

SUMMARY
The etiology of non-alcoholic fatty liver disease (NAFLD), the most common form of chronic liver disease, is poorly understood. To understand the causal mechanisms underlying NAFLD, we conducted a multi-omics, multi-tissue integrative study using the Hybrid Mouse Diversity Panel (HMDP), consisting of ~100 strains of mice with various degrees of NAFLD. We identified both tissue-specific biological processes as well as processes that were shared between adipose and liver tissues. We then used gene network modeling to predict candidate regulatory genes of these NAFLD processes, including Fasn, Thrsp, Pklr, and Chchd6. In vivo knockdown experiments of the candidate genes improved both steatosis and insulin resistance. Further in vitro testing demonstrated that down regulation of both Pklr and Chchd6 lowered mitochondrial respiration and led to a shift towards glycolytic metabolism, thus highlighting mitochondria dysfunction as a key mechanistic driver of NAFLD.
Keywords: Non-alcoholic fatty liver disease, multi-omics integration, network modeling, key drivers, mitochondrial dysfunction, oxidative phosphorylation, glycolysis
eTOC BLURB
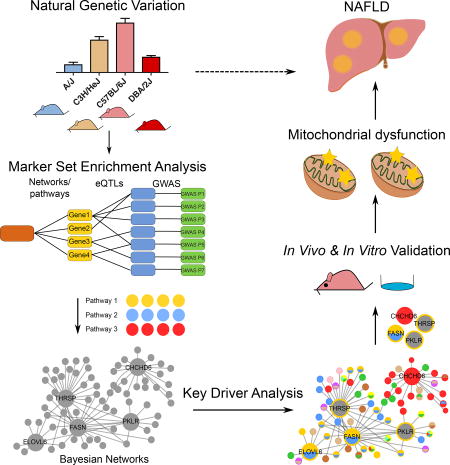
Chella Krishnan and Kurt et al. apply integrative genetics approaches to delineate “key driver” genes regulating NAFLD using multi-omics data from ~100 mouse strains. In vivo modulation of these genes rescued animals from steatosis and insulin resistance. Follow-up bioenergetics studies highlight mitochondrial dysfunction as a key mechanistic driver of NAFLD.
INTRODUCTION
Non-alcoholic fatty liver disease (NAFLD) has become a very common chronic liver disease, affecting 20–30% of Western populations (Hui et al., 2015; Ratziu et al., 2010; Vernon et al., 2011). It can progress through different stages of hepatic abnormalities, from hepatocellular lipid accumulation (steatosis) to non-alcoholic steatohepatitis (NASH) involving inflammation and fibrosis, to cirrhosis and hepatocellular carcinoma (Adams et al., 2005; Browning et al., 2004; Kopec and Burns, 2011). The disease is highly associated with metabolic disorders such as obesity and insulin resistance (de Alwis and Day, 2008; Browning et al., 2004; Marchesini et al., 2003; McCullough, 2004), and there are no directly established pharmacological treatments for NAFLD other than reducing these risk factors such as weight reduction, using insulin sensitizers and lipid-lowering agents.
Thus far, only a handful of genes (PNPLA3, SAMM50, PARVB, GCKR, LCP1, LYPLAL1, PPP1R3B, TM6SF2, TRIB1) have been identified by human genome-wide association studies (GWAS) (Chambers et al., 2011; Kitamoto et al., 2013; Kozlitina et al., 2014). An important complication in human genetic studies is the very significant role of NAFLD environmental factors in the disorder. Rodent models allow a better control of environmental factors when examining the genetic contributors of NAFLD and also enable the collection of molecular traits from the relevant tissues to help elucidate tissue-specific mechanisms. To this end, we recently conducted a study (Hui et al., 2015) to examine hepatic steatosis and its relevant clinical and molecular traits in more than 100 distinct inbred mouse strains belonging to the Hybrid Mouse Diversity Panel (HMDP). The HMDP mice were fed a high fat and high sucrose (HF/HS) diet to induce hepatic triglyceride (TG) accumulation, a hallmark of NAFLD.
Using the HMDP, we produced a rich multi-omics data resource for NAFLD including dense genotyping of common genetic variants, liver and adipose transcriptome data, and the corresponding tissue-specific expression quantitative trait loci (eQTLs) that reflect genetic regulation of gene expression. In the present study, we applied an integrative genomics approach to fully incorporate the whole spectrum of NAFLD genetic association with functional genomics information from liver and adipose eQTLs, and from gene networks constructed using liver and adipose transcriptome data from HMDP as well as from a multitude of additional existing genomic studies. This multi-omics integration revealed coordinated gene-gene interactions in liver and adipose tissues that are perturbed by polygenic risks of NAFLD and uncovered hidden biology missed by traditional genomic analysis. This data-driven integrative approach further predicted potential regulators of the NAFLD processes, leading to the identification of both known (e.g., Fasn and Thrsp) and novel (e.g., Pklr and Chchd6) regulators. Further in vivo and in vitro experimental validation of the predicted novel regulators revealed mitochondria dysfunction as a key driving mechanism in NAFLD.
RESULTS
Integrative genomics framework
As detailed in Figure 1, we modeled NAFLD gene networks using the multi-omics HMDP data along with additional public gene expression datasets to identify pathways and predict potential “key driver” genes underlying hepatic TG accumulation. Briefly, we first constructed gene co-expression networks based on liver and adipose expression data across the HMDP strains and then integrated these networks with GWAS analyses of hepatic TG levels as well as liver and adipose eQTL information using the Mergeomics platform (Arneson et al., 2016; Shu et al., 2016). This integration led to the identification of tissue-specific co-expression modules (groups of co-expressed genes) and biological pathways that are enriched for NAFLD GWAS signals. Subsequently, we mapped the NAFLD-associated network modules and pathways to gene regulatory Bayesian networks of liver and adipose tissues that are based on numerous genetic and gene expression datasets (details in Method Details section) to predict potential key regulators, termed key drivers (KDs), of the NAFLD processes. We then prioritized the resulting predicted KD genes for experimental validation and mechanistic studies in mice. The details of these operations are described below.
Figure 1. Schematic diagram of the methodology.

Liver and adipose tissue gene expression data, genotype, and hepatic TG phenotypic data of the Hybrid Mouse Diversity Panel (HMDP) mice were integrated to identify putative causal mechanisms for the NAFLD. TG: Triglyceride, eQTL: expression Quantitative Trait Loci; GWAS: genome-wide association studies.
Modeling of functional gene-gene relationships in the HMDP using tissue-specific co-expression networks
To capture gene-gene relationships in NAFLD-relevant tissues in a data-driven manner, we first constructed gene co-expression networks of liver and adipose tissues using the Weighted Gene Co-expression Network Analysis (WGCNA) (Langfelder and Horvath, 2008) and the Multiscale Embedded Gene Co-expression Network Analysis (MEGENA) (Song and Zhang, 2015) (details in Method Details). The networks were constructed based on the transcriptome data from liver and adipose tissue samples of 228 HMDP mice from 113 mouse strains that were fed an 8-week HF/HS diet to induce hepatic steatosis (Hui et al., 2015). The two network modeling methods are complementary in that WGCNA generates networks comprised of larger modules, which together fit a scale-free topology at a single scale, whereas MEGENA produces multi-scale networks comprised of more compact modules (detailed comparison in Methods). Using WGCNA, we identified a total of 40 and 26 co-expression modules in liver and adipose tissue, respectively. Using MEGENA, we identified 204 and 79 co-expression modules in liver and adipose tissue, respectively. We listed all member genes of each module for each tissue in Table S1. These co-expression modules from WGCNA and MEGENA mainly serve to group genes based on their coordinated co-regulation patterns, which have been shown to be a powerful data-driven way to define functionally related genes (Langfelder and Horvath, 2008; Song and Zhang, 2015). Indeed, the majority of modules showed enrichment of specific pathways or biological processes (Table S2 and Figure S1), indicating functional coordination of genes in the co-expression modules identified.
We evaluated the preservation of the modules between the MEGENA and WGCNA methods (details in Method Details). We observed that all of the WGCNA liver and adipose modules were conserved in the MEGENA liver and adipose modules (Figure S2A–D). Similarly, 97% and 86% of MEGENA liver and adipose modules, respectively, were conserved within the corresponding liver and adipose WGCNA modules. Hence, these two methods capture largely conserved co-expression patterns while the multi-scale algorithm in MEGENA can reveal additional modules. Additionally, the varying module sizes from the two methods confer a broad range of statistical power and various degrees of functional coherence among the module genes, which allows for comprehensive screening in the downstream analyses to capture biologically relevant pathways and genes in NAFLD.
Co-regulation of genes is most likely attributable to the sharing of common regulators such as transcription factors or microRNAs, which act in trans to regulate large numbers of downstream target genes. Therefore, we would expect that the tissue-specific modules to reflect tissue-specific trans-eQTLs, in a manner that the module genes share trans-eQTLs to loci that control the activities of their upstream regulators. Such trans-eQTLs, however, are intrinsically weak and are difficult to reliably detect in practice (Van Nas et al., 2010). To date, the eQTLs that can be reproducibly identified are mainly cis-eQTLs (including those used in the current study). These are responsible for local control of individual genes and, when the genes under cis-regulation encode transcriptional regulators expressed in a tissue-specific manner, can contribute to tissue-specific co-expression by perturbing the expression of sets of genes in trans. Because of the difficulty in capturing trans-eQTLs, we are not able to directly assess whether the tissue-specific modules reflect tissue-specific eQTLs, although in theory tissue-specific trans-eQTLs underlie the tissue-specific modules.
Identification of co-expression modules that show correlation with NAFLD phenotypes
To investigate which co-expression modules are associated with NAFLD, we correlated the module eigen-gene of each module with hepatic TG (see Method Details). We found 105 MEGENA modules (65 from liver and 40 from adipose) and 17 WGCNA modules (14 from liver and 3 from adipose) to be correlated with hepatic TG at Pearson correlation P<1E-3, which is determined based on a permutation test (see Method Details). The hepatic-TG associated liver co-expression modules are mainly related to lipid metabolism, cell cycle, and peroxisomal pathways. The hepatic TG associated adipose co-expression modules are mainly related to immune system, cell cycle, citric acid (TCA) cycle, and insulin signaling pathways (Table S2).
Biological pathways and co-expression modules that show genetic association with NAFLD
To infer potential causal processes in NAFLD, we incorporated genetic information into our analysis to retrieve groups of genes that together show evidence of genetic association with NAFLD. This is in contrast to conventional GWAS, which focus on individual genetic variants and have limited statistical power to detect moderate and subtle genetic signals as well as gene-gene interactions. Specifically, we integrated the full spectrum of GWAS results (not just the top genome-wide significant hits) of hepatic TG levels (Hui et al., 2015), with the liver and adipose co-expression modules defined above as well as canonical pathways taken from several pathway databases (BIOCARTA, KEGG, REACTOME). To connect the GWAS signals in the form of single nucleotide polymorphisms (SNPs) to genes in the modules or pathways, we used the liver and adipose eQTLs (details in Methods), each containing one or more expression SNPs (eSNPs) associated with gene expression, from the same HMDP cohort (Hui et al., 2015). eSNPs capture the potential functional relationships between GWAS SNPs and expressed genes in a tissue-specific manner. Integration of GWAS, eQTLs (in the form of eSNPs), and networks/pathways was performed using the Marker Set Enrichment Analysis (MSEA) from the Mergeomics pipeline (Shu et al., 2016) (Figure 1; details in Method Details) to test for over-representation of stronger GWAS signals among the eSNPs mapped to individual pathways or co-expression modules compared to random sets of genes. We provide a detailed Table S3 to list the member genes in each module from each tissue, the corresponding eSNPs of each member gene that have at least one eQTL in the matched tissue, and P-value of each eSNP in disease GWAS.
MSEA was conducted using the eQTLs and networks from liver and adipose tissues separately to infer tissue-specific perturbations in NAFLD. Among the 1823 pathways and 122 TG-correlated co-expression modules (79 from liver and 43 from adipose) tested, at false discovery rate (FDR) <5%, we found 35 pathways and 9 co-expression modules from the liver-specific analysis and 24 pathways and 8 co-expression modules from the adipose-specific analysis to be significantly enriched for GWAS signals of NAFLD in the HMDP (Table S4). Among the NAFLD-associated pathways and co-expression modules identified from each tissue, we found that certain modules or pathways had genes highly overlapping with other modules or pathways (e.g., a liver co-expression module contained mostly the same set of genes captured in the “lipid metabolism” pathway from KEGG). To reduce the redundancy, we merged the overlapping gene sets to derive “supersets” each comprised of one or more overlapping pathways/modules associated with NAFLD (details in Method Details), and confirmed that these supersets retained significant genetic association to NAFLD as shown by their constituents (Table S5). Comparison between tissues revealed 9 supersets to be liver-specific (e.g., peroxisome, oxidative phosphorylation, NOTCH signaling), 13 adipose-specific (e.g., innate immunity, insulin signaling, branched chain amino acid metabolism), and 8 common to both tissues (e.g., adaptive immune system, multiple lipid metabolism processes, apoptosis/cell cycle, gene expression regulation; Figure 2). Therefore, these gene sets informed by hepatic TG GWAS captured a broad range of molecular processes that are likely perturbed by genetic factors associated with NAFLD.
Figure 2. Comparison between NAFLD processes between liver and adipose tissue.

Putative causal pathways shared between tissues as well as those unique to each tissue are listed. For co-expression modules, the 5 most over-represented gene ontology terms are shown. See also Tables S1–S7 and Figure S1.
Comparison of the NAFLD-associated supersets identified in the mouse HMDP with previously known NAFLD genes and pathways
We compared our data-driven findings from the HMDP mouse study with 107 previously identified NAFLD-associated genes (listed in Table S6) from the DisGeNET database (Piñero et al., 2015), which manually curates gene-disease associations from a multitude of quality controlled databases such as the GWAS Catalog, ClinVar, Comparative Toxicogenomics Database, and Genetic Association Database (details in Method Details). The 107 known NAFLD genes contained nine human GWAS candidate genes (PNPLA3, SAMM50, PARVB, GCKR, LCP1, LYPLAL1, PPP1R3B, TM6SF2, TRIB1). Notably, 5 of the NAFLD GWAS genes (PNPLA3, GCKR, LYPLAL1, PPP1R3B, TM6SF2) and 60 of the other 97 previously studied NAFLD genes were among the pathways and co-expression modules identified in our HMDP analysis. To assess consistency at the pathway level, we used a one-tailed Fisher’s exact test to identify the canonical pathways enriched for the 107 known genes (Table S7) and compared the significant pathways to our findings from the HMDP mouse study. This analysis showed replication of numerous pathways (highlighted in Table S5 and S7) including fatty acid, lipid, and lipoprotein metabolism, apoptosis, immune system, insulin signaling, drug metabolism, and a cancer pathway. These analyses suggest that our data-driven integrative analysis of a single mouse HMDP study captures previously known NAFLD genes and processes. Moreover, our study highlighted several novel processes such as oxidative phosphorylation, extracellular matrix (ECM), branched-chain amino acid (BCAA) metabolism, and the cell cycle.
Identification of key drivers (KDs) of the NAFLD-associated gene supersets
We identified potential regulatory genes, termed KDs, within the NAFLD-associated supersets using tissue-specific gene regulatory networks and the Key Driver Analysis (KDA) in Mergeomics (Shu et al., 2016). The main concept behind the KDA is to project disease-associated gene sets onto an independently derived gene regulatory network and subsequently pinpoint KDs whose network neighbors are enriched for disease genes. To identify the KDs for the NAFLD-associated gene supersets, we leveraged liver and adipose Bayesian Networks (BNs) (Zhu et al., 2008) constructed from a number of existing human and mouse studies (Derry et al., 2010; Emilsson et al., 2008; Schadt et al., 2008; Tu et al., 2012; Wang et al., 2007; Yang et al., 2006; Zhong et al., 2010), as listed in the "key resource table" in the STAR methods section, and utilized the union BN for each tissue to capture gene-gene interactions under different pathophysiological conditions (see Method Details). As BNs are constructed based on gene expression patterns, genetic information, and causal inference, they capture causal regulatory relationships between genes and the KDs derived from such BNs are potential regulators of the NAFLD genes and the disease itself. We identified a number of KDs for each NAFLD superset at FDR<5% (Table S8), among which Fasn is a particularly consistent KD identified across multiple supersets in both liver and adipose tissues, suggesting its critical role in NAFLD. Other notable KDs include Hmgcr, the target of statin drugs, and Pparg, the target of glitazones, supporting the current therapies involving lowering metabolic risks of NAFLD. We also found that 17 of the KD genes predicted in our study were among the 107 previously reported NAFLD genes curated in DisGeNET (highlighted in bold in Table S8). As shown in Figure 3, the top KDs of each NAFLD superset orchestrate tightly connected subnetworks of NAFLD pathways and processes in a tissue-specific manner (Figure 3A for liver and 3B for adipose).
Figure 3. Bayesian gene subnetworks representative of NAFLD pathways and their key drivers.

(A) Liver Bayesian subnetwork comprised of liver NAFLD supersets and the top 3 key drivers of each superset. (B) Adipose Bayesian subnetwork comprised of adipose NAFLD supersets and the top 3 key drivers of each superset. (C) Liver Bayesian subnetworks of selected genes Fasn, Thrsp, Pklr, and Chchd6. Key Driver (KD) genes are illustrated with large node sizes, human GWAS candidate genes are represented in hexagon shapes, and the rest of the genes are represented by medium node sizes. Member genes of each NAFLD-associated superset are indicated with a distinct color. Non-member genes are represented in grey with small node sizes. Blue edges show the interactions between human GWAS candidate genes and our candidate KDs. The other interactions were shown in grey. See also Table S8.
We checked the genetic regulation of the predicted KDs. We found that the majority of the KDs did not have significant cis-eQTLs (Table S8). Among the 69 unique liver KDs, 6 genes (9%) have cis-eQTLs in liver, whereas among 88 unique adipose tissue KDs, 4 genes (5%) have cis-eQTLs in adipose tissue. As the robust eQTLs we included in our analysis are cis-eQTLs, the lack of cis-eQTLs among the KDs implies that they are likely regulated in trans.
Selection of candidate KDs for validation: Fasn, Thrsp, Pklr, and Chchd6
We selected 4 predicted liver KDs to test their potential regulatory role in NAFLD. Genes encoding fatty acid synthase (Fasn) and thyroid hormone responsive (Thrsp), and pyruvate kinase, liver and red blood cell (Pklr) are the 2nd, 3rd, and 10th KDs, respectively, predicted for the superset representing fatty acid, triacylglycerol, and ketone body metabolism pathways. However, in terms of gene-trait (hepatic TG level) correlation in liver tissue, Pklr is the most significant KD of this pathway (Pearson correlation coefficient r=0.47, P=1.36E-07). The 4th KD selected for validation encodes coiled-coil-helix-coiled-coil-helix domain containing 6 (Chchd6), chosen from a liver co-expression module related to the mitotic cell cycle, G2-M checkpoints, and DNA damage response. Also, Chchd6 was ranked as the 25th most correlated gene with hepatic TG levels (Pearson correlation coefficient r=0.54, P=1.27E-09) in the HMDP liver transcriptome data. While Fasn (Kawano and Cohen, 2013) and Thrsp (Wu et al., 2013) have been implicated in NAFLD and can serve as positive controls, Pklr and Chchd6 are novel predictions from our study.
As shown in the sub networks of the 4 select KDs (Figure 3C), they are hub genes surrounded by many genes in the NAFLD-associated pathways or co-expression modules identified in our study. Fasn, Thrsp, and Pklr subnetworks are interconnected and partner with many lipid and cholesterol metabolism genes such as Acacb, Elovl6, Hmgcr, Mvk, Pltp, Me1, Dhcr7, and Acly, whereas Chchd6 forms a separate subnetwork surrounded by cell cycle genes as well as fatty acid related genes Cd36 and Pparg. Notably, human NAFLD GWAS candidate genes PNPLA3, PPP1R3B, LCP1, TRIB1, and SAMM50 are within the subnetworks. PNPLA3 is directly connected to Fasn and indirectly connected to the other selected KDs, Thrsp, Pklr, and Chchd6, via one gene for each KD. These subnetworks support the relevance of the selected KDs in NAFLD. Interestingly, the NAFLD GWAS genes themselves were not predicted to be KDs. This agrees well with the consistent observations from us and others that human GWAS genes tend to be peripheral nodes in gene regulatory networks whereas key regulatory genes likely do not contain GWAS signals due to evolutionary constraints (Boyle et al., 2017; Mäkinen et al., 2014; Shu et al., 2017; Zhao et al., 2016).
Validation of candidate key driver genes as causal genes for hepatic steatosis
To assess the in vivo effects of the candidate KD genes, we knocked down hepatic expression of Fasn, Thrsp, Pklr, and Chchd6 by adenoviral expression of shRNA in 8-week-old C57BL/6J mice (see Method Details and Figure S3A). The control groups received adenovirus expressing an empty vector while the test groups received adenovirus expressing the respective shRNAs against each of the 4 candidate genes. Three shRNAs were tested against each candidate gene in vitro and the strongest shRNA was used in in vivo studies. One day after injection, all animals were subjected to a HF/HS diet for 14 days followed by euthanization and tissue extraction. Knockdown of these KD genes led to significant lowering of liver mass as well as the mass of certain adipose depots compared with the control group post HF/HS treatment (Figure 4A–D). Analysis of hepatic lipids revealed significant reductions in both hepatic TG and total cholesterol (TC) levels (Figure 4E–F) but no change in hepatic unesterified cholesterol and phospholipid levels between the groups (Figure S4A–B). We also noted that the plasma glucose levels were unchanged between the control and test animal groups (Figure S4C). However, plasma insulin levels were lowered in groups with candidate gene knockdown, accompanying improved insulin sensitivity as measured by HOMA-IR (Figure 4G–H). Quantitative PCR analyses of liver tissues also revealed that knockdown of these KD genes led to a reduction in genes associated with de novo lipogenesis (and lipid uptake, to a lesser extent) relative to control groups (Figure S3C). To further validate the key driver genes as regulators of the predicted gene networks, we randomly selected 11 nodes connected to either Pklr or Chchd6 in their respective networks and tested their expression via quantitative PCR analyses of liver tissues. As control, we randomly selected 3 nodes that were 3-edges away from the respective key driver genes. Compared to the control groups, knockdown of either Pklr or Chchd6 affected the expression of most of their neighborhood genes (Figure 5A and C). However, distant genes were unaffected (Figure 5B and D).
Figure 4. Effects of shRNA knockdown of KD genes on mouse phenotypes.

Eight week old C57BL/6J mice were injected with adenovirus carrying either empty vector or shRNA against respective KD genes and fed with HF/HS diet for 14 days. Comparisons of (A–D) liver and three white adipose tissue (WAT) weights, (E) hepatic triglyceride (TG) levels, (F) hepatic total cholesterol (TC) levels, (G) plasma insulin levels and (H) HOMA-IR measurements between control and shRNA animal groups. Data are represented as mean ± SEM (n = 7–12 animals per group). P values were calculated by unpaired two-sided student’s t-test. †P < 0.10, *P < 0.05, **P < 0.01, ***P < 0.001. See also Figures S3 and S4.
Figure 5. Effects of shRNA knockdown of predicted KD genes on network neighborhood genes.

Relative normalized expression values of neighborhood genes and distant genes (3-edges apart) of (A–B) Pklr and (C–D) Chchd6 network respectively, between control and shRNA animal groups after 14 days of infection. Data are represented as mean ± SEM (n = 4–5 animals per group). P values were calculated by unpaired two-sided student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001. See also Figure S3.
Knockdown of novel KD genes altered mitochondrial metabolic profile
To further explore the mechanisms underlying the phenotypic effects of the two novel candidate KD genes, Pklr and Chchd6, we focused on the mitochondria function as measured by oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) using an XF24 analyzer. We focused on mitochondrial function because both the product of Pklr (pyruvate) and fatty acids are oxidized in the mitochondria, also Chchd6 is known to affect mitochondrial cristae morphology where the electron transport chain complexes are assembled (Ding et al., 2015). We used siRNAs to knockdown each of the KDs in AML12 cells grown in the presence of exogenous oleic acid (see Method Details and Figure S3B) and observed a significant reduction in the overall OCR profile (Figure 6A). Specifically, both mitochondrial and non-mitochondrial respiration was reduced, with Chchd6 knockdown leading to lowering of ATP-linked respiration and Pklr knockdown resulting in lowered proton leak (Figure 6B–E). Conversely, we observed a significant increase in the ECAR profile (Figure 6F). Specifically, knockdown of both of these KD genes increased both basal and maximum ECAR (Figure 6G–H). Taken together, the lowered basal OCR but increased ECAR in Pklr and Chchd6 knockdown experiments suggested that these cells shifted away from oxidative metabolism to a more glycolytic metabolism (Figure 6I). Further ex vivo studies using isolated liver mitochondria from mice injected with either adenovirus carrying empty vector or respective shRNAs against Pklr and Chchd6, revealed that both complex I- and II-mediated OCR were affected by knockdown of these genes (Figure 6J).
Figure 6. Knockdown of Pklr and Chchd6 affects both mitochondrial respiration and glycolysis.

Bioenergetic studies on intact AML12 cells transfected with either scrRNA or siRNA against respective novel KD genes (Pklr and Chchd6) were analyzed. Comparisons of (A) oxygen consumption rate (OCR) profile, (B) mitochondrial (datapoint 14 subtracted from 3), (C) non-mitochondrial (datapoint 14), (D) ATP-linked (datapoint 6 subtracted from 3) and (E) proton leak (datapoint 14 subtracted from 6) associated respiration levels, (F) extracellular acidification rate (ECAR) profile, (G) basal ECAR (datapoint 3), (H) maximum ECAR (datapoint 6) levels and (I) overall metabolic profile between scrambled and siRNA groups. Data are represented as mean ± SEM. The experiment was repeated in two independent times with n = 4–8 wells per group each time. (J) Bioenergetic analyses on isolated liver mitochondria from mice injected with adenovirus carrying either empty vector or respective shRNA (n = 2 animals per group). Data are represented as mean ± SEM (n = 3–4 wells per data point). (A, F) P values were calculated by one-factor (time) repeated measures two-way ANOVA (time by treatment interaction P value). (B–E, G–H) P values were calculated by unpaired student’s t-test. *P < 0.05, **P < 0.01, ***P < 0.001. See also Figure S3.
DISCUSSION
Unlike previous studies focusing on individual genes influencing NAFLD progression (Hill-Baskin et al., 2009; Montgomery et al., 2013), our study utilized an integrative genomics approach to capture the genetically perturbed molecular processes by integrating genomic, transcriptomic, and phenotypic data derived from a large panel of inbred mouse strains. This comprehensive multi-omics integration revealed potential causal molecular mechanisms in NAFLD that are informed by aggregate actions of genetic risk factors in GWAS that affect gene expression, pathways and gene networks. The diverse and complex pathogenic pathways captured in our analysis of HMDP, a single mouse study, encompassed numerous causal NAFLD processes established through decades of research such as lipid metabolism and the immune system, and provided evidence for the causal involvement of processes such as insulin and growth factor signaling, the ECM, and the BCAA metabolism pathways. In addition, we identified candidate regulatory genes governing these NAFLD processes and experimentally validated four predicted key regulator genes, Fasn, Thrsp, Pklr, and Chchd6. These validation experiments highlight mitochondrial dysfunction as a core process in NAFLD. Of note, the current study focused on steatosis, the early stage of NAFLD captured in our HF/HS-induced fatty liver model. Later stages of NAFLD such as inflammation and liver fibrosis do not occur to a significant extent in even the most susceptible strains when fed a high fat/high sucrose diet for 8 weeks.
The use of liver and adipose eQTLs and transcriptome data in conjunction with GWAS in our integrative study revealed tissue-specific pathways as well as common processes shared between tissues. Although both adipose and liver tissues have been implicated in NAFLD pathogenesis (Hardy et al., 2016), the diverse processes revealed by our study significantly expand the scope of mechanistic drivers of this disease in these tissues. For instance, previous studies have mainly positioned the adipose tissue as a source of free fatty acid and inflammatory signals that initiate and propagate liver steatosis, inflammation, and injury (Hardy et al., 2016). In contrast, our study indicates a much broader role of adipose tissue in NAFLD, as reflected by the numerous adipose-specific processes including interferon and cytokine signaling, BCAA degradation, TCA cycle, respiratory electron transport, ECM receptor interaction, and signaling by insulin, TGF beta, Wnt, MAPK, and phosphatidylinositol. Similarly, we found numerous liver-specific pathways including oxidative phosphorylation, peroxisome, lysophospholipid, cytochrome p450s, complement and coagulation cascades, and trans-membrane transport of small molecules.
Shared pathways in both liver and adipose tissues include lipid metabolism (lipoprotein, fatty acid, triacylglycerol, and phospholipids), B cell receptor signaling, growth factor signaling, cell cycle, and apoptosis. Our finding on the B cell signaling, a component in adaptive immunity, in both liver and adipose, for NAFLD pathogenesis extends the previous notion that the innate immune system is a key process in NAFLD. Apoptosis and cell cycle pathways, previously known for NAFLD liver injury and scaring (Nolan and Larter, 2009), are also associated with steatosis. Although many of the pathways have previously been correlated with NAFLD, the use of genetic data from GWAS in our study to guide the extraction of these pathways supports their putative causal roles in NAFLD, as the expression levels of the genes in these pathways were perturbed by the same genetic variants that are associated with hepatic TG accumulation.
Previously, we also have developed an HMDP panel fed with a chow diet (Bennett et al., 2010), and co-expression modules from the chow HMDP panel may also be informative for NAFLD pathogenesis. We compared the chow and HF/HS modules and found that the chow modules were mostly preserved in the HF/HS modules and did not add additional value to inform on NAFLD processes (Figure S2E–I). Therefore, the HF/HS-modules used captured sufficient biological information.
To further prioritize genes that play critical roles in regulating these NAFLD-associated pathways/networks and thus to help identify therapeutic targets, we used a gene network-driven modeling approach and predicted potential KDs including Fasn, Thrsp, Pklr, and Chchd6. Many of the top KDs are connected to known NAFLD-associated genes such as Pnpla3, Pparg, and Cd36, supporting their relevance and importance to NAFLD. Both Fasn and Thrsp have been well supported by previous studies as critical genes involved in NAFLD pathogenesis (Chakravarthy et al., 2009; Wu et al., 2013). Their prominent consistency and high ranks in our network analysis and our subsequent gene knockdown experiments confirmed their importance in NAFLD. KDs Pklr, and Chchd6, however, represent novel predictions from the current study. Pklr is abundantly expressed only in liver, while Chchd6 is expressed in all other tissues. However, in the context of diet-induced NAFLD studied over 100 mice strains, Chchd6 was predicted and validated to be a KD only in liver but not in adipose tissue.
Pklr was a top KD of the fatty acid, triacylglycerol, and ketone body metabolism pathway and catalyzes the transphosphorylation of phosphoenolpyruvate into pyruvate and ATP. The Pklr-centered subnetwork (Figure 3C) includes many genes related to lipid metabolism, such as Acly and Acacb. The other novel candidate gene, Chchd6, is a KD of a co-expression module overrepresented with genes involved in cell cycle and “ataxia telangiectasia and rad3 (ATR)” pathways. The known function of this gene is that CHCHD6/MIC25 along with CHCHD3/MIC19 and Mitofilin/MIC60 forms the ‘core’ of the mitochondrial contact site and cristae organizing system, which is predominantly associated with the mitochondrial inner membrane (Huynen et al., 2016; van der Laan et al., 2016). Depleting Chchd6 has substantial effects on mitochondrial cristae morphology leading to fewer cristae junctions and lower cristae density (Ding et al., 2015). In our network model, genes such as Cd36 and Pparg are both linked to Chchd6 (Figure 3C), and the subnetwork centered at Chchd6 is connected to the Fasn and Pklr subnetworks via PNPLA3, a validated human GWAS gene for NAFLD. Our in vivo validation results show that the knockdown of either Pklr or Chchd6 perturbed genes predicted to be in their subnetworks and was significantly associated with NAFLD-related phenotypes including liver mass, hepatic TG, and plasma insulin levels, supporting our predictions made based on network modeling. One of the limitations in our study was using a single shRNA against each of the KDs tested as it may elicit concerns of off-target effects. However, our phenotypic and network validation analyses make this concern very unlikely. It is to be noted that shRNA validation and follow-up adenoviral packaging is a time-consuming and expensive process, thus limiting the use of multiple shRNAs for each gene in vivo (Hui et al., 2015).
To further explore the potential mechanisms underlying the connection of Pklr and Chchd6 to NAFLD, we focused on mitochondrial function. There are over 800 mitochondria in each hepatocyte. Several studies have shown that mitochondrial dysfunction is closely associated with and, in fact, precedes both insulin resistance and NAFLD (Begriche et al., 2006; Pessayre and Fromenty, 2005). Both human and animal studies have shown that during insulin resistance and NAFLD, when the liver is overwhelmed with free fatty acid flux due to increased hepatic fatty acid transport and augmented hepatic de novo lipogenesis, several mitochondrial abnormalities including ultrastructural lesions, depletion of mitochondrial DNA, decreased activity of respiratory chain complexes, and impaired mitochondrial β-oxidation occur (Begriche et al., 2006; Pessayre and Fromenty, 2005; Sobaniec-Lotowska and Lebensztejn, 2003). Genetic polymorphisms in genes encoding PPAR alpha, leptin, adiponectin, or adipokine receptors that affect the mitochondria’s ability to oxidize lipids can affect NAFLD susceptibility (Begriche et al., 2013). Our bioenergetics study shows that knockdown of either Pklr or Chchd6 reduced respiration with a shift towards glycolysis; thereby confirming the critical role of mitochondria dysfunction in NAFLD and revealing previously uncharacterized regulatory genes of mitochondrial function such as Pklr and Chchd6. This shift could be the reason for the observed in vivo insulin sensitivity in Pklr or Chchd6 knockdown animals. In fact, the four KD genes validated here, as well as numerous other KD genes found in our current study, were linked to mitochondrial and other molecular metabolic pathways leading to hepatic triglyceride accumulation in multiple ways (glycolysis, fatty acid uptake, beta oxidation, oxidative phosphorylation, de novo lipid and cholesterol biosynthesis), as summarized in Figure 7.
Figure 7. Summary figure illustrating that a number of KD genes are linked to mitochondrial and metabolic pathways leading to hepatic triglyceride accumulation.

KD genes including Fasn, Thrsp, Pklr and Chchd6 found in the current study are colored in red. I, II, III, IV and V correspond to respective electron transport chain (ETC) complexes. TCA: Tricarboxylic acid cycle; OAA: Oxaloacetate; FFA: free fatty acid.
A shift towards a more glycolytic profile in Pklr knockdown cells was somewhat unexpected and counterintuitive since pyruvate kinase was one of the key glycolytic enzymes catalyzing the final step. This phenomenon can be explained either by compensatory pyruvate production by other enzymatic reactions or through redirection of glycolytic intermediates to other pathways. Examples for the former include substrates such as glutamine and malate being used for compensatory pyruvate production via glutaminolysis and Me1 (malic enzyme) respectively. The latter could be explained by channeling glycolytic intermediates upstream of pyruvate (such as glucose-6-phosphate, fructose-6-phosphate and glyceraldehyde-3-phosphate) into biosynthesis of nucleotide, amino acid and NADPH production. As of now, we do not know the mechanism(s) of this phenomenon. A more detailed metabolic flux analyses using labelled substrates for the above-mentioned metabolic pathways (glucose, glutamine and others) combined with transcriptomics data can enable us to delineate the global metabolic alterations that contribute to the development of NAFLD.
Prior to our study, Williams et al., studied variations in metabolism, mitochondrial function, and cardiovascular phenotypes using multi-omics data (transcriptome, metabolome, proteome) from 80 BXD mouse cohorts that were exposed to a chow diet and a high-fat diet (Williams et al., 2016). Among their key findings, we found the electron transport chain, BCAA, lipid, and energy metabolism pathways to be genetically associated with NAFLD in our study. We also confirmed the regulatory roles of Bckdha and Dbt in BCAA degradation and Echdc1 and Hmgcs1 in the lipid metabolism process. Both previously known cholesterol synthesis genes (e.g. Cyp51a1, Ebp, Fdps, Nsdhl, Pmvk, Sqle, Thrsp, Tm7sf2) and novel cholesterol genes (e.g. Acot1, Acot2, Elovl6, and Gpam) that were found by (Williams et al., 2016) were also identified as KDs in our study for lipid and fatty acid metabolism and mitotic cell cycle-related pathways. Therefore, despite the differences in study design, the two studies converge on a number of key biological findings.
An important point to be considered is that all the findings presented here are based on animal studies using over 100 well-characterized inbred strains, the HMDP population. We acknowledge that mouse models have intrinsic differences from humans and not all findings from mice can be translated to humans. It is critical to compare the genes and networks derived from our study with those from human populations in the future to better assess the translational value of our findings. However, while this manuscript was being reviewed, Lee et al., have reported Pklr as one of the liver-specific targets for treating NAFLD and HCC based on human data (Lee et al., 2017). Additionally, a number of human NAFLD GWAS hits such as PNPLA3 were within our top pathways and networks. These lines of evidence support certain levels of consistency between our findings from animal models and those from human studies.
In summary, our data-driven and integrative genomics study coupling GWAS with tissue-specific genetics of gene expression and network modeling enabled a comprehensive view of the molecular processes and key regulators involved in NAFLD. The novel insights offer new avenues for developing effective therapies by targeting the key regulators and pathways.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Aldons J. Lusis (jlusis@mednet.ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Ethics statement
All animal studies were performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols (#92–169) of the University of California at Los Angeles and were housed in an IACUC-approved vivarium with daily monitoring by vivarium personnel.
Animals
The NAFLD HMDP study design was previously described in detail (Bennett et al., 2010; Hui et al., 2015). A hundred and thirteen mouse strains (listed in Table S11) were purchased from The Jackson Laboratory and bred at University of California, Los Angeles. Genotypes for 113 mouse strains were obtained from the Jackson Laboratories using the Mouse Diversity Array (Yang et al., 2009). The animals (all healthy male mice) used for this study were fed ad libitum a chow diet (Ralston Purina Company) until 8 weeks of age and then placed ad libitum on a high fat high sucrose diet (Research Diets-D12266B, New Brunswick, NJ) with 16.8% kcal protein, 51.4% kcal carbohydrate, 31.8% kcal fat for an additional 8 weeks (total 16 weeks of age). Mice were maintained on a 14-hr light/10-hr dark cycle (light is on between 6 a.m. and 8 p.m.). On the day of the experiment, the mice were sacrificed after 4 hour fasting.
Cell lines and culture media
Mouse hepatocyte cell line AML12 (male) was used for in vitro validation of shRNA and human embryonic cell line HEK293 (female) was used for adenovirus packaging and propagation. Both these lines were obtained from ATCC and were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin solution at 37°C under a humidified atmosphere of 5% CO2. Additionally, 1% non-essential amino acids (NEAA) was added for AML12, while 2.5µg/mL anti-mycoplasma reagent (Plasmocin™) was added for HEK293 cells. After reaching 70–80% confluence in plastic dishes, cells were used for experiments.
METHOD DETAILS
Liver lipid measurement
Liver lipids were extracted as described in (Folch et al., 1957). Lipids extracted from about 60 mg of liver were dissolved in 1.8% (wt/vol) Triton X-100; and colorimetric assays from Sigma (St. Louis, MO) (triglyceride, total cholesterol and unesterified cholesterol) and Wako (Richmond, VA) (phospholipids) were used to measure respective lipid concentrations according to the manufacturer’s instructions.
Adipose and liver RNA isolation and gene expression analyses
Flash-frozen liver and epididymal adipose samples extracted from 113 strains were weighed and homogenized from which RNA was isolated according to the manufacturer’s protocol using RNeasy columns (Qiagen). Global gene expression was analyzed for the isolated RNA using Affymetrix HT_MG430A arrays and microarray data was filtered as previously described (Bennett et al., 2010). Then, ComBat method from the SVA Bioconductor package (Leek et al., 2012) was used to remove known batch effects on the gene expression data.
Adenovirus generation
Recombinant adenovirus expressing shRNA against the target key driver genes (Table S9) was generated as described previously (Bennett et al., 2013). Briefly, pBluescript KS-vector containing shRNA (three constructs per gene) driven by a U6 promoter was first made. The shRNA cassette with highest knockdown efficiency (one construct per gene) was then cloned into the pAdTrack shuttle vector. This shuttle vector is then recombined into adenovirus backbone plasmid pAdEasy-1 in Escherichia coli BJ5183 cells. Linearized positive recombinants were then transfected into HEK293 cells for virus packaging and propagation. Adenoviruses were then purified by cesium chloride (CsCl) banding and stored at −80°C until use.
Animal knockdown studies
Eight-weeks old male C57BL/6J mice (7–12 mice per group pooled from a total of 2–3 independent experiments) were injected with adenovirus containing shRNA against target key driver genes (Table S9) or empty vector for control animals (~5×108 pfu in 200µL PBS). One day after the injection, the animals were kept on high fat/high sucrose (HF/HS) diet for fourteen days. The adenovirus is highly immunogenic and can evoke innate, humoral and cellular immunity in animal models (Crystal, 2014), which would limit the adenoviral-mediated shRNA expression to 1–2 weeks. For this reason, we chose to study the in vivo effects at 14 days. On the day of the experiment, the animals were fasted for 4h, followed by their sacrifice and tissue extraction. Retro-orbital blood was collected for hematology analyses; plasma for analyzing glucose and insulin analyses; liver tissues for lipid content analyses; three white adipose depots (subcutaneous, gonadal, and retroperitoneal) were collected for weight measurements. The HOMA-IR was calculated using the equation . Liver tissues were also used for quantifying expression values of genes (4–5 mice per group) associated with de novo lipogenesis and lipid uptake (Figure S3C), and for isolating mitochondria (2 mice per group) for bioenergetics studies. A separate cohort of animals (4–5 mice per group) that were kept on HF/HS diet for four days were sacrificed, liver tissues were extracted to determine percent knockdown of respective genes by quantitative PCR analyses (Figure S3A).
RNA isolation and quantitative polymerase chain reaction
Total RNA was isolated from frozen mouse liver tissues using QIAzol Lysis Reagent (Qiagen, Germantown, MD) following manufacturer’s RNA isolation protocol. First-strand complementary DNA (cDNA) was made from 2µg total RNA of each mouse according to the manufacturer’s protocol using High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Waltham, MA). Relative quantitative gene expression levels were measured by quantitative PCR using Kapa SYBR Fast qPCR kit (Kapa Biosystems, Inc., Wilmington, MA) on a LightCycler 480 II (Roche) and analyzed using the Roche LightCycler1.5.0 Software. All qPCR targets were normalized to B2M expression and percent knockdown was measured using the equation [(1 − 2−ΔΔCt) × 100%] (Figure S3A–B), while relative normalized expression was measured using the equation 2−ΔΔCt (Figure S3C). All qPCR primer sequences were designed using Primer-BLAST (Ye et al., 2012) and listed in Table S9.
Preparation of cells for bioenergetics experiments
AML12 cells previously seeded on 6-well plates were transfected with either scrambled (scr) or silencing (si) RNA against Pklr or Chchd6 purchased from Sigma (Table S9) using TransIT-X2® Dynamic Delivery System (Mirus Bio LLC, Madison, WI). After ~30h post-transfection, cells were pooled and seeded in a XF24 plate at ~40×104 cells/well in the presence of 250µM oleic acid and incubated for overnight. Separately, cells were seeded in a 6-well plate and were used to determine percent knockdown of the respective genes (Figure S3B) by quantitative PCR as described.
Cellular bioenergetics
Cells seeded in a XF24 plate were analyzed in a XF24 analyzer (Agilent) as described (Wu et al., 2007). Briefly, oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured before and after the sequential injection of 0.75µM oligomycin, 2µM FCCP, and 0.75µM of rotenone/myxothiazol. Mixing, waiting, and measurement times were 3, 2, and 3 min, respectively. Measures were normalized by total protein.
Bioenergetics of isolated mitochondria
Isolated mitochondrial respiration was measured as described (Rogers et al., 2011). Briefly, livers of mice injected with either control or shRNA adenovirus were minced, rinsed in PBS and homogenized with a glass Dounce on ice, in mitochondrial isolation buffer (MSHE) containing 0.5% BSA. Mitochondria were obtained by dual centrifugation (800 and 8000g) and resuspended in mitochondrial assay solution (MAS). Ten microgram of mitochondrial protein in MAS buffer were seeded into a XF24 Seahorse plate by centrifugation at 2000g for 20 min at 4 °C. The plate was warmed for 10 min at 37°C before the measures. For the complex I respiration, the measures were collected in presence of 10mM pyruvate, 2mM malate and 4µM FCCP. Complex II respiration was measured with 10mM succinate and 2µM rotenone.
QUANTIFICATION AND STATISTICAL ANALYSIS
GWAS of hepatic TG and eQTL analyses
Genotypes for 113 mouse strains were obtained from the Jackson Laboratories using the Mouse Diversity Array (Yang et al., 2009). As previously described (Hui et al., 2015), poor quality-flagged SNPs were removed. Then, SNPs that had a minor allele frequency (MAF) of <5% and a missing genotype rate of >10% were removed, yielding about 200,000 SNPs. Genome-wide association mapping of the hepatic TG content was previously generated in (Hui et al., 2015) and the tissue-specific eQTLs were generated in (Parks et al., 2013). GWAS and eQTLs were calculated using the Factored Spectrally Transformed Linear Mixed Models (FaST-LMM) approach, which uses a linear mixed model to correct for population structure (Listgarten et al., 2012). Cis-eQTLs were defined as those within ±1Mb region of the transcription start and end sites of the genes. False Discover Rate (FDR) estimated by the q values (Storey and Tibshirani, 2003) was used to correct for multiple testing. We included 216,611 cis-eQTL associations (75,857 unique cis-eSNPs and 1,938 cis-genes) in adipose tissue, and 258,312 cis-eQTL associations (86,336 unique cis-eSNPs and 2,261 cis-genes) in liver at P<1E-6 (FDR<0.01) in the current study.
Reconstruction of the co-expression networks from liver and adipose tissue transcriptome data
We used two methods to infer tissue-specific co-expression networks from both liver and adipose tissue samples: Weighted Gene Co-expression Network Analysis (WGCNA) (Langfelder and Horvath, 2008), and Multiscale Embedded Gene Co-expression Network Analysis (MEGENA) (Song and Zhang, 2015). WGCNA has been the most widely used method to construct gene coexpression networks and a large number of studies have demonstrated its superior power to define biologically relevant modules. However, WGCNA modules tend to be large in size, with some modules each containing thousands of genes, making it challenging to conduct downstream refinement and prioritization of genes and processes. Moreover, WGCNA only allows each gene to be assigned to a single module, which does not agree with the known biology where a gene can interact with different partners to carry out different functions. The newly developed method MEGENA, on the other hand, overcomes the main limitations of WGCNA by producing more compact and coherent modules and allowing each gene to be assigned to different modules. Therefore, the two methods complement each other and may uncover hidden biology missed by the other method. The key differences between the two methods are summarized in Table S10.
Both methods aim to identify co-regulated gene sets based on correlation of gene pairs and assign sets of co-regulated genes to coexpression modules via hierarchical clustering. MEGENA is based on divisive hierarchical clustering, which is opposite to the WGCNA approach, which uses agglomerative clustering. Agglomerative method starts with the assumption that each gene is a single cluster by itself. Then, it successively merges pair of clusters until all the genes are grouped into a single cluster (module). A pair of clusters is merged based on their distance according to a pre-defined distance measure (e.g. average {1−abs(correlation)} scores between all gene pairs, where each gene pair consists of two genes, one gene from each of the clusters). In contrast, divisive clustering starts at the top with all genes in one cluster and it requires a flat clustering procedure (e.g. k-means) to split a cluster into smaller groups. This procedure is applied recursively until each gene is in its own singleton cluster. Divisive algorithms produce more accurate clustering than agglomerative algorithms in most cases, since agglomerative methods make clustering decisions based on local patterns without taking into account the global distribution (Alpaydin, 2010; Bhatnagar, 2014). These early decisions in the agglomerative approach cannot be prevented. In contrast, divisive clustering benefits from complete information about the global distribution of the samples when making top-level partitioning decisions (Bhatnagar, 2014). However, divisive methods are more complex than the agglomerative way since a second, flat clustering algorithm is needed as a nested subroutine as mentioned above. MEGENA uses Planar Filtered Networks, which aim to reduce computational costs of multi-scale clustering. MEGENA performs a nested k-medoids clustering, which detects k optimal clusters at each iteration by minimizing the shortest path distance within each cluster to provide more compact modules. The nested clustering process goes on until no more compact child cluster can occur. Since MEGENA clusters genes into modules in a multi-scale manner, at each scale we can obtain alternative gene sets despite using the same gene expression input. This multi-scale clustering mechanism allows genes to be members of multiple modules, but at different scales. In contrast, WGCNA uses Topological Overlap Matrix (TOM) subtracted from 1 (dissTOM= 1−TOM) as a dissimilarity measure and distance between two clusters is defined by the average dissTOM value of all possible gene pairs (one gene from each cluster in a pair-wise manner). We used the average distance feature, which is the default choice. In agreement with (Song and Zhang, 2015), we observed that MEGENA, using modules obtained from all scales instead of only one scale, tends to cluster the genes into more compact and functionally coherent groups than WGCNA. The use of modules generated from these complementary methods allows us to comprehensively capture the potential gene organization patterns among genes.
Preservation analysis of the coexpression modules
We searched the preservation of the modules derived from the MEGENA and WGCNA methods using the modulePreservation procedure provided in the WGCNA R package. This procedure reports a Z-summary score, which depends on both density and connectivity statistics of the nodes in the sub network (module), to determine whether a module is conserved or non-conserved. A Z-summary score<2 means that the tested module is not conserved in the expression dataset or adjacency matrix of the second condition that is tested. A Z-summary score above 2 means there are evidences for the module preservation (Langfelder and Horvath, 2008). We applied the modulePreservation procedure to the tissue-specific WGCNA modules and searched their preservation in the MEGENA modules and then we applied this procedure to the MEGENA modules to check their preservation in the WGCNA modules. Additionally, we analyzed the reciprocal preservation of the modules obtain from the HF/HS diet expression data with those from a set of gene expression data from HMDP mice fed a chow diet (Bennett et al., 2010).
Selection of coexpression modules correlated with NAFLD
To filter the co-expression modules down to those that are relevant to the trait of interest (hepatic TG level), eigen genes of MEGENA and WGCNA modules were obtained and their associations with the trait were calculated using Pearson correlation. Correlation P<1E-3 was used as the cutoff to select NAFLD related modules. This cutoff corresponds to False Positive Rate (FPR)<0.1 based on permutation analysis. We randomly generated 1000 gene sets as our negative controls with varying member sizes ranging from 20 to 500 genes. Then, the eigen gene of each negative control is obtained and their associations with the trait were calculated using Pearson correlation. 102 gene sets among 1000 negative controls exhibited a correlation at P<1E-3. Since these sets are randomly generated, we hypothesized that these 102 sets were False Positives (FP). The remaining negative control gene sets were accepted as True Negatives (TN). Hence, FPR=FP/(FP+TN) score for the P<1E-3 cutoff is ~0.1.
Functional annotation of the NAFLD correlated coexpression modules
The TG-correlated co-expression modules were annotated with the pathways from KEGG, Reactome, Biocarta, MatrisomeDB, and PID databases collected in the MSigDB database (Subramanian et al., 2005) via the hypergeometric test (one-tailed version of Fisher Exact test). Bonferroni correction was used to obtain adjusted P-values. Pathways reaching adjusted P <0.05 and shared gene numbers ≥ 5 were assigned as significant. Overlaps with nominal P values<5E-3 and shared gene numbers ≥ 5 were considered (even if the adjusted P>=0.05) as suggestive pathways.
Curation of previously known NAFLD genes
We collected 107 NAFLD-associated genes from the DisGeNET database (Piñero et al., 2015), which curates gene-disease and variant-disease associations from databases such as UniProt, ClinVar, Comparative Toxicogenomics Database-CTD, from the GWAS Catalog, and cautiously derives genes via text mining of previous studies. Some of these studies use only mouse or rat models while others use both human and animal models in a cross-species manner. Genes are scored according to multiple measures to provide users prioritized phenotype-genotype associations. These measures encompass the number of the publications from PubMed supporting the gene-disease associations, the type of the study (animal models, human studies, or both), and the disease specificity index (DSI). DSI of a gene is inversely correlated with the number of diseases associated with this gene. If the gene is related to multiple diseases, DSI diminishes to zero; when the gene tends to be specific to one disease, DSI approaches 1. We selected the NAFLD genes with a DSI score >0.2 and those that were validated and published at least in two PubMed studies, as recommended by the method. These genes were used to compare with genes identified from the current HMDP study as a means for in silico validation. we annotated these genes with functional terms via the hypergeometric test as described above. The top functional terms (adjusted P<1E-04, as listed in Table S7) that are highly associated with these 107 genes were compared with the pathways from our HMDP analyses.
Mergeomics pipeline (MSEA and KDA procedures)
We conducted multi-omics integration using the Mergeomics (Shu et al., 2016) pipeline, a computational pipeline developed to identify pathways, gene network, and key regulators via multi-omics integration. In this study, we first used liver TG GWAS information from the HMDP male mice to guide the identification of pathways and coexpression networks that are genetically associated with TG using the Marker Set Enrichment Analysis (MSEA) module in Mergeomics. Briefly, genes within each pathway (from KEGG, Biocarta, and Reactome) or tissue-specific co-expression modules (from WGCNA or MEGENA) were mapped to eSNPs via tissue-matched eQTL of the same mouse samples (we listed all of the data sources that are leveraged in this study in the "key resource table" of the STAR methods). In this study, only the cis-eQTLs (within ±1Mb of the transcription start and end sites) with association P<1E-6, which are more reliable than trans-eQTLs, were used to map genes to the loci. The eSNP sets representing each pathway or module were then filtered for linkage disequilibrium (LD) based on the LD block information determined by the PLINK2 tool (Chang et al., 2015) and annotated with the liver TG GWAS association P values. The GWAS p values of each eSNP set was then compared against eSNPs generated from random gene sets to assess the significance of enrichment for stronger GWAS association p values, using a modified chi-square statistics which is not based on a single GWAS p value cutoff but summarized over a range of quantile-based cutoffs to avoid artifacts and produce stable enrichment scores (Shu et al., 2016). The enrichment statistics is defined as . In the formula, n denotes the number of quantile points (we used ten quantile points ranging from the top 50% to the top 99.9% based on the rank of the GWAS P values), O and E denote the observed and expected counts of positive findings (i.e. signals above the quantile point), and κ = 1 is a stability parameter to reduce artefacts from low expected counts for small SNP sets.
An FDR < 0.05 cutoff value for the MSEA process was chosen by evaluating the specificity values of 1,000 random gene-sets created using the background genes included in the original gene-sets comprised of both pathways and coexpression modules. The sizes of the random gene-sets vary between 20 and 500. These random gene-sets (negative controls) were expected not to be significantly enriched for the liver TG related GWAS data for a given FDR value. A specificity score is defined as: , where TN are true negatives, representing random gene-sets that are not enriched for the GWAS data (FDR >= given cutoff); FP are false positives, representing random gene-sets that are enriched for the GWAS data (FDR < given cutoff). We aim to have a specificity score close to one. For the FDR<0.05 cutoff, specificity reaches at S=0.829 for the adipose-specific analysis (821 TNs and 171 FPs among 1,000 random sets from the adipose data) and it reaches S=0.799 for the liver-specific analysis (799 TNs and 201 FPs among 1,000 random sets from the liver data).
Significantly enriched pathways and co-expression modules (FDR < 0.05) were merged into the supersets if the overlapping ratio between gene sets was > 0.33 and significance of overlap passed Bonferroni corrected P-value<0.05 based on Fisher’s exact test to reduce redundancy. The merging process was performed separately for the significant gene sets from each tissue. In some cases, a canonical pathway and a co-expression module, which are annotated with the same pathway name, may not be merged together and they can be represented by two separate gene sets due to their relatively small overlapping ratio. For instance, the insulin signaling term associated with a co-expression module is not merged with the insulin signaling term from the KEGG database due to the small overlap ratio.
The second step of the Mergeomics pipeline is the Key Driver Analysis (KDA) process, which identifies hub or key regulator genes within the liver TG-associated supersets by mapping the genes in each superset onto predefined tissue-specific Bayesian Networks (BNs). Liver and adipose tissue BNs were utilized for this process. BNs involve the directed causal relationships between gene pairs by considering both gene expression data and previously known regulatory relationships between genes. BNs used in our study were derived from multiple human and mouse datasets from previous studies (Derry et al., 2010; Emilsson et al., 2008; Schadt et al., 2008; Tu et al., 2012; Wang et al., 2007; Yang et al., 2006; Zhong et al., 2010), as listed in the key resource table of the STAR methods. A BN from each dataset was constructed using an established method, RIMBANET (Zhu et al., 2007, 2008). A BN from a dataset represents a consensus network in which only edges that passed a probability of >30% across 1000 BNs generated starting from different random seed genes, were kept. For each tissue, BNs from individual studies were combined without considering the edge weights (as the edges included in each BN were considered robust), to form a union network. This strategy has been successfully used previously to derive meaningful biological insights (Mäkinen et al., 2014; Shu et al., 2016, 2017; Zhao et al., 2016). Since the directions of the interactions might be conflicting in some of these previous studies, we omit the directionality in these BNs when applying KDA. Because these BNs were collected from both mouse and human studies, gene symbols in network figures are given in human orthologs. A Key Driver (KD) of a NAFLD superset was defined based on the enrichment of member genes in the superset in the candidate KD’s network neighborhood compared to that of a random gene selected from the network, using a modified chi-square based statistics as described in the MSEA section above. The Benjamini-Hochberg false discovery rate (FDR) approach was used to correct for multiple hypothesis testing and FDR < 0.05 were used to determine significant KDs of a given superset.
Statistical analysis used in experimental validation
Statistical analyses were performed using Prism v7.0a (GraphPad Software, Inc., La Jolla, CA, USA). Errors bars plotted on graphs are presented as the mean ± SEM unless reported otherwise. The critical significance value (α) was set at 0.05, and if the P values were less than α, we reported that, by rejecting the null hypothesis, the observed differences were statistically significant.
DATA AND SOFTWARE AVAILABILITY
The NCBI GEO accession number for the microarray data used in our study is GSE64770.
Raw expression data for liver tissue can be found at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE64769.
Raw expression data for adipose tissue can be found at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE64768.
Supplementary Material
Table S4. Raw MSEA results. Related to Figure 2. Left Panel shows the liver tissue results, while right panel shows the adipose tissue results.
Table S5. Tissue-specific and shared supersets between liver and adipose tissue. Related to Figure 2. A superset is a gene set merging a group of highly overlapping pathways or coexperssion modules to reduce redundancy. Original pathways are labeled with IDs from Reactome (""rctm"") or KEGG (""M""), and coexpression modules are labeled with IDs from WGCNA or MEGENA networks. See also Figure 2 and Table S2. Pathways, which are enriched by previously reported NAFLD-associated genes curated in DisGeNET, are highlighted in bold. Overlaps between the coexpression modules and GO terms with adjusted P <0.05 are assigned as significant, while overlaps with nominal P<5E-3 and shared gene numbers ≥ 5 are considered (when adjusted P>=0.05) as suggestive and denoted with an *.
Table S6. Previously annotated NAFLD-associated genes from the literature. Related to Figure 2. Genes that were identified as a KD in our analysis are highlighted in particular colors as denoted below.
Table S7. Top 50 GO terms associated with the previously annotated NAFLD-associated genes (listed in Table S6). Related to Figure 2. Pathways that were identified by our analysis are highlighted in yellow.
Table S8. Top KDs of each of the significant supersets that are genetically associated with NAFLD. Related to Figure 3. Previously reported NAFLD genes curated in DisGeNET are highlighted in bold. See also Tables S6 – S7. Besides, we notified the genes with significant cis-eQTLs in the corresponding tissue. Overlaps between the coexpression modules and GO terms with adjusted P<0.05 are assigned as significant, while overlaps with nominal P<5E-3 and shared gene numbers ≥ 5 are considered (when adjusted P>=0.05) as suggestive and denoted with an *.
Table S9. List of shRNA construct sequences, qPCR primer sequences, and siRNA. Related to STAR Methods.
Table S10. Comparison of WGCNA and MEGENA methods. Related to STAR Methods.
Table S11. HMDP strain and sample details used in the study. Related to STAR Methods.
Table S1. Tissue-specific coexpression modules from each method and each tissue. Related to STAR Methods and Figure 2.
Table S2. Top 5 GO terms associated with the tissue-specific coexpression modules. Related to STAR Methods, Figure 2, and Figure S1. Fisher exact test is used to annotate the coexpression modules (nominal P<0.005).
Table S3. Tissue-specific eQTL content of hepatic TG-related tissue-specific modules. Related to STAR Methods and Figure 2. Linkage disequilibrium is considered.
HIGHLIGHTS.
NAFLD was modeled in a population of ~100 diverse inbred strains of mice.
Network modeling was used to predict key driver genes regulating NAFLD.
In vivo knockdown of these genes rescued from steatosis and insulin resistance.
In vitro knockdown of Pklr or Chchd6 shifted towards glycolytic metabolism.
Acknowledgments
FUNDING INFORMATION
This work was supported by NIH-HL28481 (AJL), NIH-DK104363 (XY), AHA 13SDG17290032 (XY), AHA fellowship 17POST33670739 (ZK), Iris Cantor-UCLA Women's Health Center/CTSI fellowship UL1TR001881 (ZK), the Foundation Leducq 12CVD04 (LV and KR), NIH-HL28481 (KR), the National Center for Research Resources Grant S10RR026744 (KR), NIH-T32HL007895 (MMS), NIH-T32HL69766 (MMS) and the Research Council of Norway grant 240405/F20 (FN). The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
We would like to acknowledge the following undergrad students, Sonul Gupta and Sung Ho Hong, for their help in constructing the shRNAs.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
KCK, ZK, AJL and XY conceived the study, with AJL responsible for the design of the experimental components and XY responsible for the design of the multi-omics integration analysis. ZK led the integrative genomics analyses and KCK led the experimental validation studies. All authors performed experiments or analyzed the data; KCK, ZK, REB, AJL and XY drafted the manuscript; all authors read or revised the manuscript.
References
- Adams LA, Lymp JF, St Sauver J, Sanderson SO, Lindor KD, Feldstein A, Angulo P. The natural history of nonalcoholic fatty liver disease: a population-based cohort study. Gastroenterology. 2005;129:113–121. doi: 10.1053/j.gastro.2005.04.014. [DOI] [PubMed] [Google Scholar]
- Alpaydin E. Introduction to Machine Learning. The MIT Press; 2010. [Google Scholar]
- de Alwis NMW, Day CP. Non-alcoholic fatty liver disease: The mist gradually clears. J. Hepatol. 2008;48 doi: 10.1016/j.jhep.2008.01.009. [DOI] [PubMed] [Google Scholar]
- Arneson D, Bhattacharya A, Shu L, Mäkinen V-P, Yang X, Civelek M, Lusis A, Joyce A, Palsson B, Schadt E, et al. Mergeomics: a web server for identifying pathological pathways, networks, and key regulators via multidimensional data integration. BMC Genomics. 2016;17:722. doi: 10.1186/s12864-016-3057-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begriche K, Igoudjil A, Pessayre D, Fromenty B. Mitochondrial dysfunction in NASH: Causes, consequences and possible means to prevent it. Mitochondrion. 2006;6:1–38. doi: 10.1016/j.mito.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Begriche K, Massart J, Robin MA, Bonnet F, Fromenty B. Mitochondrial adaptations and dysfunctions in nonalcoholic fatty liver disease. Hepatology. 2013;58:1497–1507. doi: 10.1002/hep.26226. [DOI] [PubMed] [Google Scholar]
- Bennett BJ, Farber CR, Orozco L, Kang HM, Ghazalpour A, Siemers N, Neubauer M, Neuhaus I, Yordanova R, Guan B, et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010;20:281–290. doi: 10.1101/gr.099234.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BJ, Vallim TQDA, Wang Z, Shih DM, Meng Y, Gregory J, Allayee H, Lee R, Graham M, Crooke R, et al. Trimethylamine-N-Oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013;17:49–60. doi: 10.1016/j.cmet.2012.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatnagar V. Data Mining and Analysis in the Engineering Field. Hershey, PA, USA: IGI Global; 2014. [Google Scholar]
- Boyle EA, Li YI, Pritchard JK. An Expanded View of Complex Traits: From Polygenic to Omnigenic. Cell. 2017;169:1177–1186. doi: 10.1016/j.cell.2017.05.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, Grundy SM, Hobbs HH. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology. 2004;40:1387–1395. doi: 10.1002/hep.20466. [DOI] [PubMed] [Google Scholar]
- Chakravarthy MV, Lodhi IJ, Yin L, Malapaka RR, Xu HE, Turk J, Semenkovich CF. Identification of a physiologically relevant endogenous ligand for PPARalpha in liver. Cell. 2009;138:476–488. doi: 10.1016/j.cell.2009.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers JC, Zhang W, Sehmi J, Li X, Wass MN, Van der Harst P, Holm H, Sanna S, Kavousi M, Baumeister SE, et al. Genome-wide association study identifies loci influencing concentrations of liver enzymes in plasma. Nat. Genet. 2011;43:1131–1138. doi: 10.1038/ng.970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal RG. Adenovirus: The First Effective In Vivo Gene Delivery Vector. Hum. Gene Ther. 2014;25:3–11. doi: 10.1089/hum.2013.2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry JMJ, Zhong H, Molony C, MacNeil D, Guhathakurta D, Zhang B, Mudgett J, Small K, El Fertak L, Guimond A, et al. Identification of genes and networks driving cardiovascular and metabolic phenotypes in a mouse F2 intercross. PLoS One. 2010;5 doi: 10.1371/journal.pone.0014319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding C, Wu Z, Huang L, Wang Y, Xue J, Chen S, Deng Z, Wang L, Song Z, Chen S. Mitofilin and CHCHD6 physically interact with Sam50 to sustain cristae structure. Sci. Rep. 2015;5:16064. doi: 10.1038/srep16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emilsson V, Thorleifsson G, Zhang B, Leonardson AS, Zink F, Zhu J, Carlson S, Helgason A, Walters GB, Gunnarsdottir S, et al. Genetics of gene expression and its effect on disease. Nature. 2008;452:423–428. doi: 10.1038/nature06758. [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Stanley GHS. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957;226:497–509. [PubMed] [Google Scholar]
- Hardy T, Oakley F, Anstee QM, Day CP. Nonalcoholic Fatty Liver Disease: Pathogenesis and Disease Spectrum. Annu. Rev. Pathol. Mech. Dis. 2016;11:451–496. doi: 10.1146/annurev-pathol-012615-044224. [DOI] [PubMed] [Google Scholar]
- Hill-Baskin AE, Markiewski MM, Buchner DA, Shao H, Desantis D, Hsiao G, Subramaniam S, Berger NA, Croniger C, Lambris JD, et al. Diet-induced hepatocellular carcinoma in genetically predisposed mice. Hum. Mol. Genet. 2009;18:2975–2988. doi: 10.1093/hmg/ddp236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui ST, Parks BW, Org E, Norheim F, Che N, Pan C, Castellani LW, Charugundla S, Dirks DL, Psychogios N, et al. The genetic architecture of NAFLD among inbred strains of mice. Elife. 2015;4:e05607. doi: 10.7554/eLife.05607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynen MA, Mühlmeister M, Gotthardt K, Guerrero-Castillo S, Brandt U. Evolution and structural organization of the mitochondrial contact site (MICOS) complex and the mitochondrial intermembrane space bridging (MIB) complex. Biochim. Biophys. Acta - Mol. Cell Res. 2016;1863:91–101. doi: 10.1016/j.bbamcr.2015.10.009. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Cohen DE. Mechanisms of hepatic triglyceride accumulation in non-alcoholic fatty liver disease. J. Gastroenterol. 2013;48:434–441. doi: 10.1007/s00535-013-0758-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamoto T, Kitamoto A, Yoneda M, Hyogo H, Ochi H, Nakamura T, Teranishi H, Mizusawa S, Ueno T, Chayama K, et al. Genome-wide scan revealed that polymorphisms in the PNPLA3, SAMM50, and PARVB genes are associated with development and progression of nonalcoholic fatty liver disease in Japan. Hum. Genet. 2013;132:783–792. doi: 10.1007/s00439-013-1294-3. [DOI] [PubMed] [Google Scholar]
- Kopec KL, Burns D. Nonalcoholic fatty liver disease: a review of the spectrum of disease, diagnosis, and therapy. Nutr. Clin. Pract. 2011;26:565–576. doi: 10.1177/0884533611419668. [DOI] [PubMed] [Google Scholar]
- Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg-Hansen A, Vogt TF, Hobbs HH, Cohen JC. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014;46:352–356. doi: 10.1038/ng.2901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Laan M, Horvath SE, Pfanner N. Mitochondrial contact site and cristae organizing system. Curr. Opin. Cell Biol. 2016;41:33–42. doi: 10.1016/j.ceb.2016.03.013. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Zhang C, Liu Z, Klevstig M, Mukhopadhyay B, Bergentall M, Cinar R, Ståhlman M, Sikanic N, Park JK, et al. Network analyses identify liver- specific targets for treating liver diseases. Mol. Syst. Biol. 2017;13:938. doi: 10.15252/msb.20177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The SVA package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012;28:882–883. doi: 10.1093/bioinformatics/bts034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Listgarten J, Lippert C, Kadie CM, Davidson RI, Eskin E, Heckerman D. Improved linear mixed models for genome-wide association studies. Nat. Methods. 2012;9:525–526. doi: 10.1038/nmeth.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkinen V-P, Civelek M, Meng Q, Zhang B, Zhu J, Levian C, Huan T, Segrè AV, Ghosh S, Vivar J, et al. Integrative genomics reveals novel molecular pathways and gene networks for coronary artery disease. PLoS Genet. 2014;10:e1004502. doi: 10.1371/journal.pgen.1004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini G, Bugianesi E, Forlani G, Cerrelli F, Lenzi M, Manini R, Natale S, Vanni E, Villanova N, Melchionda N, et al. Nonalcoholic fatty liver, steatohepatitis, and the metabolic syndrome. Hepatology. 2003;37:917–923. doi: 10.1053/jhep.2003.50161. [DOI] [PubMed] [Google Scholar]
- McCullough AJ. The clinical features, diagnosis and natural history of nonalcoholic fatty liver disease. Clin Liver Dis. 2004;8:521–33. doi: 10.1016/j.cld.2004.04.004. viii. [DOI] [PubMed] [Google Scholar]
- Montgomery MK, Hallahan NL, Brown SH, Liu M, Mitchell TW, Cooney GJ, Turner N. Mouse strain-dependent variation in obesity and glucose homeostasis in response to high-fat feeding. Diabetologia. 2013;56:1129–1139. doi: 10.1007/s00125-013-2846-8. [DOI] [PubMed] [Google Scholar]
- Van Nas A, Ingram-Drake L, Sinsheimer JS, Wang SS, Schadt EE, Drake T, Lusis AJ. Expression quantitative trait loci: Replication, tissue- and sex-specificity in mice. Genetics. 2010;185:1059–1068. doi: 10.1534/genetics.110.116087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolan CJ, Larter CZ. Lipotoxicity: Why do saturated fatty acids cause and monounsaturates protect against it? J. Gastroenterol. Hepatol. 2009;24:703–706. doi: 10.1111/j.1440-1746.2009.05823.x. [DOI] [PubMed] [Google Scholar]
- Parks BW, Nam E, Org E, Kostem E, Norheim F, Hui ST, Pan C, Civelek M, Rau CD, Bennett BJ, et al. Genetic control of obesity and gut microbiota composition in response to high-fat, high-sucrose diet in mice. Cell Metab. 2013;17:141–152. doi: 10.1016/j.cmet.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessayre D, Fromenty B. NASH: a mitochondrial disease. J. Hepatol. 2005;42:928–940. doi: 10.1016/j.jhep.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Piñero J, Queralt-Rosinach N, Bravo À, Deu-Pons J, Bauer-Mehren A, Baron M, Sanz F, Furlong LI. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database. 2015;2015:bav028. doi: 10.1093/database/bav028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. Journal of Hepatology. 2010:372–384. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- Rogers GW, Brand MD, Petrosyan S, Ashok D, Elorza AA, Ferrick DA, Murphy AN. High throughput microplate respiratory measurements using minimal quantities of isolated mitochondria. PLoS One. 2011;6 doi: 10.1371/journal.pone.0021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY, Kasarskis A, Zhang B, Wang S, Suver C, et al. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008;6:1020–1032. doi: 10.1371/journal.pbio.0060107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu L, Zhao Y, Kurt Z, Byars SG, Tukiainen T, Kettunen J, Orozco LD, Pellegrini M, Lusis AJ, Ripatti S, et al. Mergeomics: multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genomics. 2016;17:874. doi: 10.1186/s12864-016-3198-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu L, Chan KHK, Zhang G, Huan T, Kurt Z, Zhao Y, Codoni V, Trégouët D-A, Yang J, Wilson JG, et al. Shared genetic regulatory networks for cardiovascular disease and type 2 diabetes in multiple populations of diverse ethnicities in the United States. PLOS Genet. 2017 doi: 10.1371/journal.pgen.1007040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobaniec-Lotowska ME, Lebensztejn DM. Ultrastructure of hepatocyte mitochondria in nonalcoholic steatohepatitis in pediatric patients: usefulness of electron microscopy in the diagnosis of the disease. Am J Gastroenterol. 2003;98:1664–1665. doi: 10.1111/j.1572-0241.2003.07561.x. [DOI] [PubMed] [Google Scholar]
- Song WM, Zhang B. Multiscale Embedded Gene Co-expression Network Analysis. PLoS Comput. Biol. 2015;11 doi: 10.1371/journal.pcbi.1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. {G}ene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu Z, Keller MP, Zhang C, Rabaglia ME, Greenawalt DM, Yang X, Wang IM, Dai H, Bruss MD, Lum PY, et al. Integrative Analysis of a Cross-Loci Regulation Network Identifies App as a Gene Regulating Insulin Secretion from Pancreatic Islets. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1003107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernon G, Baranova A, Younossi ZM. Systematic review: The epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment. Pharmacol. Ther. 2011;34:274–285. doi: 10.1111/j.1365-2036.2011.04724.x. [DOI] [PubMed] [Google Scholar]
- Wang SS, Schadt EE, Wang H, Wang X, Ingram-Drake L, Shi W, Drake TA, Lusis AJ. Identification of pathways for atherosclerosis in mice: Integration of quantitative trait locus analysis and global gene expression data. Circ. Res. 2007;101 doi: 10.1161/CIRCRESAHA.107.152975. [DOI] [PubMed] [Google Scholar]
- Williams EG, Wu Y, Jha P, Dubuis S, Blattmann P, Argmann CA, Houten SM, Amariuta T, Wolski W, Zamboni N, et al. Systems proteomics of liver mitochondria function. Science (80-.) 2016;352:aad0189-aad0189. doi: 10.1126/science.aad0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Wang C, Li S, Li S, Wang W, Li J, Chi Y, Yang H, Kong X, Zhou Y, et al. Thyroid hormone-responsive SPOT 14 homolog promotes hepatic lipogenesis, and its expression is regulated by Liver X receptor α through a sterol regulatory element-binding protein 1c–dependent mechanism in mice. Hepatology. 2013;58:617–628. doi: 10.1002/hep.26272. [DOI] [PubMed] [Google Scholar]
- Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, et al. Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am. J. Physiol. Cell Physiol. 2007;292:C125–36. doi: 10.1152/ajpcell.00247.2006. [DOI] [PubMed] [Google Scholar]
- Yang H, Ding Y, Hutchins LN, Szatkiewicz J, Bell TA, Paigen BJ, Graber JH, de Villena FP-M, Churchill GA. A customized and versatile high-density genotyping array for the mouse. Nat. Methods. 2009;6:663–666. doi: 10.1038/nmeth.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Schadt EE, Wang S, Wang H, Arnold AP, Ingram-Drake L, Drake TA, Lusis AJ. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 2006;16:995–1004. doi: 10.1101/gr.5217506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Chen J, Freudenberg JM, Meng Q, Rajpal DK, Yang X. Network-based identification and prioritization of key regulators of coronary artery disease loci. Arterioscler. Thromb. Vasc. Biol. 2016;36:928–941. doi: 10.1161/ATVBAHA.115.306725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, Beaulaurier J, Lum PY, Molony C, Yang X, MacNeil DJ, Weingarth DT, Zhang B, Greenawalt D, Dobrin R, et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 2010;6:32. doi: 10.1371/journal.pgen.1000932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Wiener MC, Zhang C, Fridman A, Minch E, Lum PY, Sachs JR, Schadt EE. Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput. Biol. 2007;3:692–703. doi: 10.1371/journal.pcbi.0030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, Bumgarner RE, Schadt EE. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet. 2008;40:854–861. doi: 10.1038/ng.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S4. Raw MSEA results. Related to Figure 2. Left Panel shows the liver tissue results, while right panel shows the adipose tissue results.
Table S5. Tissue-specific and shared supersets between liver and adipose tissue. Related to Figure 2. A superset is a gene set merging a group of highly overlapping pathways or coexperssion modules to reduce redundancy. Original pathways are labeled with IDs from Reactome (""rctm"") or KEGG (""M""), and coexpression modules are labeled with IDs from WGCNA or MEGENA networks. See also Figure 2 and Table S2. Pathways, which are enriched by previously reported NAFLD-associated genes curated in DisGeNET, are highlighted in bold. Overlaps between the coexpression modules and GO terms with adjusted P <0.05 are assigned as significant, while overlaps with nominal P<5E-3 and shared gene numbers ≥ 5 are considered (when adjusted P>=0.05) as suggestive and denoted with an *.
Table S6. Previously annotated NAFLD-associated genes from the literature. Related to Figure 2. Genes that were identified as a KD in our analysis are highlighted in particular colors as denoted below.
Table S7. Top 50 GO terms associated with the previously annotated NAFLD-associated genes (listed in Table S6). Related to Figure 2. Pathways that were identified by our analysis are highlighted in yellow.
Table S8. Top KDs of each of the significant supersets that are genetically associated with NAFLD. Related to Figure 3. Previously reported NAFLD genes curated in DisGeNET are highlighted in bold. See also Tables S6 – S7. Besides, we notified the genes with significant cis-eQTLs in the corresponding tissue. Overlaps between the coexpression modules and GO terms with adjusted P<0.05 are assigned as significant, while overlaps with nominal P<5E-3 and shared gene numbers ≥ 5 are considered (when adjusted P>=0.05) as suggestive and denoted with an *.
Table S9. List of shRNA construct sequences, qPCR primer sequences, and siRNA. Related to STAR Methods.
Table S10. Comparison of WGCNA and MEGENA methods. Related to STAR Methods.
Table S11. HMDP strain and sample details used in the study. Related to STAR Methods.
Table S1. Tissue-specific coexpression modules from each method and each tissue. Related to STAR Methods and Figure 2.
Table S2. Top 5 GO terms associated with the tissue-specific coexpression modules. Related to STAR Methods, Figure 2, and Figure S1. Fisher exact test is used to annotate the coexpression modules (nominal P<0.005).
Table S3. Tissue-specific eQTL content of hepatic TG-related tissue-specific modules. Related to STAR Methods and Figure 2. Linkage disequilibrium is considered.