

Abstract
Skyline is a freely-available, open-source Windows client application for accelerating targeted proteomics experimentation, with an emphasis on the proteomics and mass spectrometry community as users and as contributors. This review covers the informatics encompassed by the Skyline ecosystem, from computationally-assisted targeted mass spectrometry method development, to raw acquisition file data processing, and quantitative analysis and results sharing.
Keywords: targeted proteomics, quantitative mass spectrometry, informatics
I. Introduction
Since the completion of the Human Genome project (Consortium 2001; Venter et al. 2001; Consortium 2005), a wealth of functional genomic techniques have emerged as the focus of research shifts to assigning function and understanding the regulation of each of the identified gene products. The focus of these efforts is to better understand how the information stored in a genome encodes all the complexity necessary to sustain a complex multicellular organism (Lander 2011). Nothwithstanding impressive gains in these technologies, interpretation of their results is limited without corresponding data on proteins, the primary functional macromolecules encoded by the genome. This limitation is highlighted by the observation that measurements performed at the nucleic acid level tend to correlate very poorly with those performed at the protein level (Greenbaum et al. 2003; Schrimpf et al. 2009), especially in cases when experimental noise is not considered (Csardi et al. 2015). A combination of factors likely contribute to the poor protein-transcript correlation, including the variable lifetime of each protein dictated by its respective synthesis and degradation rates; the existence of multiple different forms of each transcript product due to post-translational modifications; and finally, the temporal/spatial regulation imparted by protein complexes and the highly compartmentalized nature of cellular processes. Accordingly, the direct analysis of proteins, albeit more technically challenging, is absolutely crucial to a complete understanding of gene regulation and systems biology.
A. Introduction to quantitative mass spectrometry proteomics
To meet these ends, tandem mass spectrometry (MS/MS) has emerged as the dominant analytical platform for the direct characterization of the protein fraction from complex biological matrices (Ong and Mann 2005). To date, a majority of mass spectrometry-based proteomic workflows have utilized a “bottom-up” approach in which proteins are digested with an endoprotease prior to analysis. The resulting peptide mixture is typically separated via nano-flow reverse-phase liquid chromatography, ionized, and emitted directly into a mass spectrometer for analysis.
Both absolute and relative quantitative measurements, reviewed in detail elsewhere (Ong and Mann 2005), are possible via several of the commonly applied MS acquisition methods. Targeted acquisition methods, including selected reaction monitoring (SRM) (Picotti and Aebersold 2012), also known as multiple reaction monitoring (MRM) (H. Zhang et al. 2011), and parallel reaction monitoring (PRM) (Peterson et al. 2012), quantify peptides from a preprogrammed list of precursor-fragment pairs and scheduled isolation windows based on previously-determined chromatography elution times. Data-independent acquisition (DIA) (Venable et al. 2004) such as Sequential Window Acquisition of all Theoretical Fragment ion spectra (SWATH) (Gillet et al. 2012) forgo preprogrammed precursor-fragment pairs, widening the isolation windows to activate all ions in a pre-specified mass-to-charge (m/z) range. (A detailed review of DIA methodology can be found elsewhere (Chapman, Goodlett, and Masselon 2014; Bilbao et al. 2015), including peptide-centric approaches to DIA (Ting et al. 2015).) It is also possible, through MS1 filtering informatics techniques (Schilling et al. 2012), to use data dependent acquisition (DDA) for quantitative analysis as opposed to conventional detection analysis.
The type of acquisition influences the selectivity, reproducibility, repeatability, limit of detection, dynamic range, and data density of the assay (Domon and Aebersold 2010). Additionally, acquisition type places specific requirements on assay development and influences the computational strategy for analyzing data. A variety of individual informatics tools have been developed to aid in assay development and to process the data collected with various acquisition types, reviewed elsewhere (Cham, Bianco, and Bessant 2010; Colangelo et al. 2013). Many freely available informatics tools, however, struggle with community adoption, due to issues with limited end user design, and lack a complete pipeline spanning method development through data analysis for an experiment.
B. Overview of the Skyline ecosystem for quantitative mass spectrometry informatics
Properties such as easy access, large dataset management, integration with other commonly used tools, intuitive data visualization, timely issue resolution, documentation, support, as well as facilitated sharing of data files and the methods used to collect them (Codrea et al. 2007) are important aspects that influence software adoption. With these needs in mind, the freely-available and open-source Skyline ecosystem was developed with a user-friendly interface, comprehensive file compatibility, vendor-neutral data processing, intuitive visualization, and reasonable computational requirements (MacLean et al. 2010). The original objective of the Skyline project was to create a single informatics tool to generate MS methods and to analyze the data collected for chromatography-based quantitative MS experiments. In addition to these core functions, Skyline now invites the community to share their own informatics tools through an external tool store (Broudy et al. 2014) for software tools that support point-and-click installation and can be run from the Skyline Tools menu. Furthermore, the introduction of additional software to the Skyline ecosystem such as Chorus for sharing raw MS files (http://chorusproject.org) and Panorama for sharing Skyline processed experimental results (Sharma et al. 2014), has helped facilitate large-scale MS datasets and inter-laboratory collaborations.
The Skyline ecosystem is unique among freely-available, open source mass spectrometry proteomics software in its end-to-end support of the targeted proteomic mass spectrometry workflow. A Skyline document is first used for assay development, aiding in instrument method creation for targeted and DIA experiments (Figure 1). Skyline exports the methods for use in mass spectrometry acquisition on a broad range of instruments from 6 different mass spectrometer vendors (Table 1). Without need of any file conversion, Skyline then supports importing raw data from most LCMS capable instruments, calculating peak areas in a vendor-neutral manner. Peak area data may be explored within the Skyline document using core analyses, comparing peptide retention times, peak areas, sample groups, underlying chromatograms and even mass spectra when available. Further analyses are possible, including those made available by external tools integrated into the Skyline ecosystem and through data report exports that researchers can process using their own tools, and custom code in R, MATLAB, Python, etc. Although a freely-available, open source academic project, Skyline’s engineering includes rigorous nightly testing to ensure any code changes made during the day are compatible with the program’s many other various functions. This level of thoroughness ensures the mass spectrometry community receives an informatics toolkit that is consistent and highly maintained, allowing researchers to upgrade with confidence as the software is adapted and changed.
Figure 1. Generalized workflow for quantitative MS assay development.
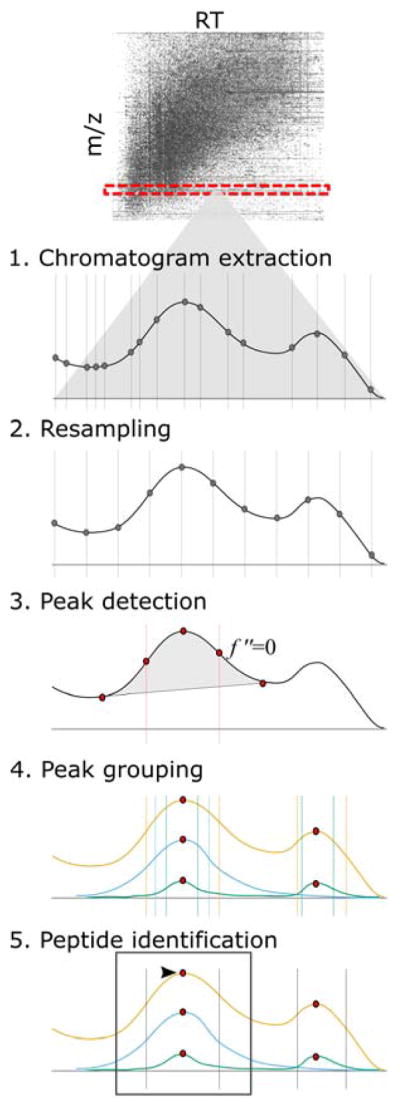
Six main steps are outlined, beginning with the development of a hypothesis and continuing through additional analyses, with examples of the associated Skyline ecosystem features.
Table 1. Proprietary file formats supported by Skyline.
Vendors and instruments supported by the Skyline ecosystem are specified along with their respective proprietary file format and general acquisition types. (QqQ: triple quadrupole; Q-TOF: quadrupole time of flight; IMS-TOF: ion mobility spectrometry-time of flight; Q-OT: quadrupole-Orbitrap; Q-LIT: quadrupole-linear ion trap)
| Vendor | File extension | Instruments Supported | Acquisitions Supported |
|---|---|---|---|
| Agilent | .d (directory) | QqQ, Q-TOF, IMS-TOF | DDA, SRM, DIA |
| Bruker | .d (directory) | Q-TOF | DDA, PRM, DIA |
| Sciex | .wiff (file) | QqQ, Q-TOF | DDA, SRM, PRM, DIA |
| Shimadzu | .qgd | QqQ | SRM |
| Thermo | .raw (file) | QqQ, Q-OT, Q-LIT | DDA, SRM, PRM, DIA |
| Waters | .raw (directory) | QqQ, Q-TOF, IMS-TOF | SRM, PRM, DIA |
Today, over 8,700 mass spectrometrists are registered Skyline users with more than 64,000 installations since first public release, and over 1,100 publications have cited the original Skyline paper. We next describe how the community uses the Skyline ecosystem, and the informatics employed by the Skyline ecosystem, from assay development, to data processing and visualization, and finally dissemination of results.
II. Assay development
The requirements for developing an effective quantitative MS proteomics assay are specific to the type of experiment and the peptide targets being assayed. For all experiments, prior to MS acquisition, it is obligatory to create a program for the instrument that defines the instrument parameters and defines how the data is to be collected by the instrument. In addition, depending on the acquisition mode of the instrument (i.e., SRM/MRM, PRM, DIA and DDA), multiple decisions must be made to optimize the acquisition of the data (Figure 1). For example, the experiments with the most intensive assay development, scheduled SRM/MRM and PRM type experiments, necessitate selection of target peptides and their transitions (SRM only) prior to acquisition, validation of transitions by MS/MS spectra, potentially optimization of individual parameters (such as collision energy - CE) and determination of retention times (RT) for optimal MS instrument scheduling. On the other hand, for DIA experiments, the only required step pre-acquisition is calculating isolation window schemes. Although this review is focused on the Skyline ecosystem for quantitative proteomics, we note that the ecosystem also works for generalized small molecules (Tang et al. 2015) and briefly describe considerations for non-peptide targets. In this section, we describe the steps required for assay development, noting which steps are necessary for which experiment types.
A. Peptide and transition selection for targeted experiments
Many proteomics hypotheses are rooted in biological observations, and so selecting proteins of interest and peptides that are exclusively representative of those proteins is often the first experimental design step in targeted bottom-up proteomic experiments, such as SRM/MRM and PRM. Selection of peptides for targeted assays is a complex process, involving consideration of (1) specific peptides or amino acid modifications of interest, (2) biological influences on the protein of interest, (3) chemical influences on peptide suitability for MS experiments, and (4) for SRM/MRM experiments, the selection of fragment ions for quantitation.
1. Specific peptides or amino acid modifications of interest
In the first case, specific amino acid modifications, especially post-translational modifications at the protein level, may dictate a peptide sequence of interest. This is especially seen in targeted phosophoproteomics assays, where the phosphosite of interest has previously been determined by prior experiments (Schilling et al. 2012, Sherrod et al. 2012, Abelin et al. 2016). In these cases, it may be easiest to manually enter the peptide sequences of interest. Skyline accepts peptides added directly to the document as lists in the Targets window. Peptides added as lists may have modifications and even charge states specified in the added sequence text. They may also be modified manually within Skyline one at a time, or in bulk by changing the Skyline modification settings.
2. Biological influences on the protein of interest
For situations where the peptide sequence is not defined by the experiment, Skyline accepts lists of proteins, either entered manually, copy-pasted, or as a FASTA file import. After proteins are added to the document, Skyline digests the proteins in silico to generate a list of peptides. The result of Skyline’s in silico digestion depends on the particular endoprotease specified in the settings of the Skyline document. The most common endoproteases used in bottom-up proteomics are Lys-C, which hydrolyzes specifically at the carboxyl side of lysine; chymotrypsin, which cleaves amide bonds on the carboxyl side; and trypsin, which cleaves the carboxyl side of lysine or arginine. Other Skyline Peptide Settings that affect results of peptide list generation are common biochemical sample preparation concerns such as missed cleavages, oxidized methionine, and peptide amino acid length (Anderson and Hunter 2005; Lange et al. 2008; Prakash et al. 2009). After endoprotease(s) are selected and biochemical considerations are defined in the Peptide Settings in the Skyline document, researchers can add proteins of interest to the Skyline target list and Skyline automatically performs in silico digestion on the proteins and the resulting peptides displayed, organized by protein of origin.
A point of consideration for proteomics research with clinical applications is the selection of peptides that may have naturally occurring amino acid variations due to individual subjects’ genetic backgrounds. Single nucleotide polymorphisms (SNPs) in the genome may give rise to amino acid changes in the final proteoform, which may alter a peptide sequence. To help guide users collecting data on clinical samples that may include SNP-related variation, Skyline provides users with access to the informatics tool Population Variation (Fujimoto et al. 2014). Population Variation reveals all human sequence variation within a set of user-specified peptides or proteins by identifying the minor allele frequency of peptide targets. The tool then filters SNP data records from dbSNP by criteria directly relevant to proteomics experiments, storing entries with minor allele frequency > 0.01, a non-null protein accession, and a protein-influencing mutation (missense, stop-gain, frameshift). The refined list is stored as a SQLite database and can be accessed through a Skyline plug-in. Running the Population Variation Skyline plug-in outputs a table listing the isoforms and peptide variants for all proteins included in the Skyline document. Researchers can use this output to consider variant peptide targets to ensure that the assay accurately measures.
3. Chemical considerations of selected peptides
Next, the hypothesis-based, biologically considered peptides must be validated for chemical considerations, namely MS signal robustness. Peptides from the same protein of interest have a range of MS signal response, with some peptides reliably responding strongly and others responding weakly or variably to MS conditions (Kuster et al. 2005). These widely ranging responses are dictated by sequence-specific physiochemical properties (e.g., length of the amino acid sequence, charge, presence of various amino acids, and hydrophobicity) and can be empirically determined using prior knowledge from MS experiments (Stergachis et al. 2011) or by using predictive algorithms.
Empirical determination of high-responding peptides requires performing preliminary MS experiments with the potential targets, often synthesized or purchased, in the intended sample matrix (Stergachis et al. 2011). The mass spectrometrist then evaluates the potential target peptide and transition pairs for signal response and chemical noise interference. Skyline facilitates this empirical evaluation with simple transition deletion and addition tools, including ability to Undo these operations, allowing researchers to easily create or modify transition lists for targeted assay development. Besides empirical determination, however, it is also possible to query past MS experiments to evaluate peptide signal response, making use of Skyline-supported online repositories like PeptideAtlas (Desiere et al. 2006), Human Proteinpedia (Mathivanan et al. 2008), GPM Proteomics Database (Craig, Cortens, and Beavis 2004), and PRIDE (Jones et al. 2008). A caveat to using repositories, as opposed to an assay-specific preliminary experiment, is that peptide response is not the same across instruments and acquisition types.
In addition to empirical determination, predictive algorithms provide an alternative or complementary method to select the target peptides most likely to be high-responding for a set of proteins (Mallick et al. 2007; Fusaro et al. 2009; Eyers et al. 2011; Muntel et al. 2015). For researchers interested in using predictive algorithms for SRM/MRM and PRM peptide selection, Skyline has implemented the publically available, open-source PREGO algorithm Searle et al. (2015) as a plug-in. PREGO (Searle et al. 2015) predicts high responding peptides using an artificial neural network on DIA experimental data. The artificial neural network was trained using 11 minimally redundant, maximally relevant physiochemical properties that describe peptide size, structure, and hydrophobicity. PREGO outperforms previous predictive algorithms, correctly predicting more high-responding peptides than other algorithms. This performance improvement is believed to stem from a more representative training set. As mentioned above, peptide signal response differs between instruments and acquisition types. PREGO, being trained on a DIA dataset, may perform better because peptide signals in DIA datasets better represent peptide signals in SRM datasets. An important note is that these predictive algorithms mentioned above do not predict transition signal response, only peptide response.
The final number of peptides required for a quantitative assay depend on the analytical rigor of the experiment, the details of the project, and the purpose. A description of these considerations and their implications on assay development is described elsewhere (Carr et al. 2014).
4. Selection of transitions for SRM/MRM experiments
By definition of the method, all transitions for a precursor are measured for a PRM experiment, and therefore PRM experiments do not require selection of fragments prior to acquisition. However, SRM/MRM experiments target only the transitions preprogrammed for acquisition. Selection of optimal transitions is critical for quantitative experiments, as poorly designed assays will suffer unreliable, inaccurate, or nonspecific quantitation (Ludwig et al. 2012).
It is common to choose y-type ion fragments, due to high ion abundance compared to the alternative, b-type ion fragments (Holstein (Sherwood), Gafken, and Martin 2011). Similar to peptide selection, transition selection must be evaluated for chemical considerations, namely transition MS signal response and transition selectivity. Transition signal response may be assessed empirically through preliminary MS experiments to evaluate potential transitions in the appropriate experimental sample matrix and under the experimental instrument conditions. The mass spectrometrist must manually confirm that the transitions are high-responding and free of interference, and remove any transitions that do not meet those criteria. Alternatively, predictive algorithms for thermodynamic peptide fragmentation (Z. Zhang 2004; Z. Zhang 2005) may provide computationally-assisted transition selection, and computational tools have been designed to aid in SRM method development (Röst et al 2012), though none have been integrated with Skyline yet.
Current standard practice (Carr et al. 2014) monitors three or more transitions per peptide to make a reliable quantitation. However, statistically, if the transition has been evaluated as high-responding and free of interference, it is possible to perform quantitative analysis on one transition, using the other monitored transitions for confirming the identity of the peptide precursor.
B. Retention time determination for scheduled MS experiments
Most quantitative mass spectrometry experiments hyphenate reversed-phase high performance liquid chromatography (RP HPLC) to separate and simplify complex proteomic samples. Coupling LC to MS adds a time dimension to the data, as peptides elute off the solid-state column at a particular time in the chromatographic gradient. As with other modes of reversed phase chromatography, LC-MS peptide RT is dependent on several experimental factors, such as the physiochemical properties of the target peptide itself; background matrix of the sample; column-specific details including stationary phase material, bed length, and temperature; and the chromatography details including gradient percentage and delivery speed (Moseley et al. 1991). In the case of liquid chromatography coupled SRM/MRM and PRM experiments (LC-SRM/MRM, LC-PRM) on triple-quadrupole mass spectrometers, the number of peptide precursor-fragment transitions to be measured may exceed the speed at which the instrument can measure them and still maintain a cycle time appropriate for quantification (2–3 seconds per cycle maximum). In these cases, “scheduling” methods enable measurements of tens to hundreds of individual peptides, by allowing only a subset of the targeted peptides to be measured in any given cycle. The acquisition schedule for these methods includes precursor m/z, transition m/z, and the RT, or time window during which the precursor peptide elutes off the LC column.
Skyline’s ecosystem incorporates several complementary tools to predict peptide RT. The first, SSRCalc (Krokhin 2006; Spicer et al. 2007), is based on calculated hydrophobicity, as determined from the peptide amino acid sequence, to predict a peptide RT. This approach is particularly useful when empirical RT is unknown for a peptide. Alternatively, when peptide RT has been previously observed, a standard set of reference peptides can be used to calibrate RT prediction for any number of target peptides of interest on new columns or chromatography methods. In this approach, termed indexed retention time (iRT) (Escher et al. 2012), the reference peptides act as anchor points across a range of hydrophobicities, allowing the HPLC run-time to be calibrated and the assay-specific peptides to be aligned to the observed iRT reference peptide anchors. The iRT method is particularly useful in interlaboratory and large-scale experiments, projects which typically necessitate use of multiple LC systems and columns. For these projects, the iRT workflow integrated into Skyline provides a simple method to transfer chromatography empirical knowledge from one system to another, or to easily transition to a new column when the previous is replaced.
After predicting peptide RT through either method, or simply by using prior measurements that have already been imported, Skyline can export an acquisition table including all relevant information for a scheduled LC-SRM/MRM or LC-PRM method, including start and end times for peptide elution. The priority for these experiments is to capture the entirety of the chromatogram peak as the peptide elutes from the column, but with as narrow a window as possible. The mass spectrometer is limited in the number of peptide precursors it can measure at any given time, as dictated by the speed of the instrument (duty cycle), and the number of transitions to measure at that time, as dictated by predicted RT and the width of the scheduling time window. In order to assay as many peptides as possible, it is necessary to adjust the scheduling windows to reflect the instrument’s speed and the number of transitions eluting at each time point. Skyline facilitates this adjustment with a visualization option in the retention time pane that displays the number of transitions eluting over the chromatographic gradient under several potential scheduling window widths.
C. Instrument parameter optimization
Determining the optimal set of MS instrument parameters for a targeted experiment is necessary in order to create an effective assay. One parameter of particular importance to targeted experiments is collision energy (CE). Optimized CE increases fragment ion intensity, which confers stronger, more reliable signal response (Sherwood et al. 2009). Computational estimation of optimal CE based on precursor m/z and a simple linear equation (Equation 1) is useful for both triple quadrupole (Picotti et al. 2010) and quadrupole time-of-flight instruments (Griffin et al. 1991; Prakash et al. 2009). An automated pipeline for optimizing CE specifically for quantitative assays is integrated in Skyline to achieve maximum fragment ion intensity (MacLean et al. 2010) and therefore strongest, most reliable signal response for the peptides in the assay. Recent versions have added the ability to store optimized parameter values in a library for future re-use and easier sharing.
D. MS/MS spectral library creation
Although not strictly required for assay development, inclusion of spectral libraries in quantitative proteomics aids in downstream data processing. In spectral library searching, spectra acquired by tandem mass spectrometry (MS/MS) are compared with previously identified reference spectra (Craig, Cortens, and Beavis 2005). The benefits to library searching as opposed to database searching, in which spectra are compared with spectra predicted from amino acid sequences (Eng, McCormack, and Yates 1994), is a more accurate comparison of fragment ion intensities and a more efficient spectra search.
The Skyline ecosystem includes a suite of software tools, Bibliospec (Frewen et al. 2006), for creating and searching MS/MS peptide spectrum libraries. The Bibliospec 2.0 software package is composed of two informatics tools: BlibBuild and BlibFilter. All Skyline installations include these tools, and Skyline itself provides user interface for creating spectral libraries. The first step in building a spectral library is creating a full redundant library of peptide MS/MS spectra matched with known peptide identifications, which is performed computationally by BlibBuild and written to sqlite3 database file. To obtain peptide identifications for this step, an assortment of available database search programs are supported by BiblioSpec 2.0 (Table 2). Second, BlibFilter refines the redundant library to choose just one representative spectrum for each peptide, preserving the original retention times of the redundant spectra, and then writes a new non-redundant sqlite3 database containing this information. BlibFilter choses the one representative spectrum by measuring the similarity between all pairs of redundant spectra for a given peptide, and selecting the spectrum with the highest average similarity score.
Table 2.
Peptide spectrum matching pipelines supported by Skyline with BiblioSpec for spectral library-building.
| Peptide spectrum matching pipeline | Type | Creator | Peptide ID file | Spectrum file |
|---|---|---|---|---|
| Mascot | Proprietary | Matrix Science (Perkins et al 1999) | .dat | |
| ByOnic | Proprietary | Protein Metrics, Inc. (Bern, Cai, and Goldberg 2007) | .mzid | .MGF, .mzXML, .mzML |
| Comet/SEQUEST/Percolator | Open source | Dept Genome Sciences, University of Washington (Eng, Jahan, and Hoopmann 2013; Eng, McCormack, and Yates 1994; Kall et al 2007) | .perc.xml (.sqt) | .cms2, .ms2 |
| ID Picker (Myrimatch) | Open source | MSRC Bioinformatics, Vanderbilt University (Tabb, Fernando, and Chambers 2007) | .idpXML | .mzXML, .mzML |
| MaxQuant Andromeda | freeware | Max Planck Institute (Cox et al 2011) | msms.txt | |
| Morpheus | Open source | Coon lab, University of Wisconsin-Madison (Wenger and Coon 2013) | .pep.xml, .pep.XML, .pepXML | .mzXML, .mzML |
| MS-GF+ | freeware | Pevzner lab, UCSD (Kim et al 2010) | .mzid, .pepXML | .MGF, .mzXML, .mzML |
| OMSSA | Open source | NCBI (Geer et al 2004) | .pep.xml, .pep.XML, .pepXML | .mzXML, .mzML |
| PEAKS DB | Proprietary | Bioinformatics Solutions, Inc. (Zhang et al 2012) | .pep.xml, .pep.XML, .pepXML | .mzXML, .mzML |
| Proteomics Identifications (PRIDE) | EMBL-EBI (Martens et al 2005) | .pride.xml | ||
| Protein Pilot | Proprietary | SCIEX (Shilov et al 2007) | .group.xml | |
| Protein Prospector | Open source | UCSF Mass Spectrometry Facility (Baker and Clauser) | pepXML/mzXML | |
| Proteome Discoverer | Proprietary | Thermo | .msf | |
| Scaffold | Proprietary | Proteome Software (Searle 2010) | .mzid | .MGF, .mzXML, .mzML |
| Spectrum Mill | Proprietary | Agilent | .pep.xml, .pep.XML, .pepXML | .mzXML, .mzML |
| Trans-Proteomic Pipeline (TPP) | Open source | Aebersold lab, Institute for Systems Biology (Deutsch et al 2015) | pepXML/mzXML | |
| X! Tandem | Open source | Global Proteome Machine Organization (Craig and Beavis 2004) | .xtan.xml | |
| ProteinLynx Global SERVER (PLGS) - MSe | Proprietary | Waters | final_fragment.csv | |
| Custom | .ssl |
The Skyline GUI also supports MS/MS spectral library creation. To do so, it takes the best scoring PSM from a variety of supported search engines (Table 2) as a reference spectrum, picking the most intense in the event of a tie. In addition to creation of spectral libraries, Skyline supports several sources of reference libraries, including Peptide Atlas (Desiere et al. 2006), the National Institute of Standards and Technology (NIST), and the Global Proteome Machine (GPM) (Craig, Cortens, and Beavis 2004). Most Skyline users will choose to use their spectral libraries, once created, for targeted method creation and data extraction.
E. Skyline for small molecule research
Although this review is focused on the Skyline ecosystem for quantitative proteomics, the ecosystem also works for generalized small molecules (Tang et al. 2015), such as lipidomics, glycomics, and metabolomics. While some functions do not yet work for non-proteomic data, online tutorials detailing with how to make use of the Skyline ecosystem for small molecule research, including assay development, are available on-line with the Skyline software documentation.
Generally, the Skyline informatics for small molecule assay development mirrors that proteomic experiments described above. A notable difference, however, is the way Skyline treats ionization. For proteomics data, typically only sequence and charge state are required to describe a charged peptide. As such, Skyline assumes ionization by protonation, the most typical ionization for these experiments. Ionization of small molecules occurs through many means, including sodium addition and hydrogen loss. Therefore, Skyline’s informatics work best with manually entered charges states and either generalized ion formulas or manually entered m/z values for precursors and products.
F. Isolation window determination for DIA experiments
Unlike targeted experiments, DIA experiments do not require selection of proteins, peptides, or transitions prior to acquisition. There are multiple data collection strategies for DIA experiments with associated advantages and disadvantages that have been evaluated elsewhere (Chapman, Goodlett, and Masselon 2014). The most basic method used with Skyline (Egertson et al. 2015) acquires MS and MS/MS data for all molecular species between a certain predefined precursor m/z range in specified fragment m/z isolation windows. Determining the most appropriate MS/MS isolation scheme requires consideration of the particular instrument’s scan rate, resolving power, dynamic range, and sensitivity of the mass analyzer (Zhang et al. 2015). For many DIA experiments analyzed with Skyline, our lab monitors a precursor m/z range of 500–900 m/z as this m/z range reflects most proteotypic peptides. Restricting the total range can allow for smaller, more selective precursor isolation windows or shorter cycle times. Skyline is extremely flexible and currently supports all commonly used isolation schemes.
For the precursor m/z isolation scheme, window placement is calculated one of two ways: integer or optimized. Simple arithmetic division is used for integer window placement. For example, a 20 window isolation scheme with each window covering 5 m/z (20 × 5) for a 500–600 m/z range are placed at 500–505 m/z, 505–510 m/z, etc. This method requires a margin (usually 0.5 m/z) added to the instrument method but ignored during extraction, e.g. 499.5–505.5 m/z, 504.5–510.5 m/z, etc. Alternatively, optimized window placement considers peptide mass distribution and calculates isolation windows that encompass “allowable regions” (Egertson et al. 2013). By placing window edges at “forbidden zones” where peptide masses do not occur and windows over “allowable regions”, the resulting window width and position is optimized for m/z ranges where peptides are most likely occur. This algorithm for calculating optimized isolation window placement is integrated into Skyline, facilitating quick generation of isolation lists for DIA methods.
G. Final method export and refinement
Once a Skyline document is built with the settings and optimizations described above, the final developed assay is exported either as a native method for triple quadrupole instruments or as scheduled isolation lists for certain Q-TOF and the Thermo Q-Exactive instruments. After acquiring mass spectrometry data, the acquisition files are imported into the Skyline document for method refinement such as peptide and transition validation. The cycle of export, acquisition, and refinement is repeated until the assay is considered effective, at which point final acquisition and quantitative analysis begins.
III. Data processing: Peak detection and integration
Skyline’s targeted data analysis strategy begins when the researcher selects raw mass spectrometer acquisition files to import. Skyline derives information from the native, vendor-specific file formats or from portable files like mzXML (Pedrioli et al. 2004) or mzML and caches the information into a single, high-performance data file. The caching step is critical to Skyline’s ability to quickly load large experiments with many data files, allowing researchers to process multiple MS runs at the same time. Skyline handles files sequentially or in parallel, performing the operations described below on each data file. The end result of Skyline’s data processing is a calculated peak area, or area under the curve (AUC), for each peptide ion (modified peptide plus charge state) in the Skyline Target list, visualizations of the data, and cached chromatogram information for quick recall.
A. Chromatogram extraction
Mass spectrometry data contains three dimensions: m/z, retention time, and intensity. In the first step of data processing, Skyline extracts the retention time and intensity information for a given m/z (Figure 2, Step 1). For PRM or DIA experiments, this information is calculated from the measured spectra as extracted-ion chromatograms (XIC), and for SRM/MRM experiments, the measured chromatograms are themselves imported. No file conversion is necessary prior to this step; raw files from the instrument are directly imported. It should be noted, however, that several settings in Skyline affect the chromatogram extraction process, such as retention time window width and parameters for instrument resolving power for profile spectra or mass accuracy for centroided spectra, therefore researchers should be sure that the Skyline document is prepared with the appropriate instrument and experimental details before importing data. These settings can be exported and imported from other Skyline documents, aiding repeatability in data processing and ensuring the proper instrument and experimental details are preserved across laboratory sites and experiments.
Figure 2. Data processing pipeline in Skyline.
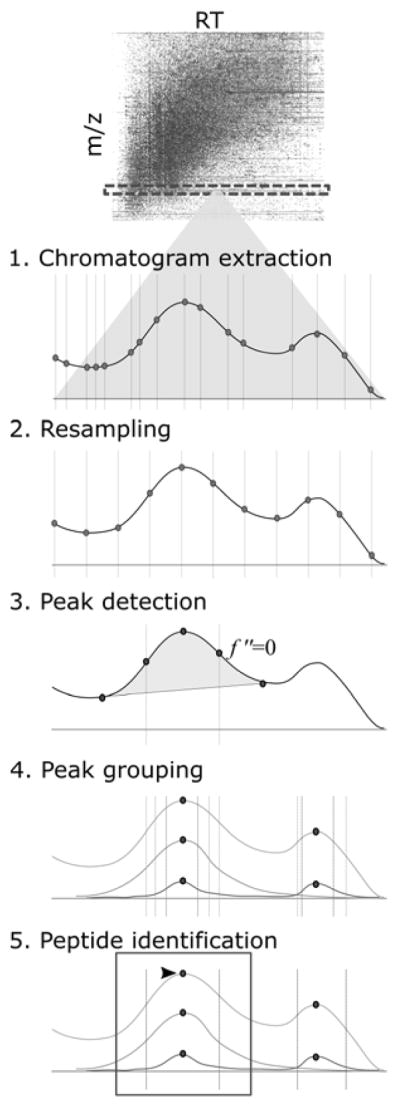
Skyline derives information from native, vendor-specific file formats or from portable files, producing peak area calculations, and visualizations of the data.
B. Resampling
For all tandem mass spectrometry data acquisition types, the time intervals between MS2 scans are irregular. For example, in an SRM/MRM experiment, the rate of MS2 scans depends on the number of transitions scheduled for collection at a given time and the dwell time for each. For its purposes, Skyline requires all chromatogram time, intensity points for a peptide to be placed on a uniform scale with a consistent interval. Even for DIA, this requires some adjustment of MS1 with MS2 scans and ions for multiple charge states or isotope labeling. To place these points, a linear interpolation of each raw chromatogram is performed. Skyline calculates an interval that captures as much information about the peak as possible (Figure 2, Step 3). Intervals placed too wide distort the shape of the peak, while intervals too narrow are costly in storage and processing time. The end product of resampling is an interval width that works best for the dataset, avoiding as much distortion as possible.
C. Peak detection
The resampled data is then searched for areas that represent peaks. Peak detection is performed by the Chromatogram Retention time Alignment and Warping for Differential Analysis of Data (CRAWDAD) Peaks algorithm. (Finney et al. 2008, Finney 2012) CRAWDAD finds the maxima and minima by points were the first derivative is equal to zero, then takes the second derivative in the retention time dimension, noting the point at which the second derivative is equal to zero in order to find inflection points. This set of points (local maxima, local minima, and inflection points) define a detected peak. In the absence of spectral library retention time information for peptide spectrum matches (IDs) within the files being analyzed (usually for DDA, PRM or DIA - with initial processing by tools like DIA-Umpire (Tsou et al 2015)), Skyline takes only the 20 most intense peaks for each transition from CRAWDAD. When ID times are present, Skyline also includes all CRAWDAD detected peaks containing IDs, or aligned IDs in runs which do not contain any IDs for the target being analyzed. This results in an initial set of raw peak detections for each individual chromatogram with boundaries set at the inflection points and peak areas in interval units.
D. Peak grouping
Next skyline creates peak groups for each targeted modified peptide or molecular structure, combining the raw peaks for its chromatograms and grouping them by retention time overlap. Peak grouping is based on elution profile similarity (Figure 2, Step 4), with apex RT, start RT, and end RT drawn from the local maxima and inflection points from the previous step. It should be noted that different charge states and isotopes (heavy labeled peptides, medium labeled peptides, endogenous or light peptides) are each considered together. After grouping, the individual peak boundaries are replaced with a single boundary for each entire peak group. This boundary may be adjusted outward from the original 2D inflection point boundary, using Savitzky-Golay smoothing and combined information of all chromatograms contributing to the peak group. Peak statistics are also recalculated to reflect the new agreed-upon boundary values and interval unit areas are multiplied by the number of seconds in the chosen interval to yield an ion count estimate (ions/second * seconds = ions).
E. Peptide identification
During the peptide identification step, commonly called “peak picking”, the top 10 results from peak grouping are evaluated for probability that they represent the peptide. For each of the 10 considered peak groups, a number of peak group features are calculated. These features, derived both from the CRAWDAD calculate statistics and raw chromatogram data, are weighted with particular coefficients, and summed to give a final score to the peak group. The seven scores and corresponding coefficients in Skyline’s default peak picking model are log intensity (1.0), coelution count (1.0), identified count (20.0), library intensity correlation (3.0), shape score (4.0), weighted co-elution (−0.05), and retention time delta from prediction (−0.7). The peak group with the highest score is identified (“picked”) as the peak for that peptide.
Many of these scoring features used in the Skyline default peak picking strategy are similar to those used in the mProphet method (Reiter et al. 2011). Researchers also have the ability to use other peak picking algorithms, such as the mProphet model itself, after initial data import by using a Re-integrate command to generate and apply these models, using decoys and semi-supervised machine learning. As evident from the exceptionally high weight given to the identification count feature, if external tools for peptide identification are used to identify a time of peptide elution within the data, Skyline will give very high priority to finding a peak at that time, using retention time alignment between runs to propagate ID times between runs.
F. Peak area calculation
In Skyline, the peak area, or area under the curve (AUC), refers to the total integrated area within the peak boundaries, minus the background area (in intensity for seconds of time units - or ion count where intensity is ions per second). Background area is defined as the total integrated area of the minimum of background height and intensity at each point, where background height is the minimum intensity of the two points where the chromatogram crosses the integration boundaries, which is assumed to be the level of intensity contributed not by the transitions themselves but from chemical noise (background) in the measurement. The background area is subtracted from the total integrated area within the peak boundaries to return the final reported peak area. Although Skyline allows display of chromatograms with various smoothing options (2D, 1D, Savitzky-Golay) applied, it uses the interpolated points displayed in the unsmoothed graphs to calculate peak area. Total area values sum the AUC values of individual chromatograms, rather than performing a separate AUC calculation on a summed chromatogram.
IV. Core analyses and visualizations
Once raw acquisitions are processed, Skyline creates visual displays of the data. Chromatograms for each peptide in the Skyline document are displayed with visualizations of the boundaries and indicators for the retention time and dot product of each picked peak. Retention times for the top 10 peaks detected in the RT window are also shown, allowing researchers to see other candidates that were considered in peak picking.
A. Data curation and quality assessment
Visualizations in Skyline allow researchers to quickly identify issues in data, explore causes, and evaluate solutions to resolve the issues. One common example of this functionality of data visualization in Skyline is “peak picking”. Although automated peak detection and boundary setting are generally reliable, it is important to manually curate data to ensure reliable quantification (Bereman et al. 2012). Here, Skyline’s visualizations facilitate determination of which peptides can be robustly measured in a specific target matrix, which transitions for a peptide are the best transitions for the measurable peptides, and whether a given peak actually measures the peptide of interest. The picked peak is marked by a solid black arrowhead in Skyline’s chromatogram window (Figure 3a). Evaluation of peptide identification (“peak picking”) is computationally-aided by display of iRT-predicted RT, relative transition intensities compared to library intensities. Dot product values are calculated (Stein and Scott 1994; Tabb et al. 2003), correlating peak intensities of the transitions with the library spectrum for that peptide (dotp - and between precursor isotope peak intensities and expected isotope distribution - idotp - and between analyte peak intensities and those for matching isotope labeled reference peptides - rdotp) and establishing a measure of confidence in peak detection (Prakash et al. 2009; Sherwood et al. 2010). Peak boundaries are also displayed as dashed vertical lines, shown in Figure 3a, and researchers are able to adjust the boundaries as they deem appropriate. Skyline recalculates peak statistics, including peak area integration, with the new boundaries or peak picking.
Figure 3. Real-time updating visualizations natively embedded in Skyline.

(a) Skyline chromatogram visualizations show the intensity at each resampled retention time point for all fragment ions (displayed as different colored lines identified in the legend) of a precursor, enabling researchers to assess Skyline’s automated peak picking or adjust integration boundaries if necessary. (b) Calculation of coefficient of variation (CV) informs researchers of the reproducibility of peptide peak areas (shown here as the peak area ratio to a global standard) over multiple acquisitions or custom-annotated groups of acquisitions. (c) Real-time updating visualization of precursor retention time across acquisitions enables quick identification of mis-picked peaks over many MS acquisition runs. Out of 42 replicates, the peptide shown here appears to elute three minutes late in one replicate (eighth from the left, marked with arrow) compared to all other replicates, an observation that may prompt the researcher to evaluate that picked peak in the chromatogram visualization pane. (d) Peak area is displayed here as the percentage contributed by each fragment ion of the precursor which allows the researcher to quickly evaluate data quality. For example, the boxed replicate (eighth run from the left, marked with arrow) displays a noticeably different distribution of contributed fragment peak areas, indicating that the picked peak group for this replicate may require further examination.
Critically for quantification, Skyline allows convenient evaluation of transitions. Skyline gives the option to display for each peptide all transitions included in the document, precursors-only (M, M+1, M+2, etc.), products-only, a single transition, or a total ion chromatogram, summing all transitions, for each precursor. The individual fragments measured for a peptide are visualized as different colored chromatograms (Figure 3a). The ability to simply delete or add transitions for a peptide precursor in the Target window, and easily undo such changes, lets researchers visually evaluate transitions for characteristics such as intensity, co-elution with interference, shouldering, and other qualities undesirable for accurate, robust quantification. For MRM experiments with heavy-paired peptide targets, the Automated Detection of Inaccurate and Imprecise Transitions in Peptide Quantification (AuDIT) algorithm (Abbatiello et al. 2010) employed by the Skyline External Tool QuaSAR automatically suggests transitions for removal based on similar criteria. In addition to the chromatogram view, clicking on a chromatogram opens a Full-Scan view of normal 2D spectra (intensity by m/z).
B. Native, real-time updating visualizations
Statistics for data are shown as visual graphs in embeddable live plot windows. Statistics include plots of retention time, peak area, mass error, and group comparisons. The retention time display is user-defined to show a floating column chart by replicate or peptide, a linear regression plot of the peptide elution times by SSRCalc or iRT score, or a scheduling window with the number of expected transitions over time for multiple scheduling window widths. Retention time data can be plotted by Replicate Comparison or Peptide Comparison, allowing researchers to evaluate various aspects of their data. Specifically, replicate comparisons can be sorted as they are in the document or by acquired time helping to make the impact of instrument run order more easily understood. For example, when the retention times are displayed as Replicate Comparison for an experiment, it is clear if a particular run deviates significantly from others (Figure 3c), which may indicate a potentially mis-picked peak. Options for display of peak areas allow the researcher to specify between displaying a bar chart of total peak areas, peak areas normalized to heavy peptide isotope pairs, user-specified global standards (Figure 3d), maximums, or the total peak area; or to view bar graphs of coefficient of variance (CV) (Figure 3b). Similar to notably deviating retention time values, an outlying peak area may prompt a researcher to visually examine that replicate or peptide.
In addition to retention time and peak area data, mass error graphs are available for inspecting mass error summary information. Mass error is calculated in Skyline as a weighted mean of the mass error in all the integrated points across the annotated chromatogram. When visualized as a Replicate Comparison, this data is helpful for detecting interference at the transition level. As a Peptide Comparison, researchers may sort by mass error to get an overview of all targeted peptides. Unique to the mass error visualization options are a histogram (for display of mass error at the full document scope or each replicate for detecting calibration issues and a 2D histogram with m/z and retention time dimensions available for increased visibility of instrument calibration issues.
For instances where displaying data in the form of Replicate or Peptide Comparison is inadequate, Skyline offers options for grouping and ordering of peptides by a number of characteristics, including custom annotations that researchers can add based on experimental details or sample characteristics. The Group Comparisons feature natively calculates differential statistics for proteins in a table or graph view within Skyline. For many proteomics studies, correcting for multiple hypothesis testing is required. To calculate statistically significant differential expression, Group Comparisons employs a user-specified cut-off for the Benjamini-Hochberg adjusted p-value to account for false discovery rate (Benjamini and Hochberg 1995).
In experiments where absolute quantification of the analyte target is necessary, Skyline allows for internal single point calibration to a reference and also multiple point calibration curves via the Calibration Curve feature. The Calibration Curve feature works with data from a dilution series of isotope-labeled reference peptides. This external calibration curve is used to regress the known concentration of each reference peptide target against the intensity measured for that target, allowing conversion of intensity measurements into absolute quantitative values. Although this method requires multiple injections to gather the external calibration curve data, the Calibration Curve feature accounts for linear peptide responses that have nonstandard slopes or intercepts. At this time, the feature provides conversion of measured intensity values to absolute quantitation values like concentration, not for determining limits of detection or limits of quantitation.
C. Skyline informatics considerations for ion mobility spectrometry
In experiments involving gas-phase ion mobility spectrometry (IMS) separations in place of or hyphenated with LC, the additional dimension of drift-time is introduced to the data (Baker et al. 2015). For these datasets, as a single LC RT has multiple associated drift-times, Skyline considers drift-time data in processing, allowing chromatogram extraction to be limited to specified drift time ranges, and visualization. Spectra from which chromatogram points are extracted can be visualized in a 3D heat map plot (intensity by m/z and drift time), displayed when the chromatogram is clicked on. As fragments have the same drift-time as their precursor (potentially slightly offset by a constant fragmentation factor) a drift time value and extraction range allow Skyline to ignore signal outside a targeted drift range, improving selectivity. Skyline’s incorporation of IMS considerations and continuing optimization of IMS informatics holds promise for analysis of large, multi-dimensional datasets involving IMS.
V. Additional analyses: external tools
A. Goals for External Tools
One distinguishing aspect of the Skyline ecosystem is the ability for researchers to contribute their data processing software packages through the external tools framework (Broudy et al. 2014). Through this framework, researchers can conveniently and quickly distribute their programs to the community. The ultimate goal is to provide a common, convenient hub that connects the data found in a Skyline document with the community’s many informatics methods. Although Skyline itself is built from the C# programming language, the installable tools framework includes extra support for tools using the R or Python programming languages. To date, nine external tools from community researchers are integrated in the Skyline ecosystem with applications ranging from assay development to biological inference (Table 3).
Table 3.
Community-built informatics tools integrated into the Skyline ecosystem.
| Tool | Creator | Integration Date | Purpose |
|---|---|---|---|
| BiodiversityPlugin | Computational Proteomics Group, Pacific Northwest National Laboratory (Payne et al 2015) | 2015 Jun 10 | mass spectrometry data retrieval by organism and biological pathway |
| MPPReport | Agilent Technologies | 2014 Sep 9 | data export for use in Agilent’s Mass Profiler Professional multivariate statistics software |
| MS1Probe | Gibson Lab, The Buck Institute for Research on Aging (Schilling et al 2012) | 2014 Apr 16 | high throughput statistical quantification of MS1 Filtering datasets |
| MSstats | Vitek Lab, Purdue University (Choi et al 2014) | 2015 Jul 30 | statistical relative quantification of proteins and peptides in global, targeted, and data-independent proteomics |
| Population Variation | Computational Proteomics Group, Pacific Northwest National Laboratory (Fujimoto et al 2014) | 2013 Dec 20 | protein variant lookup from dbSNP and the 1000 Genome project |
| Prego | MacCoss lab, University of Washington (Searle et al 2015) | 2015 Jun 23 | peptide SRM response prediction |
| Protter | Wollscheid Lab, ETH Zurich (Omasits et al 2014) | 2015 Dec 19 | transmembrane protein topology visualization |
| QuaSAR | Carr Lab, Broad Institute of MIT and Harvard (Mani, Abbatiello, and Carr 2012) | 2014 Oct 23 | QC, statistical analysis, and visualization of data from quantitative MRM-MS |
| SProCoP | Bereman Lab, North Carolina State University (Bereman et al 2014) | 2014 Dec 3 | Visualization, detection, and identification of assignable causes of variation in LC-MS |
B. External tools for assay development
Generating a specific hypothesis for a quantitative MS experiment often begins with prior knowledge from previous proteomics experiments. The Biodiversity Library Plugin (Payne et al. 2015) enables fast, convenient survey and retrieval of existing proteomics data for an organism and biological pathway of interest. Researchers can query spectra for over 3 million peptides and 230,000 proteins, annotated with KEGG pathways, from 118 organisms. These functionalities allow researchers to quickly compile a list of potential assay proteins on the basis of a biological function.
As mentioned in the computationally-assisted assay development section, selecting target peptides for an SRM/MRM or PRM poses a significant challenge. One such challenge in clinical applications is natural genetic variation, which may confound MS experiments that attempt to measure a specific protein. The Population Variation external tool (Fujimoto et al. 2014) enables researchers to explore possible variants for their protein of interest by surveying the dbSNP and 1000 Genome project for mutations. The PREGO external tool (Searle et al. 2015) is an algorithm that ranks peptides by their predicted response level, intended to facilitate the selection of peptides that will produce the most intense MS signal.
C. External tools for acquisition monitoring
It is necessary to control for LC-MS performance variations during acquisition in order to ensure accurate, reproducible measurements. Aspects such as retention time, chromatographic peak width, mass measurement, and ion intensity all influence the robustness of an assay and are affected run to run by minor, necessary adjustments like column changing. The external tool Statistical Process Control in Proteomics (SProCoP) (Bereman et al. 2014) allows for semi-automated real time evaluation of an assay, including both chromatographic and mass spectrometric performance. SProCop assesses metrics such as retention time reproducibility, peak asymmetry, targeted peptide ion intensity, and mass measurement accuracy, constructing control charts and boxplots that a researcher monitors throughout the lifetime of an experiment to ensure reproducibility between LC-MS runs.
D. External tools for quantitative statistical analysis
The experimental workflow used to generate samples for mass spectrometry each require specialized data analysis strategies. The combination of sample generation method (labeled versus label-free) and the spectral acquisition method (DDA, SRM/MRM and PRM, or DIA) require different informatics approaches. The external tool MSstats (Choi et al. 2014) considers these data properties to calculate the relative quantification of proteins and peptides. MSstats begins with data processing and visualization of the identified and quantified spectral peaks. It then performs statistical modeling and inference using linear mixed models, customized to the method of sample generation and MS acquisition. Finally, researchers can specify a particular statistical power for their experiment, and MSstats determines the minimal number of replicates required to achieve that statistical power by considering the dataset as a pilot experiment.
Other external tools are designed for use with specific acquisition methods. For DDA analyses, an MS1 filtering approach through the external tool MS1Probe (Schilling et al. 2012) enables high throughput statistical quantification of peptide analytes. The external tool QuaSAR (Mani, Abbatiello, and Carr 2012) produces figures of merit (limit of detection, LOD; limit of quantitation, LOQ) for statistical characterization of stable isotope dilution MRM-MS assays (SID-MRM-MS) generated with heavy labeled stable-isotope peptide standards. Within the QuaSAR external tool, AuDIT (Abbatiello et al. 2010) performs automated filtering of transition validation, improving sensitivity and specificity for peptide quantitation by SID-MRM-MS. For label-free quantitative DIA analyses, Skyline exported custom reports can be used to optimize fragment selection and detect interferences using the nonoutlier fragment ion (NOFI) ranking algorithm (Bilbao et al 2015).
In addition to the tools described above, Skyline also enables the export of results for analysis in other software suites. The MPPReport tool, for example, creates a results file designed for import into Agilent’s Mass Profiler Professional multivariate statistics software package. Researchers can create their own custom reports with a wide range of values to view, edit, and export. Exported custom reports enable researchers to perform their own statistical analyses in Excel, R, Matlab, Java, C++, and other languages, and formats of custom reports can be saved as templates to share and re-use in future analyses.
E. External tools for biological inference
The ultimate goal of many MS proteomics experiments is deriving biological information. Towards this end, researchers have developed several tools to facilitate the visualization and biological importance of peptide and protein measurements. The external tool Protter (Omasits et al. 2014) combines known annotations of protein structure and function with experimental MS data to give researchers an interactive visualization of protein topology. Protter is especially powerful for visualization of membrane protein topology.
VI. Methods and results sharing
Skyline, being designed for the mass spectrometry proteomics community, is ideal for interlaboratory collaborations and experimental results comparisons in a vendor-neutral manner. With these types of collaborations in mind, the Skyline ecosystem grew to include storage and sharing applications.
A. Panorama and CHORUS projects for raw and Skyline file storage and sharing
Panorama (Sharma et al. 2014), a web-based application for storing, sharing, analyzing and reusing targeted Skyline assays, allows laboratories to communicate the details for replicating or reproducing targeted Skyline experiments. To this end, during the development of Panorama, data integrity, security, and scalability were stressed. Storing Skyline documents in Panorama does not confer any loss of information and data can be made public or kept private at the discretion of the researcher.
It is possible to automate entire informatics pipelines, from acquisition to Panorama publishing, using the command-line version of Skyline, called SkylineRunner. An exemplary case of informatics automation is AutoQC, a completely automated pipeline designed to monitor system suitability in bottom-up proteomics (Bereman et al 2016). As a mass spectrometer runs, AutoQC imports quality control acquisitions into Skyline, extracts multiple identification-free metrics, and uploads the data to a Panorama Skyline document repository. Users can view system suitability metrics in the web-based interface, including Levey-Jennings and Pareto plots.
In addition to the Panorama module, the CHORUS platform was developed to provide storage, analysis, and sharing function for raw mass spectrometry files with a simple user interface. When raw data is placed into CHORUS, it is uploaded to the Amazon Web Services (AWS) cloud and translated into a distributed data structure. By utilizing AWS cloud computing and the unique distributed file format, accessing DIA data remotely from CHORUS is faster than from the local hard drive. When researchers wish to request data from the cloud, Skyline requests the extracted ion chromatograms, CHORUS generates the chromatograms, and then returns a Skyline cache. In addition to this scalable data access and remote extraction of chromatograms, CHORUS also provides a browser-based vendor-neutral spectrum and chromatogram viewer, integrated protein database searching and quantitative analysis tools. CHORUS is intended to facilitate community-driven mass spectrometry proteomics, and is therefore a not-for-profit public/private partnership.
B. CPTAC: an exemplary use case scenario
The Clinical Proteomic Technologies Assessment for Cancer (CPTAC) program (Abbatiello et al. 2015) exemplifies the strengths of Skyline for methods and results sharing in large, multi-site collaborations. As part of the CPTAC efforts to improve cancer diagnosis, treatment, and prevention with LC-MRM-MS methodologies, the Skyline ecosystem has been utilized to develop targeted proteomics assays that are precise, accurate, reproducible, and transferable between laboratories, across expertise levels, and over instrument platforms. CPTAC scientists utilized the Skyline ecosystem for computationally-assisted methods development, taking ease of simple transition evaluation, retention time scheduling, and method export. Additionally, because Skyline’s analysis pipeline is instrument-independent, the CPTAC researchers were able to integrate data across LC-MS platforms. Further, informatics tools developed by the CPTAC team to quantitatively analyze the data, namely QuaSAR, have been integrated into the Skyline ecosystem as external tools. From assay development to quantitative data analysis, the Skyline ecosystem helped to enable scientists of the CPTAC consortium accomplish their goals for a robust, sensitive absolute quantification assay across laboratory sites, instrument platforms, and operators.
VII. Perspectives
The Skyline informatics ecosystem described above has become a powerful tool in the quantitative measurement and analysis of peptides by mass spectrometry. Skyline’s generalized, vendor-neutral design provides the base for an informatics toolkit that expands to fit the needs of the community. As new needs arise from the community, Skyline frequently releases software developments in the form updates for Skyline-daily, the beta release version of Skyline. Areas of orthogonal interest such as small molecule research, analytical methods for rigorous quantitation, and statistical techniques are inspiring new Skyline developments. Important future goals are adapting Skyline’s informatics for big data mass spectrometry proteomics through parallelization of file processing. These developments will be vital in obtaining the robust, sensitive quantitative measurements required to better understand the systems biology of cells, organisms, and disease states.
Supplementary Material
Acknowledgments
We thank the members of the MacCoss laboratory and Skyline team, especially Nat Brace, Brian Pratt, and Nicholas Shulman, for helpful discussion of manuscript material.
References
- Abbatiello SE, Mani DR, Keshishian H, Carr SA. Automated Detection of Inaccurate and Imprecise Transitions in Peptide Quantification by Multiple Reaction Monitoring Mass Spectrometry. Clinical Chemistry. 2010;56(2):291–305. doi: 10.1373/clinchem.2009.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbatiello SE, Schilling B, Mani DR, Zimmerman LJ, Hall SC, MacLean B, Albertolle M, Allen S, Burgess M, Cusack MP, Ghosh M, Hedrick V, Held JM, Inerowicz HD, Jackson A, Keshishian H, Kinsinger CR, Lyssand J, Makowski L, Mesri M, Rodriguez H, Rudnick P, Sadowski P, Sedransk N, Shaddox K, Skates SJ, Kuhn E, Smith D, Whiteaker JR, Whitwell C, Zhang S, Borchers CH, Fisher SJ, Gibson BW, Liebler DC, MacCoss MJ, Neubert TA, Paulovich AG, Regnier FE, Tempst P, Carr SA. Large-Scale Inter-Laboratory Study to Develop, Analytically Validate and Apply Highly Multiplexed, Quantitative Peptide Assays to Measure Cancer-Relevant Proteins in Plasma. Molecular & Cellular Proteomics. 2015;14(9):2357–74. doi: 10.1074/mcp.M114.047050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abelin JG, Patel J, Lu X, Feeney CM, Fagbami L, Creech AL, Hu R, Lam D, Davison D, Pino L, Qiao JW, Kuhn E, Officer A, Li J, Abbatiello S, Subramanian A, Sidman R, Snyder E, Carr SA, Jaffe JD. Reduced-representation Phosphosignatures Measured by Quantitative Targeted MS Capture Cellular States and Enable Large-scale Comparison of Drug-induced Phenotypes. Molecular & Cellular Proteomics. 2016;15(5):1622–41. doi: 10.1074/mcp.M116.058354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson L, Hunter CL. Quantitative Mass Spectrometric Multiple Reaction Monitoring Assays for Major Plasma Proteins. Molecular & Cellular Proteomics. 2005;5(4):573–588. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- Baker ES, Burnum-Johnson KE, Ibrahim YM, Orton DJ, Monroe ME, Kelly RT, Moore RJ, Zhang X, Theberge R, Costello CE, Smith RD. Enhancing Bottom-up and Top-down Proteomic Measurements with Ion Mobility Separations. Proteomics. 2015;15(16):2766–2776. doi: 10.1002/pmic.201500048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PR, Clauser KR. http://prospector.ucsf.edu.
- Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 1995;57:289–300. [Google Scholar]
- Bereman MS, Johnson R, Bollinger J, Boss Y, Shulman N, Maclean B, Hoofnagle AN, MacCoss MJ. Implementation of Statistical Process Control for Proteomic Experiments Via LC MS/MS. Journal of the American Society for Mass Spectrometry. 2014;25(4):581–7. doi: 10.1007/s13361-013-0824-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereman MS, MacLean B, Tomazela DM, Liebler DC, MacCoss MJ. The Development of Selected Reaction Monitoring Methods for Targeted Proteomics via Empirical Refinement. Proteomics. 2012;12(8):1134–1141. doi: 10.1002/pmic.201200042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereman MS, Beri J, Sharma V, Nathe C, Eckels J, MacLean B, MacCoss MJ. An Automated Pipeline to Monitor System Performance in Liquid Chromatography-Tandem Mass Spectrometry Proteomic Experiments. Journal of Proteome Research. 2016;15(12):4763–4769. doi: 10.1021/acs.jproteome.6b00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bern M, Cai Y, Goldberg D. Lookup peaks: A hybrid of de novo sequencing and database search for protein identification by tandem mass spectrometry. Anal Chem. 2007;79(4):1393–1400. doi: 10.1021/ac0617013. [DOI] [PubMed] [Google Scholar]
- Bilbao A, Varesio E, Luban J, Strambio-De-Castillia C, Hopfgartner G, Muller M, Lisacek F. Processing strategies and software solutions for data-independent acquisition in mass spectrometry. Proteomic. 2015;15(5–6):964–980. doi: 10.1002/pmic.201400323. [DOI] [PubMed] [Google Scholar]
- Bilbao A, Zhang Y, Varesio E, Luban J, Strambio-De-Castilla C, Lisacek F, Hopfgartner G. Ranking Fragment Ions Based on Outlier Detection for Improved Label-Free Quantification in Data-Independent Acquisition LC-MS/MS. Journal of Proteome Research. 2015;14(11):4581–4593. doi: 10.1021/acs.jproteome.5b00394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broudy D, Killeen T, Choi M, Shulman N, Mani DR, Abbatiello SE, Mani D, Ahmad R, Sahu AK, Schilling B, Tamura K, Boss Y, Sharma V, Gibson BW, Carr SA, Vitek O, MacCoss MJ, MacLean B. A Framework for Installable External Tools in Skyline. Bioinformatics. 2014;30(17):1–26. doi: 10.1093/bioinformatics/btu148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, Grant RP, Hoofnagle AN, Huttenhain R, Koomen JM, Liebler DC, Liu T, MacLean B, Mani DR, Mansfield E, Neubert H, Paulovich AG, Reiter L, Vitek O, Aebersold R, Anderson L, Bethem R, Blonder J, Boja E, Botelho J, Boyne M, Bradshaw RA, Burlingame AL, Chan D, Keshishian H, Kuhn E, Kinsinger C, Lee JSH, Lee SW, Moritz R, Oses-Prieto J, Rifai N, Ritchie J, Rodriguez H, Srinivas PR, Townsend RR, Van Eyk J, Whiteley G, Wiita A, Weintraub S. Targeted Peptide Measurements in Biology and Medicine: Best Practices for Mass Spectrometry-Based Assay Development Using a Fit-for-Purpose Approach. Molecular & Cellular Proteomics. 2014;13(3):907–917. doi: 10.1074/mcp.M113.036095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cham JA, Bianco L, Bessant C. Free Computational Resources for Designing Selected Reaction Monitoring Transitions. Proteomics. 2010;10(6):1106–26. doi: 10.1002/pmic.200900396. [DOI] [PubMed] [Google Scholar]
- Chapman JD, Goodlett DR, Masselon CD. Multiplexed and Data-Independent Tandem Mass Spectrometry for Global Proteome Profiling. Mass Spectrometry Reviews. 2014;33:452–470. doi: 10.1002/mas.21400. [DOI] [PubMed] [Google Scholar]
- Choi M, Chang CY, Clough T, Broudy D, Killeen T, MacLean B, Vitek O. MSstats: An R Package for Statistical Analysis of Quantitative Mass Spectrometry-Based Proteomic Experiments. Bioinformatics. 2014;30(17):1–2. doi: 10.1093/bioinformatics/btu305. [DOI] [PubMed] [Google Scholar]
- Codrea MC, Jiménez CR, Heringa J, Marchiori E. Tools for Computational Processing of LC-MS Datasets: A User’s Perspective. Computer Methods and Programs in Biomedicine. 2007;86:281–290. doi: 10.1016/j.cmpb.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Colangelo CM, Lisa Chung L, Can Bruce C, Kei-Hoi Cheung KH. Review of Software Tools for Design and Analysis of Large Scale MRM Proteomic Datasets. Methods. 2013;61(3):287–98. doi: 10.1016/j.ymeth.2013.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium International Human Genome Sequencing. Initial Sequencing and Analysis of the Human Genome. Nature. 2001;409(6822):860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Consortium International Human Genome Sequencing. Finishing the Euchromatic Sequence of the Human Genome. Nature. 2005;50(2):162–168. [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. Andromeda: A peptide search engine integrated into the MaxQuant environment. Journal of Proteome Research. 2011;10(4):1794–1805. doi: 10.1021/pr101065j. [DOI] [PubMed] [Google Scholar]
- Craig R, Cortens JP, Beavis RC. Open Source System for Analyzing, Validating, and Storing Protein Identification Data. Journal of Proteome Research. 2004;3(6):1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- Craig R, Beavis RC. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics. 2004;20(9):1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Craig R, Cortens JP, Beavis RC. The Use of Proteotypic Peptide Libraries for Protein Identification. Rapid Communications in Mass Spectrometry. 2005;19(13):1844–1850. doi: 10.1002/rcm.1992. [DOI] [PubMed] [Google Scholar]
- Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN, Aebersold R. The Peptide Atlas Project. Nucleic Acids Research. 2006;34:D655–D658. doi: 10.1093/nar/gkj040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutsch EW, Mendoza L, Shteynberg D, Slagel J, Sun Z, Moritz RL. Trans-Proteomic Pipeline, a standardized data processing pipeline for large-scale reproducible proteomics informatics. Proteomics Clin Appl. 2015;9(0):745–754. doi: 10.1002/prca.201400164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domon B, Aebersold R. Options and Considerations When Selecting a Quantitative Proteomics Strategy. Nature Biotechnology. 2010 Jul;28(7):710–21. doi: 10.1038/nbt.1661. [DOI] [PubMed] [Google Scholar]
- Egertson JD, Kuehn A, Merrihew GE, Bateman NW, MacLean B, Ting YS, Canterbury JD, Marsh DM, Kellmann M, Zabrouskov V, Wu CC, MacCoss MJ. Multiplexed MS/MS for Improved Data-Independent Acquisition. Nature Methods. 2013;10(8):744–6. doi: 10.1038/nmeth.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egertson JD, MacLean M, Johnson R, Xuan Y, MacCoss MJ. Multiplexed Peptide Analysis Using Data-Independent Acquisition and Skyline. Nature Protocols. 2015;10(6):887–903. doi: 10.1038/nprot.2015.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR. An Approach to Correlate Tandem Mass Spectral Data of Peptides with Amino Acid Sequences in a Protein Database. Journal of the American Society for Mass Spectrometry. 1994;5(11):976–89. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Eng JK, Jahan TA, Hoopmann MR. Comet: An open-source MS/MS sequence database search tool. Proteomics. 2013;13(1):22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- Escher C, Reiter L, MacLean B, Ossola R, Herzog F, Chilton J, MacCoss MJ, Rinner O. Using iRT, a Normalized Retention Time for More Targeted Measurement of Peptides. Proteomics. 2012;12(8):1111–1121. doi: 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyers CE, Lawless C, Wedge DC, Lau KW, Gaskell SJ, Hubbard SJ. CONSeQuence: Prediction of Reference Peptides for Absolute Quantitative Proteomics Using Consensus Machine Learning Approaches. Molecular & Cellular Proteomics. 2011;10(11):M110003384–M110.003384. doi: 10.1074/mcp.M110.003384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney GL, Blackler AR, Hoopmann MR, Canterbury JD, Wu CC, MacCoss MJ. Label-Free Comparative Analysis of Proteomics Mixtures Using Chromatographic Alignment of High-Resolution μLC-MS Data. Analytical Chemistry. 2008;80(4):961–971. doi: 10.1021/ac701649e. [DOI] [PubMed] [Google Scholar]
- Frewen BE, Merrihew GE, Wu CC, WS, MacCoss MJ. Analysis of Peptide MS/MS Spectra from Large-Scale Proteomics Experiments Using Spectrum Libraries. Analytical Chemistry. 2006;78(16):5678–5684. doi: 10.1021/ac060279n. [DOI] [PubMed] [Google Scholar]
- Fujimoto GM, Matthew E, Monroe ME, Rodriguez L, Wu C, MacLean B, Smith RD, MacCoss MJ, Payne SH. Accounting for Population Variation in Targeted Proteomics. Journal of Proteome Research. 2014;13(1):321–3. doi: 10.1021/pr4011052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusaro VA, Mani DR, Mesirov JP, Carr SA. Prediction of High-Responding Peptides for Targeted Protein Assays by Mass Spectrometry. Nature Biotechnology. 2009;27(2):190–198. doi: 10.1038/nbt.1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geer LY, Markey SP, Kowalak JA, Wagner L, Xu M, Maynard DM, Yang X, Shi W, Bryant SH. Open mass spectrometry search algorithm. Journal of Proteome Research. 2004;3(5):958–964. doi: 10.1021/pr0499491. [DOI] [PubMed] [Google Scholar]
- Gillet LC, Navarro P, Tate S, Röst H, Selevsek N, Luks Reiter L, Bonner R, Aebersold R. Targeted Data Extraction of the MS/MS Spectra Generated by Data-Independent Acquisition: A New Concept for Consistent and Accurate Proteome Analysis. Molecular & Cellular Proteomics. 2012;11(6):O111016717. doi: 10.1074/mcp.O111.016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing Protein Abundance and mRNA Expression Levels on a Genomic Scale. Genome Biology. 2003;4(9):117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin PR, Coffman JA, Hood LE, Yates JR. Structural Analysis of Proteins by Capillary HPLC Electrospray Tandem Mass Spectrometry. International Journal of Mass Spectrometry and Ion Processes. 1991;111:131–149. [Google Scholar]
- Holstein CA, Gafken PR, Martin DB. Collision Energy Optimization of B- and Y-Ions for Multiple Reaction Monitoring Mass Spectrometry. Journal of Proteome Research. 2011;10:231–240. doi: 10.1021/pr1004289. [DOI] [PubMed] [Google Scholar]
- Jones P, Côté RG, Cho SY, Klie S, Martens L, Quinn AF, Thorneycroft D, Hermjakob H. PRIDE: New Developments and New Datasets. Nucleic Acids Research. 2008;36(SUPPL 1):878–883. doi: 10.1093/nar/gkm1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käll L, Canterbury JD, Weston J, Noble WS, MacCoss MJ. Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nature Methods. 2007;4(11):923–925. doi: 10.1038/nmeth1113. [DOI] [PubMed] [Google Scholar]
- Kim S, Mischerikow N, Bandeira N, Navarro JD, Wich L, Mohammed S, Heck AJR, Pevzner PA. The generating function of CID, ETD, and CID/ETD pairs of tandem mass spectra: applications to database search. Molecular & Cellular Proteomics. 2010;9(12):2840–2852. doi: 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krokhin OV. Sequence-Specific Retention Calculator. Algorithm for Peptide Retention Prediction in Ion-Pair RP-HPLC: Application to 300- and 100-A Pore Size C18 Sorbents. Analytical Chemistry. 2006;78(22):7785–7795. doi: 10.1021/ac060777w. [DOI] [PubMed] [Google Scholar]
- Kuster B, Schirle M, Mallick M, Aebersold R. Scoring Proteomes with Proteotypic Peptide Probes. Nature Reviews Molecular Cell Biology. 2005;6(7):577–83. doi: 10.1038/nrm1683. [DOI] [PubMed] [Google Scholar]
- Lander ES. Initial Impact of the Sequencing of the Human Genome. Nature. 2011;470(7333):187–197. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- Lange V, Picotti P, Domon B, Aebersold R. Selected Reaction Monitoring for Quantitative Proteomics: A Tutorial. Molecular Systems Biology. 2008;4(222):222. doi: 10.1038/msb.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig C, Claassen M, Schmidt A, Aebersold R. Estimation of Absolute Protein Quantities of Unlabeled Samples by Selected Reaction Monitoring Mass Spectrometry. Molecular & Cellular Proteomics. 2012;11(3):M111013987–M111.013987. doi: 10.1074/mcp.M111.013987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Tomazela DM, Shulman N, Chambers M, Finney GL, Frewen B, Kern R, Tabb DL, Liebler DC, MacCoss MJ. Skyline: An Open Source Document Editor for Creating and Analyzing Targeted Proteomics Experiments. Bioinformatics. 2010;26(7):966–8. doi: 10.1093/bioinformatics/btq054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean B, Daniela M, Tomazela DM, Susan E, Abbatiello SE, Shucha Zhang S, Whiteaker JR, Paulovich AG, Carr SA, MacCoss MJ. Effect of Collision Energy Optimization on the Measurement of Peptides by Selected Reaction Monitoring (SRM) Mass Spectrometry. Analytical Chemistry. 2010;82(24):10116–10124. doi: 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallick P, Schirle M, Chen SS, Flory MR, Lee H, Martin D, Ranish J, Raught B, Schmitt R, Werner T, Kuster B, Aebersold R. Computational Prediction of Proteotypic Peptides for Quantitative Proteomics. Nature Biotechnology. 2007;25(1):125–31. doi: 10.1038/nbt1275. [DOI] [PubMed] [Google Scholar]
- Mani DR, Abbatiello SE, Carr SA. Statistical Characterization of Multiple-Reaction Monitoring Mass Spectrometry (MRM-MS) Assays for Quantitative Proteomics. BMC Bioinformatics. 2012;13(Suppl 16):S9. doi: 10.1186/1471-2105-13-S16-S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens L, Hermjakob H, Jones P, Adamsk M, Taylor C, States D, Gevaert K, Vandekerckhove J, Apweiler R. PRIDE: The proteomics identifications database. Proteomics. 2005;5(13):3537–3545. doi: 10.1002/pmic.200401303. [DOI] [PubMed] [Google Scholar]
- Mathivanan S, Ahmed M, Ahn NG, Alexandre H, Amanchy R, Andrews PC, Bader JS, Balgley BM, Bantscheff M, Bennett KL, Björling E, Blagoev B, Bose R, Brahmachari SK, Burlingame AS, Bustelo XR, Cagney G, Cantin GT, Cardasis HL, Celis JE, Chaerkady R, Chu F, Cole PA, Costello CE, Cotter RJ, Crockett D, DeLany JP, De Marzo AM, DeSouza LV, Deutsch EW, Dransfield E, Drewes G, Droit A, Dunn MJ, Elenitoba-Johnson K, Ewing RM, Van Eyk J, Faca V, Falkner J, Fang X, Fenselau C, Figeys D, Gagné P, Gelfi C, Gevaert K, Gimble JM, Gnad F, Goel R, Gromov P, Hanash SM, Hancock WS, Harsha HC, Hart G, Hays F, He F, Hebbar P, Helsens K, Hermeking H, Hide W, Hjernø K, Hochstrasser DF, Hofmann O, Horn DM, Hruban RH, Ibarrola N, James P, Jensen ON, Jensen PH, Jung P, Kandasamy K, Kheterpal I, Kikuno RF, Korf U, Körner R, Kuster B, Kwon MS, Lee HJ, Lee YJ, Lefevre M, Lehvaslaiho M, Lescuyer P, Levander F, Lim MS, Löbke C, Loo JA, Mann M, Martens L, Martinez-Heredia J, McComb M, McRedmond J, Mehrle A, Menon R, Miller CA, Mischak H, Mohan SS, Mohmood R, Molina H, Moran MF, Morgan JD, Moritz R, Morzel M, Muddiman DC, Nalli A, Navarro JD, Neubert TA, Ohara O, Oliva R, Omenn GS, Oyama M, Paik YK, Pennington K, Pepperkok R, Periaswamy B, Petricoin EF, Poirier GG, Prasad TS, Purvine SO, Rahiman BA, Ramachandran P, Ramachandra YL, Rice RH, Rick J, Ronnholm RH, Salonen J, Sanchez JC, Sayd T, Seshi B, Shankari K, Sheng SJ, Shetty V, Shivakumar K, Simpson RJ, Sirdeshmukh R, Siu KW, Smith JC, Smith RD, States DJ, Sugano S, Sullivan M, Superti-Furga G, Takatalo M, Thongboonkerd V, Trinidad JC, Uhlen M, Vandekerckhove J, Vasilescu J, Veenstra TD, Vidal-Taboada JM, Vihinen M, Wait R, Wang X, Wiemann S, Wu B, Xu T, Yates JR, Zhong J, Zhou M, Zhu Y, Zurbig P, Pandey A. Human Proteinpedia Enables Sharing of Human Protein Data. Nature Biotechnology. 2008;26(2):164–167. doi: 10.1038/nbt0208-164. [DOI] [PubMed] [Google Scholar]
- Moseley MA, Deterding LJ, Tomer KB, Jorgenson JW. Nanoscale Packed-Capillary Liquid Chromatography Coupled with Mass Spectrometry Using a Coaxial Continuous-Flow Fast Atom Bombardment Interface. Analytical Chemistry. 1991;1:1467–1473. doi: 10.1021/ac00014a023. [DOI] [PubMed] [Google Scholar]
- Muntel J, Boswell SA, Tang S, Ahmed S, Wapinski I, Foley G, Steen H, Springer M. Abundance-Based Classifier for the Prediction of Mass Spectrometric Peptide Detectability Upon Enrichment (PPA) Molecular & Cellular Proteomics. 2015;14(2):430–440. doi: 10.1074/mcp.M114.044321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omasits U, Ahrens CH, Müller S, Wollscheid B. Protter: Interactive Protein Feature Visualization and Integration with Experimental Proteomic Data. Bioinformatics. 2014;30(6):884–886. doi: 10.1093/bioinformatics/btt607. [DOI] [PubMed] [Google Scholar]
- Ong SE, Mann M. Mass Spectrometry-Based Proteomics Turns Quantitative. Nature Chemical Biology. 2005;1(5):252–62. doi: 10.1038/nchembio736. [DOI] [PubMed] [Google Scholar]
- Payne SH, Monroe ME, Overall CC, Kiebel GR, Degan M, Gibbons BC, Fujimoto GM, Purvine SO, Adkins JN, Lipton MS, Smith RD. The Pacific Northwest National Laboratory Library of Bacterial and Archaeal Proteomic Biodiversity. Scientific Data. 2015;2:150041. doi: 10.1038/sdata.2015.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrioli PG, Eng JK, Hubley R, Vogelzang M, Deutsch EW, Raught B, Pratt B, Nilsson E, Angeletti RH, Apweiler R, Cheung K, Costello CE, Hermjakob H, Huang S, Julian RK, Kapp E, McComb ME, Oliver SG, Omenn G, Paton NW, Simpson R, Smith R, Taylor CF, Zhu W, Aebersold R. A Common Open Representation of Mass Spectrometry Data and Its Application to Proteomics Research. Nature Biotechnology. 2004;22(11):1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Peterson AC, Russell JD, Bailey DJ, Westphall MS, Coon JJ. Parallel Reaction Monitoring for High Resolution and High Mass Accuracy Quantitative, Targeted Proteomics. Molecular & Cellular Proteomics. 2012;11(11):1475–88. doi: 10.1074/mcp.O112.020131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Aebersold R. Selected Reaction Monitoring–based Proteomics: Workflows, Potential, Pitfalls and Future Directions. Nature Methods. 2012;9(6):555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Picotti P, Bodenmiller B, Mueller LN, Bruno Domon B. Full Dynamic Range Proteome Analysis of S. Cerevisiae by Targeted Proteomics. Cell. 2010;138(4):795–806. doi: 10.1016/j.cell.2009.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash A, Tomazela D, Frewen B, MacLean B, Peterman S, MacCoss MJ. Expediting the development of targeted SRM assays: using data from shotgun proteomics to automate method development. J Proteome Res. 2009 Jun;8(6):2733–9. doi: 10.1021/pr801028b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter L, Rinner O, Picotti P, Hüttenhain R, Beck M, Brusniak MY, Hengartner MO, Aebersold R. mProphet: Automated Data Processing and Statistical Validation for Large-Scale SRM Experiments. Nature Methods. 2011;8(5):430–435. doi: 10.1038/nmeth.1584. [DOI] [PubMed] [Google Scholar]
- Rost H, Malmstrom L, Aebersold R. A Computational Tool to Detect and Avoid Redundancy in Selected Reaction Monitoring. Molecular & Cellular Proteomics. 2012;11(8):540–549. doi: 10.1074/mcp.M111.013045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling B, Rardin MJ, MacLean BX, Zawadzka AM, Frewen BE, Cusack MP, Sorensen DJ, Bereman MS, Jing E, Wu CC, Verdin E, Kahn CR, Maccoss MJ, Gibson BW. Platform-Independent and Label-Free Quantitation of Proteomic Data Using MS1 Extracted Ion Chromatograms in Skyline: application to protein acetylation and phosphorylation. Molecular & Cellular Proteomics. 2012;11(5):202–214. doi: 10.1074/mcp.M112.017707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrimpf SP, Weiss M, Reiter L, Ahrens CH, Jovanovic M, Malmström J, Brunner E, Mohanty S, Lercher MJ, Hunziker PE, Aebersold R, von Mering C, Hengartner MO. Comparative Functional Analysis of the Caenorhabditis Elegans and Drosophila Melanogaster Proteomes. PLoS Biology. 2009;7(3):e48. doi: 10.1371/journal.pbio.1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle BC, Egertson JD, Bollinger J, Stergachis AB, MacCoss MJ. Using Data Independent Acquisition to Model High-Responding Peptides for Targeted Proteomics Experiments. Molecular & Cellular Proteomics. 2015;(206):1–40. doi: 10.1074/mcp.M115.051300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V, Eckels J, Taylor GK, Shulman NJ, Stergachis AB, Joyner SA, Yan P, Whiteaker JR, Halusa GN, Schilling B, Gibson BW, Colangelo CM, Paulovich AG, Carr SA, Jaffe JD, MacCoss MJ, MacLean B. Panorama: A Targeted Proteomics Knowledge Base. Journal of Proteome Research. 2014;13(9):4205–10. doi: 10.1021/pr5006636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherrod SD, Myers MV, Li M, Myers JS, Carpenter KL, MacLean B, MacCoss MJ, Liebler DC, Ham AJL. Label-Free Quantitation of Protein Modifications by Pseudo Selected Reaction Monitoring with Internal Reference Peptides. Journal of Proteome Research. 2012;11(6):3467–3479. doi: 10.1021/pr201240a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood CA, Eastham A, Lee LW, Risler J, Mirzaei H, Falkner JA, Martin DB. Rapid Optimization of MRM-MS Instrument Parameters by Subtle Alteration of Precursor and Product M/z Targets. Proteome. 2009;8(7):3746–3751. doi: 10.1021/pr801122b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherwood CA, Eastham A, Lee LW, Risler J, Vitek O, Martin DB. Correlation between Y-Type Ions Observed in Ion Trap and Triple Quadrupole Mass Spectrometers. Journal of Proteome Research. 2010;8(9):4243–4251. doi: 10.1021/pr900298b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shilov IV, Seymour SL, Patel AA, Loboda A, Tang WH, Keating SP, Hunter CL, Nuwaysir LM, Schaeffer DA. The Paragon Algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Molecular & Cellular Proteomics. 2007;6(9):1638–1655. doi: 10.1074/mcp.T600050-MCP200. [DOI] [PubMed] [Google Scholar]
- Spicer V, Yamchuk A, Cortens J, Sousa S, Ens W, Standing KG, Wilkins JA, Krokhin OV. Sequence-Specific Retention Calculator. A Family of Peptide Retention Time Prediction Algorithms in Reversed-Phase HPLC: Applicability to Various Chromatographic Conditions and Columns. Analytical Chemistry. 2007;79(22):8762–8768. doi: 10.1021/ac071474k. [DOI] [PubMed] [Google Scholar]
- Stein SE, Scott DR. Optimization and Testing of Mass Spectral Library Search Algorithms for Compound Identification. Journal of the American Society for Mass Spectrometry. 1994;5(9):859–66. doi: 10.1016/1044-0305(94)87009-8. [DOI] [PubMed] [Google Scholar]
- Stergachis AB, MacLean B, Lee K, Stamatoyannopoulos JA, MacCoss MJ. Rapid Empirical Discovery of Optimal Peptides for Targeted Proteomics. Nature Methods. 2011;8(12):1041–3. doi: 10.1038/nmeth.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabb DL, MacCoss MJ, Wu CC, Anderson SD, Yates JR. Similarity among Tandem Mass Spectra from Proteomic Experiments: Detection, Significance, and Utility. Analytical Chemistry. 2003;75(10):2470–7. doi: 10.1021/ac026424o. [DOI] [PubMed] [Google Scholar]
- Tabb DL, Fernando CG, Chambers MC. MyriMatch: highly accurate tandem mass spectral peptide identificaiton by multivariate hypergeometric analysis. J Proteome Res. 2007;6(2):654–661. doi: 10.1021/pr0604054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X, Melissa M, Keenan MM, Wu J, Lin CA, Dubois L, Thompson JW, Freedland SJ, Murphy SK, Chi JT. Comprehensive Profiling of Amino Acid Response Uncovers Unique Methionine-Deprived Response Dependent on Intact Creatine Biosynthesis. PLOS Genetics. 2015;11:e1005158. doi: 10.1371/journal.pgen.1005158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ting YS, Egertson JD, Payne SH, Kim S, Maclean B, Aebersold R, Smith RD, Noble WS, Maccoss MJ. Peptide-Centric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data. Molecular & Cellular Proteomics. 2015:1–23. doi: 10.1074/mcp.O114.047035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venable JD, Dong MQ, Wohlschlegel J, Dillin A, Yates JR. Automated Approach for Quantitative Analysis of Complex Peptide Mixtures from Tandem Mass Spectra. Nature Methods. 2004;1(1):39–45. doi: 10.1038/nmeth705. [DOI] [PubMed] [Google Scholar]
- Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG, Smith HO, Yandell M, Evans CA, Holt RA, Gocayne JD, Amanatides P, Ballew RM, Huson DH, Wortman JR, Zhang Q, Kodira CD, Zheng XH, Chen L, Skupski M, Subramanian G, Thomas PD, Zhang J, Gabor Miklos GL, Nelson C, Broder S, Clark AG, Nadeau J, McKusick VA, Zinder N, Levine AJ, Roberts RJ, Simon M, Slayman C, Hunkapiller M, Bolanos R, Delcher A, Dew I, Fasulo D, Flanigan M, Florea L, Halpern A, Hannenhalli S, Kravitz S, Levy S, Mobarry C, Reinert K, Remington K, Abu-Threideh J, Beasley E, Biddick K, Bonazzi V, Brandon R, Cargill M, Chandramouliswaran I, Charlab R, Chaturvedi K, Deng Z, Di Francesco V, Dunn P, Eilbeck K, Evangelista C, Gabrielian AE, Gan W, Ge W, Gong F, Gu Z, Guan P, Heiman TJ, Higgins ME, Ji RR, Ke Z, Ketchum KA, Lai Z, Lei Y, Li Z, Li J, Liang Y, Lin X, Lu F, Merkulov GV, Milshina N, Moore HM, Naik AK, Narayan VA, Neelam B, Nusskern D, Rusch DB, Salzberg S, Shao W, Shue B, Sun J, Wang Z, Wang A, Wang X, Wang J, Wei M, Wides R, Xiao C, Yan C, Yao A, Ye J, Zhan M, Zhang W, Zhang H, Zhao Q, Zheng L, Zhong F, Zhong W, Zhu S, Zhao S, Gilbert D, Baumhueter S, Spier G, Carter C, Cravchik A, Woodage T, Ali F, An H, Awe A, Baldwin D, Baden H, Barnstead M, Barrow I, Beeson K, Busam D, Carver A, Center A, Cheng ML, Curry L, Danaher S, Davenport L, Desilets R, Dietz S, Dodson K, Doup L, Ferriera S, Garg N, Gluecksmann A, Hart B, Haynes J, Haynes C, Heiner C, Hladun S, Hostin D, Houck J, Howland T, Ibegwam C, Johnson J, Kalush F, Kline L, Koduru S, Love A, Mann F, May D, McCawley S, McIntosh T, McMullen I, Moy M, Moy L, Murphy B, Nelson K, Pfannkoch C, Pratts E, Puri V, Qureshi H, Reardon M, Rodriguez R, Rogers YH, Romblad D, Ruhfel B, Scott R, Sitter C, Smallwood M, Stewart E, Strong R, Suh E, Thomas R, Tint NN, Tse S, Vech C, Wang G, Wetter J, Williams S, Williams M, Windsor S, Winn-Deen E, Wolfe K, Zaveri J, Zaveri K, Abril JF, Guigó R, Campbell MJ, Sjolander KV, Karlak B, Kejariwal A, Mi H, Lazareva B, Hatton T, Narechania A, Diemer K, Muruganujan A, Guo N, Sato S, Bafna V, Istrail S, Lippert R, Schwartz R, Walenz B, Yooseph S, Allen D, Basu A, Baxendale J, Blick L, Caminha M, Carnes-Stine J, Caulk P, Chiang YH, Coyne M, Dahlke C, Mays A, Dombroski M, Donnelly M, Ely D, Esparham S, Fosler C, Gire H, Glanowski S, Glasser K, Glodek A, Gorokhov M, Graham K, Gropman B, Harris M, Heil J, Henderson S, Hoover J, Jennings D, Jordan C, Jordan J, Kasha J, Kagan L, Kraft C, Levitsky A, Lewis M, Liu X, Lopez J, Ma D, Majoros W, McDaniel J, Murphy S, Newman M, Nguyen T, Nguyen N, Nodell M, Pan S, Peck J, Peterson M, Rowe W, Sanders R, Scott J, Simpson M, Smith T, Sprague A, Stockwell T, Turner R, Venter E, Wang M, Wen M, Wu D, Wu M, Xia A, Zandieh A, Zhu X. The Sequence of the Human Genome. Science. 2001;291(5507):1304–51. doi: 10.1126/science.1058040. [DOI] [PubMed] [Google Scholar]
- Wenger CD, Coon JJ. A Proteomics Search Algorithm Specifically Designed for High-Resolution Tandem Mass Spectra. J Proteome Res. 2013;12(3):1377–1386. doi: 10.1021/pr301024c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Liu Q, Zimmerman LJ, Ham AJ, Slebos RJ, Rahman J, Kikuchi T, Massion PP, Carbone DP, Billheimer D, Liebler DC. Methods for Peptide and Protein Quantitation by Liquid Chromatography-Multiple Reaction Monitoring Mass Spectrometry. Molecular & Cellular Proteomics. 2011;10(6):M110006593. doi: 10.1074/mcp.M110.006593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z. Prediction of Low-Energy Collision-Induced Dissociation Spectra of Peptides. Anal Chem. 2004;76:3908–3922. doi: 10.1021/ac049951b. [DOI] [PubMed] [Google Scholar]
- Zhang Z. Prediction of Low-Energy Collision-Induced Dissociation Spectra of Peptides with Three or More Charges. Anal Chem. 2005;77:6364–6373. doi: 10.1021/ac050857k. [DOI] [PubMed] [Google Scholar]
- Zhang J, Xin L, Shan B, Chen W, Xie M, Yuen D, Zhang W, Zhang Z, Lajoie GA, Ma B. PEAKS DB: De Novo Sequencing Assisted Database Search for Sensitive and Accurate Peptide Identification. Molecular & Cellular Proteomics. 2012;11(4) doi: 10.1074/mcp.M111.010587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Bilbao A, Bruderer T, Luban J, Strambio-De-Castillia C, Lisacek F, Hopfgartner G, Varesio E. The Use of Variable Q1 Isolation Windows Improves Selectivity in LC-SWATH-MS Acquisition. Journal of Proteome Research. 2015;14(10):4359–4371. doi: 10.1021/acs.jproteome.5b00543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.