

Abstract
Informed consent for randomized trials often causes significant and persistent anxiety, distress and confusion to patients. Where an experimental treatment is compared to a standard care control, much of this burden is potentially avoidable in the control group. We propose a “just-in-time” consent in which consent discussions take place in two stages: an initial consent to research from all participants, and a later specific consent to randomized treatment only from those assigned to the experimental intervention. All patients are first approached and informed about research procedures, such as questionnaires or tests. They are also informed that they might be randomly selected to receive an experimental treatment, and that, if selected, they can learn more about the treatment and decide whether or not to accept it at that time. After randomization, control patients undergo standard clinical consent whereas patients randomized to the experimental procedure undergo a second consent discussion. Analysis would be by intent-to-treat, which protects the trial from selection bias, although not from poor acceptance of experimental treatment. The advantages of just-in-time consent stem from the fact that only patients randomized to the experimental treatment are subject to a discussion of that intervention. We hypothesize that this will reduce much of the patient’s burden associated with the consent process, such as decisional burden, confusion and information overload. We recommend well-controlled studies to compare just-in-time and traditional consent, with endpoints to include characteristics of participants, distress and anxiety and participants’ understanding of research procedures.
Keywords: Randomized controlled trials, informed consent, pragmatic trials, anxiety, decisional burden, decisional conflict
Introduction: Subject burdens related to informed consent
In many randomized trials, an experimental intervention is compared against the current standard of care. Such trials include those of drugs (e.g. third line chemotherapy vs. best supportive care), surgery (e.g. standard closure vs. use of mesh), organization of care (e.g. in-patient versus out-patient treatment) and adjunctive therapies (e.g. physical therapy vs. physical therapy plus exercise). In standard interpretations of the Federal Common Rule (45 CFR 46), the traditional informed consent discussion for such trials involves explaining to the patient at least three key points: first, the purpose of the study; second, trial procedures such as randomization as well as any questionnaires, additional visits or tests; third, risks and potential benefits of research interventions, including those of the experimental treatment.1
Researchers often find obtaining informed consent difficult, and observe that it can cause significant and persistent anxiety, distress and confusion to patients. The act of decision-making itself can cause adverse emotional consequences, often described in terms of decisional burden,2 decisional conflict3 and decisional regret.4 Informed consent can create anxiety and confusion over and above that directly associated with decision-making. Consent often takes place at a difficult time in a patient’s journey – such as just before surgery, or just after a cancer diagnosis – and may involve huge amounts of information, with 15 pages being far from an unusual length for a consent form. These two factors commonly lead to information overload, especially since receiving the non-research related information about usual care is already difficult to understand. Consent discussions also require that patients focus intently on risks over and above what would be typical in routine care.
In standard informed consent discussions, patients are told about usual care and about the experimental alternative. Possible benefits of the experimental intervention are then highlighted, while also explaining that the treatment could turn out to be less effective. It is generally accepted that most patients who agree to randomization after receiving information about the trial do so because they like the theoretical benefits of the experimental treatment. Patients who are then randomized to the usual care control group often experience considerable disappointment and frustration: disappointment about not receiving an appealing-sounding experimental intervention and frustration about receiving a great deal of extra information that now turned out to be irrelevant. This disappointment need not imply that a patient has failed to understand research, since many people understand the purpose of research but are personally motivated by benefit. Vignettes illustrating the different types of distress caused by informed consent are given in the online appendix, along with some data from studies that have measured anxiety in patients undergoing consent.
The ethical principle that guides informed consent is that of patient autonomy. Implicit in much of contemporary practice is the view that autonomy is optimized when as much information as possible is given as early as is practicable. We challenge this assumption. Indeed, information overload is defined as the state in which increasing the amount of information decreases the ability to make a rational decision. We hypothesize that this unexamined premise of early, maximum information as a requirement of autonomy not only poses an obstacle to autonomous decision-making, but also fails to minimize avoidable harm—as is required by research regulations—because it is associated with unnecessary burden and distress. We therefore propose an alternative to traditional consent for those randomized trials in which an experimental treatment is compared to usual care control. This approach, which we term “just-in-time” consent, is designed to both reduce patient distress (thus minimizing harm and burdens to subjects) and to enhance patient autonomy by providing information that is more timely and salient in the consent process.
Just-in-time consent
We propose that consent to participating in research can be split from consent to receiving experimental treatment so that consent takes place in two separate stages.
In just-in-time consent, researchers approach potential participants and explain that the clinic or hospital regularly conducts studies to test the value of different treatments. Patients are told that they are being invited to take part in research and that they will be informed about the elements that are relevant to them at each stage of the research. At this first stage, they would be informed about any research procedures, such as answering questionnaires and the use of their medical records, for eligibility determinations or trial endpoints. They are then informed that, if determined to be eligible, they might be randomly selected for an experimental treatment and, if selected for the experimental treatment, they will receive further information, and can make a decision at that time whether to accept this option or receive usual care.
Patients who consent during this first stage of consent and who are found to be eligible are then randomized. If assigned to usual care, there are no further consent discussions related to research, and patients undergo standard clinical consent for usual care. Patients assigned to the experimental treatment have a second consent discussion, in which the intervention is described along with its potential risks and benefits. This second discussion is also an opportunity to emphasize the experimental nature of the allocated treatment in order to ensure that patients understand and appreciate that the intervention is not the “best new” treatment but as yet unproven intervention that is being tested (as a measure to counteract what is sometimes described as the “therapeutic misconception”).5 Patients who decline the experimental treatment receive usual care, although due to the intent-to-treat principle, they are analyzed in the experimental arm.
Figure 1 provides a graphical representation of the study design. Assessment of eligibility occurs before randomization. Depending on the trial, eligibility might require a second visit, but would more commonly be based on the medical record and information received from the patient during the first stage of consent. As eligibility assessment occurs before the second stage consent to experimental treatment, it will not include items specific to experimental treatment, such as an exclusion for patients with an allergy to an experimental drug. However, such exclusions would be rare, and could be incorporated as post randomization exclusions.
Figure 1.
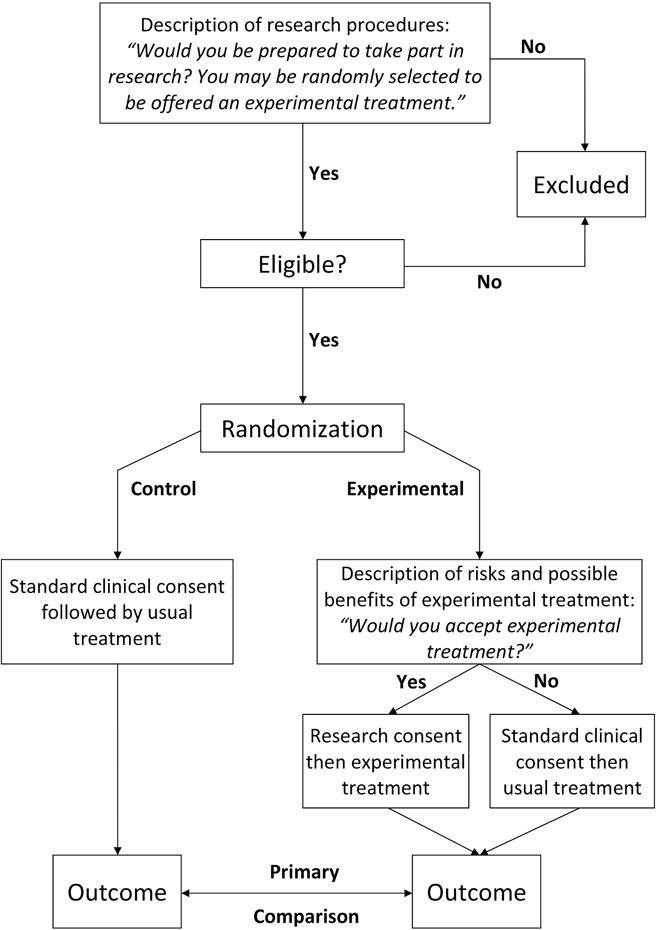
Overview of trial methodology
The advantages of just-in-time consent stem from the fact that only patients randomized to the experimental treatment are subject to a discussion of that intervention. We hypothesize that just-in-time consent will therefore reduce the problems of decisional burden, conflict and regret, and reduce anxiety caused by increased focus on risks. Just-in-time consent may also dramatically reduce two other sources of distress: confusion as to lack of access to the novel treatment and disappointment after assignment to the control group. The control group subjects will be spared a lengthy consent process that covers material that will, for them, be irrelevant and may well cause confusion, frustration and distress.
Note that trialists may choose to inform patients about their assignment and the results of the trial after it is complete. In such case, patients in the control group may well experience some disappointment after learning about their assignment. However, it is reasonable to assume that learning post hoc about a treatment not received several months previously is more tolerable than being told up front about a treatment that could be administered, only to be immediately informed that it will not be available.
A key advantage of just-in-time consent is that, for both groups, it avoids patients being overwhelmed with a large amount of information at one time. Instead of needing to explain research procedures, randomization and the experimental treatment all at once, investigators can start by introducing research procedures and randomization and then at a separate time, and for only half the patients, discuss the experimental treatment. We hypothesize that, by ensuring that the information patients receive is more salient and focused, and by avoiding information overload, just-in-time consent will increase patient autonomy.
The information disclosure for the initial consent to randomization will vary depending on the details of the trial. In the text box we present some generic language to illustrate our approach. Note that the just-in-time consent model does comply with the required “elements of informed consent” in the U.S. Federal Regulations (e.g., 45CFR46.116).6
Just-in-time consent and usual clinical care
One of the key ethical arguments for just-in-time consent is that it follows the good communication procedures generally used in everyday medical practice. It is routine for doctors to make statements to patients such as: “There is nothing to worry about now, but I’ve seen some things that concern me. Let’s run some tests and then we can discuss the results and decide what to do then”. Doctors do not typically lay out all the possibilities up front: you might have disease A, in which case you would need treatment B, which has these side-effects C through K; but you might also have disease X, for which there is treatment Y or Z, and here are the risks and benefits of each; or it might just be nothing”.
Consider the following analogy. Imagine that a new patient presents to a primary care physician, and after a preliminary history and examination, the doctor thinks that the patient has about a 50:50 chance of having high cholesterol. The doctor orders a blood test: if the test comes back negative, the doctor will continue with routine care; if positive, the doctor will explain the result, talk to the patient about treatment options – including a discussion of side effects – and then ask the patient to make a decision about whether to receive treatment or not. In this scenario, the doctor is practicing just-in-time consent. When asked why they practice medicine in this way, doctors will generally talk about avoiding unnecessary anxiety, confusion and information overload.
Comparison with prior proposals
Just-in-time consent has similarities with several prior proposals. Perhaps the most well known is the “Zelen” design,7 which involves consent only for patients randomized to the experimental arm. A modern variant of the Zelen design - but without its ethical problems6 - is the cohort multiple randomized controlled trial (cmRCT) approach8 - now more commonly referred to as “Trials within Cohorts” (TWiCs) design - in which patients are asked to join a longitudinal cohort to provide research data and then randomized to any one of a series of rolling trials that take place sequentially over time. A recent modification has been to include an explicit consent to randomization as part of cohort enrollment.9 The Zelen design has been used in trials of screening10 and supportive care11 published in major journals, and numerous TWiCs cohorts have been established.12
There are several differences between our proposal and those in the prior literature. First, unlike the Zelen design, all patients must consent to the relevant research components that they will be involved in (such as providing data, agreeing to be contacted if randomly selected) before randomization. Second, our approach is not restricted to longitudinal cohorts and an intention to perform multiple trials as in TWiCs: it can be implemented for both TWiCs and single, stand-alone studies. It is also the case that TWiCs can be implemented with a variety of different approaches to consent, with just-in-time consent being just one option. Third, our emphasis is different from the traditional justifications and motivations for Zelen or TWiCs, which have largely been proposed to enhance researchers’ goals. For instance, in the paper introducing the TWiCs design, Relton et al. describe how such a design might solve problems that include low accrual, contamination and the need to compare multiple treatments.8 Our emphasis with the just-in-time consent design is not just that it is an ethically acceptable approach to improving research efficiency, but may also be an ethically superior approach in terms of enhancing patient autonomy and reducing patient burdens. Although some may see the essential components of some TwiCs procedures and the “just-in-time” model as the same, we believe that this difference in emphasis and justification will be important when specific operational details are developed for particular RCTs.
Finally, our focus on the research participants’ autonomy and welfare, unlike the other models, makes the just-in-time consent an empirically testable model. Research should be conducted on the patient-experience of informed consent with a view to determining whether just-in-time consent would reduce participants’ burden and distress while also enhancing their autonomy. For instance, trials utilizing just-in-time consent could be initiated for trials where stakes are relatively low (e.g. usual surgical care vs. usual surgical plus post-operative counseling and support). Endpoints would include the proportion of patients who decline the experimental treatment and the quality of informed consent as assessed by a questionnaire such as the QuIC,13 that measures patient knowledge of research procedures. Additional questionnaires might include items specific to the just-in-time consent design, such as whether patients in the control group felt misled by the two-stage design or whether patients in the experimental arm felt pressured into accepting the experimental treatment because they had already consented to the trial in general. We also recommend well-controlled studies to determine whether just-in-time consent for research is superior to traditional consent, using endpoints such as patient distress and knowledge. It would also be important to compare the characteristics of patients consenting to traditional vs. just-in-time consent.
Applicability of the just-in-time consent design
Just-in-time consent is suitable for many, but by no means all, trials involving experimental treatment. It cannot be used, for instance, where neither comparison arm is standard of care, or where blinding is sought. But a very large number of trials are conducted in which a novel treatment is compared to standard care control, and blinding is either not possible or unwarranted. All of the examples we give in the introduction - third line chemotherapy vs. best supportive care; standard surgical wound closure vs. use of mesh; in-patient versus out-patient treatment; physical therapy vs. physical therapy plus exercise – could be implemented using a just-in-time consent approach.
Internal and external validity
An obvious potential drawback to just-in-time consent would be if a high proportion of patients refused the experimental treatment at the second stage of consent. For instance, if, say, an experimental surgical treatment improves the chance of recovery from chronic back pain from 20 – 30%, but only half of trial patients randomized to the experimental arm agree to surgery, the apparent effect would be half as great, 20% vs. 25%. On the other hand, this could be seen as an advantage of just-in-time consent, as it allows greater insight into the generalizability of a trial results to clinical care. In a trial with traditional informed consent, only patients willing to consider the experimental treatment would be randomized. Such a trial would not provide information that patient acceptance of the new surgical treatment was poor. Moreover, statistical techniques are available to estimate the effects of treatments in clinical trials that take into account non-compliance.14 As such, trials with just-in-time consent could be used to address the practical question of the effects of a treatment if offered at the population level and the more clinical question of how the treatment affects a patient who decides to accept it.
Conclusion
To be clear, our recommendation is not to abandon traditional consent entirely and move to a just-in-time consent instead. We propose research to determine the value or otherwise of our proposed modification to consent.
Patient autonomy remains a key principle in medicine and with few exceptions, researchers must receive consent from patients before they undergo research procedures. However, we need to think creatively as to how we might enhance patient autonomy while—or perhaps by means of—reducing patients’ decisional distress and burdens. In trials of experimental treatments with usual care control groups, it may just be time to explore just-in-time consent.
Example language for initial consent.
You are in our hospital to discuss treatment for [condition/disease]. To improve care for future patients, we try to learn from our patients’ experiences by conducting studies. We would like to learn from your experiences as well. Therefore, we would like to invite you to take part in a study.
If you agree to participate in this study, we will use information in your medical record. Researchers can then look at how your outcomes compare to others with similar or different treatments. Your information will be treated confidentially by the research team.
[Optional when additional measurements take place:]
If you decide to take part, you will receive questionnaires about your [quality of life and symptoms]. You will receive a questionnaire [today], and [3 and 12 months later]. This questionnaire will take [20] minutes to complete and you will be asked to fill it out on [a website].
In this hospital, we also study new, experimental treatments that we think might have benefits compared to treatment as usual, but we do not know if this is really true. We also do not fully understand their risks. To get a fair answer when comparing results, we let chance decide who will be offered experimental treatments. Therefore, you might be selected at random to be offered an experimental treatment within the next [XX days/weeks/months].
If you have been selected, you will receive all relevant further information about this experimental treatment – including potential risks and benefits – from a researcher or your physician. It is then up to you to decide whether to accept the alternative approach or continue with usual care.
If you are not offered the experimental treatment, this may be because you did not meet all the criteria or because you were not randomly selected. You will continue to receive treatment as usual and you will not automatically receive further information about what kind of experimental treatment other patients may have been offered.
Results will be compared between those receiving treatment as usual and those being offered an alternative approach.
In this study you will receive information in stages. This is done to avoid information overload, such as when hearing about the risk and benefits of treatments that you will not be able to receive. However, you will never receive an experimental treatment without receiving information about it first and making an active decision to receive this treatment.
Acknowledgments
Funding
Supported by funds from David H. Koch provided through the Prostate Cancer Foundation, the Sidney Kimmel Center for Prostate and Urologic Cancers, P50-CA92629 SPORE grant from the National Cancer Institute to Dr. H Scher, the P30-CA008748 NIH/NCI Cancer Center Support Grant to MSKCC.
Appendix
1. Examples of types of distress caused by informed consent
Decisional conflict, Decisional burden, Decisional regret
A 52-year old woman with three school-age children has metastatic breast cancer. She previously finished several rounds of chemotherapy, with limited response. Her current life expectancy is about one year. There are no other treatment options for her and she is receiving the best available supportive care, consisting of pain management and bi-weekly consults with a psychologist.
Her pain is currently well-managed, and despite daily fatigue and occasional pain flares, she is feeling relatively healthy. Her physician tells her that she is eligible to participate in a randomized trial, where an experimental chemotherapy drug is compared to best supportive care. Her physician explains that previous smaller studies have suggested that the experimental drug might prolong life by several months, but that it remains unclear how many patients would actually benefit. Also, reported acute side effects such as nausea might be quite severe. With her children in mind, any form of hope triggers her interest.
-
-
Decisional conflict – state of uncertainty about which course of action take:
The patient had previously promised herself to remain realistic, and was working hard with her psychologist to accept her prognosis. She starts to feel anxious while thinking “What should I do? Should I put myself through this again? Should I allow myself – and my family – to get hope again, while I may spend most of the time in the hospital and feeling sick at home after each chemotherapy session?” The patient is generally feeling well, and this will likely change if she were to start on the experimental agent. However, turning down the opportunity to have a few extra months with her children also feels wrong. “Should I enjoy these months with my family now that I am only experiencing minor symptoms, or should I start treatment in the hopes of spending more time with my children? Will it be worth it, if these extra months consist of frequent hospital visits for the chemotherapy and possibly feelings nauseous all day, like I did during previous rounds of chemotherapy?” She is conflicted and starts to have trouble sleeping. Conversations with her family and friends suddenly seem to consist only of talking about the trial.
-
-
Decisional burden – emotional and cognitive stress required to make a decision:
If she participates in this trial, she will be randomly allocated either to the experimental drug or to continuing with her current supportive care. She has to decide within 7 days whether she wants to participate. While trying to find answers to all these difficult questions, she also realizes that even if she consents to randomization, she might not receive the experimental drug. She starts to wish that she never heard about this experimental option, as she was doing just fine without it and had just started to accept that she would probably not be alive much more than one more year. She feels that if she does not get allocated to receiving the drug, all this anxiety, distress and difficult conversations with family, friends and herself will not have been worth it. However, not thinking about it is no longer an option, because the deadline for deciding whether or not she wants to take part in the trial is approaching fast.
-
-
Decisional regret – the feeling that the wrong decision was or could have been made:
The patient decides not to participate in the trial. The thought of spending several weeks to months with side effects from chemotherapy, for a treatment that might not prolong her life, is not something she wants, especially now that she is still feeling relatively well. However, as weeks pass by, she starts to worry whether or not she made the right decision. She starts to feel guilty that she has denied her children a chance to spend more time with their mother. She finds herself regularly looking for information about the experimental drug on the Internet. Now that several months have passed, she realizes how valuable extra months are to her and she regrets her choice. She knows that she might still have been randomly allocated to supportive care, and that the treatment may not even have worked, but not having taken a chance weighs heavily.
Confusion, Disappointment, Anxiety
A 60-year old man is scheduled to undergo abdominal surgery for an aortic aneurysm. The surgeon explains that incisional hernia is one of the potential complications of this surgery, and it can cause long-term problems. The patient is eligible for participation in a randomized controlled trial where standard suture closure is compared with suture augmented with a mesh. The surgeon tells the patient that there is pilot data suggesting that the mesh could reduce the risk of hernia, but explains that a large clinical trial is needed to know for sure. She also explains that the trial is randomized to make sure that it is fair.
-
-
Confusion
The patient is confused after discussing the trial. He received a large amount of new information, and he does not fully understand his options. There was information about the standard treatment but also information about the mesh, with different sets of risks. He has trouble remembering which risk was associated with which surgical procedure. Also, “If there is a risk of hernia, and there is a potential way to reduce it, how come I’m not going to receive it? Maybe I should go to a surgeon who actually knows what the best treatment is, instead of one who needs to flip a coin to make a decision.”
-
-
Disappointment
The patient reads the study information and likes the thought of a mesh. Even though the folder explains the mesh might not show actual benefits, to him it seems logical that adding an extra barrier will reduce the risk of hernia. He decides to participate in the trial in the hope of receiving the mesh. On the day of the surgery, he learns that he has been allocated to the standard treatment and immediately feels disappointed. It feels as if he has been consigned to receive a second-class treatment. Three months later, he has to complete a questionnaire about his recovery, quality of life and the cosmetic outcome. He thinks his recovery and cosmetic outcome could have been much better had he received the mesh.
-
-
Anxiety
The patient had been generally aware that surgery can cause complications, but the long discussion about the trial has made him focus on the risk of hernia. He is worried before surgery, and in the weeks and months after discharge, he inspects his abdomen regularly to see if he might be developing a hernia. This is especially because the trial information included extensive descriptions about hernia and its dangers.
2. Quantitative data on consent-related distress
There have been relatively few systematic evaluations of consent-related distress. We hypothesize this is because patient anxiety and distress is seen as an ordinary and unremarkable aspect of clinical trials. Simes et al.1 compared consent involving “full disclosure”, what is typical for US clinical trials, with “individualized disclosure”, when the doctor decides what to tell a patient about the trial. The study was conducted with patients involved in 16 randomized trials conducted at an Australian hospital. Mean Spielberger State Anxiety Inventory (STAI) scores were 49 and 42 respectively, with standard deviations close to 10. Given that 55 is one cut-off given for clinical anxiety in older patients2, we can estimate that about 25% of patients experienced clinical anxiety in the “full disclosure” group compared to 10% in the “individualized disclosure” group. Hence about 1 in 7 patients had clinical levels of anxiety caused by giving additional information during consent. Unless the informed consent in the “individualized disclosure” group caused zero anxiety, that is, all anxiety reported in that group was background levels, then we can infer that more than 1 in 7 patients have clinical levels of anxiety caused by informed consent.
Aaronson et al.3 randomized 180 Dutch cancer patients being approached for participation in Phase II or III trials to either standard informed consent procedures or standard procedures followed by a nurse phone call. Mean STAI scores were similar in both groups, and very close to those reported in the Simes trial. Of note, the mean score of 49 was reported to be at the 85th centile of a Dutch normative sample, again suggesting very high rates of distress.
Evidence that these high levels of distress are related directly to the consent process itself, rather than say, anxiety about a cancer diagnosis, come from a study of US cancer patients involved in clinical trials4. STAI scores were measured one week after consent and were close to 40, elevated, but not as high as anxiety scores measured close to the time of consent itself.
Given the relatively weak data on distress associated with consent, we propose that more systematic evaluations are undertaken, involving a broad range of outcomes, including anxiety, decisional conflict and regret.
- 1.Simes RJ, Tattersall MH, Coates AS, Raghavan D, Solomon HJ, Smartt H. Randomised comparison of procedures for obtaining informed consent in clinical trials of treatment for cancer. British medical journal (Clinical research ed) 1986;293:1065–8. doi: 10.1136/bmj.293.6554.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kvaal K, Ulstein I, Nordhus IH, Engedal K. The Spielberger State-Trait Anxiety Inventory (STAI): the state scale in detecting mental disorders in geriatric patients. International journal of geriatric psychiatry. 2005;20:629–34. doi: 10.1002/gps.1330. [DOI] [PubMed] [Google Scholar]
- 3.Aaronson NK, Visser-Pol E, Leenhouts GH, et al. Telephone-based nursing intervention improves the effectiveness of the informed consent process in cancer clinical trials. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1996;14:984–96. doi: 10.1200/JCO.1996.14.3.984. [DOI] [PubMed] [Google Scholar]
- 4.Coyne CA, Xu R, Raich P, et al. Randomized, controlled trial of an easy-to-read informed consent statement for clinical trial participation: a study of the Eastern Cooperative Oncology Group. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2003;21:836–42. doi: 10.1200/JCO.2003.07.022. [DOI] [PubMed] [Google Scholar]
Footnotes
The opinions expressed are the author’s and do not represent the views of any part of the US government
Declaration of conflicting interests
The Authors declare that there is no conflict of interest.
References
- 1.World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310:2191–2194. doi: 10.1001/jama.2013.281053. [DOI] [PubMed] [Google Scholar]
- 2.Hickman RL, Jr, Pinto MD. Advance directives lessen the decisional burden of surrogate decision-making for the chronically critically ill. J Clin Nurs. 2014;23:756–765. doi: 10.1111/jocn.12427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Connor AM. Validation of a decisional conflict scale. Med Decis Making. 1995;15:25–30. doi: 10.1177/0272989X9501500105. [DOI] [PubMed] [Google Scholar]
- 4.Becerra Perez MM, Menear M, Brehaut JC, et al. Extent and predictors of decision regret about health care decisions: a systematic review. Med Decis Making. 2016;36:777–790. doi: 10.1177/0272989X16636113. [DOI] [PubMed] [Google Scholar]
- 5.Appelbaum PS, Roth LH, Lidz CW, et al. False hopes and best data: consent to research and the therapeutic misconception. Hastings Cent Rep. 1987;17:20–24. [PubMed] [Google Scholar]
- 6.Kim SYH, Flory J, Relton C. Ethics and practice of Trials within Cohorts (TwiCs): An emerging pragmatic trial design. Clin Trials. doi: 10.1177/1740774517746620. IN PRESS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zelen M. A new design for randomized clinical trials. N Engl J Med. 1979;300:1242–1245. doi: 10.1056/NEJM197905313002203. [DOI] [PubMed] [Google Scholar]
- 8.Relton C, Torgerson D, O’Cathain A, et al. Rethinking pragmatic randomised controlled trials: introducing the “cohort multiple randomised controlled trial” design. BMJ. 2010;340:c1066. doi: 10.1136/bmj.c1066. [DOI] [PubMed] [Google Scholar]
- 9.Young-Afat DA, Verkooijen HA, van Gils CH, et al. Brief report: Staged-informed consent in the cohort multiple randomized controlled trial design. Epidemiology. 2016;27:389–392. doi: 10.1097/EDE.0000000000000435. [DOI] [PubMed] [Google Scholar]
- 10.Hugosson J, Carlsson S, Aus G, et al. Mortality results from the Göteborg randomised population-based prostate-cancer screening trial. Lancet Oncol. 2010;11:725–732. doi: 10.1016/S1470-2045(10)70146-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hinman RS, McCrory P, Pirotta M, et al. Acupuncture for chronic knee pain: a randomized clinical trial. JAMA. 2014;312:1313–1322. doi: 10.1001/jama.2014.12660. [DOI] [PubMed] [Google Scholar]
- 12.Trials within Cohorts. Use of the TwiCs design. 2016 https://www.twics.global/use-of-the-design. accessed 15 March 2017.
- 13.Joffe S, Cook EF, Cleary PD, et al. Quality of informed consent: a new measure of understanding among research subjects. J Natl Cancer Inst. 2001;93:139–147. doi: 10.1093/jnci/93.2.139. [DOI] [PubMed] [Google Scholar]
- 14.Angrist JD, Imbens GW, Rubin DB. Identification of causal effects using instrumental variables. J Am Stat Assoc. 1996;91:444–455. [Google Scholar]