

ABSTRACT
The expression patterns of endogenous circular RNA (circRNA) molecules during epidermal stem cell (EpSC) differentiation have not previously been explored. Here, we show that circRNAs are abundantly expressed in EpSCs and that their expression change dramatically during differentiation in a coordinated manner. Overall, circRNAs are expressed at higher levels in the differentiated cells, and many upregulated circRNAs are derived from developmental genes, including four different circRNAs from DLG1. The observed changes in circRNA expression were largely independent of host gene expression, and circRNAs independently upregulated upon differentiation are more prone to AGO2 binding and have more predicted miRNA binding sites compared to stably expressed circRNAs. In particular, upregulated circRNAs from the HECTD1 and ZNF91 genes have exceptionally high numbers of AGO2 binding sites and predicted miRNA target sites, and circZNF91 contains 24 target sites for miR-23b-3p, which is known to play important roles in keratinocyte differentiation. We also observed that upregulated circRNAs are less likely to be flanked by homologues inverted Alu repeats compared to stably expressed circRNAs. This coincide with DHX9 being upregulated in the differentiated keratinocytes. Finally, none of the circRNAs upregulated upon differentiation were also upregulated upon DNMT3A or DNMT3B knockdown, making it unlikely that epigenetic mechanisms are governing the observed circRNA expression changes. Together, we provide a map of circRNA expression in EpSCs and their differentiated counterparts and shed light on potential function and regulation of differentially expressed circRNAs.
KEYWORDS: AGO2, circular RNA, DNA methylation, differentiation, epidermal stem cells, keratinocytes, microRNA, gene regulation, RNA sequencing, epigenetics
Introduction
Circular RNAs (circRNAs) are covalently closed natural RNA circles resulting from a back-splicing event. Most often, they are derived from annotated host gene exons, which may give rise to alternative circRNAs depending on the exact exons involved in the back-splicing, and it has now become apparent that circRNAs comprise a large class of animal RNAs with complex developmental and tissue-specific expression patterns [1],[2]. Recently, we and others have shown a gene-regulatory function for some of the most abundantly expressed circRNAs in human tissues [1],[3-6]. In particular, a circular RNA located on the X chromosome contains more than 70 conserved binding sites for miRNA-7 and can act as a sponge. Accordingly, we named it circular RNA sponge for miR-7 (ciRS-7) [3]. However, the function of most circRNAs and what regulates their expression during cellular differentiation remain largely unknown. At the genetic level it has been shown that reverse complementary Alu repeats flanking the circularized exons can facilitate their biogenesis [7-11], and back-splicing can also be regulated by general splicing factors through binding to cis-acting splicing regulatory elements, but with regulatory rules distinct from canonical splicing [12],[13]. In addition, the transacting RNA binding factors ADAR, Quaking, FUS, HNRNPL and DHX9 have been shown to affect the biogenesis of some but not all circRNAs [9],[14-17]. In particular, DHX9 was recently shown to bind and resolve double stranded RNA formations caused by nearby homologues inverted Alu repeats leading to specific suppression of circRNAs with Alu mediated biogenesis [15] suggesting that DHX9 counteracts the back-splicing events that the Alu invasion of the human genome initiated [15].
CircRNAs have mostly been studied in brain tissues and are dynamically expressed in a spatio-temporal manner during mammalian brain development [2],[7],[18]. They are likely to be involved in neurodegenerative disorders [19],[20] and are deeply involved in human cancer [21], for instance by modulating tumor growth by affecting the Wnt/β-catenin pathway [22],[23], and some are promising as diagnostic and prognostic biomarkers [24],[25].
On the contrary, no data on circRNA expression in human epidermal stem cell (EpSC) homeostasis and differentiation are available. Therefore, we explored the expression of endogenous circRNAs during epidermal stem cell differentiation using previously published high-throughput RNA sequencing (RNA-seq) data [26]. Importantly, the sequencing libraries were prepared from total RNA after ribosomal RNA removal [26] as circRNAs lack polyA tails. Characterization of circRNAs was performed using two independent well-established bioinformatics pipelines [7],[11], and circRNAs supported by at least 10 reads per replicate in EpSCs or in the differentiated keratinocytes were further characterized bioinformatically. Finally, we analyzed RNA-seq data from individual knockdowns of DNMT3A and DNMT3B, respectively, in the EpSCs to investigate if DNA methylation may play a role in regulating circRNA expression.
Results
Circular RNAs are abundantly expressed in EpSCs and differentiated keratinocytes
Total ribosomal depleted RNA from EpSCs and differentiated keratinocytes was deep sequenced in triplicates and analyzed using a stringent version of the find_circ pipeline [7] and the CIRCexplorer pipeline [11]. The find_circ pipeline detected 13.851 unique circRNA candidates supported by at least two head-to-tail junction-spanning reads in a single replicate within the entire dataset. The two circRNA candidates supported by most reads were clearly artifacts due to sequence homology between exon 8 of KRT6A and exon 8 of KRT6C and between exon 2 of KRT5 and exon 2 of KRT6A, respectively (Figure S1A and S1B). These were not detected by CIRCexplorer and excluded from further analyses. Likewise, many of the circRNAs supported by very few reads are potentially reverse transcriptase- or sequencing artifacts. Therefore, we decided to analyze, in detail, only circRNA candidates supported by at least ten head-to-tail junction-spanning reads on average per replicate as detected by the find_circ pipeline. Among these, we manually inspected all circRNA candidates not detected by CIRCexplorer, to exclude obvious artifacts due to sequence homology, and ended up with 402 and 563 circRNA species in the EpSCs and differentiated keratinocytes, respectively (Fig. 1A and B). The reproducibility between replicates was good both when considering reads per million (RPM) across the back-splicing junction and circular-to-linear (CTL) ratios (R2>0.66 for all analyses, Figure S2A and S2B) and 365 (90.8%) and 510 (90.6%) of the circRNAs were also detected by CIRCexplorer in the EpSCs and differentiated keratinocytes, respectively. Among the top 100 most highly expressed circRNAs in the EpSCs and differentiated keratinocytes, respectively, we only detected a total of three, which are not present in circBase [27], namely circRNAs produced from C1orf116, TNS4 (CTEN) and KRT5 (Figure S3A and S3B). Interestingly, the circRNA from the KRT5 gene is derived from cryptic splice sites found within the first exon. Therefore, it was only detected by the find_circ pipeline (Figure S3A) which, in contrast to CIRCexplorer, also detects circRNAs derived from nonannotated splice sites [28]. The overlap between the circRNAs detected in the EpSCs and the differentiated keratinocytes was substantial and many circRNAs were unique, mainly to the differentiated cells (Fig 1C). The 402 and 563 circRNAs were generated from 334 and 452 host genes, respectively, and of these 24 and 42, respectively, were expressed at higher levels than their linear host genes (Fig. 1A and B). In total, 64 and 101, respectively, of the circRNAs were generated from a single exon and circRNAs composed of two exons were most frequently observed in both cell types (Fig. 1A and B). We successfully validated the RNA-seq data for selected circRNAs by amplification of cDNA with divergent primers followed by Sanger sequencing across the back-splicing junction (Fig. 1D). However, we were unable to validate the presence of circRNA from exon 1 of KRT5, potentially due to its small size (138 bp) and sequence homology with other loci making it difficult to design specific PCR primers.
Figure 1.
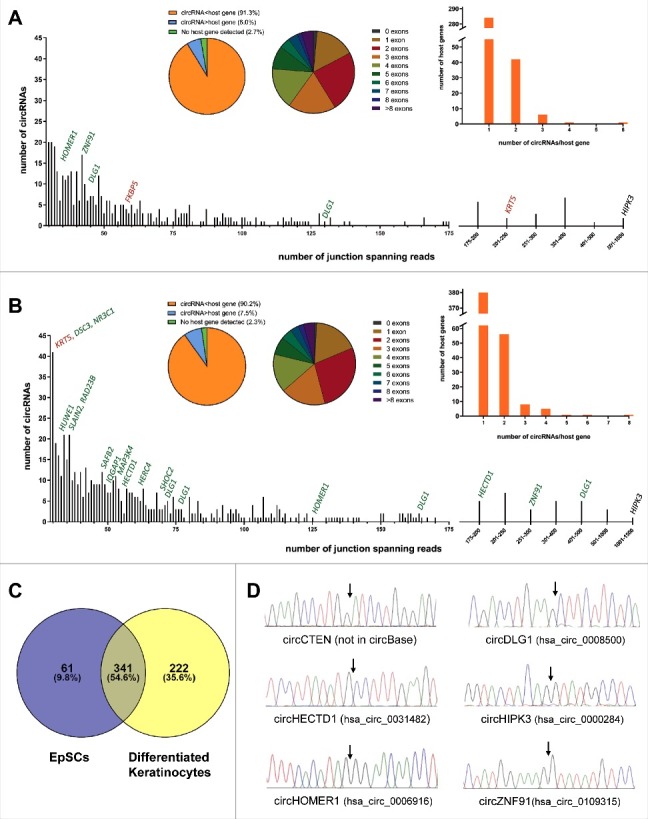
circRNAs are highly abundant in EpSCs and differentiated keratinocytes. (A) In the EpSCs 402 circRNA species were supported by an average of 10 reads per replicate. The circRNA derived from exon 3 of HIPK3 was the most abundant. Selected host genes with upregulated (green) or downregulated (red) circRNAs upon differentiation are indicated. (B) In the differentiated keratinocytes 563 circRNA species were supported by an average of 10 reads per replicate. The circRNA derived from exon 3 of HIPK3 was the most abundant. Selected host genes with upregulated (green) or downregulated (red) circRNAs upon differentiation are indicated. (C) Venn diagram illustrating the overlap of circRNAs detected in the EpSCs and the differentiated keratinocytes. (D) Validation of RNA-seq data by Sanger sequencing across back-splicing junctions of selected circRNAs.
Many circRNAs are upregulated upon differentiation of EpSCs to keratinocytes
In total, 624 unique circRNAs were detected in the EpSCs and differentiated keratinocytes combined (Fig. 1C). The expression of many of these circRNAs were changed during EpSC differentiation (Fig. 2A-C). The most up- and downregulated circRNAs are listed in Table 1 and Table 2, respectively. Overall, circRNAs were higher expressed in the differentiated cells, both when considering RPM and CTL ratios (P<0.0001 for both analyses). Thirteen circRNAs were not detected in the EpSCs (Table 1) while being expressed in the differentiated keratinocytes and could, therefore, not be represented in Fig. 2A-C. Likewise, circ-RNAs with host gene expression below the detection limit (Table 1) are not displayed in Fig. 2B and C. Interestingly, when considering CTL ratios (Fig. 2B), some of the most upregulated circRNAs are derived from epidermal developmental genes, including RAB6A [29], DSC3 [30] and IQGAP1 [31] and two of the most upregulated circRNAs are derived from HECTD1, which has recently been shown to regulate the protein levels of IQGAP1 through ubiquitination[32]. circRNAs derived from the MAP3K4, CTEN, and ESRP1 genes, which are involved in stemness, epidermal growth factor signaling and epithelial to mesenchymal transition [33-36], were also upregulated during differentiation. This also applied to circRNAs from the two interacting genes SHOC2 and HUWE1. SHOC2 is a scaffold protein that acts as a positive modulator of ERK1/2 signaling and it is directly regulated by its binding partner, the E3 ubiquitin ligase, HUWE1 [37]. One of the most upregulated circRNAs was derived from the SAFB2 gene, which is likely to be involved in transcription initiation and alternative mRNA splicing [33],[38], and its homolog SAFB1 has been shown to facilitate alternative splicing of neurogenesis genes [39] (Fig. 2B). Finally, the developmental gene DLG1 produced four different circRNAs, which were all upregulated more than three fold (RPM) in the differentiated keratinocytes (Table 1). DLG1 has previously been shown to produce several alternatively spliced variants during keratinocyte differentiation [40].
Figure 2.
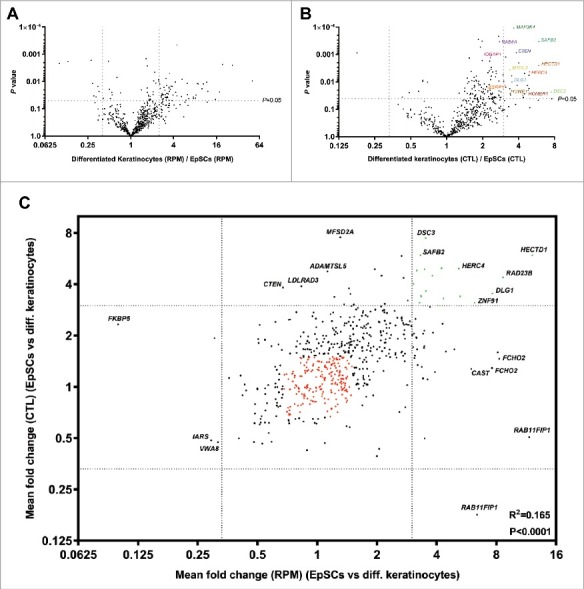
Many circRNAs are upregulated during EpSCs differentiation. (A) Volcano plot of fold changes in RPM upon differentiation. (B) Volcano plot of fold changes in CTL ratios upon differentiation. (C) Many upregulated circRNAs were upregulated independent of their respective host genes and a positive correlation between fold change in RPM and fold change in CTL ratios was observed upon differentiation. Upregulated circRNAs (RPM and CTL fold change >3) are indicated in green and stably expressed circRNAs (RPM and CTL fold change <1.5 and >0.67) are indicated in red.
Table 1.
Upregulated circRNAs during differentiation of EpSCs.
| Location (HG19) | Host gene | Fold change (CTL) | P-value | Fold change (RPM) | P-value | Δ junction spanning reads |
|---|---|---|---|---|---|---|
| Upregulated circRNAs (Fold change in RPM>3) | ||||||
| chr18:28586872-28598766 | DSC3 | 7.481 | 0.023 | 3.519 | 0.075 | 22 |
| chr19:5604593-5604947 | SAFB2 | 5.936 | 0.000 | 3.312 | 0.038 | 35 |
| chr14:31596990-31641328 | HECTD1 | 5.927 | 0.003 | 12.131 | 0.023 | 166 |
| chr4:48371865-48385801 | SLAIN2 | 4.977 | 0.087 | 4.234 | 0.010 | 28 |
| chr10:69785302-69804320 | HERC4 | 4.944 | 0.004 | 5.165 | 0.035 | 50 |
| chr5:142779220-142780417 | NR3C1 | 4.905 | 0.006 | 3.473 | 0.063 | 22 |
| chr5:78734832-78752841 | HOMER1 | 4.823 | 0.020 | 3.174 | 0.037 | 88 |
| chr12:116668237-116675510 | MED13L | 4.497 | 0.005 | 3.810 | 0.049 | 39 |
| chr9:110062421-110074018 | RAD23B | 4.398 | 0.036 | 8.644 | 0.012 | 32 |
| chr1:19244855-19246267 | IFFO2 | 4.025 | 0.002 | 3.056 | 0.037 | 34 |
| chr6:161455290-161471011 | MAP3K4 | 3.650 | 0.000 | 3.529 | 0.005 | 38 |
| chr3:196817782-196846401 | DLG1 | 3.526 | 0.010 | 7.631 | 0.041 | 63 |
| chr21:17205666-17214859 | USP25 | 3.420 | 0.030 | 3.333 | 0.090 | 27 |
| chr4:87622393-87622954 | PTPN13 | 3.407 | 0.027 | 5.247 | 0.008 | 36 |
| chr10:112723882-112745523 | SHOC2 | 3.397 | 0.004 | 3.344 | 0.028 | 50 |
| chr6:160467529-160469575 | IGF2R | 3.306 | 0.079 | 4.336 | 0.096 | 32 |
| chr19:23541231-23545527 | ZNF91 | 3.120 | 0.015 | 6.212 | 0.012 | 231 |
| chr7:99090662-99092254 | ZNF394 | 3.120 | 0.001 | 3.278 | 0.036 | 37 |
| chr2:148653869-148657467 | ACVR2A | 3.006 | 0.009 | 4.052 | 0.072 | 45 |
| chr4:87614739-87622954 | PTPN13 | 2.956 | 0.098 | 3.696 | 0.014 | 43 |
| chr16:85667519-85667738 | GSE1 | 2.514 | 0.038 | 3.128 | 0.008 | 43 |
| chr2:9419445-9437574 | ASAP2 | 2.350 | 0.029 | 4.280 | 0.024 | 24 |
| chr7:65705311-65751696 | TPST1 | 2.139 | 0.155 | 3.458 | 0.017 | 26 |
| chr3:196842797-196846401 | DLG1 | 2.085 | 0.108 | 3.833 | 0.015 | 58 |
| chr12:122825299-122826244 | CLIP1 | 2.058 | 0.073 | 3.071 | 0.073 | 64 |
| chr1:173961892-173962273 | RC3H1 | 2.054 | 0.133 | 3.418 | 0.090 | 39 |
| chrY:2829114-2829687 | ZFY | 2.018 | 0.038 | 3.010 | 0.021 | 32 |
| chr11:85707868-85714494 | PICALM | 1.956 | 0.016 | 3.154 | 0.048 | 84 |
| chr2:9083315-9098771 | MBOAT2 | 1.929 | 0.041 | 3.170 | 0.073 | 62 |
| chr3:196807921-196846401 | DLG1 | 1.735 | 0.032 | 3.463 | 0.048 | 119 |
| chr12:69214104-69218431 | MDM2 | 1.723 | 0.308 | 3.449 | 0.123 | 22 |
| chr7:22306582-22357656 | RAPGEF5 | 1.714 | 0.199 | 3.049 | 0.030 | 24 |
| chr3:196831773-196846401 | DLG1 | 1.696 | 0.007 | 3.449 | 0.016 | 346 |
| chr7:90376995-90419967 | CDK14 | 1.665 | 0.300 | 4.914 | 0.070 | 28 |
| chr10:126631025-126631876 | ZRANB1 | 1.664 | 0.099 | 3.189 | 0.014 | 57 |
| chr17:45695715-45696530 | NPEPPS | 1.614 | 0.069 | 4.457 | 0.000 | 129 |
| chr19:13919649-13920034 | ZSWIM4 | 1.596 | 0.250 | 8.119 | 0.040 | 31 |
| chr21:37619814-37620866 | DOPEY2 | 1.527 | 0.494 | 3.107 | 0.195 | 36 |
| chr2:9083315-9114564 | MBOAT2 | 1.476 | 0.540 | 3.093 | 0.339 | 21 |
| chr5:72354259-72373320 | FCHO2 | 1.462 | 0.307 | 8.266 | 0.031 | 47 |
| chr10:105767934-105778666 | SLK | 1.345 | 0.267 | 3.209 | 0.027 | 94 |
| chr11:65198239-65205128 | NEAT1 | 1.311 | 0.421 | 3.366 | 0.029 | 30 |
| chr5:72285253-72286691 | FCHO2 | 1.291 | 0.287 | 7.594 | 0.036 | 28 |
| chr5:96058342-96066564 | CAST | 1.277 | 0.343 | 5.966 | 0.048 | 64 |
| chr1:207200838-207201024 | C1orf116 | 1.223 | 0.266 | 3.919 | 0.031 | 168 |
| chr2:11905658-11907984 | LPIN1 | 1.121 | 0.689 | 3.412 | 0.076 | 31 |
| chr8:71071739-71075089 | NCOA2 | 1.107 | 0.788 | 3.597 | 0.019 | 37 |
| chr8:37734626-37735069 | RAB11FIP1 | 0.507 | 0.193 | 11.725 | 0.011 | 172 |
| chr7:20193815-20201493 | LOC101927668 | 0.498 | 0.106 | 3.478 | 0.023 | 29 |
| chr8:37727937-37735069 | RAB11FIP1 | 0.178 | 0.001 | 6.383 | 0.075 | 111 |
| Upregulated circRNAs (Fold change in RPM>3, host gene below detection limit) | ||||||
| chr12:9020437-9021799 | A2ML1 | 50.75378 | 0.009406 | 105 | ||
| chr1:12638745-12639440 | DHRS3 | 26.73041 | 0.002491 | 56 | ||
| chr3:141231004-141259451 | RASA2 | 19.34944 | 0.002329 | 40 | ||
| chr14:31596990-31638674 | HECTD1 | 17.70115 | 0.02394 | 53 | ||
| chr2:192557928-192558280 | LOC105747689 | 15.43353 | 0.044381 | 29 | ||
| chr6:168271062-168303066 | MLLT4 | 11.83506 | 0.056949 | 33 | ||
| chr17:57430575-57430887 | YPEL2 | 10.51028 | 0.0121 | 52 | ||
| chr12:27521194-27529349 | ARNTL2 | 10.02607 | 0.018472 | 30 | ||
| chr2:29344239-29404563 | CLIP4 | 8.868869 | 0.012166 | 32 | ||
| chr2:202010100-202014558 | CFLAR | 6.553219 | 0.041388 | 37 | ||
| chr21:16386664-16415895 | NRIP1 | 6.276586 | 0.02987 | 64 | ||
| chr16:21973780-21987564 | UQCRC2 | 5.770587 | 0.056439 | 42 | ||
| chr7:20198596-20198714 | LOC101927668 | 5.538243 | 0.19624 | 54 | ||
| chr16:48581917-48596355 | N4BP1 | 4.441751 | 0.033021 | 27 | ||
| chr3:152882554-152965320 | 3.173289 | 0.039495 | 21 | |||
| Upregulated circRNAs (No junction spanning reads in the EpSCs) | ||||||
| chr6:47675349-47684712 | 80 | |||||
| chr9:99797007-99797975 | CTSV | 74 | ||||
| chr4:15008928-15034835 | CPEB2 | 74 | ||||
| chr12:8991708-8995957 | A2ML1 | 66 | ||||
| chr4:87869649-87968746 | AFF1 | 60 | ||||
| chr1:6659106-6659512 | KLHL21 | 55 | ||||
| chr19:9004868-9006766 | MUC16 | 45 | ||||
| chr11:18722479-18722744 | TMEM86A | 42 | ||||
| chr6:168271062-168299147 | MLLT4 | 37 | ||||
| chr16:25066139-25123317 | 34 | |||||
| chr4:40596274-40598817 | RBM47 | 33 | ||||
| chr18:47455881-47463767 | MYO5B | 33 | ||||
| chr18:47563219-47566684 | MYO5B | 30 | ||||
Table 2.
Downregulated circRNAs during differentiation of EpSCs.
| Location (HG19) | Host gene | Fold change (CTL) | P-value | Fold change (RPM) | P-value | Δ junction spanning reads |
|---|---|---|---|---|---|---|
| Downregulated circRNAs (Fold change in RPM<0.33) | ||||||
| chr6:35586872-35610620 | FKBP5 | 2.329 | 0.174 | 0.099 | 0.002 | −52 |
| chr9:95018961-95048121 | IARS | 0.486 | 0.024 | 0.292 | 0.012 | −25 |
| chr2:10559859-10560261 | HPCAL1 | 1.935 | 0.303 | 0.303 | 0.033 | −21 |
| chr13:42385360-42407667 | VWA8 | 0.474 | 0.171 | 0.316 | 0.059 | −20 |
| Downregulated circRNAs (Fold change in RPM<0.4, host gene below detection limit) | ||||||
| chr12:52913665-52913803 | KRT5 | 0.131 | 0.002 | −187 | ||
| chr6:56482926-56483031 | 0.278 | 0.002 | −34 | |||
| chr3:114069120-114070725 | ZBTB20 | 0.325 | 0.036 | −25 | ||
| chr7:6505750-6505876 | KDELR2 | 0.346 | 0.015 | −21 | ||
| chr14:80963811-80997230 | CEP128 | 0.348 | 0.254 | −20 | ||
| chr14:105413222-105413717 | 0.349 | 0.101 | −29 | |||
| chr11:62299899-62299998 | AHNAK | 0.360 | 0.179 | −27 | ||
There was a significant correlation between fold change in RPM and fold change in CTL ratio indicating that many circRNAs are up-/downregulated independent of host gene expression (Fig. 2C). However, it was also observed that some circRNAs with an increased CTL ratio were not upregulated per se. Instead, the increased CTL ratio was due to their respective host genes being downregulated (e.g. MFSD2A, CTEN, LDLRAD3, and ADAMTSL5). Likewise, some circRNAs were upregulated along with their linear host genes (e.g. RAB11FIP1, CAST and FCHO2) (Fig. 2C).
Upregulated circRNAs may function as miRNA sponges
Next, we compared the number of AGO2 binding sites between circRNAs upregulated independent of their linear host genes (RPM- and CTL fold change >3) and stably expressed circRNAs (RPM- and CTL fold change <1.5 and >0.67) using the web tool CircInteractome [41]. This tool integrates 93 independently reported CLIP datasets from various RNA binding proteins (RBPs), including AGO2. We found that upregulated circRNAs (indicated by green in Fig. 2C) had significantly more AGO2 binding sites than stably expressed circRNAs (indicated by red in Fig. 2C) (Fig. 3A). Among the upregulated circRNAs the following seven had more than 10 AGO2 binding sites: hsa_circ_0109315 (ZNF91), hsa_circ_0031482 (HECDT1), hsa_circ_0078617 (MAP3K4), hsa_circ_0020028 (SHOC2), hsa_circ_0008812 (RAD23B), hsa_circ_0001543 (NR3C1) and hsa_circ_0126525 (SLAIN2). Among the many more stably expressed circRNAs only the following 12 had more than 10 AGO2 binding sites: hsa_circ_0000039 (YTHDF2), hsa_circ_0072732 (ERBIN) hsa_circ_0001461 (FAT1), hsa_circ_0001021 (AFTPH), hsa_circ_0061394 (BACH1), hsa_circ_0001551 (RARS), hsa_circ_0027491 (MDM2), hsa_circ_0001550 (RARS), hsa_circ_0001741 (TNPO3), hsa_circ_0001247 (ATXN10) and hsa_circ_0000620 (AAGAB). Therefore, the ratio of circRNAs with more than 10 AGO2 binding sites was significantly higher for the upregulated circRNAs (P = 0.0002).
Figure 3.
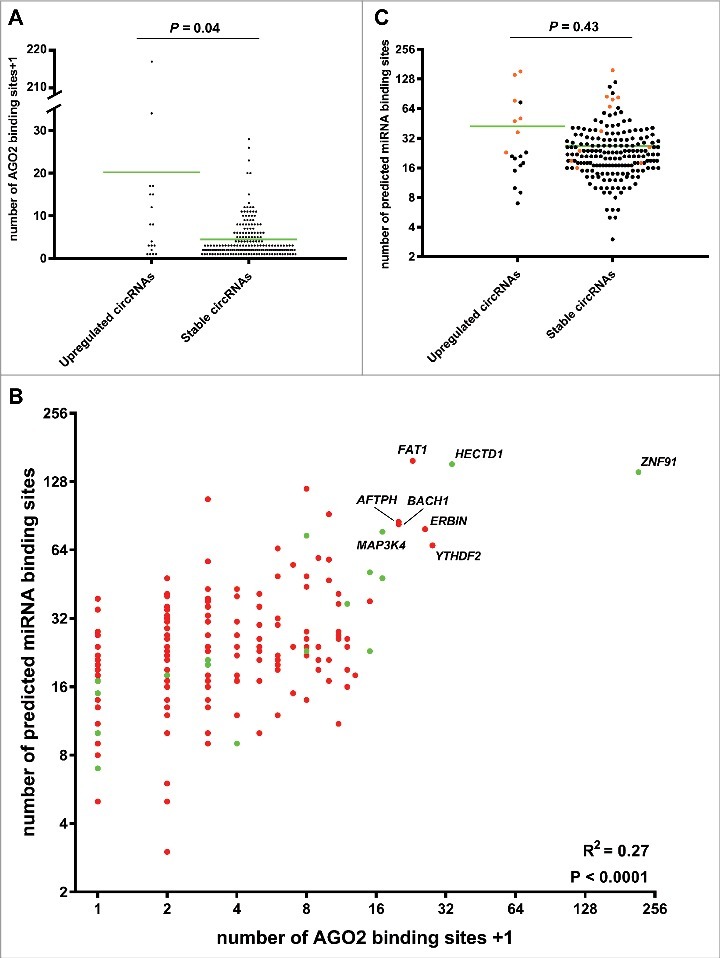
Upregulated circRNAs may function as miRNA sponges. (A) Column scatter plot comparing the number of AGO2 binding sites in the upregulated- and stably expressed circRNAs, respectively. The mean number of AGO2 binding sites was 19.2 for the upregulated circRNAs and 3.44 for the stably expressed circRNAs. (B) Scatter plot comparing the number of AGO2 binding sites with the number of predicted miRNA binding sites in the upregulated circRNAs (green dots) and stably expressed circRNAs (red dots). A positive correlation between AGO2 binding and predicted miRNA binding sites was observed. Host genes of the best miRNA sponge candidates are indicated. (C) Column scatter plot comparing the number of miRNA binding sites in the upregulated- and stably expressed circRNAs, respectively. The mean number of miRNA binding sites was 42.5 for the upregulated circRNAs and 26.8 for the stably expressed circRNAs. circRNAs with more than 10 AGO2 binding sites are indicated in orange.
We also compared the number of predicted miRNA binding sites between upregulated- and stably expressed circRNAs using CircInteractome [41], which use the TargetScan 7.0 algorithm for this purpose. There was a significant correlation between AGO2 binding and predicted miRNA binding sites (Fig. 3B). The fraction of circRNAs with more than 10 AGO2 binding sites and a high number of predicted miRNA binding sites (>30, indicated by orange in Fig. 3C) was significantly larger for the upregulated circRNAs (P<0.0001). Among all, the upregulated circRNAs from the HECTD1 and ZNF91 genes were the best candidates for being miRNA sponges when considering both AGO2 binding and predicted miRNA binding sites (Fig. 3B). Most notably, circZNF91 (hsa_circ_0109315) contain 24 miR23b-3p binding sites, of which most have strong binding also outside of the seed sequence. Most of these sites overlap with the seed sequence of miR766-3p for which 23 binding sites are present (Figure S4). miR23b-3p is an important regulator of keratinocyte differentiation making it likely that circZNF91 exerts its function in differentiation by sponging this miR [42]. The binding sites for miR23b-3p were not predicted by CircInteractome, but have previously also been found by Guo and colleagues [6].
Global perturbation of host gene expression by knockdown of DNMT3A and DNMT3B
DNMT3A epigenetically promotes the expression of most genes with active enhancers in human EpSCs, through hydroxymethylation of the center of these enhancers, in a TET2 dependent manner [26]. Furthermore, DNMT3A inhibits other genes through methylation of their promoters [43]. Therefore, to compare our results (Fig. 2C) with a setting where most changes in circRNA expression would be expected to be indirect, we analyzed RNA-seq data from human EpSCs treated with short-hairpin RNAs (shRNAs) against DNMT3A in triplicate using the same strategy for circRNA detection as described above. It was observed that upon DNMT3A knockdown many circRNAs were significantly upregulated (Fig. 4A and B), and the mean RPM as well as the mean CTL ratios were significantly higher for the knockdown cells compared to control cells (P<0.0001 for both RPM and for CTL, respectively). The correlation between fold change in RPM and fold change in CTL ratios (Pearson's correlation coefficient, r2 = 0.057) was much weaker than what was observed upon differentiation of the EpSCs, indicating that these changes were largely depended on changes in host gene expression (Fig. 4C).
Figure 4.
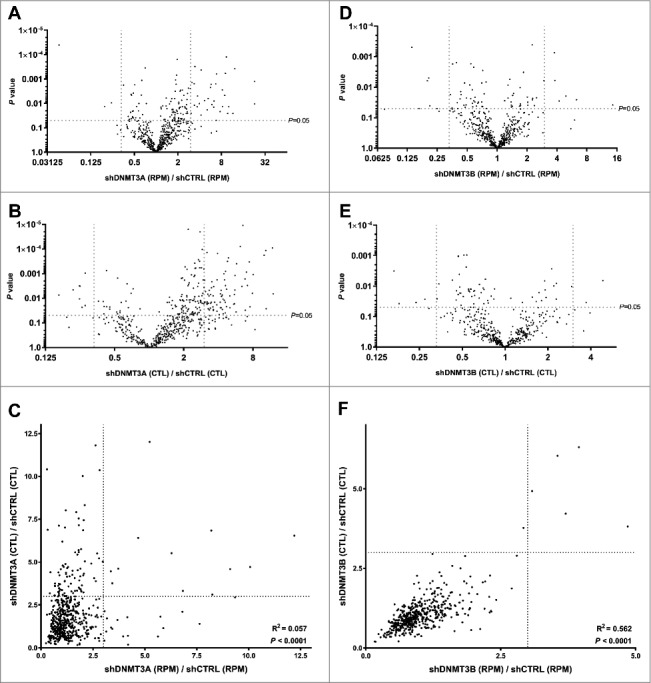
Knockdown of DNMT3A, but not DNMT3B, induce circRNA expression in a host gene dependent manner. (A) Many circRNAs are upregulated upon DNMT3A knockdown. (B) Many circRNAs are upregulated relative to their respective host genes upon DNMT3A knockdown. (C) The changes in circRNA expression are largely related to changes in host gene expression (only few circRNAs are on the diagonal). (D) Only a few circRNAs are up/downregulated upon DNMT3B knockdown. (E) Only a few circRNAs are up/downregulated relative to their respective host genes upon DNMT3B knockdown. (F) The changes in circRNA expression are largely independent of changes in host gene expression (most circRNAs are on the diagonal).
DNMT3A and DNMT3B regulate gene expression through non-overlapping functions in EpSCs [26], and DNMT3B has a known role in gene body methylation [26],[44],[45]. Thus, it was not surprising that DNMT3B knockdown had opposite effects, relative to DNMT3A knockdown, on circRNA expression. Here, only few circRNAs were changed by more than three fold (Fig. 4D and 4E), and the mean RPM was not significantly changed (P = 0.05), while the mean CTL ratio was slightly higher for the control cells compared to the knockdown cells (P<0.001). Moreover, a very strong correlation was observed between fold change in RPM and fold change in CTL ratios (Pearson's correlation coefficient, r2 = 0.562) indicating that the small changes observed were largely independent of changes in host gene expression (Fig. 4F).
Upregulated circRNAs are unlikely to be regulated by DNA methylation
The overlap between circRNAs upregulated independent of their linear host genes upon differentiation (upper right corner in Fig. 2C), circRNAs upregulated independent of their linear host genes upon DNMT3A knockdown (upper right corner in Fig. 4C) and circRNAs upregulated independent of their linear host genes upon DNMT3B knockdown (upper right corner in Fig. 4F) was very limited (Fig. 5). None of the 19 circRNAs upregulated independent of their linear host genes upon differentiation were also upregulated independent of their linear host genes upon DNMT3A- or DNMT3B knockdown, and only one circRNA, derived from the ALS2 gene (hsa_circ_0001093), was common between DNMT3A- and DNMT3B knockdown (Fig. 5).
Figure 5.
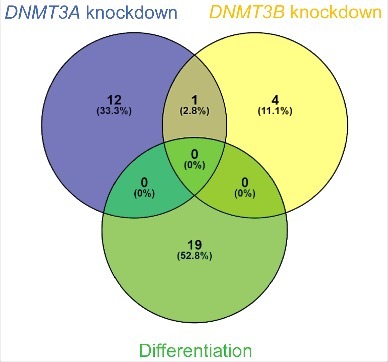
Upregulated circRNAs upon differentiation is unlikely to be regulated by DNA methylation. Venn diagram illustrating the overlap of circRNAs upregulated independent of their linear host genes upon differentiation, -upon DNMT3A knockdown and – upon DNMT3B knockdown.
Upregulated circRNAs have less Alu-mediated biogenesis
We searched for inverted homologous Alu repeats within a 20 kbp window around the back-splicing junction of the circRNAs. The fraction of circRNAs without inverted homologous Alu repeats within this window was 0.42 (8/19) and 0.18 (35/192) for upregulated circRNAs and stably expressed circRNAs, respectively. This difference was statistically significant (P = 0.01). The fraction of circRNAs without inverted homologous Alu repeats within a 10 kbp window was 0.53 and 0.27 for upregulated circRNAs and stably expressed circRNAs, respectively. This difference was also statistically significant (P = 0.02). Together these analyses indicate that the upregulated circRNAs are less prone to undergo Alu-mediated biogenesis.
Upregulated circRNAs are deregulated in cutaneous squamous cell carcinoma
The malignant counterpart of keratinocytes has previously been explored for circRNA expression using a microarray approach [46]. Among the nineteen circRNAs upregulated independent of their linear host genes upon differentiation circMAP3K4 (hsa_circ_0078617) and circDLG1 (hsa_circ_0008583) were also shown to be upregulated in cutaneous squamous cell carcinoma, while circACVR2A (hsa_circ_0001073) were shown to be downregulated [46].
Discussion
In this study, we found that circRNAs are abundantly expressed and upregulated during EpSC differentiation. This was not surprising, as it has previously been shown that circRNAs are upregulated during neuronal differentiation [2] and that non-proliferating cells have higher levels of circRNA expression compared to proliferating cells [47]. These observations are likely to be explained, in part, by circRNAs being passively diluted during cell proliferation and accumulating in terminally differentiated cells due to their high stability. However, our results also point towards a coordinated regulation of a subset of circRNAs during differentiation of EpSCs.
First, many of the up- and downregulated circRNAs were changed independently of their respective host genes and a positive correlation between fold change in RPM and fold change in CTL ratios was observed upon differentiation (Pearson's correlation coefficient, r2 = 0.165). On the other hand, artificial manipulation of host gene expression by inhibition of DNMT3A, which epigenetically regulate most human genes through methylation of promotor and enhancer regions, did not confer a strong correlation between fold change in RPM and fold change in CTL ratios (Pearson's correlation coefficient, r2 = 0.057). In particular, a large number of circRNAs had increased CTL ratios without an increase in RPM. The expression of these host genes is decreased while the circRNAs remain at similar levels, possibly due to their higher stability relative to mRNA [48]. These genes are likely downregulated due to loss of enhancer hydroxymethylation, as DNMT3A cooperate with TET2 to maintain high levels of hydroxymethylation at the center of enhancers of actively transcribed genes [26]. Likewise, some of the host genes, which were strongly upregulated, produced strongly upregulated circRNAs. These genes are likely upregulated due to loss of promoter methylation. In line with this, it has previously been shown that circRNA-producing genes have significantly higher levels of H3K27Ac and lower levels of DNA methylation in their promoter regions relative to genes that do not [48], indicating that actively transcribed genes produce more circRNA. Interestingly, the opposite effects on circRNA expression were observed upon DNMT3B knockdown. Here, only few circRNAs were significantly changed and a very strong correlation was observed between fold change in RPM and fold change in CTL ratios (Pearson's correlation coefficient, r2 = 0.562). It has previously been shown that DNMT3A and DNMT3B regulate gene expression through non-overlapping mechanisms in EpSCs [26], and DNMT3B is more susceptible to bind and methylate gene bodies [26],[44],[45]. Therefore, our results points towards that gene body methylation may play a role in regulating the expression of some circRNAs. However, there was no overlap between circRNAs upregulated independently of their respective host genes upon DNMT3A or DNMT3B knockdown and upon differentiation. Therefore, these results indicate that circRNAs are generally not directly regulated by DNA methylation during EpSC differentiation.
Second, circRNAs upregulated independently of their host genes are significantly more prone to bind AGO2 compared to stably expressed circRNAs. circRNAs prone to AGO2 binding also contained high numbers of predicted miRNA binding sites. If the observed upregulation of circRNAs in the differentiated cells is entirely passive this would not have been expected. Instead, our results are in line with the previously proposed hypothesis that differentiated cells should not react to weak autocrine and paracrine cellular signals with strong regulatory activities, and that this may be achieved by differentiated cells being less prone to miRNA regulation due to higher levels of circRNAs with miRNA sponging function compared to proliferating cells [47]. In particular, the upregulated circRNAs derived from ZNF91 and HECTD1 are strong candidates for being miRNA sponges, and circZNF91 has previously been suggested to function as a miRNA sponge in human cells [6]. circZNF91 contains most of the host gene's 3’ UTR and has 24 and 23 individual binding sites for miR-23b-3p and miR-766-3p, respectively. miR-23b-3p has been directly implicated in differentiation of EpSCs to keratinocytes [42], whereas miR-766 targets DNMT3B [49] and is upregulated in cutaneous squamous cell carcinoma [50].
Third, the upregulated circRNAs have less Alu-mediated biogenesis coinciding with DHX9 being upregulated in the differentiated cells [26]. Since DHX9 specifically suppress the expression of circRNAs with flanking reverse complementary Alu repeats [15], we propose that the cells actively suppress the expression of circRNAs with no or unwanted functions, which would otherwise accumulate in non-proliferating cells, through upregulation of DHX9.
Finally, many of the independently upregulated circRNAs are derived from epidermal developmental genes and genes involved in stemness and epidermal growth factor signaling. These circRNAs may have other functions unrelated to their host genes, but it has been shown for several circRNAs that they can regulate the expression of their own host genes [8],[22],[51]. However, it remains to be elucidated if this is a general mechanism among circRNAs.
Future studies should aim at uncovering the specific roles and functions of circRNAs, like circZNF91 and circHECDT1, in EpSC homeostasis and differentiation and to further explore the mechanisms responsible for their regulation.
In conclusion, we have shown that circRNAs are abundantly expressed in EpSCs and upregulated during differentiation to keratinocytes in a coordinated manner. Independently upregulated circRNAs are generally less prone to Alu-mediated biogenesis and unlikely to be directly regulated by DNA methylation during differentiation. Finally, several upregulated circRNAs, including two derived from the HECTD1 gene and one from ZNF91, are likely to have miRNA sponging functions.
Materials and methods
Ethics
No primary human samples or animal models were used in this study and experimental procedures were previously evaluated and approved by CEEA (Ethical Committee for Animal Experimentation) of the Government of Catalonia [26].
circRNA detection in RNA sequencing data
We analyzed RNA-seq data generated in triplicates from EpSCs and differentiated keratinocytes, as well as DNMT3A and DNMT3B knockdowns in EpSCs, for circRNA expression. This was done using a more stringent version of the Find_circ pipeline, which we have previously described [7], and the CircExplorer pipeline [11]. Libraries were prepared with the TruSeq®Stranded Total Sample Preparation kit (Illumina Inc.) according to the manufacturer's protocol. Ribosomal RNA was depleted from 0.5 µg of total RNA using the Ribo-Zero Gold Kit (Illumina Inc.). The RNA-seq data has previously been published [26], but not previously analyzed for circRNA expression.
All circRNA data analyses were based on the Find_circ pipeline. However, all circRNA candidates not detected by CIRCexplorer were manually inspected to exclude obvious artifacts due to sequence homology. Reads per million (RPM) refers to sequencing reads aligning across the particular back-splicing junction and circular-to-linear (CTL) ratios were defined as the average number of linear reads spanning the splice donor- and splice acceptor sites of the back-splicing junction divided by the number of reads spanning the back-splice junction. circRNAs with less than 15 linear splice donor and splice acceptor reads in the replicates of the EpSCs and differentiated keratinocytes, respectively, were excluded from the CTL analyses. In addition, circRNAs with no splice donor and -acceptor reads in two out of three replicates, of either the EpSCs or the differentiated keratinocytes, were excluded from CTL analyses. The same bioinformatics pipelines and criteria were used when analyzing the DNMT3A and DNMT3B knockdown RNA-seq data.
Accession numbers
The genomic data used in this paper have previously been deposited in the NCBI Gene Expression Omnibus (GEO) database with accession number: GSE65838.
Analyses of AGO2 binding sites and predicted miRNA target sites
We used CircInteractome [41] for the AGO2 binding and miRNA binding site prediction analyses of stably expressed- and upregulated circRNAs upon differentiation of EpSCs. For these analyses circRNAs for which internal splicing cannot be predicted (i.e. most of the circRNA is intronic due to usage of a cryptic splice site) were not included in the analyses. For instance, the upregulated circRNA from the MED13L gene (hsa_circ_0000442) was predicted to have 277 miRNA binding sites. However, as it is largely intronic (Figure S5), and could be subject to internal splicing, it was removed from the analyses.
Analyses of flanking Alu repeats
The Alu repeats were obtained from the UCSC Browser RepeatMasker track [52] and analyzed as described [53]. In brief, flanking regions (20 kb or 10kb) around circRNAs were intersected with Alu repeats. Inverted homologous Alu repeats in separate flanks of circRNAs were annotated along with their closest distance. Alu repeats within the same subfamily, e.g. AluJ or AluS, were considered homologous.
Sanger sequencing across back-splicing junctions of circRNA candidates
cDNA synthesis was performed on 500 ng total RNA from EpSCs using the M-MLV reverse-transcriptase (Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer's instructions using random primers. The cDNA was diluted fivefold in PCR grade water and used as template for PCR. The reaction mixtures consisted of 2–5 µL template in a total volume of 20 µL using a 1x final concentration of the LC480 HRM Scanning Master (Roche Diagnostics, Mannheim, Germany), and a final MgCl2 concentration of 2 mM. Primers (Table S1) were used at a final concentration of 300 nM. The cycling protocol was initiated by one cycle at 95°C for 10 minutes, followed by 40 PCR cycles at 95°C for 10 seconds, 60°C for 20 seconds, and 72°C for 20 seconds. Five µL of each PCR product was loaded on 2% agarose gels stained with SYBR™ Safe DNA Gel Stain (Thermo Fisher Scientific) and visualized under UV light after electrophoresis. The remaining 15 µL of each PCR product was cleaned up using the QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) and Sanger sequenced in both forward and reverse directions using the service of GATC (GATC Biotech, Konstanz, Germany).
Statistical analyses
The reproducibility between RNA-seq replicates was compared using linear regression and comparisons of circRNA expression (RPM and CTL ratios) between groups was done using the Wilcoxon signed-rank tests. For generation of volcano plots, t-tests were employed to calculate P-values. The P-values were not used for dividing circRNAs into groups of stably expressed and upregulated circRNAs. This was based solely on fold changes in expression. Comparison of AGO2 binding and predicted miRNA binding sites between groups were done using Mann-Whitney tests as the data were not normally distributed according to the D'Agostino & Pearson normality test. Analyses of 2 × 2 tables were done using Chi-square tests. All statistical tests were performed using Prism 7 (GraphPad, La Jolla, CA, USA).
Supplementary Material
Funding Statement
The Lundbeck Foundation. (grant number. R191-2015-1515). The Villum Foundation, the Independent Research Fund Denmark, and the Carlsberg Foundation. (Grant number. CF16-0087).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We are grateful to Lorenzo Rinaldi and Salvador Aznar Benitah for producing the RNA-seq data analyzed here and for sending us the RNA used for circRNA validation.
References
- [1].Memczak S, Jens M, Elefsinioti A, et al.. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature. 2013;495:333–8. doi: 10.1038/nature11928. [DOI] [PubMed] [Google Scholar]
- [2].Rybak-Wolf A, Stottmeister C, Glazar P, et al.. Circular RNAs in the mammalian brain are highly abundant, conserved, and dynamically expressed. Mol Cell. 2015;58:870–85. doi: 10.1016/j.molcel.2015.03.027. [DOI] [PubMed] [Google Scholar]
- [3].Hansen TB, Jensen TI, Clausen BH, et al.. Natural RNA circles function as efficient microRNA sponges. Nature. 2013;495:384–8. doi: 10.1038/nature11993. [DOI] [PubMed] [Google Scholar]
- [4].Yang W, Du WW, Li X, et al.. Foxo3 activity promoted by non-coding effects of circular RNA and Foxo3 pseudogene in the inhibition of tumor growth and angiogenesis. Oncogene. 2016;35:3919–31. doi: 10.1038/onc.2015.460. [DOI] [PubMed] [Google Scholar]
- [5].Zheng Q, Bao C, Guo W, et al.. Circular RNA profiling reveals an abundant circHIPK3 that regulates cell growth by sponging multiple miRNAs. Nat Communications. 2016;7:11215. doi: 10.1038/ncomms11215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Guo JU, Agarwal V, Guo H, et al.. Expanded identification and characterization of mammalian circular RNAs. Genome Biol. 2014;15:409. doi: 10.1186/s13059-014-0409-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Veno MT, Hansen TB, Veno ST, et al.. Spatio-temporal regulation of circular RNA expression during porcine embryonic brain development. Genome Biol. 2015;16:245. doi: 10.1186/s13059-015-0801-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ashwal-Fluss R, Meyer M, Pamudurti NR, et al.. circRNA biogenesis competes with pre-mRNA splicing. Mol Cell. 2014;56:55–66. doi: 10.1016/j.molcel.2014.08.019. [DOI] [PubMed] [Google Scholar]
- [9].Ivanov A, Memczak S, Wyler E, et al.. Analysis of intron sequences reveals hallmarks of circular RNA biogenesis in animals. Cell Reports. 2015;10:170–7. doi: 10.1016/j.celrep.2014.12.019. [DOI] [PubMed] [Google Scholar]
- [10].Liang D, Wilusz JE. Short intronic repeat sequences facilitate circular RNA production. Genes Dev. 2014;28:2233–47. doi: 10.1101/gad.251926.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang XO, Wang HB, Zhang Y, et al.. Complementary sequence-mediated exon circularization. Cell. 2014;159:134–47. doi: 10.1016/j.cell.2014.09.001. [DOI] [PubMed] [Google Scholar]
- [12].Wang Y, Wang Z. Efficient backsplicing produces translatable circular mRNAs. RNA. 2015;21:172–9. doi: 10.1261/rna.048272.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Starke S, Jost I, Rossbach O, et al.. Exon circularization requires canonical splice signals. Cell Reports. 2015;10:103–11. doi: 10.1016/j.celrep.2014.12.002. [DOI] [PubMed] [Google Scholar]
- [14].Conn SJ, Pillman KA, Toubia J, et al.. The RNA binding protein quaking regulates formation of circRNAs. Cell. 2015;160:1125–34. doi: 10.1016/j.cell.2015.02.014. [DOI] [PubMed] [Google Scholar]
- [15].Aktas T, Avsar Ilik I, Maticzka D, et al.. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 2017;544(7648):115–119. doi: 10.1038/nature21715. [DOI] [PubMed] [Google Scholar]
- [16].Errichelli L, Dini Modigliani S, Laneve P, et al.. FUS affects circular RNA expression in murine embryonic stem cell-derived motor neurons. Nat Commun. 2017;8:14741. doi: 10.1038/ncomms14741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fei T, Chen Y, Xiao T, et al.. Genome-wide CRISPR screen identifies HNRNPL as a prostate cancer dependency regulating RNA splicing. Proc Natl Acad Sci U S A. 2017;114(26):E5207–E5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].You X, Vlatkovic I, Babic A, et al.. Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci. 2015;18:603–10. doi: 10.1038/nn.3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ghosal S, Das S, Sen R, et al.. Circ2Traits: a comprehensive database for circular RNA potentially associated with disease and traits. Frontiers Genetics. 2013;4:283. doi: 10.3389/fgene.2013.00283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lukiw WJ. Circular RNA (circRNA) in Alzheimer's disease (AD). Frontiers Genetics. 2013;4:307. doi: 10.3389/fgene.2013.00307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kristensen LS, Hansen TB, Venø MT, et al.. Circular RNAs in cancer: opportunities and challenges in the field. Oncogene. 2017. doi: 10.1038/onc.2017.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li F, Zhang L, Li W, et al.. Circular RNA ITCH has inhibitory effect on ESCC by suppressing the Wnt/beta-catenin pathway. Oncotarget. 2015;6:6001–13. doi: 10.18632/oncotarget.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yang P, Qiu Z, Jiang Y, et al.. Silencing of cZNF292 circular RNA suppresses human glioma tube formation via the Wnt/beta-catenin signaling pathway. Oncotarget. 2016;7(39):63449–63455. doi: 10.18632/oncotarget.11523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hsiao KY, Lin YC, Gupta SK, et al.. Non-coding effects of circular RNA CCDC66 promote colon cancer growth and metastasis. Cancer Res. 2017;77(9):2339–2350. doi: 10.1158/0008-5472.CAN-16-1883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Weng W, Wei Q, Toden S, et al.. Circular RNA ciRS-7 – A promising prognostic biomarker and a potential therapeutic target in colorectal cancer. Clin Cancer Res. 2017;23(14):3918–3928. doi: 10.1158/1078-0432.CCR-16-2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rinaldi L, Datta D, Serrat J, et al.. Dnmt3a and Dnmt3b Associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell. 2016;19:491–501. doi: 10.1016/j.stem.2016.06.020. [DOI] [PubMed] [Google Scholar]
- [27].Glazar P, Papavasileiou P, Rajewsky N. circBase: a database for circular RNAs. RNA. 2014;20:1666–70. doi: 10.1261/rna.043687.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Hansen TB, Veno MT, Damgaard CK, et al.. Comparison of circular RNA prediction tools. Nucleic Acids Res. 2016;44:e58. doi: 10.1093/nar/gkv1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Bardin S, Miserey-Lenkei S, Hurbain I, et al.. Phenotypic characterisation of RAB6A knockout mouse embryonic fibroblasts. Biol Cell. 2015;107:427–39. doi: 10.1111/boc.201400083. [DOI] [PubMed] [Google Scholar]
- [30].Chen J, Den Z, Koch PJ. Loss of desmocollin 3 in mice leads to epidermal blistering. J Cell Sci. 2008;121:2844–9. doi: 10.1242/jcs.031518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Monteleon CL, McNeal A, Duperret EK, et al.. IQGAP1 and IQGAP3 serve individually essential roles in normal epidermal homeostasis and tumor progression. J Invest Dermatol. 2015;135:2258–65. doi: 10.1038/jid.2015.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shen X, Jia Z, D'Alonzo D, et al.. HECTD1 controls the protein level of IQGAP1 to regulate the dynamics of adhesive structures. Cell Communication Signal. 2017;15:2. doi: 10.1186/s12964-016-0156-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Abell AN, Jordan NV, Huang W, et al.. MAP3K4/CBP-regulated H2B acetylation controls epithelial-mesenchymal transition in trophoblast stem cells. Cell Stem Cell. 2011;8:525–37. doi: 10.1016/j.stem.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yang Y, Park JW, Bebee TW, et al.. Determination of a comprehensive alternative splicing regulatory network and combinatorial regulation by key factors during the epithelial-to-mesenchymal transition. Mol Cell Biol. 2016;36:1704–19. doi: 10.1128/MCB.00019-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Hong SY, Shih YP, Li T, et al.. CTEN prolongs signaling by EGFR through reducing its ligand-induced degradation. Cancer Res. 2013;73:5266–76. doi: 10.1158/0008-5472.CAN-12-4441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].De Luca I, Di Salle A, Alessio N, et al.. Positively charged polymers modulate the fate of human mesenchymal stromal cells via ephrinB2/EphB4 signaling. Stem Cell Res. 2016;17:248–55. doi: 10.1016/j.scr.2016.07.005. [DOI] [PubMed] [Google Scholar]
- [37].Jang ER, Shi P, Bryant J, et al.. HUWE1 is a molecular link controlling RAF-1 activity supported by the Shoc2 scaffold. Mol Cell Biol. 2014;34:3579–93. doi: 10.1128/MCB.00811-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Liu HW, Banerjee T, Guan X, et al.. The chromatin scaffold protein SAFB1 localizes SUMO-1 to the promoters of ribosomal protein genes to facilitate transcription initiation and splicing. Nucleic Acids Res. 2015;43:3605–13. doi: 10.1093/nar/gkv246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Rivers C, Idris J, Scott H, et al.. iCLIP identifies novel roles for SAFB1 in regulating RNA processing and neuronal function. BMC Biol. 2015;13:111. doi: 10.1186/s12915-015-0220-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Roberts S, Calautti E, Vanderweil S, et al.. Changes in localization of human discs large (hDlg) during keratinocyte differentiation are [corrected] associated with expression of alternatively spliced hDlg variants. Exp Cell Res. 2007;313:2521–30. doi: 10.1016/j.yexcr.2007.05.017. [DOI] [PubMed] [Google Scholar]
- [41].Dudekula DB, Panda AC, Grammatikakis I, et al.. CircInteractome: A web tool for exploring circular RNAs and their interacting proteins and microRNAs. RNA Biol. 2016;13:34–42. doi: 10.1080/15476286.2015.1128065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Barbollat-Boutrand L, Joly-Tonetti N, Dos Santos M, et al.. MicroRNA-23b-3p regulates human keratinocyte differentiation through repression of TGIF1 and activation of the TGF-ss-SMAD2 signalling pathway. Exp Dermatol. 2017;26:51–7. doi: 10.1111/exd.13119. [DOI] [PubMed] [Google Scholar]
- [43].Rinaldi L, Avgustinova A, Martin M, et al.. Loss of Dnmt3a and Dnmt3b does not affect epidermal homeostasis but promotes squamous transformation through PPAR-gamma. Elife. 2017;6:e21697. doi: 10.7554/eLife.21697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yang X, Han H, De Carvalho , et al.. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26:577–90. doi: 10.1016/j.ccr.2014.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Aran D, Toperoff G, Rosenberg M, et al.. Replication timing-related and gene body-specific methylation of active human genes. Hum Mol Genetics. 2011;20:670–80. doi: 10.1093/hmg/ddq513. [DOI] [PubMed] [Google Scholar]
- [46].Sand M, Bechara FG, Gambichler T, et al.. Circular RNA expression in cutaneous squamous cell carcinoma. J Dermatological Sci. 2016;83:210–8. doi: 10.1016/j.jdermsci.2016.05.012. [DOI] [PubMed] [Google Scholar]
- [47].Bachmayr-Heyda A, Reiner AT, Auer K, et al.. Correlation of circular RNA abundance with proliferation–exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis, and normal human tissues. Scientific Reports. 2015;5:8057. doi: 10.1038/srep08057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Enuka Y, Lauriola M, Feldman ME, et al.. Circular RNAs are long-lived and display only minimal early alterations in response to a growth factor. Nucleic Acids Res. 2016;44:1370–83. doi: 10.1093/nar/gkv1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Afgar A, Fard-Esfahani P, Mehrtash A, et al.. MiR-339 and especially miR-766 reactivate the expression of tumor suppressor genes in colorectal cancer cell lines through DNA methyltransferase 3B gene inhibition. Cancer Biol Therapy. 2016;17:1126–38. doi: 10.1080/15384047.2016.1235657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Sand M, Skrygan M, Georgas D, et al.. Microarray analysis of microRNA expression in cutaneous squamous cell carcinoma. J Dermatological Sci. 2012;68:119–26. doi: 10.1016/j.jdermsci.2012.09.004. [DOI] [PubMed] [Google Scholar]
- [51].Du WW, Fang L, Yang W, et al.. Induction of tumor apoptosis through a circular RNA enhancing Foxo3 activity. Cell Death Differ. 2017;24:357–70. doi: 10.1038/cdd.2016.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Smit AF. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr Opinion Genetics Dev. 1999;9:657–63. doi: 10.1016/S0959-437X(99)00031-3. [DOI] [PubMed] [Google Scholar]
- [53].Okholm TLH, Nielsen MM, Hamilton MP, Christensen L-L, Vang S, Hedegaard J, et al. Circular RNA expression is abundant and correlated to aggressiveness in early-stage bladder cancer. npj Genomic Medicine. 2017;2:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.