes underlies development of the exocrine gland autoimmune disease, Sjögren’s syndrome. Proc Natl Acad Sci USA. 2012;109:14544–14549. doi: 10.1073/pnas.1207354109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gallo A, Jang SI, Ong HL, Perez P, Tandon M, Ambudkar I, Illei G, Alevizos I. Targeting the Ca2+ sensor STIM1 by exosomal transfer of Ebv-miR-BART13-3p is associated with Sjögren’s syndrome. EBioMedicine. 2016;10:216–226. doi: 10.1016/j.ebiom.2016.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mortadza SAS, Wang L, Li D, Jiang LH. TRPM2 channel-mediated ROS-sensitive Ca2+ signaling mechanisms in immune cells. Front Immunol. 2015;6:407. doi: 10.3389/fimmu.2015.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun. 2013;4:1611. doi: 10.1038/ncomms2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhan K-y, Yu P-l, Liu C-h, Luo J-h, Yang W. Detrimental or beneficial: The role of TRPM2 in ischemia/reperfusion injury. Acta Pharmacol Sin. 2016;37:4–12. doi: 10.1038/aps.2015.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Becker TC, Noel RJ, Coats WS, Gomez-Foix AM, Alam T, Gerard RD, Newgard CB. Use of recombinant adenovirus for metabolic engineering of mammalian cells. Methods Cell Biol. 1994;43:161–189. doi: 10.1016/s0091-679x(08)60603-2. [DOI] [PubMed] [Google Scholar]
- 45.Baracca A, Sgarbi G, Soliani G, Lenaz G. Rhodamine 123 as a probe of mitochondrial membrane potential: Evaluation of proton flux through F0 during ATP synthesis. Biochim Biophys Acta. 2003;1606:137–146. doi: 10.1016/s0005-2728(03)00110-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.