

ABSTRACT
During the biogenesis of U1 small nuclear ribonucleoprotein, a small population of U1 snRNA molecules acquires an extra methylation at the first transcribed nucleotide and a nucleolytic cleavage to remove the 3′ structured region including the Sm protein–binding site and stem-loop 4. These modifications occur before hypermethylation of the monomethylated 5′ cap, whereby producing truncated forms of U1 snRNA (U1-tfs) that are diverted from the normal pathway to a processing body–associated degradation pathway. Here, we demonstrate that a small population of U2 snRNA molecules receives post-transcriptional modifications similar to those of U1 to yield U2-tfs. Like U1-tfs, U2-tfs molecules were produced from transcripts of the U2 snRNA gene having all cis-elements or lacking the 3′ box. Unlike U1-tfs, however, a portion of U2-tfs received additional uridylylation of up to 5 nucleotides in length at position 87 (designated as U2-tfs-polyU) and formed an Sm protein–binding site–like structure that was stabilized by the small nuclear ribonucleoprotein SmB/B' probably as a part of heptameric Sm core complex that associates to the RNA. Both U2-tfs and U2-tfs-polyU were degraded by a nuclease distinct from the canonical Dis3L2 by a process catalyzed by terminal uridylyltransferase 7. Collectively, our data suggest that U2 snRNA biogenesis is regulated, at least in part, by a novel degradation pathway to ensure that defective U2 molecules are not incorporated into the spliceosome.
KEYWORDS: RNA surveillance, uridine-rich small nuclear RNA, splicing, spliceosome, snRNP biogenesis, uridylylation, terminal uridylyltransferase 7, RNA processing
Introduction
Uridine-rich small nuclear ribonucleoproteins (U snRNPs) are essential components of the spliceosome, which facilitates the splicing of cellular pre-mRNAs. The spliceosome is composed of at least five canonical U snRNPs, each of which contains an snRNA (U1, U2, U4, U5 or U6), a common heptameric ring of Sm proteins (SmB/B′, SmD1, SmD2, SmD3, SmE, SmF and SmG), and unique proteins that are specific to each U snRNP [1]. There is also a minor spliceosome composed of similar but different snRNPs [1].
During biogenesis of U snRNPs, transcripts from the genes for U snRNAs are modified with 7-monometylguanosine (m7G) on the 5′ side of the first transcribed nucleotide to yield a 5′-monomethylguanosine cap (m7G-cap). This process involves a series of enzymes including RNA triphosphatase, RNA guanylyltransferase and mRNA cap (guanine-N7-) methyltransferase; the exception is that U6 snRNA (U6) carries a γ-methyl triphosphate cap [2]. The U snRNAs are then methylated at the O-2 position of the ribose of the first transcribed nucleotide and of the second transcribed nucleotide, probably by cap-specific mRNA (nucleoside-2′-O-)-methyltransferase 1 and 2, respectively [3]. Pseudouridine is one of the other major post-transcriptionally modified nucleosides found often in U snRNAs (http://modomics.genesilico.pl/sequences/list/). These modifications are also found in many mRNAs, which enhances their stability, transport between the nucleus and the cytoplasm, and initiation of translation [4-7]. For U snRNAs, the heptameric ring of Sm proteins forms around the Sm protein–binding site (Sm-site) of each m7G-capped snRNA. A complex composed of the survival motor neuron protein SMN and other proteins (Gemin2–8 and Unrip) carries out the assembly of the heptameric ring on each U snRNA in the cytoplasm [1,8-13]. Successful heptameric ring formation is followed by hypermethylation of the m7G-cap (m2,2,7G-cap or m3G-cap) that is required for subsequent Snurportin-1-mediated import of each snRNP into the nucleus [1,8-13]. Final 3′ end processing of U snRNAs is believed to occur after hypermethylation [14].
Uridylylation is another post-transcriptional modification that is unique to U6. In the nucleus, the U6-specific terminal uridylyltransferase (TUT) adds uridine residues (up to 20 in mammalian cells) at the 3′ end of U6 [15-19]. This addition stabilizes U6 by protecting it from attack by exonucleases, thereby ensuring adequate production of U6 snRNP. U6 also receives 3′ adenylation in competition with uridylylation, and this adenylation also helps to control degradation of U6 [20]. Recently, other U snRNAs, including U1, are proposed to be uridylylated via TUTs and processed further with the 3′-5′ exoribonuclease Dis3L2, as shown by large-scale immunoprecipitation- sequencing analyses [21-23].
In U1, an additional ribose methylation takes place at transcribed nucleotide 70 of mature U1, but its role is unknown. We previously showed that a small population of m7G-capped U1 molecules lacking the methylation at nucleotide 70 receives an extra base methylation at the first transcribed nucleotide. Those molecules also lack Sm-site and stem-loop 4 (SL4) region without m3G cap formation [24]. This produces truncated forms of U1 (U1-tfs) [24], which are stabilized by binding of Gemin5 to each m7G-cap for subsequent degradation of the RNA [25], in cellular processing bodies (P-bodies). U1-tfs probably are degraded because they lack the ability to form a heptameric Sm protein ring [24].
In this study, we demonstrate that, in human cells, U2 snRNA (U2) receives post-transcriptional modifications similar to U1-tfs and produces truncated forms of m7G-capped U2 molecules (U2-tfs) that lack the Sm-site and SL3/4. In addition, we demonstrate the existence of polyuridylylated forms of U2-tfs (U2-tfs-polyU) that are stabilized by SmB/B’ probably as a part of heptameric Sm core complex.
Results
Identification of two distinct truncated forms of U2—U2-tfs and U2-tfs-polyU
Given a report by Ishikawa et al. that the knockdown of SmB/B’ led to the localization of U1 in P-bodies with consequent reduction of the cellular levels of not only U1 but also U2 [24], we assumed first that, like U1-tfs, the formation of truncated forms of U2 (U2-tfs) might contribute to the control of the cellular level of U2. To examine whether U2-tfs are actually present in human cells, we used four DNA probes (#1, #2, #3 and #4) that were complementary to regions within U2 snRNA containing SL1, SL2a-b, Sm-site, and SL4, respectively (Fig. 1A, Table S1). Because cells contain less U2 than U1, we considered that the level of U2-tfs might be much lower than that of U1-tfs. Under the assumption that U2-tfs have a m7G-cap (as do U1-tfs), we took advantage of the ability of Gemin5 to bind the m7G-cap structure to capture U2-tfs [25]. In this way, we detected U2-tfs among the Gemin5–associated RNAs in a northern blot; U2-tfs were detected as two bands [large U2-tfs (U2-tfsL) and U2-tfs] with the probes #1 and #2 but were not detected with probe #3 or #4 (Fig. 1B), suggesting that U2-tfsL and U2-tfs lack the Sm-site and the 3′ end region.
Figure 1.
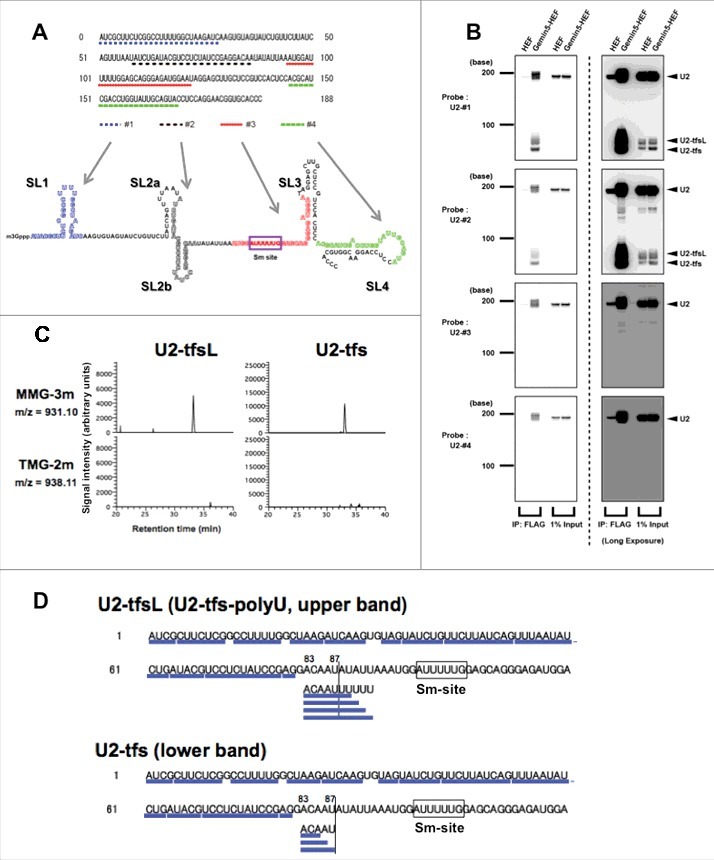
Identification of truncated forms of U2 snRNA pulled down with Gemin5. (A) Probes #1, #2, #3, and #4, which correspond to the regions shown under the sequence of U2. The secondary structure of mature U2 is shown. (B) Gemin5- hemagglutinin/TEV protease cleavage site/FLAG (HEF)–associated RNA or pull-down input RNA was separated by denaturing urea-PAGE and analyzed by northern blotting with probes #1–4. (C) Reverse-phase LC separation of RNase T1 digest products of U2-tfsL or U2-tfs. The effluent was monitored at m/z 931.10 (m7G-capped AUCGp + three methyl residues, MMG-3m; top) or m/z 938.11 (m3G-capped AUCGp + two methyl residues, TMG-2m; bottom). MMG was present in both digests. (D) The oligonucleotides identified by LC-MS are underlined in the partial sequence of U2 (1–120). The Sm-site is boxed.
Given that U1-tfs receives extra base methylation at the first transcribed nucleotide in addition to two ribose methylations at the first and the second transcribed nucleotides (m7G-mAmUmAC…), we examined whether U2-tfsL and U2-tfs had a m7G-cap and an extra methyl group at the first transcribed nucleotide. We applied in-gel RNase T1 digestion to SYBR gold–stained bands corresponding to those of U2-tfsL and U2-tfs (Fig. S1A) and analyzed the digest with liquid chromatography–coupled mass spectrometry (LC-MS). Among the detected oligonucleotides, we expected that, if an oligonucleotide had an extra methyl group and two 2′-O-methyled riboses with the structure m7G-mAmUmCG…, RNase T1 digestion would produce the doubly charged anion of m7G-capped AUCG from the 5′ end region of U2-tfs with three methyl groups (MMG-3m), which has 3′ phosphate (m/z 931.10) or 2′,3′-cyclic phosphate (m/z = 922.10) at the 3′ end. Therefore, in the LC-MS analysis, we monitored first the doubly de-protonated anion at m/z 931.10 and detected the expected oligonucleotide in the RNase T1 digests of both U2-tfsL and U2-tfs (Fig. 1C), and we detected three methyl groups in the oligonucleotide (Tables S2 and S3). In addition, we detected MMG-3 m with 2′, 3′-cyclic phosphate (m/z 922.10) in both digests (Tables S2 and S3). As a negative control, we also monitored m/z 938.11 (3′-phosphate) or m/z 929.11 (2′, 3′-cyclic phosphate), corresponding to data for a trimethylated G (m3G)-capped AUCG with two ribose methyl groups (m3G-AmUmCG, TMG-2m) that was expected not to bind Gemin5 [25]. Indeed, we did not detect such oligonucleotides in either RNase T1 digest (Fig. 1C and Tables S2 and S3), whereas both TMG-2 m with 3′ phosphate and 2′ 3′ cyclic phosphate were detected exclusively in the mature U2 digest as reported by Chao et al [25]. Collectively, these experiments confirmed our previous report that Gemin5 binds specifically to the m7G-cap structure [25], and demonstrated that U2-tfsL or U2-tfs have an extra methyl group at the first transcribed nucleotide, as seen for U1-tfs. In these analyses, we also monitored the other possible m7G-capped AUCG ions with one or two methyl groups, i.e., m7G-AmUCG or doubly charged anion of m7G-mAUCG at m/z 917.09 (MMG-1m, 3′-phosphate) and m/z 908.08 (MMG-1m, 2′ 3′-cyclic phosphate), or m7G-AmUmCG or m7m-Gm AUCG with m/z 924.10 (MMG-2m, 3′-phosphate) and m/z 915.09 (MMG-2m, 2′ 3′-cyclic phosphate). Although we did not detect such molecular species in the RNase T1 digest of U1-tfs [24], we detected them in the digests of U2-tfsL and U2-tfs (Fig. S1B, Tables S2 and S3). Thus, the 5′ end structure of U2-tfs is more heterogeneous than that of U1-tfs.
The oligonucleotides identified from U2-tfs covered the region from nucleotides 1 to 85–87 in the U2 sequence, whereas those of U2-tfsL had an additional 2 to 5 uridine residues at the 3′ end of nucleotide 87 (Fig. 1D and Tables S2 and S3); i.e., we detected four oligonucleotide species (ACAAUUU, ACAAUUUU, ACAAUUUUU, ACAAUUUUUU) from U2-tfsL and three species (ACA, ACAA, ACAAU) from U2-tfs (Fig. 1D and Tables S2 and S3). All these oligonucleotides had a hydroxyl group (-OH) at the 3′ end of the terminal nucleotide, suggesting that those oligonucleotides originated from the 3′ terminus of U2-tfsL (Table S3) and U2-tfs (Table S2). RNase T1 cleavage should generate a 2′, 3′-cyclic phosphate or a phosphate at the 3′ end of the terminal nucleotide [24,26-29]. Obviously, distinguishing the presence or absence of a phosphate or hydroxyl group at the 3′ terminus of oligonucleotide is an advantage of the MS-based method we used. Collectively, these data indicated that U2-tfsL are polyuridylylated forms of U2-tfs that have an extra 2 to 5 uridine residues at the 3′ side of nucleotide 87. Therefore, we designated U2-tfsL as U2-tfs-polyU. We noted that U2-tfs-polyU have an AUUUUU or AUUUUUU sequence at the 3′ end, which is similar to the Sm-site sequence (AUUUUUG)(Fig. 1D).
U2-tfs and U2-tfs-polyU are derived from transcripts of the U2 gene having all cis-elements or lacking the 3′ box element
The human U2 gene contains a 188-base U2-coding region (SL1, SL2a, SL2b, Sm, SL3, SL4 in Fig. 1A) and three cis-acting elements—Oct1-binding site (Oct), proximal sequence element (PSE) and 3′ box [30]. To compare the formation of U2-tfs with that of U1-tfs [24], we used vector pcDNA3.1 to construct various U2 genes fused with an y18Sn (yeast 18S neutral) tag that distinguishes exogenously expressed U2 from endogenous U2; namely we constructed y18Sn-U2, y18Sn-U2-ΔOct, y18Sn-U2-ΔPSE and y18Sn-U2-Δ3′box (Fig. 2A and Fig. S2A), similar to those constructed for the study of U1-tfs [24]. We also used the RAT (RNA Affinity in Tandem) tag (shown as the complementary sequence in Table S1) for transfection control. Vector y18Sn-U2 contained all of the cis-acting elements as well as the region containing the 779-nt sequence upstream of the Oct and the 829-nt sequence downstream of the 3′ box reported in the U2 gene and was used as a control to monitor the expression of full-length U2 (Fig. S2B). This gene construct indeed produced the expected mature y18Sn-U2 (223 nt) and y18Sn-U2-tfs as the two forms corresponding to the expected sizes of U2-tfs-polyU (y18Sn-U2-tfs-polyU) and U2-tfs (y18Sn-U2-tfs) (Fig. S2C and Fig. 2B). Similar to the production of U1-tfs from the U1 gene constructs reported by Ishikawa et al [24], U2-tfs were produced from the gene construct lacking the 3′ box in addition to that containing all of the cis-elements (Fig. 2B). In this experiment, we again detected two truncated forms of U2 corresponding to y-18Sn-U2-tfs-polyU and y18Sn-U2-tfs, consistent with the results obtained with northern blotting and LC-MS. The same experiment also revealed that the gene lacking the PSE did not express U2 at all, whereas that lacking Oct expressed U2 at a level lower than that of the full U2 gene and that lacking the 3′ box (Fig. 2B). These results were consistent with results for the U1 gene construct in terms of the roles of the cis-elements in their expression reported by Ishikawa et al [24].
Figure 2.
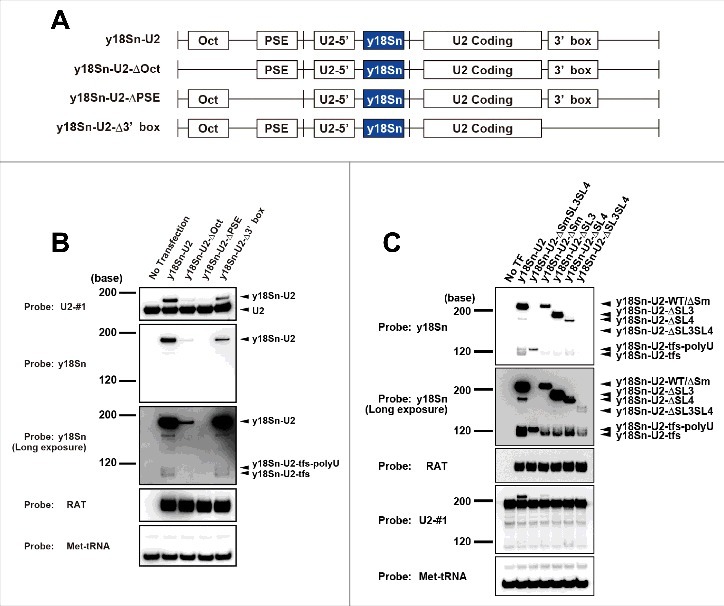
Formation of U2-tfs depends on transcription from the U2 gene. (A) Schematic diagram of the U2 gene construct or its cis-element deleted forms expressing y18Sn-tagged U2 snRNA. See the text for the explanation of each construct (y18Sn-U2, y18Sn-U2-ΔOct, y18Sn-U2-ΔPSE and y18Sn-U2-Δ3’ box). (B) RNA extracted from cells expressing each of the constructs shown was analyzed by northern blotting using the probes shown to the left of the blot. A long-exposure image is shown for probe y18Sn. The RAT tag was used for transfection control. (C) Northern blotting was carried out with total RNA extracted from cells expressing the individual constructs shown above the blot.
We next examined the possibility that U2-tfs/U2-tfs-polyU arises as a consequence of a structural defect(s) in U2 transcripts. We constructed an additional five expression vectors [y18Sn-U2ΔSmSL3SL4 (U2-tfs), 119 nt; y18Sn-U2ΔSm, 216 nt; y18Sn-ΔSL3, 190 nt; y18Sn-ΔSL4, 179 nt; y18Sn-ΔSL3SL4, 146 nt] (Fig. S2D). Although expression of each of these five vectors resulted in stained bands corresponding to y18Sn-U2-tfs-polyU and/or y18Sn-U2-tfs (Fig. 2C), their yields were not as high as those from similar constructs of U1 mutant genes, which produced almost exclusively U1-tfs [24]. Thus, it seems that defects in the 3’-side regions (Sm-site, SL3, SL4) of the U2 transcript are tolerated more so than those of U1 in terms of cellular surveillance, yet this could only be a consequence of much lower cellular abundance of U2 snRNP than U1 snRNP. On the other hand, the expression of construct y18Sn-U2ΔSmSL3SL4, which was expected to form y-18Sn-U2-tfs, resulted in a stained band similar in size to that of y18Sn-U2-tfs-polyU. The construct y18Sn-ΔSL3SL4, which has an Sm-site, produced mostly bands corresponding to y18Sn-U2-tfs (Fig. 2C). The other constructs produced bands corresponding to y-18Sn-U2-tfs and y-18Sn-U2-tfs-polyU at varying proportions (Fig. 2C). Collectively, these data suggested that both U2-tfs-polyU and U2-tfs are bona fide products in the cell.
TUT7-dependent degradation of U2-tfs and U2-tfs-polyU
It has been proposed that Dis3L2 participates in the degradation of uridylylated U snRNAs [21,22]. To examine whether Dis3L2 affects the degradation of U2-tfs-polyU, we knocked down Dis3L2 and analyzed the cellular level of U2-tfs-polyU. We found steady-state levels of U2-tfs-polyU in the Dis3L2 knockdown cells (Fig. 3A, lanes 1 and 3), indicating that U2-tfs-polyU is not a substrate of Dis3L2. Because each U2-tfs-polyU has an Sm site–like structure, we also examined whether knockdown of SmB/B' could affect the level of U2-tfs-polyU. Indeed, SmB/B' knockdown reduced the level of not only U2-tfs-polyU but also U2-tfs (Fig. 3A, lanes 1 and 2). This suggested that U2-tfs-polyU is stabilized by SmB/B' probably as a part of heptameric Sm core complex and that U2-tfs and U2-tfs-polyU share a common degradation pathway. Additional knockdown of Dis3L2 did not affect the cellular levels of U2-tfs-polyU and U2-tfs in SmB/B' knockdown cells (Fig. 3A, lanes 1, 2 and 4), suggesting that Dis3L2 does not participate in their degradation.
Figure 3.
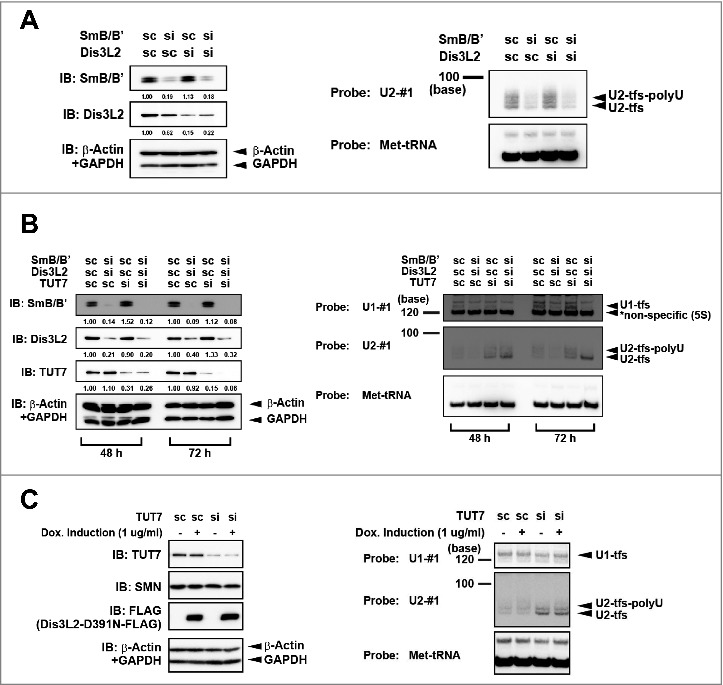
Reciprocal effects of SmB/B' and TUT7 on the cellular level of U2-tfs/U2-tfs-polyU. (A) SmB/B' stabilizes U2-tfs/U2-tfs-polyU. RNA extracted from cells transfected with a small interfering RNA (siRNA, si) or scrambled-sequence RNA (scRNA, sc, control) for the knockdown of SmB/B', Dis3L2, or both was analyzed with northern blotting using the probes shown to the left of the blot. An immunoblot (IB) is shown for SmB/B' or Dis3L2. Values shown under the protein blot indicates the staining intensity of each protein band relative to that the control (sc SmB/B', sc Dis3L2). (B) Deficiency of TUT7 increases the level of U2-tfs/U2-tfs-polyU. RNA extracted from the cells at 48 h and 72 h post-transfection with siRNA (or scRNA control) for knockdown of SmB/B', Dis3L2 or TUT7, or a combination of those siRNAs, was analyzed by northern blotting using the probes shown to the left side in right panel. Stained bands corresponding to U1-tfs, U2-tfs, and U2-tfs-polyU are shown to the right. Proteins were analyzed by immunoblotting with antibodies against the proteins indicated to the left side in left panel. (C) Northern blotting with probe U1-#1, U2-#1 or Met-tRNA (loading control) detected RNAs extracted from scRNA- or TUT7 siRNA–transfected cells treated with (+) or without (–) doxycycline inducing Dis3L2-D391N-FLAG expression. Protein was detected by immunoblotting with antibodies against the proteins indicated to the left.
Given that TUT7 (ZCCHC6) and TUT4 (ZCCHC11) reportedly are responsible for uridylylation events in human cells [31], we knocked down TUT7 or TUT4 to examine possible uridylylation-dependent degradation of U2-tfs/U2-tfs-polyU. We expected that, if they were indeed involved in the degradation, then knockdown would result in the accumulation of U2-tfs/U2-tfs-polyU. We found that the knockdown of TUT7, but not of TUT4, resulted in the accumulation of U2-tfs/U2-tfs-polyU (Fig. 3B and Fig. S3A and S3B), suggesting that TUT7 is involved in the degradation of U2-tfs/U2-tfs-polyU. We also found that TUT7 knockdown in combination with the double knockdown of SmB/B’ and Dis3L2 or single knockdown of SmB/B’ resulted in the preferential accumulation of U2-tfs, supporting again the aforementioned notion that SmB/B' stabilizes U2-tfs-polyU probably as a part of heptameric Sm core complex (Fig. 3B and Fig. S3B). The knockdown of TUT7 alone or triple knockdown of TUT7, SmB/B’ and Dis3L2 did not cause notable accumulation of U1-tfs (Fig. 3B). Conversely, expression of an enzymatically dead mutant of Dis3L2 (Dis3L2-D391N) did not affect the level of U2-tfs/U2-tfs-polyU regardless of the TUT7 knockdown (Fig. 3C), implying that Dis3L2 is not involved in the degradation of U2-tfs/U2-tfs-polyU.
We next examined weather the heptameric Sm core complex is formed on U2-tfs-polyU by pulldown analysis using HEF (HA-TEV site-FLAG)-SmB/B’ and HF (HA-Snurportin-1 (SPN1) as affinity baits. We postulated that if U2-tfs-polyU is incorporated into heptameric Sm core complex and stably exists in the later steps of U2 snRNP biogenesis, it could be pulled down not only with SmB/B’ but also with SPN1, a component associated with U snRNP after the m3G-cap formation. Indeed, both proteins pulled down the truncated U2 molecules but had the sizes slightly larger than U2-tfs-polyU (Fig. S3C), implying that U2-tfs-polyU with slightly longer polyU tails might be assembled into heptameric Sm core complex. We finally tried to demonstrate that the U2-tfs-polyU-loaded Sm core complex localizes in the nucleus by in situ hybridization utilizing y18Sn-U2-ΔSm that produce y18Sn-U2-tfs and y18-Sn-U2-tfs-polyU; however, our attempts failed to detect such a complex presumably due to its extremely low abundance.
Discussion
Our results demonstrate that human cells produce truncated forms of U2 (U2-tfs) that appear to be equivalent to U1-tfs, having a m7G-cap and an extra methyl group at the first transcribed nucleotide but lacking the 3’ structured region downstream of the Sm site [24]. Unlike U1-tfs, however, we identified additional forms of U2-tfs having an Sm site–like sequence (i.e., AUUUUU or AUUUUUU) that should be formed by polyuridylylation of the 3’-terminus (U2-tfs-polyU). Our proposed scenario to explain the formation and fate of U2-tfs-polyU is that 1) a small population of U2 transcripts forms U2-tfs after methylation of the first transcribed nucleotide, 2) although U2-tfs may be delivered to the degradation pathway shared with U1-tfs because of the inability to form the Sm ring, a portion of U2-tfs that end at residue A86 or U87 are subsequently polyuridylylated by a uridylyl transferase different from TUT7 to generate U2-tfs-polyU, 3) U2-tfs-polyU are stabilized by SmB/B’ binding probably as a part of heptameric Sm core complex to an Sm site–like structure of U2-tfs-polyU, which may yield a “pseudo” heptameric Sm ring, and/or 4) U2-tfs-polyU are uridylylated by TUT7 for further processing by an enzyme different from Dis3L2, resulting in the degradation of U2-tfs-polyU or the reformation of U2-tfs that are subsequently degraded via the degradation pathway shared with U1-tfs (Fig. 4). Although we cannot exclude the possibility that U-2tfs-polyU-like molecules observed upon expression of y18Sn-U2-ΔSmSL3SL4 (Fig. 2C) may be a result of failure of 3′ end processing, this may reflect a pathway in which all U2-tfs molecules undergo uridylylation before their degradation in P-bodies and/or via the TUT7 uridylylation pathway(s). It remains to be studied how U2-tfs-polyU is generated in cells. Nonetheless, we speculate that U2-tfs-polyU may have roles in regulating splicing or other cellular processes because the sequences of regions required for pre-mRNA branch point recognition and complementary to U6 are present in U2-tfs-polyU molecules. U2-tfs-polyU may play roles in splicing events as an antagonist to U2 snRNP. From this viewpoint, the pulldown analysis using SmB/B’ or SPN1 supports this notion by suggesting the presence of the SmB/B’- and SPN1-associated U2-tfs molecules larger than U2-tfs-polyU (Fig. S3C). We predict that this molecule might have an extended polyU tail, but its detailed structural characterization remains to be studied.
Figure 4.
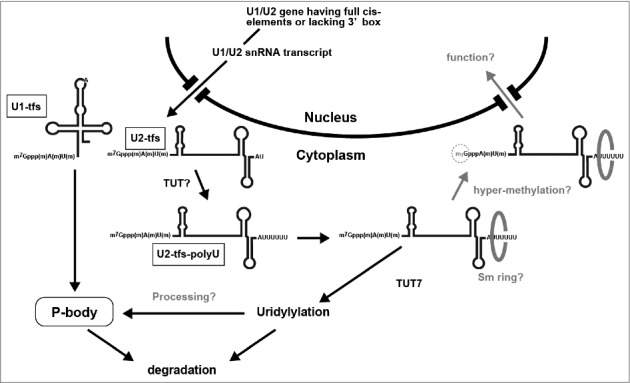
Proposed uridylylation pathway for U2-tfs that are diverted from the degradation pathway for U1-tfs. U1-tfs and U2-tfs are formed from a small population of U1 and U2 transcripts having all cis-elements or lacking the 3’ box, respectively. Whereas U1-tfs are delivered to P-bodies for degradation, U2-tfs are uridylylated by a TUT other than TUT7 to form U2-tfs-polyU, which is stabilized by SmB/B’ possibly by forming an Sm ring, or is degraded in a manner that is dependent on TUT7.
Besides the 3′-uridylylated U2-tfs, we identified three additional forms of U2-tfs that have 1 to 3 methyl groups at the 5′-terminus, i.e., MMG-1m, MMG-2m, and MMG-3m, whereas we detected only the MMG-3m form for U1-tfs. Given that U1-tfs with MMG-3m localize to P-bodies [24], we assume that U2-tfs having MMG-3m may also be delivered to the degradation pathway shared by U1-tfs; however, this awaits further investigation. These methylated forms were found in both U2-tfs and U2-tfs-polyU, suggesting that the uridylylation of U2-tfs is independent of 5′ methylation. Given the reported order of methylation events at the 5′ end [3], U2-tfs might be formed at earlier stages than U1-tfs during snRNP biogenesis. On the other hand, we found that the expression profile of U2-tfs/U2-tfs-polyU is very similar to that of U1-tfs—their expression appears to be regulated by cis-elements within their genes, suggesting that truncated forms of U1 and U2 are formed by a common mechanism that is closely linked to transcription. To understand the mechanism in further detail, it is critical to identify the nuclease(s) responsible for the formation of U1-tfs and U2-tfs. Since fragments of U2 snRNA containing Sm-site sequence known as RNU2-1f are highly preserved and abundant in serum and plasma from many cancer patients including those in lung, pancreatic and colorectal adenocarcinoma [32-37], it is intriguing to speculate that the nuclease(s) responsible for the formation of U2-tfs may also participate in the formation of RNU2-1f in those cancer patients.
Finally, our results demonstrate that current MS-based technology, coupled with the genome-wide search engine Ariadne, is a powerful means for characterizing RNAs, including the U snRNAs reported here, which carry a variety of post-transcriptional modifications. This approach allows unbiased identification of RNAs isolated from natural sources and determination of their chemical structures in detail, and thus the approach is suitable for the analysis of the cellular dynamics and fate of RNAs. Clearly, this approach complements current genomics-based technologies that rely on high-throughput sequencing of RNA or cDNA amplified by RT-PCR.
Materials and methods
LC-MS based RNA analyses
Preparation of RNase T1 digests, direct nanoflow LC-MS/MS of RNA fragments and database searching with interpretation of MS/MS spectra were described previously [24,26-29].
Construction of y18Sn RNA tagged U2 and its mutant expression vectors
U2 gene cloning was performed with standard cloning method with genomic DNA extracted from 293T cells; i.e., a cDNA fragment containing U2 gene sequence that included all cis-elements required for U2 transcription and their extended 5′/3′ flanking regions (2071 bp) (Homo sapiens chromosome 17, GRCh38.p7 Primary Assembly, NC_000017.11, Range: 43232907 to 43234977), was amplified by PCR using primer set HIW497 and HIW498. The nucleotide sequences of all primers used in this section are summarized in Table S4. The PCR product was cloned into the BamH I / Xho I sites of pDNA3.1(+) vector. DNA sequencing verified the cloned U2 gene sequence. A fragment containing 5′ flaking region of U2 gene including Oct1-binding site (Oct) and proximal sequence element (PSE) was amplified by PCR with the cloned U2 gene as template using primers set HIW497 and HIW517 that contained Bgl II site in the 3′ side. Another fragment containing 3′ flanking region of U2 gene that included 3′ box element, was amplified from the cloned U2 gene by PCR using primers set HIW498 and HIW538; the latter covered the first 11 nucleotide sequence of the 5′ side of U2 (U2-5′) and y18Sn RNA tag sequence (shown as the complementary sequence in Table S1) and contained Bgl II site in the 5′ side (Fig. S2A). Those two fragments were ligated at Bgl II sites and the ligated fragment was amplified with PCR using primer set HIW497 and HIW498. The amplified PCR fragment was cut with BamH I / Xho I sites and ligated into Bgl II / Xho I sites of pcDNA3.1(+) vector to remove CMV promoter. DNA sequencing verified the sequence of the final construct (2086 bp) (Fig. S2B). We named the resulted vector as pcDdCMV-y18Sn-U2 and used it as template for construction of U2 vectors lacking cis element after the confirmation of y18Sn-U2 RNA (y18Sn-U2) expression (Fig. S2C). Primer sets used for the construction of the vectors lacking cis elements by inverse PCR were; HIW553 and HIW554 for the construction of pcDdCMV-y18Sn-U2-ΔOct that lacked Oct element, HIW555 and HIW556 for pcDdCMV-y18Sn-U2-ΔPSE lacking PSE element and HIW557 and HIW558 for pcDdCMV-y18Sn-U2-Δ3′box lacking 3′ box element (Table S4). The PCR products having blunt end were ligated after phosphorylation by T4 polynucleotide kinase, amplified in E. coli and verified their sequences by DNA sequencing for the deletion of desired cis element.
For the construction of vectors expressing U2 mutants that have the defects in Sm-site, SL3 and/or SL4 (U2-ΔSmSL3SL4, U2-ΔSm, U2-ΔSL3, U2-ΔSL4 and U2-ΔSL3SL4, Fig. S2D), we first prepared U2 gene construct shorter than the one described above (short U2 gene); this gene contained all of the cis-acting elements with total length of 908 bp that included the region containing the 190-nt sequence upstream of the Oct and the 240-nt sequence downstream of the 3′ box. The short U2 gene was amplified by primer sets HIW559 and HIW560 using pcDdCMV-y18Sn-U2 (2086 bp) as template. The resulted PCR product was cut with BamH I / Xho I sites, ligated into Bgl II / Xho I sites of pcDNA3.1(+) vector and verified by DNA sequencing. We used this short U2 gene construct for the preparation of vectors expressing U2 mutants further after confirming that the expression products were indistinguishable between those constructs in terms of the role of cis elements and their sub-products (Fig. S2E). Primer sets used for the preparation of vectors expressing U2 mutants by inverse PCR were; HIW602 and HIW603 for pcDdCMV-y18Sn-U2-ΔSmSL3SL4 expressing y18Sn-U2-ΔSmSL3SL4, HIW604 and HIW605 for pcDdCMV-y18Sn-U2-ΔSm expressing y18Sn-U2-ΔSm, HIW606 and HIW607 for pcDdCMV-y18Sn-U2-ΔSL3 expressing y18Sn-U2-ΔSL3, HIW608 and HIW603 for pcDdCMV-y18Sn-U2-ΔSL4 expressing y18Sn-U2-ΔSL4 and HIW606 and HIW603 for pcDdCMV-y18Sn-U2-ΔSL3SL4 expressing y18Sn-ΔSL3SL4.
Supplementary Material
Funding Statement
This work was funded by a grant for Core Research for Evolutionary Science and Technology (CREST) from Japan Science and Technology Agency (JPMJCR13M2), and a Grant-in-Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, & Technology of Japan (MEXT), and JSPS KAKENHI Grant Number 16K18489. Funding for open access charge: Core Research for Evolutionary Science and Technology (CREST), Japan Science and Technology Agency.
Abbreviations
- LC-MS
liquid chromatography–coupled mass spectrometry
- m7G/MMG7
monomethylguanosine
- m3G/TMG
2,2,7-trimethylguanosine
- poly-U
polyuridylylation
- RAT
RNA affinity in tandem
- SL
stem loop
- Sm-site
Sm-protein binding site
- TUT
terminal uridylyltransferase
- U2-tfs
truncated forms of U2 snRNA
- y18Sn tag
yeast 18 S neutral tag
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Sally Nakamura-Fujiyama for her technical assistance.
References
- [1].Patel SB, Bellini M. The assembly of a spliceosomal small nuclear ribonucleoprotein particle. Nucleic Acids Res. 2008;36:6482–93. doi: 10.1093/nar/gkn658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Singh R, Reddy R. Gamma-monomethyl phosphate: a cap structure in spliceosomal U6 small nuclear RNA. Proc Natl Acad Sci U S A. 1989;86:8280–3. doi: 10.1073/pnas.86.21.8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Langberg SR, Moss B. Post-transcriptional modifications of mRNA. Purification and characterization of cap I and cap II RNA (nucleoside-2′-)-methyltransferases from HeLa cells. J Biol Chem. 1981;256:10054–60 [PubMed] [Google Scholar]
- [4].Schroeder SC, Zorio DA, Schwer B, et al.. A function of yeast mRNA cap methyltransferase, Abd1, in transcription by RNA polymerase II. Mol Cell. 2004;13:377–87. doi: 10.1016/S1097-2765(04)00007-3 [DOI] [PubMed] [Google Scholar]
- [5].Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annu Rev Biochem. 1999;68:913–63. doi: 10.1146/annurev.biochem.68.1.913 [DOI] [PubMed] [Google Scholar]
- [6].Kuge H, Brownlee GG, Gershon PD, et al.. Cap ribose methylation of c-mos mRNA stimulates translation and oocyte maturation in Xenopus laevis. Nucleic Acids Res. 1998;26:3208–14. doi: 10.1093/nar/26.13.3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Izaurralde E, Lewis J, McGuigan C, et al.. A nuclear cap binding protein complex involved in pre-mRNA splicing. Cell. 1994;78:657–68. doi: 10.1016/0092-8674(94)90530-4 [DOI] [PubMed] [Google Scholar]
- [8].Meister G, Eggert C, Fischer U. SMN-mediated assembly of RNPs: a complex story. Trends Cell Biol. 2002;12:472–8. doi: 10.1016/S0962-8924(02)02371-1 [DOI] [PubMed] [Google Scholar]
- [9].Fischer U, Liu Q, Dreyfuss G. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell. 1997;90:1023–9. doi: 10.1016/S0092-8674(00)80368-2 [DOI] [PubMed] [Google Scholar]
- [10].Mattaj IW. Cap trimethylation of U snRNA is cytoplasmic and dependent on U snRNP protein binding. Cell. 1986;46:905–11. doi: 10.1016/0092-8674(86)90072-3 [DOI] [PubMed] [Google Scholar]
- [11].Chari A, Paknia E, Fischer U. The role of RNP biogenesis in spinal muscular atrophy. Curr Opin Cell Biol. 2009;21:387–93. doi: 10.1016/j.ceb.2009.02.004 [DOI] [PubMed] [Google Scholar]
- [12].Huber J, Dickmanns A, Luhrmann R. The importin-beta binding domain of snurportin1 is responsible for the Ran- and energy-independent nuclear import of spliceosomal U snRNPs in vitro. J Cell Biol. 2002;156:467–79. doi: 10.1083/jcb.200108114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ohno M, Segref A, Bachi A, et al.. PHAX, a mediator of U snRNA nuclear export whose activity is regulated by phosphorylation. Cell. 2000;101:187–98. doi: 10.1016/S0092-8674(00)80829-6 [DOI] [PubMed] [Google Scholar]
- [14].Kleinschmidt AM, Pederson T. RNA processing and ribonucleoprotein assembly studied in vivo by RNA transfection. Proc Natl Acad Sci U S A. 1990;87:1283–7. doi: 10.1073/pnas.87.4.1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Munoz-Tello P, Rajappa L, Coquille S, et al.. Polyuridylation in Eukaryotes: A 3′-End Modification Regulating RNA Life. Biomed Res Int. 2015;2015:968127. doi: 10.1155/2015/968127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Trippe R, Guschina E, Hossbach M, et al.. Identification, cloning, and functional analysis of the human U6 snRNA-specific terminal uridylyl transferase. RNA. 2006;12:1494–504. doi: 10.1261/rna.87706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lund E, Dahlberg JE. Cyclic 2′,3′-phosphates and nontemplated nucleotides at the 3′ end of spliceosomal U6 small nuclear RNA's. Science. 1992;255:327–30. doi: 10.1126/science.1549778 [DOI] [PubMed] [Google Scholar]
- [18].Hirai H, Lee DI, Natori S, et al.. Uridylation of U6 RNA in a nuclear extract in Ehrlich ascites tumor cells. J Biochem. 1988;104:991–4. doi: 10.1093/oxfordjournals.jbchem.a122597 [DOI] [PubMed] [Google Scholar]
- [19].Rinke J, Steitz JA. Association of the lupus antigen La with a subset of U6 snRNA molecules. Nucleic Acids Res. 1985;13:2617–29. doi: 10.1093/nar/13.7.2617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chen Y, Sinha K, Perumal K, et al.. Effect of 3′ terminal adenylic acid residue on the uridylation of human small RNAs in vitro and in frog oocytes. RNA. 2000;6:1277–88. doi: 10.1017/S1355838200000285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ustianenko D, Pasulka J, Feketova Z, et al.. TUT-DIS3L2 is a mammalian surveillance pathway for aberrant structured non-coding RNAs. EMBO J. 2016;35:2179–2191. doi: 10.15252/embj.201694857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Pirouz M, Du P, Munafo M, et al.. Dis3l2-Mediated Decay Is a Quality Control Pathway for Noncoding RNAs. Cell Rep. 2016;16:1861–73. doi: 10.1016/j.celrep.2016.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Labno A, Warkocki Z, Kulinski T, et al.. Perlman syndrome nuclease DIS3L2 controls cytoplasmic non-coding RNAs and provides surveillance pathway for maturing snRNAs. Nucleic Acids Res. 2016;44:10437–53. doi.org/ 10.1093/nar/gkw649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ishikawa H, Nobe Y, Izumikawa K, et al.. Identification of truncated forms of U1 snRNA reveals a novel RNA degradation pathway during snRNP biogenesis. Nucleic Acids Res. 2014;42:2708–24. doi: 10.1093/nar/gkt1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Xu C, Ishikawa H, Izumikawa K, et al.. Structural insights into Gemin5-guided selection of pre-snRNAs for snRNP assembly. Genes Dev. 2016;30:2376–2390. doi: 10.1101/gad.288340.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Taoka M, Nobe Y, Hori M, et al.. A mass spectrometry-based method for comprehensive quantitative determination of post-transcriptional RNA modifications: the complete chemical structure of Schizosaccharomyces pombe ribosomal RNAs. Nucleic Acids Res. 2015;43:e115. doi: 10.1093/nar/gkv560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Taoka M, Ikumi M, Nakayama H, et al.. In-gel digestion for mass spectrometric characterization of RNA from fluorescently stained polyacrylamide gels. Anal Chem. 2010;82:7795–803. doi: 10.1021/ac101623j [DOI] [PubMed] [Google Scholar]
- [28].Taoka M, Yamauchi Y, Nobe Y, et al.. An analytical platform for mass spectrometry-based identification and chemical analysis of RNA in ribonucleoprotein complexes. Nucleic Acids Res. 2009;37:e140. doi: 10.1093/nar/gkp732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Nakayama H, Akiyama M, Taoka M, et al.. Ariadne: a database search engine for identification and chemical analysis of RNA using tandem mass spectrometry data. Nucleic Acids Res. 2009;37:e47. doi: 10.1093/nar/gkp099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Strom AC, Forsberg M, Lillhager P, et al.. The transcription factors Sp1 and Oct-1 interact physically to regulate human U2 snRNA gene expression. Nucleic Acids Res. 1996;24:1981–6. doi: 10.1093/nar/24.11.1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Thornton JE, Chang HM, Piskounova E, et al.. Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA. 2012;18:1875–85. doi: 10.1261/rna.034538.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kohler J, Schuler M, Gauler TC, et al.. Circulating U2 small nuclear RNA fragments as a diagnostic and prognostic biomarker in lung cancer patients. J Cancer Res Clin Oncol. 2016;142:795–805. doi: 10.1007/s00432-015-2095-y [DOI] [PubMed] [Google Scholar]
- [33].Baraniskin A, Zaslavska E, Nopel-Dunnebacke S, et al.. Circulating U2 small nuclear RNA fragments as a novel diagnostic biomarker for primary central nervous system lymphoma. Neuro Oncol. 2016;18:361–7. doi: 10.1093/neuonc/nov144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kuhlmann JD, Wimberger P, Wilsch K, et al.. Increased level of circulating U2 small nuclear RNA fragments indicates metastasis in melanoma patients. Clin Chem Lab Med. 2015;53:605–11. doi: 10.1515/cclm-2014-1064 [DOI] [PubMed] [Google Scholar]
- [35].Kuhlmann JD, Baraniskin A, Hahn SA, et al.. Circulating U2 small nuclear RNA fragments as a novel diagnostic tool for patients with epithelial ovarian cancer. Clin Chem. 2014;60:206–13. doi: 10.1373/clinchem.2013.213066 [DOI] [PubMed] [Google Scholar]
- [36].Baraniskin A, Nopel-Dunnebacke S, Schumacher B, et al.. Analysis of U2 small nuclear RNA fragments in the bile differentiates cholangiocarcinoma from primary sclerosing cholangitis and other benign biliary disorders. Dig Dis Sci. 2014;59:1436–41. doi: 10.1007/s10620-014-3034-5 [DOI] [PubMed] [Google Scholar]
- [37].Baraniskin A, Nopel-Dunnebacke S, Ahrens M, et al.. Circulating U2 small nuclear RNA fragments as a novel diagnostic biomarker for pancreatic and colorectal adenocarcinoma. Int J Cancer. 2013;132:E48–57. doi: 10.1002/ijc.27791 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.