

ABSTRACT
Nuclear multisubunit RNA polymerases IV and V (Pol IV and Pol V) evolved in plants as specialized forms of Pol II. Their functions are best understood in the context of RNA-directed DNA methylation (RdDM), a process in which Pol IV-dependent 24 nt siRNAs direct the de novo cytosine methylation of regions transcribed by Pol V. Pol V has additional functions, independent of Pol IV and 24 nt siRNA biogenesis, in maintaining the repression of transposons and genomic repeats whose silencing depends on maintenance cytosine methylation. Here we report that Pol IV and Pol V play unexpected roles in defining the 3′ boundaries of Pol II transcription units. Nuclear run-on assays reveal that in the absence of Pol IV or Pol V, Pol II occupancy downstream of poly A sites increases for approximately 12% of protein-coding genes. This effect is most pronounced for convergently transcribed gene pairs. Although Pols IV and V are detected near transcript ends of the affected Pol II – transcribed genes, their role in limiting Pol II read-through is independent of siRNA biogenesis or cytosine methylation for the majority of these genes. Interestingly, we observed that splicing was less efficient in pol IV or pol V mutant plants, compared to wild-type plants, suggesting that Pol IV or Pol V might affect pre-mRNA processing. We speculate that Pols IV and V (and/or their associated factors) play roles in Pol II transcription termination and pre-mRNA splicing by influencing polymerase elongation rates and/or release at collision sites for convergent genes.
KEYWORDS: Plant, nuclear multisubunit RNA polymerases IV and V, Pol IV, Pol V, RNA directed DNA Methylation, RdDM, transcription termination, nuclear run-on assay
Introduction
Nuclear multisubunit RNA polymerases IV and V (Pol IV and Pol V) play distinct roles in the transcriptional silencing of genes and transposons via RNA-directed DNA methylation (RdDM) [1]. Pol IV is thought to initiate the major RdDM pathway, transcribing DNA to generate relatively short RNA transcripts that are then copied into double-stranded RNAs by RNA-DEPENDENT RNA POLYMERASE 2 (RDR2) [2-9]. Resulting double-stranded RNAs are diced by DICER-LIKE 3 (DCL3) into 24 nt siRNAs that are loaded into ARGONAUTE 4 (AGO4), or a related AGO protein [10-13]. In parallel, Pol V generates transcripts and recruits AGO4-siRNA complexes to RdDM loci via its C-terminal AGO4 hook motifs [14-16]. As Pol V elongates, AGO4-siRNA interaction with either complementary bases in lncRNA or DNA guides de novo cytosine methyltransferase, DRM2, to bring about repressive cytosine methylation (in CG, CHG or CHH contexts) [2,16-19]. There is evidence that Pol V has functions independent of RdDM, helping maintain repression of transposons and genomic repeats whose silencing depends on maintenance methylation by MET1 and the chromatin remodeling ATPase, DDM1 [20-22]. First, it has been reported that heterochromatin organization and silencing of pericentromeric transcription is affected in pol V mutant plants independently of the 24-nt siRNA biogenesis pathway [20]. Also, Pol V has been implicated in at least one alternative TE silencing pathway, the RDR6-dependent DNA methylation pathway (RDR6-RdDM) [12,21-24]. In this Pol IV- and 24-nt siRNA- independent pathway, Pol II-derived RNAs from TE regions are processed into 21- and 22-nt siRNAs in a RDR6, DCL2, and DCL4-dependent manner, loaded into AGO6 and targeted to Pol V scaffold transcripts [21,22,24]. Thus, Pol V can function as a downstream component in both 24-nt siRNA-dependent (canonical RdDM) and 21- and 22-nt siRNA-dependent (RDR6-RdDM) TE silencing pathways.
Steady-state levels of mRNAs transcribed by Pol II in Arabidopsis thaliana are minimally affected in pol IV or pol V mutant plants [25]. However, several observations have suggested that Pol IV and V function in proximity to Pol II transcription units. At long retrotransposons, 24-nt siRNA biogenesis and RdDM occur primarily at the ends of the elements, where promoter sequences recognized by the Pol II transcription machinery are located[21,26]. Moreover, chromatin immunoprecipitation studies have shown that approximately 30% of Pol V occupied positions are proximal to the promoters of Pol II – transcribed genes [17]. Likewise, 21–24 nt sRNAs are enriched within 1 kb of either the 5′ or 3′ ends of transcription units for approximately 20% of protein-coding genes, with studies suggesting that sRNA abundance correlates with mRNA abundance at these genes [27,28]. However, unlike Pol V ChIP peaks that correspond to transposons, Pol V peaks that overlap protein-coding genes typically do not correlate with 24-nt siRNAs or CHH DNA methylation [17]. which are hallmarks of RdDM, suggesting the possibility of an alternative function of Pol V in regulation of protein-coding genes.
RNA Pol II transcription termination in eukaryotes is a complex process. Termination by Pol II occurs stochastically within a region of several hundred bases downstream of the polyA site and is functionally connected to the cleavage and polyadenylation of nascent transcript 3′ ends [29-32]. Transcription pause sites, binding sites for protein termination factors, the presence of RNA-DNA hybrids, or chromatin modifications within the termination region can affect termination efficiency [33-35].
We conducted nuclear run-on assays combined with high-throughput sequencing (NRO-Seq) [36], to identify genomic regions where nascent RNAs are synthesized, comparing wild-type plants to pol IV or pol V mutant plants, similar to an analogous study conducted in maize [37]. Interestingly, as in maize, we detect increased RNA Pol II abundance downstream from the cleavage and polyadenylation sites of protein coding genes in pol IV or pol V null mutants. By contrast, nascent transcript abundance in the region from the initiation site to the PolyA site is unaffected, as are steady-state mRNA levels. This suggests that 3′ end processing of pre-mRNAs is not affected in pol IV or pol V mutants, but termination and Pol II release is impaired or delayed. Convergently-transcribed protein coding genes whose 3′ regions overlap show the greatest increase in nascent RNA signals downstream of PolyA addition sites when Pol IV or Pol V are mutated. Those gene pairs also have a higher tendency to overlap with sites of Pol IV and Pol V occupancy, as measured by ChIP. However, these regions are not characterized by abundant 24-nt siRNAs or CHH methylation, suggesting that Pol II transcription termination does not require RdDM modification within termination zones for the majority of these genes. Collectively, our results suggest a methylation independent role for Pols IV and V in shaping the boundaries of Pol II transcription units.
Results
Nascent transcript levels increase downstream of mRNA polyA addition sites in pol IV or pol V mutants
Nascent RNA transcripts produced by template-engaged RNA polymerases in purified nuclei were generated by nuclear run-on (NRO) in the presence of biotin-UTP, allowing capture of the labeled RNAs using streptavidin beads. Quantitative PCR (qPCR) assays, comparing reactions conducted using biotin-UTP versus unmodified UTP, were then used to calculate the fold enrichment achieved for biotinylated RNAs versus background for a set of highly-expressed Arabidopsis thaliana genes (Fig. S1). This analysis allowed us to estimate that nascent biotinylated RNA accounts for more than 99.7% of the RNA captured on the streptavidin beads, with background binding, attributable to unlabeled RNAs, accounting for the remaining 0.3%.
We performed nuclear run-on using nuclei of wild-type plants as well as pol IV and pol V mutant plants and constructed sequencing libraries for resulting RNAs (NRO-Seq library), as well as for purified total nuclear RNAs (RNA-Seq library), thus allowing comparisons between nascent and steady-state RNA levels (Fig. S2). To examine the distribution of RNA polymerase II activity we focused our analysis on 1 kb regions surrounding RefSeq transcription start sites (TSSs) or transcription end sites (TESs) for 27859 protein-coding genes (Fig. 1). Both nascent RNA (Fig. 1A) and steady-state RNA (Fig. 1B) profiles show a sharp increase in RNA sequence reads starting at the TSS, followed by a plateau that begins ∼200 bps downstream. This pattern contrasts with nascent RNA profiles reported for yeast and other eukaryotic organisms, in which peaks are often observed downstream of the TSS, reflecting promoter-proximal pausing of Pol II [36,38-42]. The absence of a promoter-proximal peak in the nascent RNA profile in Arabidopsis suggests that promoter-proximal pausing is not a general feature of Arabidopsis genes transcribed by Pol II. Similarly, no evidence of promoter-proximal pausing was observed in maize NRO data [37].
Figure 1.
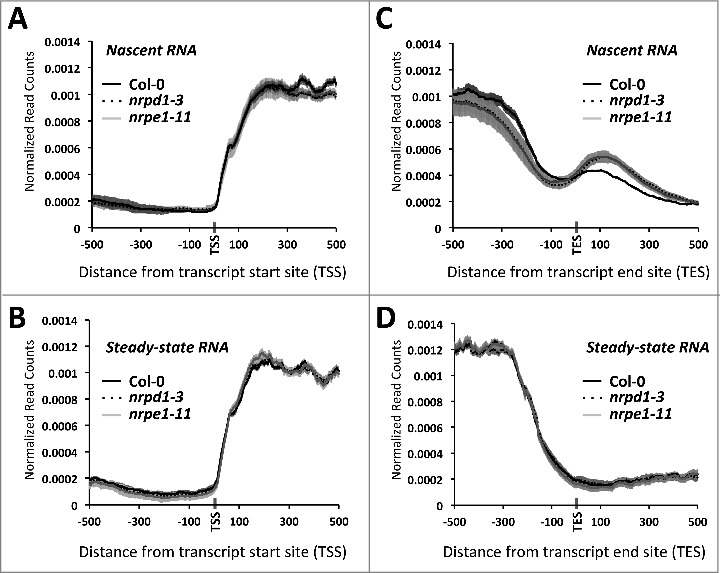
Loss of Pol IV or Pol V results in increased NRO-Seq read density downstream of protein-coding gene transcript end sites. (A,B) Average read densities are plotted relative to the transcription start sites (TSS) of 27,859 genes for nascent RNA (A) or steady-state RNA (B) isolated from Col-0, pol IV or pol V mutant plants. (C) Nascent RNA read-density plotted relative to transcript end sites (TES). (D) As in (C) but for steady-state RNA. Shadings represent 95% confidence intervals.
Loss of Pol IV or V activity does not affect RNA Pol II read density profiles near TSSs for either nascent RNA (Fig. 1A) or steady-state RNA (Fig. 1B), suggesting that RNA polymerases IV and V exert no detectable influence on Pol II transcription initiation at protein-coding genes on a genome-wide scale.
Analysis of the nascent RNA (NRO) read distribution in the 1 kb region surrounding the transcript end sites (TESs) revealed a gradual decrease in read abundance prior to the TES, followed by a small increase in reads ∼100 bp post-TES (Fig. 1C). This read density peak just downstream of TES in the nascent RNA population is not observed for steady-state RNA (Fig. 1D), suggesting that post-TES RNA is removed by cleavage at the polyA addition site and thus absent from mature mRNAs. Interestingly, a further increase in nascent transcript abundance occurs downstream of TESs in pol IV or pol V mutant plants, suggesting increased in Pol II occupancy, possibly due to delayed termination or Pol II release (Fig. 1C).
To determine if specific genes have dramatically altered Pol II occupancy downstream of TESs in pol IV or pol V mutants, we calculated a transcript end site – associated termination index (TES-TI) for each gene as the number of total reads in the first 500 bp downstream of the TES (interval 2) divided by the number of total reads in the 500 bp preceding the TES (interval 1) (Fig. 2A). Approximately 12% of the 18,419 protein coding genes analyzed exhibit a two fold or greater increase in their termination index in pol IV (nrpd1–3) or pol V (nrpe1–11) mutants, compared to wild-type (Fig. 2B). Profiles for these genes, plotted as in Fig. 1, are shown in Fig. 2C. A strong concordance (∼80% overlap) is observed for genes affected in pol IV or pol V mutant plants (Fig. 2B, Venn diagram). A set of 1312 genes, affected at least two fold by both pol IV and pol V, was chosen for all subsequent analyses. Loss of Pol IV or Pol V activities did not cause detectable lengthening of mature mRNAs for these genes, suggesting that RNA processing still occurs (Fig. 2D). Examples of protein-coding genes with increased read density downstream of TES are shown in Fig. 2E.
Figure 2.
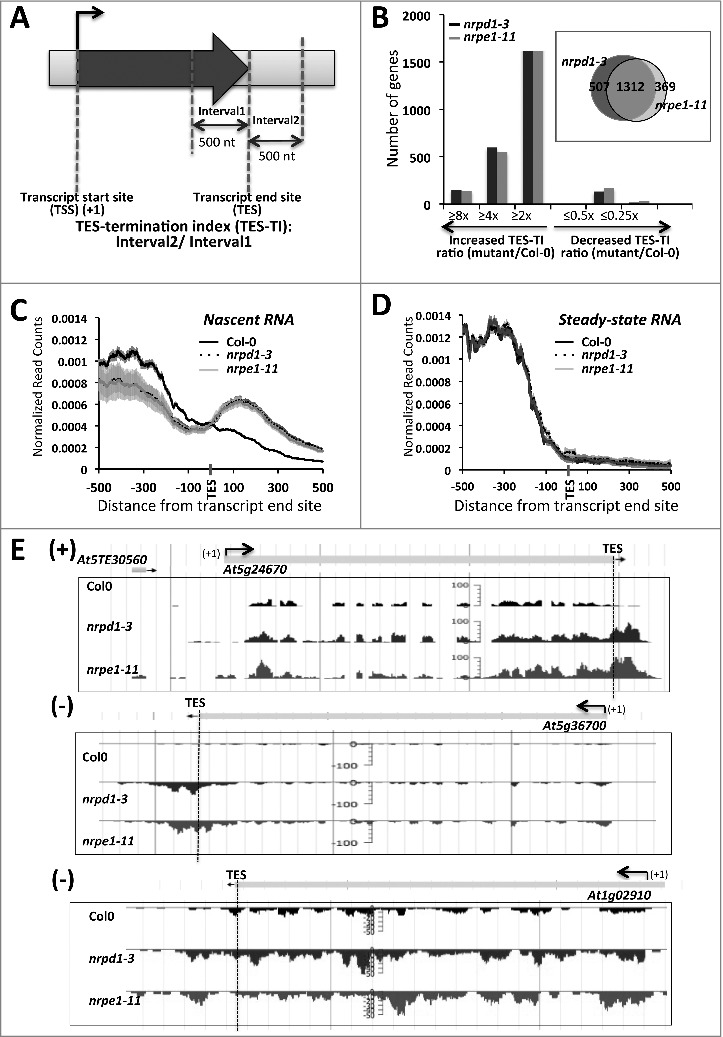
Genes with increased read density downstream of their transcript end sites (TES) in pol IV or pol V mutant plants. (A)Schematic for calculating the TES-termination index (TES-TI). (B) Histogram showing the number of genes with increased or decreased TES-TI values in pol IV or pol V mutant plants, relative to wild-type (Col-0) plants. (Insert in B) Venn diagram showing the overlap between genes with 2 fold or more increase in post-TES read density in pol IV (nrpd1–3) or pol V (nrpe1–11) plants relative to Col-0 plants. (C) Nascent RNA read-density plotted relative to TESs for the 1312 genes affected by both Pol IV and Pol V (see Venn diagram of panel B). (D) As in (C) but for steady-state RNA. For (C) and (D) shadings represent 95% confidence intervals. (E) Examples of protein-coding genes with increased read density downstream of the TES (marked with blue dashed vertical line) in pol IV (nrpd1–3) or pol V (nrpe1–11) mutants, displayed using JBrowse.
To further examine whether changes in pol IV or pol V mutants affect qualitative or temporal aspects of 3′ end processing, we performed circular RT-PCR analyses at five loci, using primers that allow discrimination between full-length transcripts and transcripts that extend post-TES (Fig. 3A). Prior to circularization total RNA from Col-0, pol IV or pol V mutant plants was subjected to decapping by Tobacco Acid Pyrophosphatase (TAP) to provide 5′-monophosphorylated terminus. Longer circularized transcripts were detected in pol IV and pol V mutant plants, compared to wild-type Col-0 plants (Fig. 3B, Fig. S3). Cloning and sequencing of these PCR products revealed that the majority of clones support the annotated gene models, but for the At5g24670 transcript 14% of the cloned cDNAs in nrpd1 and 24% of the cDNA clones in nrpe1 include sequences downstream of the annotated TES (Fig. 3C), suggesting that 3′ end processing may be less efficient in pol IV or pol V mutants. However, we speculate that the impaired RNA processing might not be the major factor that causes increase in post-TES reads in pol IV or pol V mutant plants as loss of Pol IV or Pol V activities did not result in detectable lengthening of mature mRNAs for these genes in RNA-Seq experiment (Fig. 2D), suggesting that RNA processing still occurs.
Figure 3.
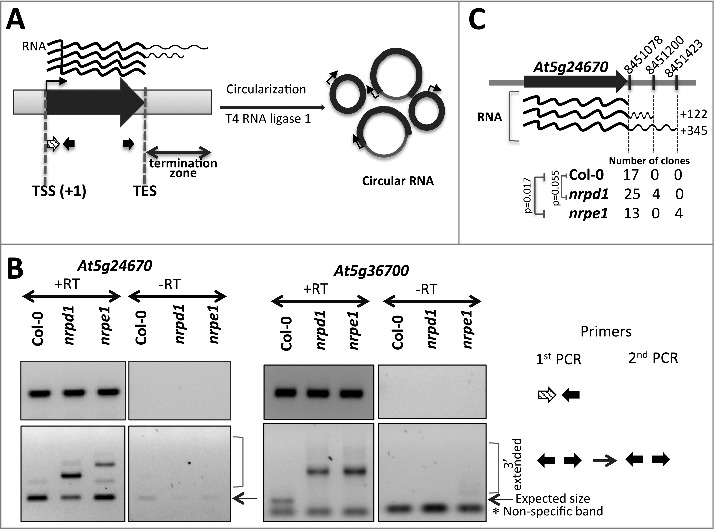
Transcripts with increased read density post-TES in pol IV or pol V mutant plants exhibit a longer termination zone. (A) Schematics of the circular RT-PCR approach used for independent confirmation of transcript ends. (B) Circular RT-PCR results for At5g24670 and At5g36700. Top panels show PCR products obtained using primers placed near transcription start sites (TSS). Bottom panels show both mature and 3'-extended PCR products. Primers for each PCR reactions are color-coded as in (A) and are shown on the right. (C) Sequencing results for cloned At5g24670 PCR products (from B, bottom panel). Number of clones supporting transcript ends at the marked (vertical dashed lines) genomic positions are shown for Col-0, nrpd1, and nrpe1 plants. Numbers on the right represent observed increase (in nt) in the length of the transcripts relatively to mature transcripts. Significance has been tested using one-tailed Chi-square test.
Pol II occupancy correlates with nuclear run-on signal intensity
Using chromatin immunoprecipitation (ChIP) of exonuclease-trimmed chromatin-protein complexes (ChIP-exo), we examined Pol II association with genes displaying increased (>2x) post-TES nascent RNA-seq read density in pol IV or pol V mutants (Fig. 4). The ChIP-exo method allows high resolution mapping of DNA binding factors due to the exonuclease trimming of sequences not protected by the bound protein [43]. Consistent with NRO-seq results, the ChIP-exo sequence data revealed increased Pol II association with gene 3′ ends in pol IV mutants (Fig. 4A), compared to Col-0 wild-type, with a strong peak downstream of the TESs. In contrast, no differences in RNA Pol II occupancy was observed near the transcription start sites for the same set of genes (Fig. 4B), suggesting an asymmetric affect at gene 3′ ends.
Figure 4.
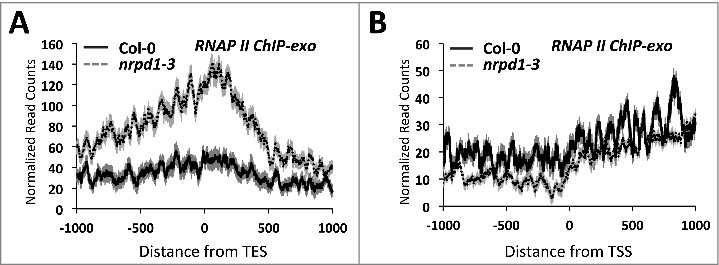
Pol II accumulates near TESs in pol IV mutant plants. (A) RNA Pol II ChIP-exo reads are plotted relative to transcript end sites (TES) for genes with increased post-TES read density in pol IV or pol V mutants. (B) As in (A) but reads are plotted relative to TSSs. Shadings represent 95% confidence intervals.
Genes with Pol IV and V – affected transcription termination associate with RdDM pathway components
We examined published Pol IV [44], Pol V [17], and AGO4 [45] ChIP-seq datasets to see if these proteins are enriched near the 3′ ends of genes that show increased Pol II occupancy post-TES in pol IV or V mutant plants. Interestingly, 60% of 1312 genes with increased TES-proximal read density have the 1 kb region surrounding their TESs occupied by at least one of the above-mentioned proteins, with Pol V being present at 50% of these genes (Fig. 5A). Analysis of Pol V ChIP-seq [17], read distribution in wild-type (Col-0) plants revealed Pol V association primarily upstream of TESs, with an additional peak, compared to background (nrpe1–11), downstream of the TES (Fig. 5B). Analysis of Pol IV ChIP-seq [44], data did not reveal evidence for a similar pattern of enrichment (Fig. S4).
Figure 5.
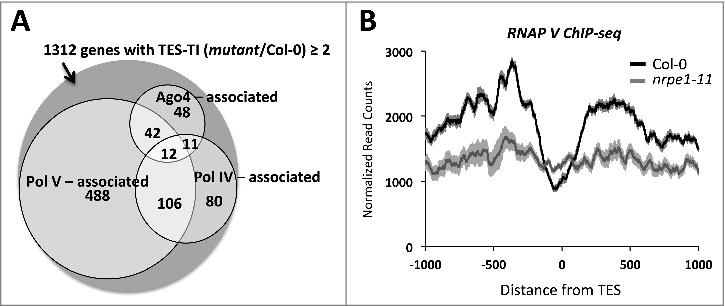
Correspondence between genes with increased read density post-TES and RdDM pathway proteins. (A) Venn diagram showing number of genes with TES-TI (mutant/Col-0) ≥ 2 that overlap sites enriched by at least 1.5 fold over the corresponding background signal for Pol IV, Pol V or AGO4 proteins (Pol IV [44], Pol V [17], and AGO4 [45] ChIP-seq datasets) within 2 kb regions centered at their TES. (B) Pol V ChIP-seq read density for Col-0 and pol V mutant plants (Wierzbicki et al., 2012) (SRX156079 and SRX156080)) were plotted relative to TESs for the 1312 genes affected by both Pol IV and Pol V. Shadings represent 95% confidence intervals.
Only 113 genes (8.6% of 1312 genes with increased post-TES read density in pol IV or pol V mutant plants) show enrichment of AGO4 near their TESs and only 54 (4%) of these genes also exhibit increased Pol V occupancy (Fig. 5A). Thus, only a small percent of analyzed genes could potentially have a transcriptionally active Pol V that could recruit AGO4 protein to target loci either via AGO hook motifs (present in Pol V C-terminal domain) or by base-paring between siRNA of the AGO4-siRNA complex with Pol V transcripts [16,46] Alternatively (or in addition), physical interaction between Pol II and AGO4 could play a role in recruiting AGO4 to these loci [47].
Fewer than 1% (12 out of 1312) of the analyzed genes were occupied by all three proteins, suggesting that loci with increased post-TES NRO signal are not likely subjects of RNA-directed DNA methylation. To further confirm this statement we examine whether 23–24 nt siRNAs map near TESs of genes affected by the loss of Pol IV or Pol V. Only 4.2% of affected genes overlap with 23–24 nt and 3.5% with 21–22 nt siRNA reads within a 1 kb region surrounding the TESs (Fig. S5A). To exclude these outliers, we sorted genes with TES-TI based on the sRNA read density within 1 kb region surrounding their TESs and removed 130 genes (top 10%). The remaining 1,182 genes were used in sRNA and DNA methylation metagene profiles (Figs. S6 & S7). 21 nt and 24 nt sRNA read distribution, as well as cytosine methylation (in CG, CHG, and CHH sequence contexts (H indicates non-G nucleotides)), near the TESs for genes with increased read density post-TES were comparable to profiles obtained for random set of genes (Figs. S6 & S7). This analysis detected some reduction in CHH methylation just downstream of the TES in pol V mutant plants suggesting that Pol V might function at least on some of these loci (Fig. S7G). Additional analysis of the 500 bp region immediately downstream of TES revealed that ratios of DNA methylated sites (Col-0/mt) for genes with TES-TI were similar (and with smaller median values) as compared to two random sets of genes (Fig. S7C, F, & I). Consistently, no significant overlap was observed between 1312 genes with increased read density post-TES and CHH or CHG hypo-methylated regions reported for pol IV [48], and pol V [49], mutants (Fig. S5C, D), as well as no correlation between post-TES read density and DNA methylation was detected in rdd (ros1–3 dml2–1 dml3–1) triple mutant [50], that lacks nearly all DNA demethylation activity (Figs. S5B & S8). This suggests that for the majority of genes the effect of Pol IV and Pol V on Pol II transcription termination is unlikely to be due to RNA-directed DNA methylation. Consistently, we found that TEs are not more prevalent among 1312 genes with increased post-TES reads as compared to random set of genes of the same size, suggesting that increased read density post-TES does not result from de-repression of transposable elements (TEs) that could be present within these regions (Fig. S9A).
Consistent with previous report [46] lack of CHH methylation and siRNA accumulation at these sites suggests that Pol V is not transcriptionally engaged. Indeed, analysis of Pol V RIP-seq data [46] revealed that only 3.5% of genes with increased Pol II occupancy post TES in pol IV or V mutants overlap with Pol V-associated transcripts (Fig. S9B).
Pol II accumulation near the TES region is increased for convergent gene pairs
Interestingly, convergent gene pairs were significantly enriched among genes with increased post-TES read density in pol IV or pol V mutant plants (Fig. 6A). Furthermore, these genes tend to have neighboring genes that initiate or end within 100 bps downstream or 100 bps upstream of their TESs (Fig. 6B).
Figure 6.
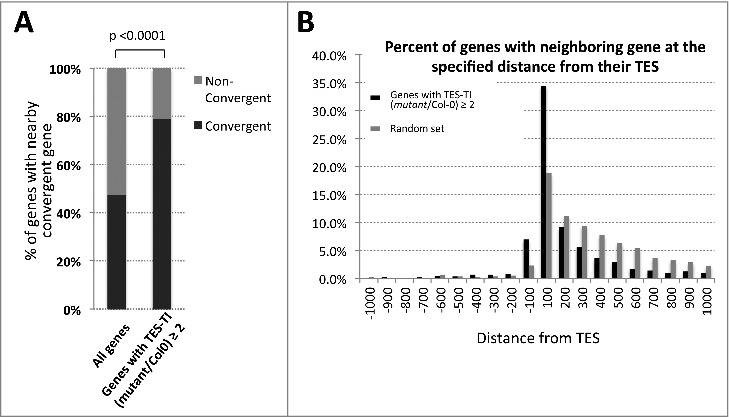
post-TES read accumulation is more prevalent among convergent gene pairs. (A) Histogram showing the percent of convergent genes among all protein-coding genes in Arabidopsis thaliana versus genes displaying increased post-TES read density in pol IV or pol V mutant plants. P-value was calculated using Fisher's exact test. (B) Histogram showing the percent of genes with increased post-TES read density in pol IV or pol V mutant plants that have the nearest neighbor gene initiating or ending at various distances (x-axes) from their transcript end sites (TES).
Regulation of transcription termination of convergent gene pairs might be an important mechanism to modulate levels of natural anti-sense transcripts (NATs) in cells as the base-pairing between sense and antisense transcripts can form dsRNAs, which can be then processed into cis-NAT-derived siRNAs (nat-siRNAs) via unique Pol IV-dependent pathway and lead to down-regulation of transcript levels though mRNA cleavage [51,52]. Analysis of antisense and sense reads revealed that both were increased within 1 kb region surrounding TESs for genes with increased Pol II occupancy post-TES in pol IV or V mutant plants (Fig. S10A). This increase of sense and antisense reads near TESs likely reflects Pol II activity as it coincides with increase in Pol II occupancy within this region (Fig. 4). However, we did not detect the expected increase in numbers of neither 21–22 nt nor 23–24 nt sRNA clusters (Fig. S10B) nor did we detect statistically significant changes in transcript abundance for either gene in the convergent gene pairs (Fig. S10C), although it is possible that a specific abiotic or biotic stress condition might be required. Indeed, gene ontology analysis revealed that genes with increased post-TES read density are enriched for transcripts involved in response to abiotic stress (P = 8.0 × 10−6) (Fig S11).
Discussion
Nuclear run-on combined with high-throughput sequencing (NRO-Seq)[36] and RNA-Seq [53] revealed that Pol IV and Pol V affect Pol II occupancy downstream of polyA addition sites (TESs) for approximately 12% of protein-coding genes. Consistently, Pol IV or Pol V interaction sites include the TES regions for genes that display increased NRO signals downstream of TESs in pol IV or pol V mutant plants.
A similar study in Zea mays (maize) examined nascent RNA transcripts in wild-type and pol IV mutant plants and found a similar effect of Pol IV on RNA Pol II transcription termination [37]. RDR2-dependent 24-nt siRNAs were enriched at maize 3′ gene ends immediately downstream of TESs, suggesting a presence of transcriptionally active Pol IV near TESs of Pol II-transcribed genes in maize. In that study the authors speculated that Pol IV might play a role in the proper termination of Pol II transcription and in the prevention of inappropriate read-through transcription into downstream neighboring genes or TE. We found that in Arabidopsis 24-nt siRNAs and CHH methylation were enriched on a small subset of genes that exhibit longer termination zone in pol IV or pol V mutant plants. However, for the majority of these genes there was no enrichment for 24-nt siRNAs and/or changes in CHH methylation, a hallmark of Pol IV and Pol V – dependent RdDM. Thus, for the majority of genes any influence of Pol IV or Pol V on Pol II transcription termination is likely independent of RNA-directed DNA methylation.
Pol II elongation rates can affect co-transcriptional processes such as transcription termination resulting in many RNA polymerase molecules continue transcribing post-TES [54]. Interestingly, genes with high post-TES NRO signals in pol IV or pol V mutant plants exhibit not only higher Pol II occupancy near TES (Fig. 4), but also significantly higher rates of transcription, compared to all protein-coding genes (Fig S12 & Fig. 2E). Elongation rate could be influenced by various factors that can modulate Pol II transcription efficiency directly (e.g. elongation factors) or indirectly, by changing chromatin organization (chromatin remodelers). Pol V transcription can recruit the SWI/SNF chromatin remodeling complex, such that a loss of Pol V transcripts correlates with changes in nucleosome positioning [55]. Whether or not Pol IV can similarly affect chromatin states has not yet been determined. However, CLASSY1, a putative ATP-dependent DNA translocase, has been implicated in Pol IV function and speculated to affect nucleosome repositioning, similar to SWI/SNF [56,57]. Thus, Pol IV or Pol V – mediated chromatin organization near Pol II TESs may influence Pol II elongation rates and its subsequent termination or release from the template (Fig. 7). Interestingly, a recent study by Rowley et al., (2017) [58] discovered a role of Pol V and AGO4 in long range chromosomal interactions. Thus, it is possible that effect of Pol IV or Pol V on Pol II elongation rate and/or termination efficiency is influenced by their role in 3D chromatin organization.
Figure 7.
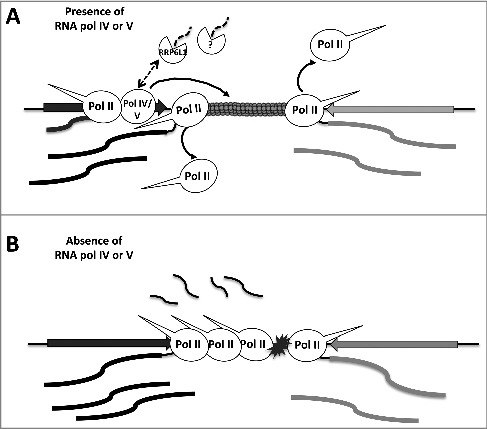
Model for RNA Pol IV or Pol V involvement in RNA Pol II transcription termination. Pol IV or Pol V may impede Pol II to promote transcript cleavage and release at Pol II convergent genes. In the absence of Pol IV or Pol V, Pol II undergoes head-to-head collision and pausing.
Interestingly, convergent gene pairs tend to show the highest post-TES NRO signals in pol IV or pol V mutant plants. Convergent transcription can result in head-to-head collisions of transcribing Pol II enzymes [59]. Though such events are expected to be rare, Pol II collision doesn't result in polymerase dissociation from the template but leads to transcriptional arrest and gene blockage [59]. To date, not much is known about how cells deal with collision-induced transcription blocks. However, Pol II ubiquitination-mediated degradation has been shown to be involved in clearing Pol II from the region between convergent genes, such that lack of the yeast RNA Pol II ubiquitination/degradation protein ELC1 results in a peak of RNA Pol II density between convergent genes [59]. A possibility is that, similar to transcription factor for ribosomal protein coding genes, Reb1,[60] Pol IV or Pol V occupancy at intergenic DNA may reduce RNA polymerase II collisions by slowing or dissociating Pol II, thus allowing more efficient transcription termination. Analysis of non-convergent gene models with increased read density post-TES revealed that Pol V association is predominantly upstream of TESs (Fig. S13). Consistently, we observed lower Pol II occupancy just upstream of TES (Fig. 2C) in pol IV or pol V mutant plants.
Alternatively, or in addition, Pol IV or Pol V can recruit factors that ensure proper processing of 3′ ends of RNA or degradation of the read-through post-TES fragments of the original transcripts (Fig. 7). Indeed, Pol V has been shown to interact with putative 3′ to 5′ exoribonuclease, RRP6L1 [57]. In yeast, deletion of RRP6 results in improperly processed 3′ RNA ends and faulty transcript termination at specific target genes [60,61].
Pausing or slowing down of Pol II near the TESs during transcription termination is thought to facilitate co-transcriptional splicing [54,62,63]. Interestingly, splicing was much less efficient in pol IV or pol V mutant plants, compared to wild-type (Fig. S14), suggesting that Pol IV or Pol V affect pre-mRNA processing at the level of transcription. Alternatively, or in addition, Pol IV or Pol V might facilitate recruitment of splicing factors [64]. For example, a U4/U6 snRNP-associated protein, RDM16, is enriched at Pol V target loci, suggesting that RDM16 might be recruited by Pol V and/or by the nascent Pol V transcripts [65].
Materials and methods
Plant material
A. thaliana Col-0 and nrpd1–3 and nrpe1–11 have been described previously [2,15]. Plants were cultivated at 22°C under long-day conditions (16 h day/8 h night).
Nuclei isolation
Nuclei were isolated from 3 week old Col-0, nrpd1–3 and nrpe1–11 plant seedlings as described previously [66] with a few modifications. Briefly, 3 week-old plants were harvested and washed in ice-cold water. Then plants were submerged in cold diethyl ether for 5 min and washed twice in ice-cold water. Homogenization of plant material was performed in 100 mL of nuclei isolation buffer (1M sucrose, 50 mM Tris-HCl [pH 7.2], 5 mM MgCl, 5 mM KCl, 10 mM 2-mercaptoethnol, 0.2 mM PMSF) with a motorized homogenizer (Fisher Scientific Powergen 700). Homogenate was filtered through 1 layer of Miracloth and 1 layer of 50 um of nylon mesh. After centrifugation at 14,000 g for 15 min at 4°C the pellet was resuspended in 10 min of nuclei isolation buffer and prepared for loading into discontinuous Percoll gradient (15%, 30%, 45%, and 60%) by adding one volume of “100% Percoll solution” (34.23 g sucrose, 5 mL 1 M Tris-HCl [pH 7.2], 0.5 mL 1 M MgCl2, 0.5 mL 1 M KCl, 34 uL 2-mercaptoethnol, 100 uL 2 mM PMSF and Percoll to 100 mL) to 19 volumes of nuclei. Gradients were centrifuged at 500 g for 10 min, then 7,500 rpm for 20 min. Nuclei were harvested from 30%/45% and 45%/60% boundaries and pooled. Nuclei were diluted with 5 volumes of nuclei isolation buffer, mixed by inversion and collected by centrifugation at 1,500 g, 10 min, 4°C. Second wash of nuclei was performed with 25 volumes of nuclei isolation buffer. Final resuspension of nuclei was in 1 mL of nuclei storage buffer (50 mM HEPES [pH 7.2], 5 mM MgCl2, 5 mM KCl, 2 mM DTT, 0.2 mM PMSF, 50% glycerol). Aliquots were frozen in liquid nitrogen and stored at −80°C.
Nuclear run-on (NRO)
NRO reactions were performed as described previously [36]with a few modifications. Briefly, 10 in vitro transcription reactions (each containing 190 uL of nuclei, 60 uL 5x Txn buffer (100 mM Tris-HCl, [pH 7.7], 6 uL 0.1 M DTT, 5 uL RiboLock, 12 uL Biotin-16-UTP (10 mM), 16 uL AGC (10 mM each)) were set up in parallel for each plant line examined and incubated at 30°C for 60 min. Total RNA was extracted using Trizol method, treated with Turbo DNase I. An aliquot of total RNA was used for total RNA library contruction (see Materials and Methods). Biotinylated RNA was precipitated from total RNA with Dynabeads MyOne Streptavidin C1 (Invitrogen, Calsbad, CA). Binding was performed in 5 mM Tris-HCl [pH 7.7], 0.5 mM EDTA and 1 M NaCl for 20 min at 42°C and 2 hours at room temperature with rotation. After washes, RNA was eluted from beads by adding 50 uL of 10 uM EDTA/95% formamide and incubation at 65°C for 5 min.
RNA library construction
Libraries from total and eluted biotinylated RNA were prepared using the Illumina TruSeq Stranded mRNA Sample Prep Kit. No rRNA depletion steps were performed. Libraries were subjected to 50 bp paired-end sequencing.
exoChIP library construction
Chromatin cross-linking, isolation and shearing were carried out as previously described with slight modifications [15]. Briefly, three grams of three-week old leaf tissue was suspended in 35 ml SH Buffer (100 mM sucrose, 20 mM HEPES-NaOH) containing 1% formaldehyde and placed under vacuum for 55 minutes. Cross-linking was stopped by the addition of 1.25 ml 2 M Glycine. Leaf tissue was washed three times with water. Tissue was ground in mortar with pestle and suspended in 15 ml Honda buffer (0.44 M Sucrose, 1.25% Ficoll, 2.5% Dextran T40, 20 mM HEPES-NaOH pH7.4, 10 mM MgCl2, 0.5% Triton X-100, 5 mM DTT, 1 mM PMSF, 1% Plant Protease Inhibitor Cocktail). Slurry was passed through Miracloth and centrifuged at 2,000 x g for 15 minutes at 4 C. The pellet was resuspended in 1 ml Honda buffer and centrifuged as above. Again, the pellet was resuspended in Honda buffer, centrifuged as above and the pellet was resuspended in 575 microliters of Nuclei Lysis Buffer (50 mM Tris-HCl pH8, 10 mM EDTA, 1% SDS, 1 mM PMSF, 1% Plant Protease Inhibitor Cocktail). Chromatin was sheared using a Bioruptor sonicator (Diagenode) to generate fragment sizes between 200 and 600 bp. Sheared chromatin was centrifuged for 10 minutes at 4 C at 12,000 x g. The supernatant was removed and diluted ten fold with ChIP Dilution Buffer (1.1% Triton X-100, 1.2 mM EDTA, 16.7 mM NaCl, 16.7 mM Tris-HCl pH8). Aliquots containing approximately 6 mg of chromatin were shipped to Peconic, LLC for ChIP-exo sequencing. ChIP-exo was carried out as described in [67]. Five microliters of the 4H8 monoclonal antibody (Cell Signaling Technology), which recognizes the Pol II CTD with a preference towards the phosphoS5 form, was used for immunoprecipitation.
Circular RT-PCR
Total RNA was extracted using Trizol reagent. 10 ug of RNA was decapped by treatment with Tobacco Acid Pyrophosphatase (TAP) (Epicentre) to provide 5′-monophosphorylated terminus that was ligated to a 3′-hydrohylated terminus by T4 RNA ligase 1 (NEB, 10,000 U/uL) according to manufacture instructions. After incubation at 37°C for 1 hour, enzymes were heat inactivated at 65°C for 15 min and reactions were cleaned up and concentrated with Zymogen RNA Clean and Concentrator kit. The resulted RNA was treated with Turbo DNase I, reverse-transcribed with gene-specific reverse primer. Two rounds of PCR amplification was followed by PCR clean up (Qiagen PCR clean up kit) and DNA cloning into pGEMT easy vector according to manufacturer's instructions. Individual colonies were grown and used for DNA extraction and sequencing.
Primers
| AT5G36700_F1 | ccaaaagaagcggaaacaga |
| AT5G36700_R | ccgccacagaaagaagaaga |
| AT5G36700_F2 | atttgcagtgcctttgtgtg |
| AT5G24670_F1 | tcatcattgttgtttccttctca |
| AT5G24670_R | atgagcggagcagctactgt |
| AT5G24670_F2 | gcgcttgtgcatcaaagaat |
Sequence processing
Reads were adapter trimmed and quality filtered (q20) using trimmomatic and then mapped to TAIR10 genome reference using tophat. All transposable elements, tRNAs, and ribosomal RNAs, as well as genes in the mitochondria and chloroplast genomes were excluded from analysis. The reads were further filtered to remove low complexity reads (with the entropy of 2 or less) using custom Perl scripts. Only read alignments mapped concordantly (e.g. with correct orientation and separated by less than 7,000 bps from each other) were selected for subsequent analysis. The 7,000 bp distance cut off was based on the 99.9% intron length in the TAIR10 annotation. Read data was converted into coverage information in the form of strand specific bed files. Cumulative coverage data was determined in a strand-specific way from the 5′ and 3′ ends of genes as defined in the TAIR10 gene file. The read counts between samples were normalized based on the total number of reads mapped for each sample. AT2G16586 was excluded from analysis due to it having an extremely high and variable coverage between samples.
Read-through analysis
TES-termination index (TES-TI) was determined in the NRO and total RNA libraries by calculating ratios of read density 500 bp downstream of the TES to read density in the body of the gene 500 bp upstream of TES. For each gene, TES-TI was calculated in Col-0, nrpd1–3 and nrpe1–11 backgrounds.
ChIP data processing
Among the annotated TAIR10 nuclear protein coding genes, pairs of consecutive genes with their 3′ ends facing each other and separated by at least 1 kb were identified as convergent genes. Among these, any gene pairs overlapping with miRNA or tRNA or snRNA or snoRNA or rRNA or other non-coding RNA were excluded, resulting in 1,453 gene pairs (2,906 genes) that were used in the analysis. For each of these 2,906 genes, coverage information at each base position starting from 1000 nt upstream of the 3′ end into the gene body to 1000 nt downstream of the 3′ end was obtained and total coverage contributed by all 2,906 genes at base position was computed and plotted.
Bisulfite sequencing analyses
Bisulfite sequencing reads were quality processed (q >/ = 25) and adapter trimmed using Cutadapt version 1.9.1 [68]. Cleaned reads were mapped to the Arabidopsis thaliana TAIR10 genome using Bismark version 0.16.1 default settings [69]. PCR duplicates were removed and methylation information for cytosines in the CHH context with a minimum 5 read coverage were used for further analyses. Differentially methylated regions (DMRs) that showed a minimum 10% increase in methylation levels in rdd relative to Col-0 were identified using the R package methylKit version 0.9.5 [70]. Regions were defined using a 300 base pair sliding windows with a step size of 200 base pairs. The minimum coverage requirement for each window was 10 cytosines with at least 5 read coverage each and significance was defined as a q value less than 0.01. Overlapping DMRs were merged into a single region and genomic coordinates of rdd hyper-methylated regions (relative to Col-0) were used to assess overlap with regions flanking the 3′ end of genes of interest 500 basepairs upstream and 500 basepairs downstream. Accession numbers for Col-0 and rdd bisulfite sequencing data are GSM276809 and GSM276812, respectively [71].
sRNA sequencing analyses
Raw reads were adapter and quality trimmed and size selected using Cutadapt version 1.9.1. Reads were further filtered of all structural RNAs (tRNAs, rRNAs, snRNAs, and snoRNAs) and mapped to the Arabidopsis TAIR10 genome using ShortStack version 3.4 default settings [72]. sRNA counts for regions of interest were extracted from bam files using the ShortStack –locifile file function and 21–24nt siRNA clusters were defined using a minimum read coverage of 25 reads. The accession number for Col-0, nrpd1–3, and nrpe1–11 sRNA sequencing data are SRR2075819, SRR2505369, and GSM2451982, respectively [6].
Data availability
Raw Illumina reads are deposited to the NCBI Gene Expression Omnibus (GSE101543) and are available through http://www.ncbi.nlm.nih.gov/geo/.
Supplementary Material
Funding Statement
This work was supported by funds to Prof. Craig Pikaard as an Investigator of the Howard Hughes Medical Institute, the Gordon and Betty Moore Foundation, and from grant GM077590 from the National Institutes of Health. J.M.W. was supported by NIH training Grant, T32GM007757, and predoctoral fellowship Award F31GM116346. The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of our sponsors.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
We are grateful to Prof. Craig Pikaard for financial support of this work, critical discussions, and constructive manuscript editing. We thank Dr. Feng Wang for guidance with DNA methylation data analysis and Prof. James McKinlay for helpful discussions. We thank present and former Pikaard lab members for valuable discussions and Indiana University Center for Genomics and Bioinformatics for help with library preparation and sequencing.
Author contributions
A.M. designed and performed the research; R.C. performed ChIP-exo experiments; D.B.R., R.P., J.M.W., and A.M. analyzed the data; A.M. wrote the paper; A.M., D.B.R. revised manuscript and figures.
References
- [1].Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16:71–84. doi: 10.1038/nrg3863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Onodera Y, Haag JR, Ream T, et al.. Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell. 2005;120:613–22. doi: 10.1016/j.cell.2005.02.007 [DOI] [PubMed] [Google Scholar]
- [3].Herr AJ, Jensen MB, Dalmay T, et al.. RNA polymerase IV directs silencing of endogenous DNA. Science. 2005;308:118–20. doi: 10.1126/science.1106910 [DOI] [PubMed] [Google Scholar]
- [4].Zhai J, Bischof S, Wang H, et al.. A One Precursor One siRNA Model for Pol IV-Dependent siRNA Biogenesis. Cell. 2015;163:445–55. doi: 10.1016/j.cell.2015.09.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Li S, Vandivier LE, Tu B, et al.. Detection of Pol IV/RDR2-dependent transcripts at the genomic scale in Arabidopsis reveals features and regulation of siRNA biogenesis. Genome Res. 2015;25:235–45. doi: 10.1101/gr.182238.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Blevins T, Podicheti R, Mishra V, et al.. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in Arabidopsis. Elife. 2015;4:e09591. doi: 10.7554/eLife.09591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Haag JR, Ream TS, Marasco M, et al.. In vitro transcription activities of Pol IV, Pol V, and RDR2 reveal coupling of Pol IV and RDR2 for dsRNA synthesis in plant RNA silencing. Mol Cell. 2012;48:811–8. doi: 10.1016/j.molcel.2012.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Pontier D, Yahubyan G, Vega D, et al.. Reinforcement of silencing at transposons and highly repeated sequences requires the concerted action of two distinct RNA polymerases IV in Arabidopsis. Genes Dev. 2005;19:2030–40. doi: 10.1101/gad.348405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Lahmy S, Pontier D, Cavel E, et al.. PolV(PolIVb) function in RNA-directed DNA methylation requires the conserved active site and an additional plant-specific subunit. Proc Natl Acad Sci U S A. 2009;106:941–6. doi: 10.1073/pnas.0810310106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zilberman D, Cao X, Jacobsen SE. ARGONAUTE4 control of locus-specific siRNA accumulation and DNA and histone methylation. Science. 2003;299:716–9. doi: 10.1126/science.1079695 [DOI] [PubMed] [Google Scholar]
- [11].Zheng X, Zhu J, Kapoor A, et al.. Role of Arabidopsis AGO6 in siRNA accumulation, DNA methylation and transcriptional gene silencing. EMBO J. 2007;26:1691–701. doi: 10.1038/sj.emboj.7601603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].McCue AD, Panda K, Nuthikattu S, et al.. ARGONAUTE 6 bridges transposable element mRNA-derived siRNAs to the establishment of DNA methylation. EMBO J. 2015;34:20–35. doi: 10.15252/embj.201489499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Duan C-G, Zhang H, Tang K, et al.. Specific but interdependent functions for Arabidopsis AGO4 and AGO6 in RNA-directed DNA methylation. EMBO J. 2015;34:581–92. doi: 10.15252/embj.201489453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wierzbicki AT, Haag JR, Pikaard CS. Noncoding transcription by RNA polymerase Pol IVb/Pol V mediates transcriptional silencing of overlapping and adjacent genes. Cell. 2008;135:635–48. doi: 10.1016/j.cell.2008.09.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wierzbicki AT, Ream TS, Haag JR, et al.. RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat Genet. 2009;41:630–4. doi: 10.1038/ng.365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lahmy S, Pontier D, Bies-Etheve N, et al.. Evidence for ARGONAUTE4-DNA interactions in RNA-directed DNA methylation in plants. Genes Dev. 2016;30:2565–70. doi: 10.1101/gad.289553.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wierzbicki AT, Cocklin R, Mayampurath A, et al.. Spatial and functional relationships among Pol V-associated loci, Pol IV-dependent siRNAs, and cytosine methylation in the Arabidopsis epigenome. Genes Dev. 2012;26:1825–36. doi: 10.1101/gad.197772.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Pikaard CS, Haag JR, Pontes OMF, et al.. A transcription fork model for Pol IV and Pol V-dependent RNA-directed DNA methylation. Cold Spring Harb Symp Quant Biol. 2012;77:205–12. doi: 10.1101/sqb.2013.77.014803 [DOI] [PubMed] [Google Scholar]
- [19].Bohmdorfer G, Rowley MJ, Kucinski J, et al.. RNA-directed DNA methylation requires stepwise binding of silencing factors to long non-coding RNA. Plant J. 2014;79:181–91. doi: 10.1111/tpj.12563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pontes O, Costa-Nunes P, Vithayathil P, et al.. RNA polymerase V functions in Arabidopsis interphase heterochromatin organization independently of the 24-nt siRNA-directed DNA methylation pathway. Mol Plant. 2009;2:700–10. doi: 10.1093/mp/ssp006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Fultz D, Choudury SG, Slotkin RK. Silencing of active transposable elements in plants. Curr Opin Plant Biol. 2015;27:67–76. doi: 10.1016/j.pbi.2015.05.027 [DOI] [PubMed] [Google Scholar]
- [22].Nuthikattu S, McCue AD, Panda K, et al.. The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21–22 nucleotide small interfering RNAs. Plant Physiol. 2013;162:116–31. doi: 10.1104/pp.113.216481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Panda K, Slotkin RK. Proposed mechanism for the initiation of transposable element silencing by the RDR6-directed DNA methylation pathway. Plant Signal Behav. 2013;8:e25206. doi: 10.4161/psb.25206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wu L, Mao L, Qi Y. Roles of dicer-like and argonaute proteins in TAS-derived small interfering RNA-triggered DNA methylation. Plant Physiol. 2012;160:990–9. doi: 10.1104/pp.112.200279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kasschau KD, Fahlgren N, Chapman EJ, et al.. Genome-wide profiling and analysis of Arabidopsis siRNAs. PLoS Biol. 2007;5:e57. doi: 10.1371/journal.pbio.0050057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Stroud H, Do T, Du J, et al.. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat Struct Mol Biol. 2014;21:64–72. doi: 10.1038/nsmb.2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hollister JD, Smith LM, Guo Y-L, et al.. Transposable elements and small RNAs contribute to gene expression divergence between Arabidopsis thaliana and Arabidopsis lyrata. Proc Natl Acad Sci U S A. 2011;108:2322–7. doi: 10.1073/pnas.1018222108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Greaves IK, Groszmann M, Ying H, et al.. Trans chromosomal methylation in Arabidopsis hybrids. Proc Natl Acad Sci U S A. 2012;109:3570–5. doi: 10.1073/pnas.1201043109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Proudfoot NJ, Furger A, Dye MJ. Integrating mRNA processing with transcription. Cell. 2002;108:501–12. doi: 10.1016/S0092-8674(02)00617-7 [DOI] [PubMed] [Google Scholar]
- [30].Connelly S, Manley JL. A functional mRNA polyadenylation signal is required for transcription termination by RNA polymerase II. Genes Dev 1988;2:440–52. doi: 10.1101/gad.2.4.440 [DOI] [PubMed] [Google Scholar]
- [31].Buratowski S. Connections between mRNA 3′ end processing and transcription termination. Curr Opin Cell Biol. 2005;17:257–61. doi: 10.1016/j.ceb.2005.04.003 [DOI] [PubMed] [Google Scholar]
- [32].Logan J, Falck-Pedersen E, Darnell JEJ, et al.. A poly(A) addition site and a downstream termination region are required for efficient cessation of transcription by RNA polymerase II in the mouse beta maj-globin gene. Proc Natl Acad Sci U S A 1987;84:8306–10. doi: 10.1073/pnas.84.23.8306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Proudfoot NJ. Ending the message: poly(A) signals then and now. Genes Dev. 2011;25:1770–82. doi: 10.1101/gad.17268411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Richard P, Manley JL. Transcription termination by nuclear RNA polymerases. Genes Dev. 2009;23:1247–69. doi: 10.1101/gad.1792809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Rosonina E, Kaneko S, Manley JL. Terminating the transcript: breaking up is hard to do. Genes Dev. 2006;20:1050–6. doi: 10.1101/gad.1431606 [DOI] [PubMed] [Google Scholar]
- [36].McKinlay A, Araya CL, Fields S. Genome-Wide Analysis of Nascent Transcription in Saccharomyces cerevisiae. G3 (Bethesda). 2011;1:549–58. doi: 10.1534/g3.111.000810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Erhard KFJ, Talbot J-ERB, Deans NC, et al.. Nascent transcription affected by RNA polymerase IV in Zea mays. Genetics. 2015;199:1107–25. doi: 10.1534/genetics.115.174714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–8. doi: 10.1126/science.1162228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Churchman LS, Weissman JS Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469(7330):368–73. doi: 10.1038/nature09652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Nechaev S, Fargo DC, dos Santos G, et al.. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science. 2010;327:335–8. doi: 10.1126/science.1181421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lis JT. Imaging Drosophila gene activation and polymerase pausing in vivo. Nature. 2007;450:198–202. doi: 10.1038/nature06324 [DOI] [PubMed] [Google Scholar]
- [42].Core LJ, Lis JT. Transcription regulation through promoter-proximal pausing of RNA polymerase II. Science. 2008;319:1791–2. doi: 10.1126/science.1150843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rhee HS, Pugh BF. Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell. 2011;147:1408–19. doi: 10.1016/j.cell.2011.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Law JA, Du J, Hale CJ, et al.. Polymerase IV occupancy at RNA-directed DNA methylation sites requires SHH1. Nature. 2013;498:385–9. doi: 10.1038/nature12178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zheng Q, Rowley MJ, Bohmdorfer G, et al.. RNA polymerase V targets transcriptional silencing components to promoters of protein-coding genes. Plant J. 2013;73:179–89. doi: 10.1111/tpj.12034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Bohmdorfer G, Sethuraman S, Rowley MJ, et al.. Long non-coding RNA produced by RNA polymerase V determines boundaries of heterochromatin. Elife. 2016;5:e1909. doi: 10.7554/eLife.19092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Zheng B, Wang Z, Li S, et al.. Intergenic transcription by RNA polymerase II coordinates Pol IV and Pol V in siRNA-directed transcriptional gene silencing in Arabidopsis. Genes Dev. 2009;23:2850–60. doi: 10.1101/gad.1868009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yang D-L, Zhang G, Tang K, et al.. Dicer-independent RNA-directed DNA methylation in Arabidopsis. Cell Res. 2016;26:66–82. doi: 10.1038/cr.2015.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wendte JM, Haag JR, Singh J, et al.. Functional dissection of the Pol V largest Subunit CTD in RNA-Directed DNA methylation. Cell Rep. 2017;19:2796–808. doi: 10.1016/j.celrep.2017.05.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Penterman J, Zilberman D, Huh JH, et al.. DNA demethylation in the Arabidopsis genome. Proc Natl Acad Sci U S A. 2007;104:6752–7. doi: 10.1073/pnas.0701861104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Borsani O, Zhu J, Verslues PE, et al.. Endogenous siRNAs derived from a pair of natural cis-antisense transcripts regulate salt tolerance in Arabidopsis. Cell. 2005;123:1279–91. doi: 10.1016/j.cell.2005.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Zhang X, Xia J, Lii YE, et al.. Genome-wide analysis of plant nat-siRNAs reveals insights into their distribution, biogenesis and function. Genome Biol. 2012;13:R20. doi: 10.1186/gb-2012-13-3-r20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Nagalakshmi U, Waern K, Snyder M. RNA-Seq: a method for comprehensive transcriptome analysis. Curr Protoc Mol Biol. 2010;Chapter 4:Unit 4.11.1–13. doi: 10.1002/0471142727.mb0411s89. [DOI] [PubMed] [Google Scholar]
- [54].Jonkers I, Lis JT. Getting up to speed with transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. 2015;16(3):167–77. doi: 10.1038/nrm3953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhu Y, Rowley MJ, Bohmdorfer G, et al.. A SWI/SNF chromatin-remodeling complex acts in noncoding RNA-mediated transcriptional silencing. Mol Cell. 2013;49:298–309. doi: 10.1016/j.molcel.2012.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Smith LM, Pontes O, Searle I, et al.. An SNF2 protein associated with nuclear RNA silencing and the spread of a silencing signal between cells in Arabidopsis. Plant Cell. 2007;19:1507–21. doi: 10.1105/tpc.107.051540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem. 2009;78:273–304. doi: 10.1146/annurev.biochem.77.062706.153223 [DOI] [PubMed] [Google Scholar]
- [58].Rowley MJ, Rothi MH, Bohmdorfer G, et al.. Long-range control of gene expression via RNA-directed DNA methylation. PLoS Genet. 2017;13:e1006749. doi: 10.1371/journal.pgen.1006749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Hobson DJ, Wei W, Steinmetz LM, et al.. RNA polymerase II collision interrupts convergent transcription. Mol Cell. 2012;48:365–74. doi: 10.1016/j.molcel.2012.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Colin J, Candelli T, Porrua O, et al.. Roadblock termination by reb1p restricts cryptic and readthrough transcription. Mol Cell. 2014;56:667–80. doi: 10.1016/j.molcel.2014.10.026 [DOI] [PubMed] [Google Scholar]
- [61].Fox MJ, Gao H, Smith-Kinnaman WR, et al.. The exosome component Rrp6 is required for RNA polymerase II termination at specific targets of the Nrd1-Nab3 pathway. PLoS Genet. 2015;11:e1004999. doi: 10.1371/journal.pgen.1004999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Carrillo Oesterreich F, Preibisch S, Neugebauer KM. Global analysis of nascent RNA reveals transcriptional pausing in terminal Exons. Mol Cell [Internet]. 2010;40:571–81. Available from: http://www.sciencedirect.com/science/article/pii/S1097276510008427 doi: 10.1016/j.molcel.2010.11.004 [DOI] [PubMed] [Google Scholar]
- [63].Ip JY, Schmidt D, Pan Q, et al.. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res. 2011;21:390–401. doi: 10.1101/gr.111070.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Huang C-F, Zhu J-K. RNA Splicing Factors and RNA-Directed DNA Methylation. Biology (Basel). 2014;3:243–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Huang C-F, Miki D, Tang K, et al.. A Pre-mRNA-splicing factor is required for RNA-directed DNA methylation in Arabidopsis. PLoS Genet. 2013;9:e1003779. doi: 10.1371/journal.pgen.1003779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Gaudino RJ, Pikaard CS. Cytokinin induction of RNA polymerase I transcription in Arabidopsis thaliana. J Biol Chem 1997;272:6799–804. doi: 10.1074/jbc.272.10.6799 [DOI] [PubMed] [Google Scholar]
- [67].Rhee HS, Pugh BF. Genome-wide structure and organization of eukaryotic pre-initiation complexes. Nature. 2012;483:295–301. doi: 10.1038/nature10799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Martin Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnetjournal. 2011;17:10–2. [Google Scholar]
- [69].Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27:1571–2. doi: 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Akalin A, Kormaksson M, Li S, , et al. methylKit: a comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 2012;13:R87. doi: 10.1186/gb-2012-13-10-r87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Lister R, O'Malley RC, Tonti-Filippini J, et al.. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–36. doi: 10.1016/j.cell.2008.03.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Johnson NR, Yeoh JM, Coruh C, et al.. Improved placement of multi-mapping Small RNAs. G3 (Bethesda). 2016;6:2103–11. doi: 10.1534/g3.116.030452 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw Illumina reads are deposited to the NCBI Gene Expression Omnibus (GSE101543) and are available through http://www.ncbi.nlm.nih.gov/geo/.