

Summary
While modern structural biology technologies have greatly expanded the size and type of protein complexes that can now be studied, the ability to derive large-scale structural information on proteins and complexes as they exist within tissues is practically non-existent. Here, we demonstrate the application of cross-linking-mass-spectrometry to identify protein structural features and interactions in tissue samples, providing systems structural biology insight on protein complexes as they exist in the mouse heart. This includes insights into multiple conformational states of sarcomere proteins, as well as interactions among OXPHOS complexes indicative of supercomplex assembly. The extension of cross-linking mass spectrometry analysis into the realm of tissues opens the door to increasing our understanding of protein structures and interactions within the context of the greater biological system.
eTOC Blub
Chemical cross-linking with mass spectrometry enabled structural systems biology measurements on proteins within heart tissue. Identified cross-links provide structural information on key protein systems of the heart including the sarcomere and OXPHOS complexes. Each identified cross-linked peptide pair provides a useful molecular probe that can be used to quantify changes in protein conformations or protein-protein interactions in tissues.
Introduction
Cardiovascular diseases are the number one cause of death globally, resulting in more than 17 million deaths in 2013(Roth et al., 2015). Complex diseases such as heart failure are not readily replicated in cell culture models and the lack of structural information on proteins and complexes that exist within the heart limits understanding of relevant disease pathways and new therapy developments. With recent advances in mass spectrometry-based proteomics and the emergence of cross-linking-mass spectrometry (XL-MS) methods, molecular structural information in the form of distance restraints between cross-linked residues has been obtained on purified proteins and protein complexes (Chen et al., 2010; Leitner et al., 2012; Tosi et al., 2013). The advent of MS cleavable cross-linkers(Back et al., 2001; Tang et al., 2005) (recently reviewed by Sinz(Sinz, 2017)) has enabled XL-MS analysis of samples of increasing complexity including cell lysates(Liu et al., 2017; Liu et al., 2015; Tan et al., 2016). However, XL-MS application to intact cellular systems offers opportunities for unique biological insight, where protein concentration is several hundred mg/mL(Elowitz et al., 1999; Malmstrom et al., 2009) and molecular crowding(Ellis, 2001) and subcellular compartmentalization influence protein structures and interactions. XL-MS methods based on Protein Interaction Reporter (PIR) technologies (Tang and Bruce, 2010) have advanced applications to enable systems-level study of protein conformations and interactions in isolated functional organelles(Schweppe et al., 2017), intact virus particles (Chavez et al., 2012; DeBlasio et al., 2015), and live cells(Chavez et al., 2013; Navare et al., 2015; Schweppe et al., 2015; Weisbrod et al., 2013). These capabilities allow systems structural biology measurements and even quantitative interactome measurements in stable phenotypic and dynamic pharmacological comparisons (Chavez et al., 2016b; Chavez et al., 2015). A key advantage of in vivo XL-MS is that structural information is obtained on proteins within native environments where cellular compartmentalization, macromolecular crowding influence on protein structures and interactions, concentrations of cofactors and interaction partners are at or near normal physiological levels. On the other hand, while cultured cells provide significant insight on many physiological properties, replication of organ-level disease states such as cardiomyopathy and heart failure in cell culture models has remained elusive. Large-scale XL-MS has not previously been applied to derive protein structural information directly from tissues, in part due to the bewildering complexity of tissue samples. Here we demonstrate the feasibility of applying XL-MS to tissue, highlighting the resulting systems-level structural analysis of protein conformations and interactions as they exist within the heart. Future quantitative XL-MS tissue analyses could help elucidate key molecular details that underpin heart failure and other pathologies.
Results
Here, we demonstrate the initial systems structural biology measurements with murine heart tissue samples using XL-MS. These efforts reveal more than 2000 cross-linked peptide pairs, each of which provides a structural constraint on protein conformations and complexes that exist within the heart (Dataset S1). Each link identifies a pair of Lys residues that must have existed in relative proximity to one another hundreds of billions of times in order to be detected by mass spectrometry. In this way each link identified here provides structural information on proteins and protein complexes as they existed within the biological system of the heart tissue, hence the term systems structural biology.
To obtain such information, heart tissue was isolated from mice, dissected into 1 mm3 cubes, and subjected to chemical cross-linking with the PIR cross-linker BDP-NHP (Fig. S1)(Chavez et al., 2013). Following the cross-linking reaction, samples were either processed as a whole heart sample or subjected to subcellular fractionation to isolate mitochondria. Both the whole heart and isolated mitochondrial samples were subjected to protein extraction, enzymatic digestion and enrichment of the in-tissue cross-linked peptide pairs before analysis using a real-time MS3 technique termed ReACT(Weisbrod et al., 2013) (Fig S2). In total, Comet searching of ReACT generated MS3 spectra identified 2663 non-redundant cross-linked peptide pairs with an estimated false discovery rate (FDR) of 2.4% (See methods, Fig S2 and Fig S3 for more details on FDR). These links contain 2026 non-redundant Lys-Lys linkages in 316 proteins, identifying 419 non-redundant protein-protein interactions (including self-interactions). (Dataset S1). The interaction network is available online in XlinkDB (Schweppe et al., 2016; Zheng et al., 2013) (network name = ChavezCellSystems2017_BruceLab) as well as in Fig. S4.
Cardiac muscle consists of specialized highly ordered cytoskeletal structures known as sarcomeres, which are composed of interleaved thick myosin filaments and thin filaments comprised of actin, tropomyosin and troponin (Fig. S5a). Because sarcomeric proteins exist as dynamic, interdependent, complexed proteins with conformations and interactions critical for normal cardiac function, structural knowledge on these proteins obtained directly from heart tissue could yield unique insight on normal and dysfunctional states. Cross-linked peptide pairs from all major sarcomeric proteins were detected, enabling site-directed model assembly of the myosin, actin, troponin, tropomyosin complex onto which 53 inter-protein Lys-Lys links were mapped (Fig. 1a). Myosin 6 (MYH6) is the primary constituent of thick filaments in the sarcomere, providing the ATP driven motor arm which binds to and pulls on actin in the thin filaments resulting in sarcomere contraction. A total of 572 cross-linked peptide pairs involving MYH6, including 103 inter-protein linkages to 22 different proteins, were identified (Fig. S5b). MYH6 is a large (224 kDa) and flexible molecule, existing in an ensemble of conformational states which are regulated in part by the myosin light chain subunits (MLRV and MYL3) changing the angles about three hinge regions in MYH6(Brown et al., 2011; Pylypenko and Houdusse, 2011). Data from fluorescence and EPR based spectroscopic studies reveal more conformational diversity of the myosin complex than is currently available in models derived from crystallography or cryo-EM(Thomas et al., 2009), highlighting the need for data acquired by complementary techniques capable of sampling the complex under dynamic and physiological conditions(Knowles et al., 2008; Shih et al., 2000).
Figure 1. Cross-linking derived model for sarcomere protein complexes.
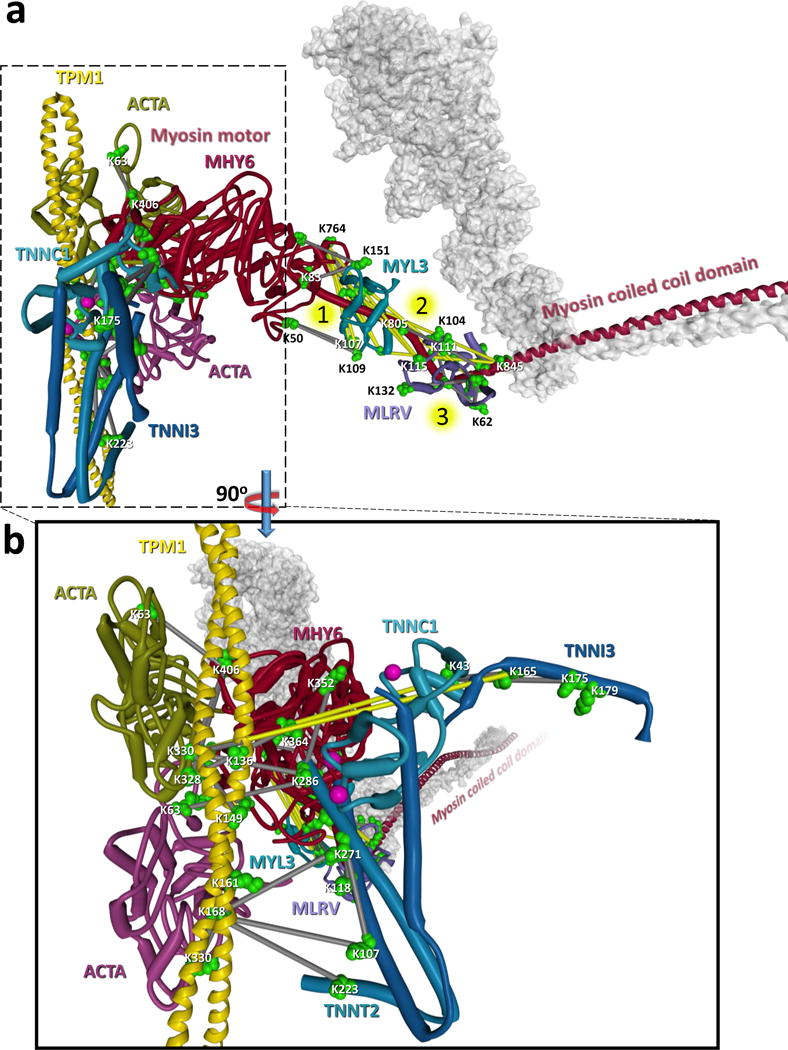
a) Structural model of sarcomere protein interactions including the thick filament proteins: myosin motor (MYH6, dark red with dimer shown as transparent grey surface), myosin essential light chain (MYL3, teal blue), myosin regulatory light chain (MLRV, light purple) and the thin filament proteins: actin (ACTA green and dark purple), tropomyosin (yellow) and the three troponin subunits (TNNC1 light blue, TNNT2, blue, TNNI3 dark blue). The model utilizes structural information from PDB structures: 5H53, 5CJ1, 4XA4, 5CHX, 5CJ0, 1J1E, and 5JLH of a homo-dimer of MYH6 indicating the motor domain (Residues 1–780, dark red) and the coiled coil tail domain (residues 842–1938, dark blue). The second MYH6 molecule is represented by a grey semi-transparent structure. Cross-linked sites are indicated by green space filled residues and links between residues are displayed as grey bars (Cα-Cα distance < 42 Å) or yellow bars (Cα-Cα distance > 42 Å). A total of 10 cross-links (2 MYH6-MLRV and 8 MYH6-MYL3) exceed 42 Å. These links are clustered around three flexible hinge regions indicated by numbered yellow circles in the MYH6 light chain binding domain. It is likely that these links are representative of the conformational flexibility of the myosin motor and formed from a different conformation than the rigor state model shown here. Forty-six homo-dimer cross-linked peptide pairs were also identified in the coiled-coil domain of MYH6 (see Dataset S1). b) Zoomed inset of a 90° rotation of the structure shown in a. Calcium ions bound to TNNC1 are shown as pink spheres. The yellow links between K165 of TNNI3 and K330 of ACTA and K43 of TNNC1 and K330 of ACTA exceed the possible cross-linkable distance and are not compatible with the calcium saturated structure of troponin (PDB: 1J1E) and are instead indicative of the calcium depleted state of troponin in which TNNI3 changes conformation to interact with ACTA effectively blocking the myosin interaction site(Cordina et al., 2014; Takeda et al., 2003). See also Dataset S1 and Figure S5.
Cross-linking captures structural information on the ensemble of conformations as evident with 10 links (colored yellow) involving MLRV or MYL3 and MYH6 clustered around the flexible three hinge region of MYH6 (Fig. 1a). These links are inconsistent (Cα-Cα > 42 Å) with the rigor state model (Fujii and Namba, 2017) in Fig. 1a, suggesting they represent a different conformation of the MYH6 machinery. Three of the over-distance links between MYH6 and MLRV do fall within an acceptable distance when mapped to a conformation of MYH6 in the OFF state. There are many known disease-related mutations that occur throughout MYH6, and troponin subunits which are linked to cardiomyopathies(Chang et al., 2008; Moore et al., 2012). In order for myosin to bind actin and induce muscle contraction, calcium is required to bind to the calcium binding subunit of troponin (TNNC1) which causes a conformational shift moving the inhibitory subunit of troponin (TNNI3) and exposing the myosin binding site on actin(Cordina et al., 2014; Takeda et al., 2003). In contrast to skeletal muscle, cardiomyocytes rely on release of intracellular calcium from the sarcoplasmic reticulum for contraction to occur. This calcium induced conformational shift is illustrated by the cross-links observed between K165 TNNI3 and K330 of alpha actin (ACTA) and K43 of TNNC1 and K330 of ACTA, which are not possible (Cα-Cα distance > 42 Å) with the calcium saturated structural model of troponin (Fig. 1b), and would only be possible in a calcium depleted state for which no structural model is available in the PDB. Furthermore, the links between the troponin subunits and K330 of ACTA are mutually exclusive with seven links identified between MYH6 and ACTA indicating the cross-linking of different conformations within the sarcomere contraction cycle. The cross-linked residues identified between these sarcomeric proteins will serve as probes for specific protein conformations and interactions in future quantitative XL-MS (qXL-MS) studies, enabling dynamic measurements between diseased and healthy heart tissue.
Due to the extraordinary energy requirements of the heart, cardiomyocytes contain the highest concentration of mitochondria of any cell in the body, comprising ~30% of the cellular volume(Schaper et al., 1985). The mitochondrial oxidative phosphorylation complexes (OXPHOS) provide chemical energy for the heart through generation of ATP. The electron transport chain, consisting of OXPHOS complexes I–IV, transfer electrons from NADH to reduce molecular oxygen and in the process, generate a proton gradient across the mitochondrial inner membrane to power ATP synthase (complex V). In total 208 cross-linked Lys residues involved in 319 site-to-site linkages were identified across the five OXHPOS complexes (Fig. S6). OXPHOS complexes can be extracted as supercomplexes comprised of various combinations of CI-CV(Milenkovic et al., 2017). Supercomplexes consisting of CI, CIII and CIV are referred to as respirasomes, as they can carry out respiration in the presence of ubiquinone and cytochrome c, and have been of intense interest in the structural biology field(Milenkovic et al., 2017).
Recently several cryo-EM structures of respirasomes consisting of CICIII2CIV have been made available(Gu et al., 2016; Letts et al., 2016; Sousa et al., 2016; Wu et al., 2016). Beyond this, respirasomes have been proposed to assemble into even higher order structures and respiratory strings(Bultema et al., 2009; Gu et al., 2016; Letts et al., 2016; Wu et al., 2016). Such higher order complexes are challenging to study as membrane extraction is likely to disrupt increasingly larger structures. However, XL-MS provides a complementary approach to study these complexes as protein interaction information is preserved through covalent bond formation on complexes within their native environment.
XL-MS experiments with murine heart tissues described here resulted in identification of multiple links between OXPHOS complexes defining the existence of larger supercomplex assemblies consistent with the circular Sus scrofa model proposed by Wu et al. (Wu et al., 2016) and single-particle EM results suggesting the presence of CI2III2 in potato(Bultema et al., 2009). For instance, lysine sites linking CI and CIII (NDUA2 K13, K75 and K98 to QCR2 K250) and between CI and CIV (NDUA9 K189 to COX5A K68) in murine heart XL-MS data were compared to homologous sites on a recent cryo-EM-derived structural model for the purified Sus scrofa CICIII2CIV supercomplex (Wu et al., 2016). The observed links between CI and CIII agree with the supercomplex model (red lines in Fig. 2 a); however, the cross-linked sites between CI and CIV are well beyond possible linking distance when mapped to the CICIII2CIV model (yellow line in Fig. 2 a). NDUA9 exists in the matrix arm of CI, whereas in all current CICIII2CIV supercomplex models, CIV is located on the opposite end of the CI membrane arm. However, cross-linked site-directed docking of CICIV with CICIII2CIV resulted in a circular model of the supercomplex comprised of CI2CIII2CIV2 with a two-fold axis of symmetry centered on the CIII dimer (Fig. 2 b). It is certainly possible and likely that a variety of supercomplex assemblies exist within heart tissue, and while the circular model shown in Fig. 2 b is consistent with all inter-complex links between CI, CIII and CIV, we cannot rule out the possibility that a different conformation resulted in the observed CI–CIV linkage. Regardless the conformation, the link between NDUA9 and COX5A can be used as a structural probe for the CI–CIV interaction in future quantitative studies aimed at supercomplex assembly. Thus, XL-MS data from murine heart tissue supports the proposed existence of higher order supercomplex assemblies consisting of two copies each of CI, CIII, and CIV within heart tissue which could function more efficiently during electron transfer due to reduced distance for diffusion of intermediate electron carriers CoQ and cytochrome c between complexes(Wu et al., 2016). Importantly, each of the more than 2000 links comprising Fig. S4 and dataset S1 can now be used to quantify supercomplex assemblies and many other interactions in tissues from normal and failing heart tissues to elucidate their roles in mitochondrial function and heart disease. As Wu et al. indicate, further investigations with non-invasive methods are required to better visualize how respiratory strings and large complex assemblies are formed on the inner mitochondrial membrane. Although the XL-MS method presented here cannot be considered non-invasive, the systems-level information derived by this approach can help fulfill this need.
Figure 2. Cross-linking derived model for respirasome supercomplex CI2CIII2CIV2.
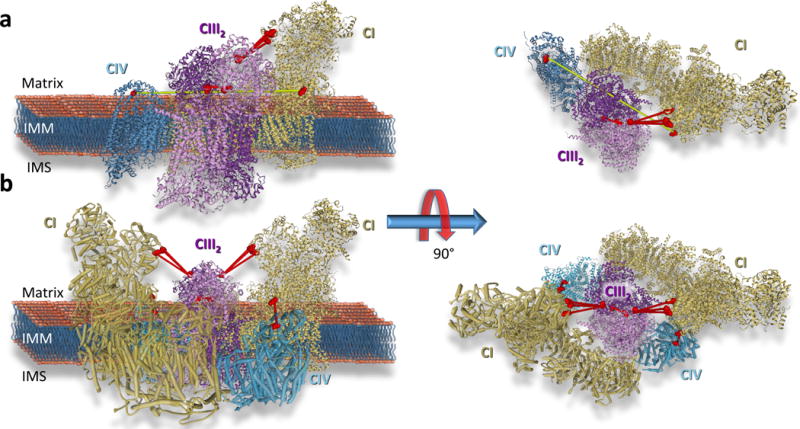
a) Cryo-EM derived structure of respirasome CICIII2CIV (PDB: 5GUP) with cross-links identifying interactions between CI (gold ribbon) and CIII (purple ribbon) (NDUA2 K13, K75 and K98 linked to QCR2 K250) CIII homodimer (QCR2 K159 linked to QCR2 K159) and CI and CIV (teal blue ribbon) (COX5A K189 linked to NDUA9 K68) displayed. Cross-linked sites are shown as space filled residues. Residues connected by red lines agree with the structure while residues connected by a yellow line exceed the maximum cross-linkable distance (42 Å). b) Structure of a circular representation of the respirasome CI2CIII2CIV2 which agrees with all observed cross-linked sites. See also Dataset S1 and Figure S6.
As was demonstrated with cultured cells, quantitative measurements of cross-linked sites can reveal conformational and interaction changes that underpin relevant functional changes in phenotypic and pharmacological comparisons (Chavez et al., 2016a; Chavez et al., 2016b; Zhong et al., 2017).
Discussion
Here we show that XL-MS enables systems structural biology studies at the tissue level. For disease states that cannot readily be replicated in cultured cells, this systems structural biology method provides unique molecular insight on relevant protein conformations and interactions in tissues. Each identified cross-linked peptide pair provides a useful molecular probe that can be used to quantify changes in protein conformations or protein-protein interactions in tissues and possibly help identify molecular interactions that can serve as new targets for mitochondrial-targeting therapies for cardiovascular or other diseases.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, James E. Bruce (jimbruce@uw.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal model
This study utilized four mice, strain C57BL/6NCrl (IMSR_CRL:27). Two female mice were harvested at 14.4 weeks of age (DOB: 10/17/2017, harvested 1/26/2017) each weighing over 24 g with no signs of illness. Two male mice were harvested at 12 weeks of age (DOB: 04/28/17, harvested 07/21/17) weighing 26 g with no signs of illness. Mice from both harvest dates were littermates housed in the same cage (group housing all C57Bl6/6Ncrl) and chosen randomly into the experimental group. Mice were maintained on a standard rodent diet and water available ad libitum in a vivarium with a 12 h light/dark cycle at 22°C. All procedures involving animal use were performed with the approval of Institutional Animal Care and Use Committee of the University of Washington.
METHOD DETAILS
Isolation of heart tissue
Mice were deeply anesthetized with pentobarbital at dose > 270mg/kg intraperitoneally. Deep anesthesia was confirmed by lack of response to toe pinch and decreased respiratory rate. The chest cavity was opened, and the heart was rapidly excised and placed into 5ml of ice-cold mitochondrial isolation medium (MIM) buffer (300 mM sucrose, 10 mM HEPES, 0.2 mM EDTA, pH 7.4). All traces of blood were rinsed away with fresh MIM buffer. The aorta, atria, and any additional tissue were removed. The heart was transferred to a pre-chilled 60mm dry petri dish and maintained on ice. The heart tissue was minced with a razor blade on the dry petri dish to achieve a homogenous tissue size distribution of approximately 1 mm cubes.
Synthesis of chemical cross-linker
The PIR cross-linker BDP-NHP(ref), is a peptide based molecule synthesized by solid phase peptide synthesis. The peptide sequence is: Succinate-Asp-Pro-Lys(Pro-Asp-Succinate)-Lys(Biotin)-Gly. For this study it was obtained from AnaSpec synthesized on Clt resin. To form the reactive cross-linker molecule the succinate groups are converted to N-hydroxyphthalamide (NHP) esters. To accomplish this, the BDP on resin is reacted with a twelve-fold molar excess of trifluoroacetate N-hydroxyphthalamide ester (TFA-NHP). TFA-NHP is synthesized by dissolving 5.86 g of NHP in a four-fold molar excess (20 mL) of trifluoroacetic anhydride in a 50 mL round bottom flask. The reaction is carried out for 1.5 h under an atmosphere of dry N2 gas with constant mixing by a magnetic stir bar. After 1.5 h, excess TFA is removed using a rotary evaporator set to 30 mbar and a water bath set to 60°C. After two hours of rotary evaporation, the water bath is exchanged for an ice bath to crystalize the TFA-NHP. The product is a white crystalline solid obtained in quantitative yield. To synthesize the active form of the cross-linker BDP-NHP, the resin with cross-linker bound is swollen with a minimal volume (~4 mL/g resin) of dimethyl formamide (DMF). A twelve-fold molar excess of NHP (3.11 g for 1 g of Clt resin containing 1 mmole BDP) is dissolved in 10 mL of dry pyridine and added to the resin. The reaction is carried out for 20 min with constant mixing at room temperature. After 20 min the reaction mixture is loaded onto a Poly-Prep chromatography column (Bio-Rad) and the liquid is removed by vacuum filtration. The resin is then washed extensively with DMF (5 × 10 mL) followed by 5 × 5 mL washes with dichloromethane (DCM). The cross-linker is then cleaved from the resin by incubating with 5 mL of 95% TFA, 5% DCM for 1h. Following cleavage, the BDP-NHP is precipitated in diethyl ether, pelleted by centrifugation and dried by vacuum centrifugation. The product is then weighed and dissolved in dimethyl sulfoxide (DMSO) at a concentration of 200–300 mM and stored at −80°C until use. The chemical structure of BDP NHP is shown in Figure S1
Chemical cross-linking
Minced mouse heart tissue was centrifuged at 1500 g for 3 min at 4°C and supernatant was removed. Tissue was suspended in 0.25 mL of 170 mM Na2HPO4 pH 8.0. The PIR cross-linker BDP-NHP (d0 light or d8 heavy isotope forms) was added to final concentration of 10 mM from a 258 mM stock solution in DMSO. The sample was mixed on a Thermomixer at 800 rpm for 30 min at room temperature. The tissue was then centrifuged at 1500 g for 3 min and the supernatant was removed. Cross-linked tissue was then either frozen at −80°C or subjected to further processing for mitochondrial isolation as described below.
Mitochondrial isolation from XL heart tissue
Cross-linked heart tissue was suspended in 4 mL of MIM buffer containing 4 mg/mL fatty acid free bovine serum albumin (BSA) and transferred to a glass homogenization tube. The tissue was homogenized using 4–6 passes at 1200–1400 rpm with a teflon pestle. The homogenate was transferred to a 15 mL tube and centrifuged at 800 g for 10 min at 4°C to pellet nuclei and tissue debris. The supernatant was transferred into two 2 mL tubes and centrifuged at 8000 g for 15 min at 4°C. The supernatant was removed by aspiration and the two mitochondrial pellets were each suspended in 1 mL MIM buffer and combined to a single tube. The sample was then centrifuged at 8000 g for 15 min at 4°C and the supernatant removed to obtain the mitochondrial pellet.
Protein extraction from XL heart tissue
Frozen cross-linked mouse heart tissue was transferred with the aid of 0.5 mL of 0.1 M NH4HCO3, to a stainless steel cryogrinding jar cooled to −196°C with liquid nitrogen. Using a Retsch MM 400 mixer mill, the sample was cryoground for five three minute cycles at 30 Hz and cooled with liquid N2 between cycles. The resulting frozen powder was transferred to a 15 mL tube and 1 mL of 8 M urea in 0.1 M tris pH 8.0 was added. Sample viscosity was reduced by sonication using a GE – 130 ultrasonic processor, followed by reduction and alkylation of cysteine residues by incubation with 5 mM tris(2-carboxyethyl)phosphine (TCEP) (Fisher Scientific) for 30 minutes followed by a 45 minute incubation with 10 mM iodoacetamide (Fisher Scientific). To reduce the urea concentration to less than 1 M the samples were diluted by a factor of 10 with fresh 0.1 M tris buffer pH 8.0. The protein concentration was measured using the Pierce Coomassie protein assay (Thermo). A fraction of the total protein content (0.37 mg) was set aside for generation of a stage 1 database (described below) while the rest of the protein in the sample was digested with a 1:200 ratio of sequencing grade modified trypsin (Promega) to protein and was incubated at 37°C for 18 hrs.
Enrichment of XL peptide pairs
Digested peptides were desalted with a C18 Sep-Pak cartridge, and the eluted peptides were dried to completion before being resuspended in SCX buffer A [7 mM KH2PO4, (pH 2.6), 30% (vol/vol) ACN]. Resuspended peptides were injected into a Phenomenex Luna SCX column and were fractionated using a 97.5-min gradient of buffer B [7 mM KH2PO4 (pH 2.6), 30% (vol/vol) ACN, 350 mM KCl] as follows: 0% buffer B at 0 min, 5% B at 7.5 min, 60% B at 47.5 min, 100% B at 67.5 min, 100% B at 77.5 min, 0% B at 77.51 min, and 0% B at 97.5 min. Fractions were taken every 5 min starting at 17.5 min and were pooled into six pools as follows: fractions 1–5, fractions 6–7, fraction 8, fraction 9, fraction 10, and fractions 11–14. Fraction pools were dried to a final volume of ∼2 mL in a vacuum centrifuge, and the pH was adjusted to 8.0 with 1.5 M NaOH. Pooled cross-linked peptides were enriched by the addition of 200 μL of monomeric avidin bead slurry (Ultralink; Pierce) and were incubated at room temperature on an orbital shaker at maximum speed. Cross-linked peptides were washed with 100 mM ammonium bicarbonate, pH 8.0, and eluted with 70% (vol/vol) ACN/1% formic acid before being dried to completion in a vacuum centrifuge and stored at −80 °C un\l analyzed.
LC-MS3 analysis of XL peptide pairs
Samples containing PIR cross-linked peptides were analyzed in technical triplicate by liquid chromatography mass spectrometry using a Waters NanoAcquity UPLC coupled to a Thermo Velos-FTICR mass spectrometer and a real-time adaptive, targeted mass spectrometry method developed for PIR cross-linked peptides (ReACT). Peptides were loaded onto a 3 cm × 100 μm inner diameter fused silica trap column packed with a stationary phase consisting of MichromMagic C8, 5 μm diameter, 200 A pore size particles (Bruker) with a flow rate of 2 μL/min of mobile phase consisting of 98% solvent A (H2O containing 0.1% formic acid) and 2% solvent B (ACN containing 0.1% formic acid) for 10 minutes. Peptides were then fractionated over a 60 cm × 75 μm inner diameter fused silica analytical column packed with Michrom Magic C8, 5 μm diameter, 100 Å pore size particles by applying a linear gradient from 95% solvent A, 5% solvent B to 60% solvent A, 40% solvent B over either 120 or 240 minutes at a flow rate of 300 nL/min. Eluting peptide ions were ionized by nano-electrospray ionization by applying a positive 2 kV potential to a laser pulled spray tip at the end of the analytical column. The Velos-FTICR mass spectrometer was operated utilizing ReACT where a high resolution MS1 scan is acquired in the ICR cell at a resolving power of 50,000 at 400 m/z. Ions with a charge state of four or greater were selected for low energy CID fragmentation using a normalized collision energy (NCE) of 25 followed by high resolution MS2 analysis in the ICR cell at a resolving power of 50,000 at 400 m/z where an “on-the-fly” check of the observed fragment ion masses against the PIR mass relationship (Mass Precursor = Mass Reporter Ion + Mass Peptide 1 + Mass Peptide 2) is performed. Masses that satisfied the PIR relationship within a tolerance of 20 ppm mass error triggered subsequent low resolution MS3 analyses of the released cross-linked peptide ions in the Velos ion trap mass analyzer operated at normal unit resolution. For details on processing of data generated on the Velos-FTICR see the section: Database searching of ReACT MS3 spectra below.
Generation of a stage 1 database of putative cross-linked proteins
100 μL of UltraLink monomeric avidin slurry was added to 0.37 mg of cross-linked protein extracted from PIR cross-linked heart tissue and incubated at room temperature for 30 min. The monomeric avidin beads were washed 5 times with 1mL of 0.1 M NH4HCO3 pH 8.0 before eluting PIR reactive proteins using 100uL of 8M urea containing 2 mM biotin in 0.1 M NH4HCO3 pH 8.0. Disulfide bonds in the eluted proteins were reduced with 5 mM TCEP for 30 minutes followed by alkylation with 10 mM iodoacetamide for 45 minutes. Samples were diluted 10× with 0.1 M NH4HCO3 pH 8.0 before overnight digestion with a 1:200 ratio of trypsin at 37°C. The peptide samples were then desalted using C18 SepPak cartridges, followed by concentration and removal of acetonitrile by vacuum centrifugation using an EZ2-Plus evaporator.
The sample volume was adjusted to 100 μL with 0.1% formic acid before being analyzed by data dependent LC-MS/MS using an Easy-nLC (Thermo) coupled to a Q-Exactive Plus mass spectrometer (Thermo). Peptides were separated by reversed-phase chromatography using in-house packed C18 columns. Peptides were loaded onto a 3 cm × 100 μm inner diameter fused silica trap column packed with a stationary phase consisting of ReproSil C18, 5 μm diameter, 200 Å pore size particles (Dr. Maisch GmbH) with a flow rate of 2 μL/min of mobile phase consisting of 98% solvent A (H2O containing 0.1% formic acid) and 2% solvent B (ACN containing 0.1% formic acid) for 10 minutes. Peptides were then fractionated over a 60 cm × 75 μm inner diameter fused silica analytical column packed with ReproSil C18, 5 μm diameter, 100 Å pore size particles (Dr. Maisch GmbH) by applying a linear gradient from 90% solvent A, 10% solvent B to 60% solvent A, 40% solvent B over 120 minutes at a flow rate of 300 nL/min. Eluting peptide ions were ionized by nano-electrospray ionization by applying a positive 2 kV potential to a laser pulled spray tip at the end of the analytical column. The Q-Exactive Plus mass spectrometer operated using a data dependent analysis method consisting of a MS1 scan at 70,000 resolving power at m/z 200 followed by MS2 scans at 17,500 resolving power on the 20 most abundant ions in the MS1. Ions selected for MS2 were isolated using a 1.6 m/z isolation window, and fragmented by higher-energy collisional dissociation (HCD) using a normalized collision energy of 27. Additional MS2 settings included an automatic gain control (AGC) setting of 50,000 ions, a maximum ion accumulation time of 50 ms, charge state exclusion settings to not select ions with charge states of one, more than eight or unassigned and a dynamic exclusion time of 30 s.
Mass spectral data acquired on the Q-Exactive plus was searched against the mouse UniProt database containing both forward and reverse protein sequences (downloaded 01/07/2017, containing 33,670 total sequences) using Comet (version 2016.01 rev. 2)(Eng et al., 2013). Default parameters were used unless stated otherwise. The mass error tolerance was set to 20 ppm for the precursor mass allowing for standard 13C error offsets (−1/0/1/2/3). Carbamidomethylated cysteine was specified as a static modification while methionine oxidation and lysine acetylation were included as variable modifications. Resulting peptide spectrum matches were filtered at 1% FDR. A total of 13,619 peptide sequences were identified corresponding to 2063 proteins. The stage 1 database used to search the ReACT generated MS3 spectra, as described below, consisted of the forward and reverse sequences of these 2063 proteins.
Database searching of ReACT MS3 spectra
Comet was used to search the MS3 spectra generated from ReACT against the stage 1 database containing the forward and reverse sequences for 2063 putatively cross-linked proteins (4126 total sequences) using the following parameters: 20 ppm mass error tolerance for the precursor mass allowing for standard 13C error offsets. Each peptide sequence was required to contain the residual PIR cross-linker mass modification on Lys residues. The residual cross-linker mass differed for the full heart sample cross-linked with the light isotope cross-linker d0-BDP (197.032422 Da) and the post-XL isolated mitochondria sample cross-linked with heavy isotope labeled d8-BDP (201.0575293 Da). Carbamidomethylated cysteine was specified as a static modification, while methionine oxidation and lysine acetylation were included as variable modifications. Fragment ion tolerance was set to 1.005 Da. Peptide spectral matches (PSMs) to the MS3 spectra were mapped back to the PIR mass relationships identified in the MS2 spectra during the ReACT analysis. Comet assigns each PSM an expectation value, or E-value, which is a statistical measure that standardizes the reporting of peptide identifications. The Comet E-value is calculated by applying a linear least-squares regression on the log transform of the cumulative distribution function of the histogram of the cross-correlation score (xcorr) and extrapolating the point at which the top scoring peptide intersects the least-squares fit line(Eng et al., 2013). The E-value can be interpreted as the number of peptides that are expected to score as well as the top scoring peptide by chance in the search(Eng et al., 2008). Based on the principle described by Trnka et al. that cross-linked peptide pair identifications are only as confident as the worst scoring of the two peptides(Trnka et al., 2014), an E-value threshold of 0.2 or less was applied to the greater E-value for each cross-linked peptide pair. This resulted in data set with an estimated FDR of 0.76% (81/10600) at the PSM level, 2.4% (64/2663) at the non-redundant peptide pair level, and (63/2026) 3.1% FDR at the non-redundant site-to-site level. Here, non-redundant cross-linked peptide pairs are defined as each pair of peptides with a unique primary sequence, including unique modifications. For example the cross-linked peptide pair AVGEK189EVR_K68GMNTLVGYDLVPEPK and AVGEK189EVR_K68GM[147.04]NTLVGYDLVPEPK are counted as two non-redundant pairs due to the site of oxidation (indicated by the mass 147.04 Da) on the third residue of the second peptide. Both of these peptide pairs identify the same Lys-Lys linkage and collapse to a single entry in the reported in the 2026 non-redundant Lys-Lys pairs. A PTM, such as methionine oxidation, results in a different mass and so the modified peptide pair analyzed and identified independently in the LC-MS workflow. Modified cross-linked peptide pairs contain unique masses, are analyzed separately, and represent unique probes that could be useful for studying the structures of the linked proteins. PTMs such as methionine oxidation could alter a protein structure or its interactions and in that sense should be listed separately so that future studies using cross-linked peptide pairs as probes for conformations or interactions can take that into account. For more details on the estimation of FDR see the Quantification and Statistical Analysis section below.
Structural analysis and modeling
For assembly of the sarcomere protein complex shown in Fig. 1, structural information from the PDB models: 5H53, 5CJ1, 4XA4, 5CHX, 5CJ0, 1J1E, and 5JLH was utilized. Clustal Omega v1.2.4 was used to align the protein sequences in the PDB structures to the homologous mouse protein sequences obtained from UniProt. A series of structural superposition and manual docking utilizing cross-link distance constraints was carried out using ICM Molsoft MolBrowser Pro v. 3.8–6. PDB 5H53 contains structural information for the myosin motor (MYH6), the essential myosin light chain (MYL3), the regulatory myosin light chain (MLRV) and two monomers of actin (ACTA). The coiled-coil domain for MYH6 was modeled by superposition of overlapping structural data from PDB files 5CJ1, 5CJ0, 5CHX and 4XA4 and aligning this with residues 836–845 of myosin in 5H53. Tropomyosin (TPM1) was incorporated into the model by aligning the actin chains of 5JLH with those in 5H53. Structural information for the troponin complex proteins TNNC1, TNNT2, and TNNI3 was obtained from PDB 1J1E. Data from cross-links (TPM1 K168- TNNT2 K223, TPM1 K168 - TNNT2 K271, TPM1 K168 - TNNI3 K107, TNNT2 K286 – K330 ACTA, TNNI3 K165 – ACTA K330, TNNC1 K43 – ACTA K330, and TNNT2 K286 – MYH6 K352) between the three troponin proteins and TPM1, ACTA and MYH6 were utilized as distance constraints to dock 1J1E onto the assembled myosin, actin, tropomyosin complex by maximizing the number links that fell below the distance constraint of 42 Å. The resulting docked structure was evaluated for consistency with published models for the interaction of troponin with the thin filament(Cordina et al., 2014; Takeda et al., 2003).
The heterodimeric complex of the trifunctional enzyme proteins ECHA and ECHB was constructed by generating homology models for ECHA and ECHB using Phyre2(Kelley et al., 2015) on intensive mode. Rigid body molecular docking of the ECHA and ECHB models was accomplished using PatchDock(Schneidman-Duhovny et al., 2005) utilizing the cross-links between ECHA and ECHB with a distance constraint of 42 Å. A total of 3 models resulted from PatchDock and the top scoring model is displayed in Fig S2.
The model of the circular CI2CIII2CIV2 supercomplex was assembled using the atomic coordinates from PDB 5GUP. CI was docked to the CIII using the three links between NDUA2 and QCR2 (NDUA2 K13, K75, and K98 to QCR2 K250). A second copy of CI was superimposed symmetrically about the CIII3 dimer and CIV was docked into the CI2CIII2 complex using the cross-link between COX5A K68 and NDUA9 K189 as a distance constraint with an upper limit of 42 Å. A total of 16 models resulted from PatchDock and the top scoring model is displayed in Fig 2b.
QUANTIFICATION AND STATISTICAL ANALYSIS
Estimation of false discovery rate for ReACT MS3 identifications
Comet generated PSMs to MS3 spectra acquired by ReACT were mapped back to the PIR relationship (Mass Precursor = Mass Reporter Ion + Mass Peptide 1 + Mass Peptide 2) identified during the LC-MS acquisition. This step is performed entirely based on scan number as MS3 spectra on released peptides are only generated following the acquisition of an MS2 spectrum that satisfies the PIR mass relationship with a mass tolerance of less than 20 ppm. Initially PSMs are filtered to only include those with a Lys residue internal to the sequences (not C-term Lys) that is modified by the cross-linker (197.032422 Da). All PSMs containing an internal cross-linked residue, irrespective of E-value, are matched to the PIR relationships detected during the MS acquisition. This results in four possibilities for cross-linked peptide pair sequences: 1) both peptides are assigned forward sequences (fwd_fwd), 2) first peptide assigned a forward sequence and second peptide assigned a reverse sequence (fwd_rev), 3) first peptide assigned a reverse sequence and second peptide assigned a forward sequence (rev_fwd), and 4) both peptides assigned reverse sequences (rev-rev). The complete unfiltered list of assignments is available in Dataset S1. Following matching of PSMs to the PIR relationships, the assigned peptide pairs are sorted in ascending order according to the worst scoring (largest E-value) of the two PSMs. This is the most conservative approach relying on the principle described by Trnka et al.(Trnka et al., 2014) that the assignment of a cross-linked peptide pair is only as good as the worst scoring PSM for the pair. With the sorted list of cross-linked peptide pair assignments, the false discovery rate is estimated by taking the ratio of the sum of decoy assignments to target assignments: (fwd_rev + rev_fwd + rev_rev)/(fwd_fwd). For the current study a maximum E-value threshold of 0.2 was applied resulting in an FDR of 0.76% at the PSM level. As recently highlighted by Fisher and Rappsilber(Fischer and Rappsilber, 2017), FDR for cross-linked peptides can be estimated at different levels, namely: PSM level, non-redundant peptide pair level, non-redundant site-site level and protein-protein level. As redundancy is removed between each level correct assignments will tend to collapse on each other while decoy assignments tend to accumulate leading to an increase in the estimated FDR at each level. One way to control for this is to calculate FDR for cross-linked peptide pairs that have multiple PSMs, as decoy assignments tend to be random and only have single PSMs, the FDR will drop while maintaining a large percentage of correct assignments. This is illustrated in Figure S1d. Beyond evaluating the FDR on the entire data set, the FDR can also be estimated for individual subsets of cross-linked peptide pairs, namely intra-protein, inter-protein, and homo-dimer cross-links. Homo-dimer cross-links are a special case where two peptides with overlapping sequences, which originate and occur only once within a given protein sequence are cross-linked together. These homo-dimer cross-links serve to unambiguously identify homo-oligomeric protein interactions. As can be seen in Figure S3a–d, all decoy assignments passing the E-value threshold occur in the inter-protein class resulting in effectively 0% FDR in the intra-protein and homo-dimer link classes at our E-value threshold of less than or equal to 0.2. While slightly enriched in decoy hits, the estimated FDR for the inter-protein linkages is also acceptably low at 3.9% at the PSM level.
Supplementary Material
Highlights.
XL-MS enables systems structural biology studies at the tissue level.
Identification of 2026 Lys-Lys linkages from mouse heart tissue.
Structural information on sarcomere proteins, indicating conformational dynamics.
Cross-links support presence of OXPHOS supercomplex assembly in heart.
Acknowledgments
We thank all members of the Bruce and Tian labs as well as the UWPR for their advice and helpful discussions. This work is supported by the NIH through grant numbers 5R01-GM086688, 2R01-HL110349, 5U19AI107775, 1R01-GM122864 and in part by the UW Proteome Resource UWPR95794.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions
Conceptualization, J.D.C, C.F.L, A.C., R.T and J.E.B; Methodology, J.D.C, C.F.L, A.C., R.T and J.E.B; Formal Analysis, J.D.C and A.K.; Investigation, J.D.C, C.F.L, A.C., and A.K.; Writing – Original Draft, J.D.C, A.C., A.K., and J.E.B; Writing – Review & Editing, J.D.C, A.K. and J.E.B; Visualization, J.D.C, J.E.B; Supervision R.T and J.E.B; Funding Acquisition, R.T and J.E.B.
DATA AND SOFTWARE AVAILABILITY
The complete set of mass spectrometry data is available through Proteome Exchange PRIDE Archive accession number PXD007673.
References
- Back JW, Hartog AF, Dekker HL, Muijsers AO, de Koning LJ, de Jong L. A new crosslinker for mass spectrometric analysis of the quaternary structure of protein complexes. J Am Soc Mass Spectrom. 2001;12:222–227. doi: 10.1016/S1044-0305(00)00212-9. [DOI] [PubMed] [Google Scholar]
- Brown JH, Kumar VS, O’Neall-Hennessey E, Reshetnikova L, Robinson H, Nguyen-McCarty M, Szent-Gyorgyi AG, Cohen C. Visualizing key hinges and a potential major source of compliance in the lever arm of myosin. Proc Natl Acad Sci U S A. 2011;108:114–119. doi: 10.1073/pnas.1016288107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bultema JB, Braun HP, Boekema EJ, Kouril R. Megacomplex organization of the oxidative phosphorylation system by structural analysis of respiratory supercomplexes from potato. Biochim Biophys Acta. 2009;1787:60–67. doi: 10.1016/j.bbabio.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Chang AN, Parvatiyar MS, Potter JD. Troponin and cardiomyopathy. Biochem Biophys Res Commun. 2008;369:74–81. doi: 10.1016/j.bbrc.2007.12.081. [DOI] [PubMed] [Google Scholar]
- Chavez JD, Cilia M, Weisbrod CR, Ju HJ, Eng JK, Gray SM, Bruce JE. Cross-linking measurements of the Potato leafroll virus reveal protein interaction topologies required for virion stability, aphid transmission, and virus-plant interactions. J Proteome Res. 2012;11:2968–2981. doi: 10.1021/pr300041t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JD, Eng JK, Schweppe DK, Cilia M, Rivera K, Zhong X, Wu X, Allen T, Khurgel M, Kumar A, et al. A General Method for Targeted Quantitative Cross-Linking Mass Spectrometry. PLoS One. 2016a;11:e0167547. doi: 10.1371/journal.pone.0167547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JD, Schweppe DK, Eng JK, Bruce JE. In Vivo Conformational Dynamics of Hsp90 and Its Interactors. Cell Chem Biol. 2016b;23:716–726. doi: 10.1016/j.chembiol.2016.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JD, Schweppe DK, Eng JK, Zheng C, Taipale A, Zhang Y, Takara K, Bruce JE. Quantitative interactome analysis reveals a chemoresistant edgotype. Nat Commun. 2015;6:7928. doi: 10.1038/ncomms8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JD, Weisbrod CR, Zheng C, Eng JK, Bruce JE. Protein interactions, post-translational modifications and topologies in human cells. Mol Cell Proteomics. 2013;12:1451–1467. doi: 10.1074/mcp.M112.024497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZA, Jawhari A, Fischer L, Buchen C, Tahir S, Kamenski T, Rasmussen M, Lariviere L, Bukowski-Wills JC, Nilges M, et al. Architecture of the RNA polymerase II-TFIIF complex revealed by cross-linking and mass spectrometry. EMBO J. 2010;29:717–726. doi: 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordina NM, Liew CK, Potluri PR, Curmi PM, Fajer PG, Logan TM, Mackay JP, Brown LJ. Ca2+-induced PRE-NMR changes in the troponin complex reveal the possessive nature of the cardiac isoform for its regulatory switch. PLoS One. 2014;9:e112976. doi: 10.1371/journal.pone.0112976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBlasio SL, Chavez JD, Alexander MM, Ramsey J, Eng JK, Mahoney J, Gray SM, Bruce JE, Cilia M. Visualization of Host-Polerovirus Interaction Topologies Using Protein Interaction Reporter Technology. J Virol. 2015;90:1973–1987. doi: 10.1128/JVI.01706-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis RJ. Macromolecular crowding: obvious but underappreciated. Trends Biochem Sci. 2001;26:597–604. doi: 10.1016/s0968-0004(01)01938-7. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Surette MG, Wolf PE, Stock JB, Leibler S. Protein mobility in the cytoplasm of Escherichia coli. J Bacteriol. 1999;181:197–203. doi: 10.1128/jb.181.1.197-203.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, Fischer B, Grossmann J, Maccoss MJ. A fast SEQUEST cross correlation algorithm. J Proteome Res. 2008;7:4598–4602. doi: 10.1021/pr800420s. [DOI] [PubMed] [Google Scholar]
- Eng JK, Jahan TA, Hoopmann MR. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Fischer L, Rappsilber J. Quirks of Error Estimation in Cross-Linking/Mass Spectrometry. Anal Chem. 2017;89:3829–3833. doi: 10.1021/acs.analchem.6b03745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Namba K. Structure of actomyosin rigour complex at 5.2 A resolution and insights into the ATPase cycle mechanism. Nat Commun. 2017;8:13969. doi: 10.1038/ncomms13969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu J, Wu M, Guo R, Yan K, Lei J, Gao N, Yang M. The architecture of the mammalian respirasome. Nature. 2016;537:639–643. doi: 10.1038/nature19359. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles AC, Ferguson RE, Brandmeier BD, Sun YB, Trentham DR, Irving M. Orientation of the essential light chain region of myosin in relaxed, active, and rigor muscle. Biophys J. 2008;95:3882–3891. doi: 10.1529/biophysj.108.131508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitner A, Joachimiak LA, Bracher A, Monkemeyer L, Walzthoeni T, Chen B, Pechmann S, Holmes S, Cong Y, Ma B, et al. The molecular architecture of the eukaryotic chaperonin TRiC/CCT. Structure. 2012;20:814–825. doi: 10.1016/j.str.2012.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letts JA, Fiedorczuk K, Sazanov LA. The architecture of respiratory supercomplexes. Nature. 2016;537:644–648. doi: 10.1038/nature19774. [DOI] [PubMed] [Google Scholar]
- Liu F, Lossl P, Scheltema R, Viner R, Heck AJR. Optimized fragmentation schemes and data analysis strategies for proteome-wide cross-link identification. Nat Commun. 2017;8:15473. doi: 10.1038/ncomms15473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Rijkers DT, Post H, Heck AJ. Proteome-wide profiling of protein assemblies by cross-linking mass spectrometry. Nat Methods. 2015;12:1179–1184. doi: 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- Malmstrom J, Beck M, Schmidt A, Lange V, Deutsch EW, Aebersold R. Proteome-wide cellular protein concentrations of the human pathogen Leptospira interrogans. Nature. 2009;460:762–765. doi: 10.1038/nature08184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milenkovic D, Blaza JN, Larsson NG, Hirst J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017;25:765–776. doi: 10.1016/j.cmet.2017.03.009. [DOI] [PubMed] [Google Scholar]
- Moore JR, Leinwand L, Warshaw DM. Understanding cardiomyopathy phenotypes based on the functional impact of mutations in the myosin motor. Circ Res. 2012;111:375–385. doi: 10.1161/CIRCRESAHA.110.223842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navare AT, Chavez JD, Zheng C, Weisbrod CR, Eng JK, Siehnel R, Singh PK, Manoil C, Bruce JE. Probing the protein interaction network of Pseudomonas aeruginosa cells by chemical cross-linking mass spectrometry. Structure. 2015;23:762–773. doi: 10.1016/j.str.2015.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pylypenko O, Houdusse AM. Essential “ankle” in the myosin lever arm. Proc Natl Acad Sci U S A. 2011;108:5–6. doi: 10.1073/pnas.1017676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth GA, Forouzanfar MH, Moran AE, Barber R, Nguyen G, Feigin VL, Naghavi M, Mensah GA, Murray CJ. Demographic and epidemiologic drivers of global cardiovascular mortality. N Engl J Med. 2015;372:1333–1341. doi: 10.1056/NEJMoa1406656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper J, Meiser E, Stammler G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res. 1985;56:377–391. doi: 10.1161/01.res.56.3.377. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33:W363–367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe DK, Chavez JD, Lee CF, Caudal A, Kruse SE, Stuppard R, Marcinek DJ, Shadel GS, Tian R, Bruce JE. Mitochondrial protein interactome elucidated by chemical cross-linking mass spectrometry. Proc Natl Acad Sci U S A. 2017 doi: 10.1073/pnas.1617220114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe DK, Harding C, Chavez JD, Wu X, Ramage E, Singh PK, Manoil C, Bruce JE. Host-Microbe Protein Interactions during Bacterial Infection. Chem Biol. 2015;22:1521–1530. doi: 10.1016/j.chembiol.2015.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweppe DK, Zheng C, Chavez JD, Navare AT, Wu X, Eng JK, Bruce JE. XLinkDB 2.0: integrated, large-scale structural analysis of protein crosslinking data. Bioinformatics. 2016;32:2716–2718. doi: 10.1093/bioinformatics/btw232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih WM, Gryczynski Z, Lakowicz JR, Spudich JA. A FRET-based sensor reveals large ATP hydrolysis-induced conformational changes and three distinct states of the molecular motor myosin. Cell. 2000;102:683–694. doi: 10.1016/s0092-8674(00)00090-8. [DOI] [PubMed] [Google Scholar]
- Sinz A. Divide and conquer: cleavable cross-linkers to study protein conformation and protein-protein interactions. Anal Bioanal Chem. 2017;409:33–44. doi: 10.1007/s00216-016-9941-x. [DOI] [PubMed] [Google Scholar]
- Sousa JS, Mills DJ, Vonck J, Kuhlbrandt W. Functional asymmetry and electron flow in the bovine respirasome. Elife. 2016;5 doi: 10.7554/eLife.21290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Yamashita A, Maeda K, Maeda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- Tan D, Li Q, Zhang MJ, Liu C, Ma C, Zhang P, Ding YH, Fan SB, Tao L, Yang B, et al. Trifunctional cross-linker for mapping protein-protein interaction networks and comparing protein conformational states. Elife. 2016;5 doi: 10.7554/eLife.12509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Bruce JE. A new cross-linking strategy: protein interaction reporter (PIR) technology for protein-protein interaction studies. Mol Biosyst. 2010;6:939–947. doi: 10.1039/b920876c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Munske GR, Siems WF, Bruce JE. Mass spectrometry identifiable cross-linking strategy for studying protein-protein interactions. Anal Chem. 2005;77:311–318. doi: 10.1021/ac0488762. [DOI] [PubMed] [Google Scholar]
- Thomas DD, Kast D, Korman VL. Site-directed spectroscopic probes of actomyosin structural dynamics. Annu Rev Biophys. 2009;38:347–369. doi: 10.1146/annurev.biophys.35.040405.102118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosi A, Haas C, Herzog F, Gilmozzi A, Berninghausen O, Ungewickell C, Gerhold CB, Lakomek K, Aebersold R, Beckmann R, et al. Structure and subunit topology of the INO80 chromatin remodeler and its nucleosome complex. Cell. 2013;154:1207–1219. doi: 10.1016/j.cell.2013.08.016. [DOI] [PubMed] [Google Scholar]
- Trnka MJ, Baker PR, Robinson PJ, Burlingame AL, Chalkley RJ. Matching cross-linked peptide spectra: only as good as the worse identification. Mol Cell Proteomics. 2014;13:420–434. doi: 10.1074/mcp.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisbrod CR, Chavez JD, Eng JK, Yang L, Zheng C, Bruce JE. In vivo protein interaction network identified with a novel real-time cross-linked peptide identification strategy. J Proteome Res. 2013;12:1569–1579. doi: 10.1021/pr3011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Gu J, Guo R, Huang Y, Yang M. Structure of Mammalian Respiratory Supercomplex I1III2IV1. Cell. 2016;167:1598–1609 e1510. doi: 10.1016/j.cell.2016.11.012. [DOI] [PubMed] [Google Scholar]
- Zheng C, Weisbrod CR, Chavez JD, Eng JK, Sharma V, Wu X, Bruce JE. XLink-DB: database and software tools for storing and visualizing protein interaction topology data. J Proteome Res. 2013;12:1989–1995. doi: 10.1021/pr301162j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong X, Navare AT, Chavez JD, Eng JK, Schweppe DK, Bruce JE. Large-Scale and Targeted Quantitative Cross-Linking MS Using Isotope-Labeled Protein Interaction Reporter (PIR) Cross-Linkers. J Proteome Res. 2017;16:720–727. doi: 10.1021/acs.jproteome.6b00752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.