

Abstract
Resuscitation with hypertonic saline (HTS) attenuates acute lung injury (ALI) and modulates postinjury hyperinflammation. TNF-α–stimulated pulmonary epithelium is a major contributor to hemorrhage-induced ALI. We hypothesized that HTS would inhibit TNF-α–induced nuclear factor (NF)–κB proinflammatory signaling in pulmonary epithelial cells. Therefore, we pretreated human pulmonary epithelial cells (A549) with hypertonic medium (180 mM NaCl) for 30 min, followed by TNF-α stimulation (10 ng/mL). Key regulatory steps and protein concentrations in this pathway were assessed for significant alterations. Hypertonic saline significantly reduced TNF-α–induced intercellular adhesion molecule 1 levels and NF-κB nuclear localization. The mechanism is attenuated phosphorylation and delayed degradation of IκBα. Hypertonic saline did not alter TNF-α–induced p38 mitogen-activated protein kinase phosphorylation or constitutive vascular endothelial growth factor expression, suggesting that the observed inhibition is not a generalized suppression of protein phosphorylation or cellular function. These results show that HTS inhibits TNF-α–induced NF-κB activation in the pulmonary epithelium and, further, our understanding of its beneficial effects in hemorrhage-induced ALI.
Keywords: Acute lung injury, IκBα, ICAM-1, VEGF, p38 MAPK
INTRODUCTION
Postinjury resuscitation with hypertonic saline (HTS) has been associated with improved clinical outcomes in select patient groups (1). Whereas the mechanism of improved outcomes was originally attributed to more efficient restoration of plasma volume, newer evidence suggests the benefit may derive from modulation of acute lung injury (ALI) and subsequent postinjury hyperinflammation (2, 3). Hemorrhage-induced ALI is mediated by activated alveolar macrophages that produce proinflammatory mediators that promote cell surface adhesion molecule and chemokine expression by the pulmonary tissues recruiting neutrophils into the lung (4–6). Hypertonic saline resuscitation has been shown to decrease alveolar macrophage activation, cell surface adhesion molecule expression, and neutrophil accumulation (7–9). Hypertonic saline alters the proinflammatory signaling that activates leukocytes (10–14), yet little is known regarding the effects of HTS on proinflammatory signaling in the pulmonary endothelium or epithelium (15). To improve our understanding of how HTS attenuates hemorrhage-induced ALI and maximize its therapeutic potential, further investigation in pulmonary endothelial and epithelial cells is needed.
TNF-α is produced in the lungs in response to hemorrhagic shock (6, 16) and is one of the dominant cytokines mediating ALI and stimulating cell surface adhesion molecule expression in the lung (17, 18). Previous work using hemorrhagic shock models with both TNF-α and TNF receptor 1 (TNF R1) knockout mice found that low levels of TNF-α signaling are sufficient and necessary to induce pulmonary neutrophil accumulation and ALI (19). The attenuated pulmonary neutrophil accumulation and ALI seen in the absence of TNF-α signaling is similar to observations after HTS resuscitation. This similarity and the growing amount of evidence that activated pulmonary epithelium is a major contributor to ALI (5, 20) led us to hypothesize that HTS inhibits TNF-α proinflammatory signaling in pulmonary epithelial cells.
The purpose of the study was to determine the effects of HTS on the classical TNF-α–induced nuclear factor (NF)–κB pathway in human pulmonary epithelial cells. The transcription factor NF-κB is responsible for the transcription of a large amount of proinflammatory genes that necessitates tight regulation of its activation (21). Within resting cells, inhibitor κB (IκB) is bound to NF-κB, sequestering it within the cytoplasm. TNF-α binding to the TNF R1 stimulates receptor complex formation and activation of the IκB kinase. Phosphorylation of IκB targets it for polyubiquination and proteosomic degradation. Once free of its inhibitor, NF-κB is free to translocate into the nucleus and initiate the transcription of proinflammatory genes. We find that, although HTS does not induce changes in TNF R1 cell surface expression or whole cell levels, it does inhibit IκB phosphorylation and delays its degradation. This explains the attenuation of NF-κB nuclear localization and decreased NF-κB–driven proinflammatory gene expression. However, we find that TNF-α–induced p38 mitogen-activated protein kinase (MAPK) phosphorylation is not decreased by HTS. This indicates that the anti-inflammatory effects are not due to a generalized attenuation of protein phosphorylation and may be specific to IκB and subsequent NF-κB activation.
MATERIALS AND METHODS
Cell culture and treatment
Human pulmonary epithelial cells (A549) were cultured in F12 medium enriched with 10% fetal bovine serum (Mediatech, Herndon, Va) and 100 IE/mL penicillin and 0.1 mg/mL streptomycin at 37°C and 5% CO2. The cells were grown to approximately 70% confluence for immunofluorescent microscopy and approximately 90% confluence for cell lysis, cell viability assay, and flow cytometry. The cells were pretreated with clinically relevant hypertonic medium (22), 180 mM NaCl (Sigma-Aldrich, St. Louis, Mo) for 30 minutes. Control cells were pretreated with fresh isotonic medium only. The cells were either lysed to measure whole cell concentration of TNF R1, harvested to measure TNF R1 cell surface expression by flow cytometry, or further incubated with isotonic medium containing 10 ng/mL TNF-α (Sigma-Aldrich) for varying times between 5 and 30 min. When measuring inter-cellular adhesion molecule 1 (ICAM-1) expression, the TNF-α medium was replaced after 30 min with fresh isotonic medium, and the cells were incubated at 37°C for an additional 3.5 h.
Cell collection and lysis
After supernatant removal, the 6-well plates were placed on ice and washed with ice-cold phosphate-buffered saline (PBS)/phosphatase inhibitor cocktail (Active Motif Inc., Carlsbad, Calif). This was followed by the addition of a lysis solution of 3% sodium dodecyl sulfate–polyacrylamide gel electrophoresis, 1 nM dithiothreitol, and 5× Halt phosphatase inhibitor cocktail (Pierce Biotechnology, Inc., Rockford Ill). The wells were scraped, and the lysates were collected in microcentrifuge tubes and immediately heated to 100°C for 15 min.
Western blot analysis
The protein concentration of the whole cell lysates was determined by bicinchoninic acid assay protein assay (Pierce Biotechnology, Inc.). Equal protein concentrations were loaded into 4% to 15% acrylamide gel, fractioned by sodium dodecyl sulfate–polyacrylamide gel electrophoresis, and then electrotransferred onto a nitrocellulose membrane (Bio-Rad Laboratories, Hercules, Calif). The membranes were stained with 1% Ponceau S (Sigma-Aldrich) and scanned for even protein loading before washing and blocking for 1 h with 5% nonfat milk/PBS solution. The membranes were probed with primary antibodies against phosphorylated IκBα (Ser32, L9241) and phosphorylated p38 MAPK (Thr180/Tyr182, L9211; Cell Signaling Technology, Danvers, Mass), total IκBα (C-21), ICAM-1 (H-108), and TNF R1 (H-5) (Santa Cruz Biotechnology, Inc., Santa Cruz, Calif) overnight at 4°C in 5% nonfat milk/PBST solution, washed, and secondarily probed with antirabbit horseradish peroxidase–coupled antibodies (Pierce Biotechnology, Inc.). Bound antibodies were detected with the enhanced chemoluminescence immunoblotting system (Pierce Biotechnology, Inc.). The Western blots provided in the figures are representative of three or more separate experiments. Relative densities were measured by National Institutes of Health ImageJ software (http://rsb.info.nih.gov/ij). Even protein loading was confirmed by probing each membrane with primary antibodies against actin (H 300; Santa Cruz Biotechnology, Inc.).
Immunofluorescent microscopy
Cells were grown on Lab-Tek Chamber Slides (Nalge Nunc International, Rochester, NY) and treated as previously mentioned. The slides were fixed with 4% paraformaldehyde in PBS at room temperature (RT), washed three times with PBS, and then permeabilized with 70% acetone/30% methanol solution at −20°C. The cells were blocked with 10% donkey serum (Jackson ImmunoResearch Laboratories, West Grove, Pa) in PBS for 60 min at RT. The slides were incubated overnight at 4°C with rabbit polyclonal anti-p65 antibody (C-20, Santa Cruz Biotechnology, Inc.) in 1% band shift assay/PBS. Negative control slides were incubated with isotype at the same concentration (Jackson ImmunoResearch Laboratories). After three PBS washes, the slides were incubated for 1 h at RT with donkey antirabbit Alexa 488–conjugated antibody (Molecular Probes Invitrogen Detection Technologies, Eugene, Ore) and bis-benzimide as a nuclear stain (Sigma-Aldrich).
Images were acquired with a Zeiss Axiovert fitted with a Cooke CCD SensiCam using a Chroma Sedat with single excitation and emission filters. The two channel images were then digitally processed using Intelligent Imaging Innovations Slidebook 4.1 software (Intelligent Imaging Innovations, Inc., Denver, Colo). Nuclear stains were masked to measure the relative p65 mean fluorescence intensity (MFI) within the nuclei. The MFIs of approximately 50 to 60 cells from each group of three separate experiments were analyzed.
Flow cytometry
The relative cell surface expression of TNF R1 was measured with immunofluorescence and flow cytometry. After pretreatment with fresh isotonic or hypertonic medium (180 mM NaCl) for 30 min, the cells were harvested by addition of trypsin (Clonetics, Cambrex Bio Science, Walersville, Md) for 5 min and then trypsin-neutralizing solution (Clonetics, Cambrex Bio Science). After centrifugation, the cells were resuspended in fresh isotonic PBS before incubation with monoclonal antihuman TNF R1–phycoerythrin labeled antibody (FAB225P; R&D Systems, Inc., Minneapolis, Minn) for 90 min at 4°C. The cells were then washed and fixed with 2% paraformaldehyde in PBS. Relative concentrations were measured by flow cytometry gated for the main cell population using forward and side scatter with greater than 90% of all events recorded (Beckman Coulter SC500; Beckman Coulter, Hialeah, Fla; 5,000 events per group with consistent voltage and gain perimeters). Flow cytometry data presented as MFI. Isotype controls were less than 0.5 MFI.
After 3 and 12 h of incubation in isotonic and hypertonic medium, the relative vascular endothelial growth factor (VEGF) concentrations in the supernatants were measured with the Bio-Plex cytokine assay (Bio Rad Laboratories, Life Science Group) according to the manufacturer’s instructions. Briefly, fluorescently dyed 5.6-μm polystyrene beads with covalently coupled anti-VEGF antibody were incubated with 50 μL supernatant in a 96-well vacuum filtration plate. After 30 min of gentle agitation at RT, the supernatant was removed, and the wells were washed. The beads were then incubated with VEGF-specific detection antibody for 30 min at RT and then washed again. Finally, the beads were incubated with streptavidin-phycoerythrin for 10 min at RT, washed, and the relative fluorescence was measured with flow cytometry and compared with standard curves (Beckman Coulter FC500 with CXP software; Beckman Coulter; and Bio-Plex Manager Luminex analysis software, Bio-Rad Laboratories).
Cell viability assay
After 18 h of incubation in isotonic and hypertonic medium, both with and without TNF-α (10 ng/mL), the cell viability assay (catalog no. 11 465 007 001; Roche Applied Science, Indianapolis, Ind) was performed according to the manufacturer’s instructions. Cells were incubated with the 3-(4,5-dimethylthiazol-2-y)-2,5-diphenyltetrazolium bromide (MTT) labeling reagent for 4 h. The solubilization solution was added, and the cells were then incubated overnight at 37°C and 5% CO2. The resulting solubilized formazan product was spectrophotometrically quantified using an enzyme-linked immunosorbent assay reader.
Statistical analysis
All data are represented as mean T SEM. The data were evaluated by either Student t test or analysis of variance with Tukey-Kramer using JMP 5.0.1 software. Statistical significance was accepted as P < 0.01.
RESULTS
HTS does not affect pulmonary epithelial cell viability or constitutive VEGF expression
The effects of HTS on pulmonary epithelial cell viability were assessed by measuring the metabolism of MTT to formazan salts. These crystals are solubilized, and the resulting colored solution is measured by enzyme-linked immunosorbent assay (Fig. 1A). Incubation for 18 h, both with and without HTS, produces similar absorbance values with the HTS group, but slightly higher. Hypertonic saline may minimally stimulate MTT metabolism but does not cause loss of cell viability.
FIG. 1. Pulmonary epithelial cell viability and constitutive VEGF expression are unchanged by HTS.
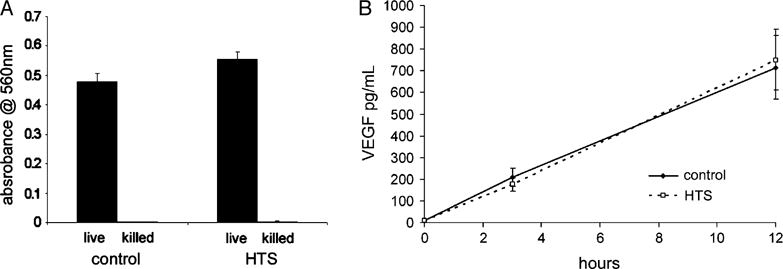
A, Metabolism of tetrasodium salt to colored formazan salt crystal is quantified by solubilization and spectrophotometry. After 18 h of incubation, with and without HTS, the measured absorbance values are not significantly different. Data from three separate experiments are averaged and presented as SEM. B, Supernatant concentrations of VEGF were measured by flow cytometry. Resting cells produce VEGF at a steady rate over 12 h of incubation. Hypertonic saline does not alter the rate of expression or the amount produced, indicating that protein synthesis and cellular function are intact. Data from three separate experiments are averaged and presented as SEM.
Vascular endothelial growth factor is abundant in the lung and highly expressed in pulmonary epithelial cells. Both in vivo and in vitro studies show that decreased VEGF expression is an indicator of increased alveolar epithelial damage and reduced epithelial cellular function (23, 24). Therefore, to determine the effect of HTS on cellular function, we assessed the effects of HTS on VEGF expression by measuring the relative concentrations in the supernatants (Fig. 1B). Resting cells produce VEGF at a steady rate over 12 h of incubation. Hypertonic saline does not alter the rate of expression or the amount produced.
HTS attenuates TNF-α–induced ICAM-1 expression
During hemorrhagic shock, ICAM-1 is expressed in pulmonary tissues in response to TNF-α stimulation (25). It interacts with cell surface adhesion molecules on the primed neutrophil and facilitates recruitment from the capillaries and tethering within the alveoli (5). We assessed the effects of HTS on TNF-α–induced ICAM-1 production by measuring relative whole cell levels with Western blot (Fig. 2, A and B) ICAM-1 is undetectable in unstimulated cells. TNF-α provokes a marked increase in ICAM-1 levels that is significantly decreased by HTS. Vascular adhesion molecule 1, another cell surface adhesion molecule, unlike ICAM-1, is unaffected by TNF-α or HTS. It is included as both a loading and an experimental control.
FIG. 2. Hypertonic saline decreases TNF-α–induced ICAM expression.
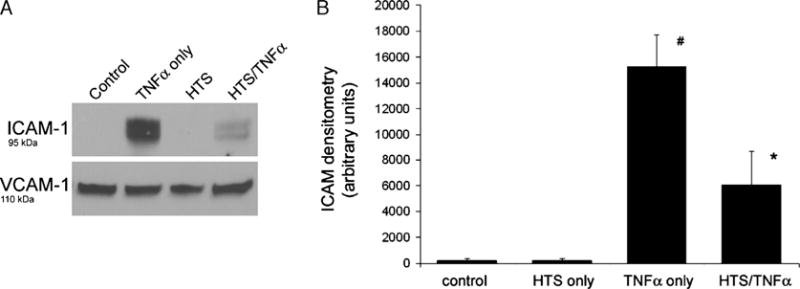
A, Western blot of whole cell lysate does not find any ICAM-1 in unstimulated cells. TNF-α markedly increases ICAM-1 amounts. Hypertonic saline pretreatment alone does not induce ICAM-1 production. Hypertonic saline largely decreases the amount of TNF-α–induced ICAM-1 production. Vascular cell adhesive molecule 1 amounts are unchanged by TNF-α or HTS. B, Hypertonic saline significantly decreases TNF-α–induced ICAM-1 production. Densitometry data from four separate experiments are presented as SEM. Both the TNF-α–only group and the HTS/TNF-α group are significantly different from all other groups (*#P < 0.001).
HTS attenuates TNF-!–induced NF-.B nuclear localization
The significant decrease in whole cell ICAM-1 expression led us to suspect that HTS attenuates NF-κB activation. We used immunofluorescence and digital microscopy to observe the cellular location and nuclear translocation of NF-κB. In unstimulated cells, we find most NF-κBs (p65 subunit) located in the cytoplasm (Fig. 3A, column 1). Using the relative nuclear MFI, we find that HTS alone causes no change in the nuclear MFI (Fig. 3B). TNF-α induces NF-κB trans-location into the nucleus with a maximal concentration at 30 min (Fig. 3A, column 3). Hypertonic saline does not completely inhibit translocation but significantly decreases nuclear localization, leaving more NF-κB within the cytoplasm. Whereas TNF-α stimulation causes a 4-fold increase, HTS reduces the NF-κB nuclear MFI by greater than 40% (Fig. 3B).
FIG. 3. Hypertonic saline significantly attenuates TNF-α–induced NF-κB nuclear localization.
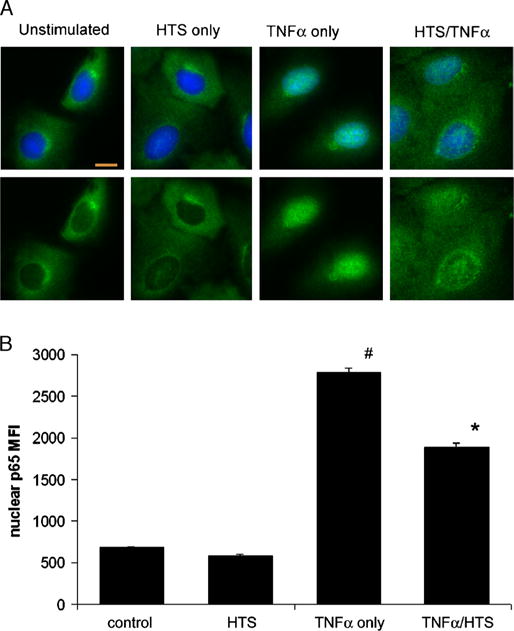
A, Immunofluorescent images show the intracellular localization of NF-κB. The nuclear stains (blue) are omitted from the bottom row. In unstimulated cells, most of the NF-κB p65 subunit (green) is sequestered in the cytoplasm (first column). Hypertonic saline pretreatment does not significantly change the intracellular location of the p65 subunit (second column). TNF-α causes the p65 subunit to accumulate in the nucleus at 30 min (third column). Hypertonic saline attenuates TNF-α– induced NF-κB nuclear translocation (fourth column) with more of the p65 subunit left within the cytoplasm. The orange bar equals 10 μm. All images acquired at 40 magnification. B, Hypertonic saline reduces nuclear NF-κB MFI. Nuclear staining was used to produce masks to measure the relative MFI of the p65 subunit within the nuclei. The MFI of HTS pretreatment–only cells are not different from controls. TNF-α causes a 4-fold increase that HTS reduces by more than 40%. Data from four separate experiments are presented as SEM. Both the TNF-α–only group and the HTS/TNF-α group are significantly different from all other groups (*#P < 0.001).
HTS alters TNF-α–induced IκBα phosphorylation and degradation dynamics
The phosphorylation and subsequent degradation of IκB is critical to regulating TNF-α–induced NF-κB activation and nuclear translocation. In the unstimulated state, IκB binds NF-κB, masking the nuclear localization sequences on the p65 subunit, thus sequestering the transcription factor in the cytoplasm (21). We measured the effects of HTS on TNF-α–induced IκBα phosphorylation and degradation dynamics by Western blot (Fig. 4). In unstimulated cells, phosphorylated IκBα is undetectable. TNF-α provokes a marked increase in whole cell levels of phosphorylated IκBα (Ser32) at 5 min that then degrades by 10 min. Hypertonic saline significantly decreases the level of TNF-α–induced phosphorylation that degrades by 10 min as in controls (Fig. 4, A and B) Furthermore, TNF-α provokes an initial IκBα degradation at 5 min that is complete at 10 min. Hypertonic saline significantly delays TNF-α–induced degradation with a high level remaining at 10 min and a small level still detectable at 30 min (Fig. 4, A and C).
FIG. 4. Hypertonic saline alters IκBα phosphorylation and degradation dynamics.
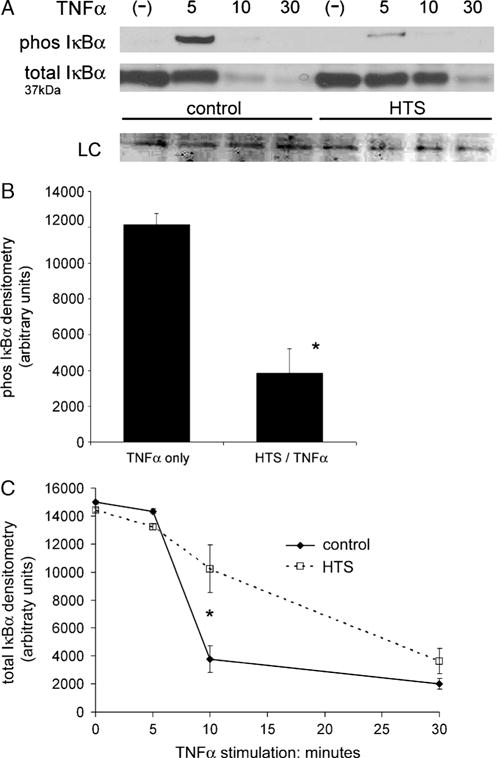
A, Western blot of whole cell lysate does not find phosphorylated IκBα in unstimulated cells. TNF-α provokes a marked increase in phosphorylated IκBα at 5 min that degrades by 10 min. Hypertonic saline significantly decreases the amount of TNF-α–induced phosphorylation that degrades by 10 min as in controls. Hypertonic saline does not change total IκBα concentration in unstimulated cells. TNF-α stimulates IκBα degradation at 5 min that is complete by 10 min. Hypertonic saline inhibits degradation with a significant amount remaining at 10 min and a small but detectable amount remaining at 30 min. B, Hypertonic saline significantly decreases the amount of phosphorylated IκBα at 5 min of TNF-α stimulation. Densitometry data from four separate experiments are presented as SEM (*P < 0.001). C, TNF-α stimulates near complete total IκBα degradation at 10 min. Hypertonic saline delays the same level of degradation to approximately 30 minutes. Hypertonic saline significantly decreases total IκBα degradation at 10 min (*P < 0.001). Densitometry data of four separate experiments are presented as SEM.
TNF R1 cell surface expression is unchanged by HTS
TNF-α activity can be regulated by the generation of extracellular TNF-α receptors that function as TNF-binding proteins (26). Even a small amount of TNF-α receptor shedding during cellular stress can significantly attenuate TNF-α signaling. To determine if HTS had any effect on receptor cell surface expression, we measured the cell surface concentration of TNF R1 with flow cytometry (Fig. 5, A and B). The relative cell surface fluorescence and MFI of cells incubated with HTS for 30 min is the same as controls. Thus, HTS does not alter TNF R1 cell surface expression. Whole cell levels of TNF R1 measured by Western blot are also unchanged (Fig. 5C).
FIG. 5. Hypertonic saline does not alter cell surface localization or whole cell expression of TNF R1.
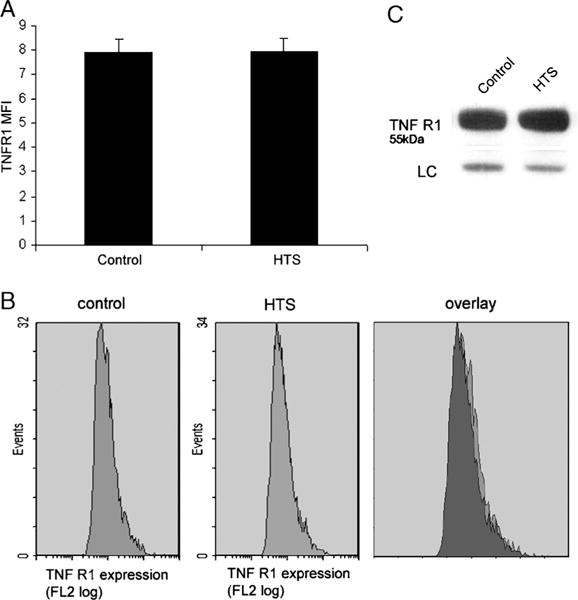
A, Flow cytometry detection of cell surface TNF R1 finds the MFI of cells incubated in HTS for 30 min unchanged from controls. Data from three separate experiments are averaged and presented as SEM. B, Flow cytometry detection of cell surface TNF R1 produces similar histograms from both controls and HTS. This similarity is confirmed by an overlay of the histograms. C, Western blot of whole cell lysate finds TNF R1 levels of cells incubated in HTS for 30 min unchanged from controls.
TNF-α –induced p38 MAPK phosphorylation is unchanged by HTS
Mitogen-activated protein assay signaling is central in the cellular stress response to hypertonicity that is in part mediated by p38 phosphorylation (27). We measured phosphorylated p38 by Western blot and confirmed that HTS alone causes p38 phosphorylation in A549 cells (Fig. 6, A and B) Phosphorylated p38 is undetectable in the control group, yet HTS only induces detectable levels.
FIG. 6. TNF-α–induced p38 MAPK phosphorylation is unchanged by HTS.
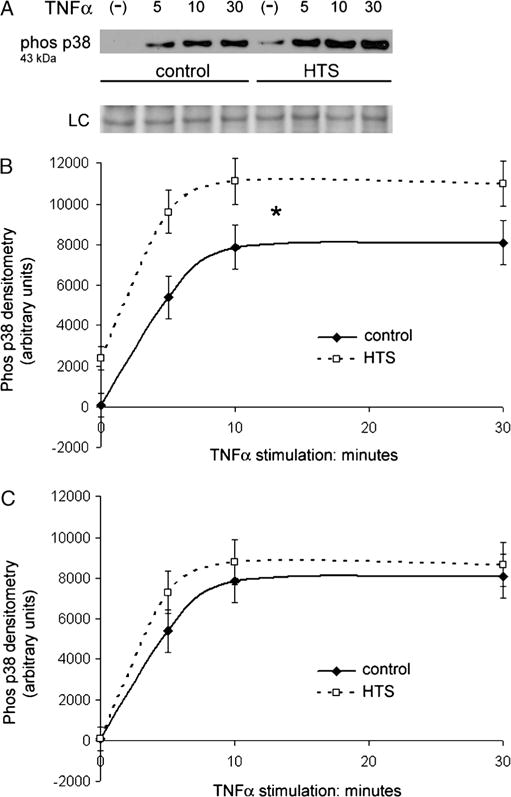
A, Western blot of whole cell lysate does not find phosphorylated p38 MAPK in unstimulated cells. TNF-α provokes a marked increase in phosphorylated p38 levels at 5 min that continues to increase and plateau at 10 and 30 min. Hypertonic saline pretreatment alone before TNF-α stimulation causes a low level of p38 phosphorylation. TNF-α provokes proportional increases in phosphorylated p38 levels at 5, 10, and 30 min while in the presence of HTS. B, Densitometry data confirm that HTS alone causes a measurable increase in phosphorylated p38. At each time point, the paired groups are significantly different. However, the similarly shaped density curves show that TNF-α induces the same relative increase in both the control and HTS groups. Densitometry data of four separate experiments are averaged and presented as SEM (*P < 0.01). C, Subtracting the average densitometry value of the initial HTS-only group from the remaining HTS groups yields a curve that is identical to that of TNF-α only. The paired time points from each curve are not significantly different with overlapping error bars. Densitometry data of four separate experiments are presented as SEM.
In addition to NF-κB activation, TNF-α induces a proinflammatory state by TNF R1–stimulated MAPK signaling and p38 phosphorylation (28). TNF-α provokes a marked increase in phosphorylated p38 levels at 5 min that continues to plateau at 10 and 30 min in controls. Although HTS induces a low level of p38 phosphorylation, making paired time point groups significantly different, it does not inhibit TNF-α–induced p38 phosphorylation as the same relative TNF-α–induced increase and plateau is observed with HTS (Fig. 6B). Subtracting the average densitometry value of the initial HTS-only group from the remaining HTS groups yields a densitometry curve that is identical to that of TNF-α only (Fig. 6C).
DISCUSSION
In this study, we find that TNF-α induces IκBα phosphorylation at 5 min, IκBα degradation occurs at 10 min, followed by NF-κB moving from the cytoplasm into the nucleus with a maximal nuclear concentration at 30 min. The presence of HTS significantly attenuates TNF-α–induced NF-κB nuclear localization at 30 min by altering IκB phosphorylation dynamics and significantly delaying its degradation. These changes result in decreased total cellular levels of ICAM-1 expression. We chose to initiate the study by measuring ICAM-1 expression because it has an absolute requirement for NF-κB mediated promoter activity (29). Any significant alteration in TNF-α–induced NF-κB signaling would likely result in decreased expression. Subsequent experiments confirmed that HTS alters the key regulatory step in the classical TNF-α–induced NF-κB signaling pathway. Considering the central role this pathway plays within innate immunity, these results give significant insight into how HTS modulates postinjury hyperinflammation.
Investigating the effects of HTS on the classical TNF-α signaling pathway narrows the search for its anti-inflammatory mechanisms. Hypertonic saline could attenuate receptor activation, protein phosphorylation, transcriptional activation, message translation, or the export of proinflammatory mediators and cell surface proteins. Our results show that VEGF expression, an indicator of epithelial cell function (23), is unchanged by 12 h of incubation in HTS, showing that constitutive protein synthesis is unaffected. Total cellular content and cell surface expression of TNF R1 are also unchanged by incubation in HTS, suggesting that HTS does not exert immediate effects on surface receptor localization. Although we did not find any change in molecular weight or cell surface expression of TNF R1, HTS could possibly alter TNF-α ligation and receptor activation. Therefore, we measured the effects of HTS on TNF-α–induced p38 phosphorylation as another readout of TNF R1 activation (28). In contrast to IκBα phosphorylation, HTS does not alter the relative amount of TNF-α–induced p38 phosphorylation. This implies that TNF R1 activation is unaffected, and that the alteration is not a generalized attenuation of protein phosphorylation. Although preliminary experiments were performed to find the most effective HTS treatment, formal experiments with differing HTS treatment time sequences were not done. This is a limitation in our study because data from simultaneous HTS and TNF-α treatments and HTS treatment after TNF-α stimulation may give additional insight into potential mechanisms.
Our results confirm that HTS significantly attenuates proinflammatory signaling in pulmonary epithelium. Continuing work shows that, in addition to gas exchange and surfactant production, the pulmonary epithelium plays a major role in the regulation of pulmonary inflammation (5, 20, 30, 31). Thorley et al. (20) recently described the differential cytokine and chemokine release from activated primary human lung macrophages and alveolar type 2 epithelial cells. In response to LPS, TNF-α and IL-1β are released in high amounts from the macrophages, whereas the alveolar epithelium produced high amounts of chemotactic chemokines (methylcyclopentane 1, IL-8, growth-related oncogene α). Blocking antibodies to TNF-α and IL-1β during LPS exposure showed that alveolar epithelial cell chemokine production depends on the autocrine effects of TNF-α and IL-1β. Further studies using in vitro chemotatic assays found that epithelial cell conditioned medium stimulated significantly more neutrophil migration than macrophage conditioned medium (20). Thus, it seems that TNF-α and IL-1β produced by activated macrophages stimulate the alveolar epithelium to orchestrate leukocyte migration into the alveoli. These results again implicate TNF-α signaling in the pulmonary epithelium as a major contributor to ALI.
Further studies by Sadikot et al. (31) and Poynter et al. (32) describe the effects of specifically altering NF-κB activation in the pulmonary epithelium. Transgenic mice expressing a mutant version of IκBα that acts to suppress NF-κB activation exclusively in airway epithelial cells showed significantly decreased epithelial NF-κB nuclear translocation, decreased chemotactic chemokine expression, and decreased neutrophil recruitment in response to intranasal LPS. However, resident alveolar macrophages in these mice were capable of activating NF-κB just as in transgene-negative mice. Moreover, constitutively active IκB kinases selectively expressed in the airway epithelium by way of intratracheal administration of adenoviral vectors were sufficient to activate NF-κB, stimulate chemotactic chemokine expression, and recruit neutrophils. Coadministration of adenoviral vectors expressing an NF-κB inhibitor abrogated the NF-κB activation, chemokine expression, and neutrophil recruitment. These studies show that pulmonary inflammation is largely mediated by NF-κB signaling in the epithelium, that NF-κB activation in the epithelium is sufficient to elicit pulmonary inflammation in the absence of exogenous stimuli, and that inhibiting NF-κB activation in the pulmonary epithelium should be a target for potential therapies. The decreased pulmonary neutrophil sequestration and markers of ALI seen with HTS resuscitation in hemorrhagic shock models have been attributed to decreased alveolar macrophage activation and neutrophil-endothelial adhesion (7, 8, 33). However, our findings, together with those previously described, suggest that decreased TNF-α–induced NF-κB signaling in the pulmonary epithelium could play a significant role.
Finding the inhibitory effects of HTS on TNF-α–induced ICAM-1 production was useful because of its promoter specificity for NF-κB (29), but understanding the role of epithelial ICAM-1 production in ALI emphasizes the therapeutic potential of HTS. Neutrophil-mediated cytotoxicity in the alveolus results in epithelial damage and loss of the permeability barrier that causes alveolar flooding and respiratory compromise (25). Tight neutrophil-epithelial cell adhesion is necessary for proteases, elastases, and oxygen metabolites produced by the activated neutrophil to mediate cytotoxicity (34). Epithelial ICAM-1 expression in large part mediates this tight interaction because anti–ICAM-1 antibodies significantly decrease the adhesion of activated neutrophils and macrophages to activated alveolar epithelium in adherence assays (35). Intratracheal instillation of the same antibodies significantly decreases the bronchoalveolar lavage fluid neutrophil count and pulmonary myeloperoxidase levels (35). In addition, soluble ICAM-1 production contributes to ALI by activating alveolar macrophages to produce TNF-α and proinflammatory chemokines (36, 37) Therapies that decrease ICAM-1 activity and production in the alveolar epithelium have potential to significantly attenuate the pathophysiology of ALI. Again, our results suggest that the beneficial effects of HTS in ALI may be in large part due to inhibition of TNF-α–induced NF-κB signaling, resulting in decreased ICAM-1 expression in the pulmonary epithelium.
The HTS concentration (180 mM NaCl) used in our studies is clinically achievable with a 250-cc intravascular bolus of 7.5% NaCl (22). The induced hypertonicity is transient but was recently shown in a clinical trial reported by Rizoli et al. (3) to have significant anti-inflammatory effects persisting for 24 h. In vivo models of hemorrhagic shock and noninfectious systemic inflammation show that alveolar macrophages recovered via bronchoalveolar lavage are affected by HTS resuscitation (33, 38). The transient hypertonicity seems to have effects within the alveolus, yet learning that HTS has direct anti-inflammatory effects on pulmonary epithelium suggests other HTS treatment methods. Repeated nebulized HTS has been shown in large trials to be safe and to improve lung function in cystic fibrosis patients (39, 40). Our results suggest that nebulized HTS, if used in the severely injured patient, could potentially attenuate hemorrhage-induced ALI by directly inhibiting alveolar epithelial activation and subsequent neutrophil sequestration.
In summary, we find that HTS attenuates TNF-α–induced NF-κB activation in pulmonary epithelial cells. IκBα phosphorylation is decreased and degradation is delayed, resulting in decreased NF-κB nuclear translocation and proinflammatory gene expression. TNF R1 activation and p38 MAPK phosphorylation are unaltered, which suggests that the inhibition may be specific to NF-κB activation. Because the significant role of the activated alveolar epithelium in ALI continues to evolve, finding that HTS inhibits TNF-α–induced NF-κB signaling in this tissue adds to our understanding of how HTS attenuates ALI and postinjury hyperinflammation. Collectively, this brings new insight in how HTS may be used more effectively as a clinical strategy to attenuate pulmonary dysfunction.
Acknowledgments
The authors thank Karen Helm, Christine Childs, and the University of Colorado Cancer Center Flow Cytometry Core for their expertise.
Supported by Grant No. P30 CA 046934, P50 GM49222, and T32 GM008315 from the National Institutes of Health.
References
- 1.Mattox KL, Maningas PA, Moore EE, Mateer JR, Marx JA, Aprahamian C, Burch JM, Pepe PE. Prehospital hypertonic saline/dextran infusion for post-traumatic hypotension. The U.S.A. Multicenter Trial. Ann Surg. 1991;213:482–491. doi: 10.1097/00000658-199105000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Junger WG, Coimbra R, Liu FC, Herdon-Remelius C, Junger W, Junger H, Loomis W, Hoyt DB, Altman A. Hypertonic saline resuscitation: a tool to modulate immune function in trauma patients? Shock. 1997;8:235–241. [PubMed] [Google Scholar]
- 3.Rizoli SB, Rhind SG, Shek PN, Inaba K, Filips D, Tien H, Brenneman F, Rotstein O. The immunomodulatory effects of hypertonic saline resuscitation in patients sustaining traumatic hemorrhagic shock: a randomized, controlled, double-blinded trial. Ann Surg. 2006;243:47–57. doi: 10.1097/01.sla.0000193608.93127.b1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage- or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1137–L1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 5.Burns AR, Smith CW, Walker DC. Unique structural features that influence neutrophil emigration into the lung. Physiol Rev. 2003;83:309–336. doi: 10.1152/physrev.00023.2002. [DOI] [PubMed] [Google Scholar]
- 6.Barsness KA, Arcaroli J, Harken AH, Abraham E, Banerjee A, Reznikov L, McIntyre RC. Hemorrhage-induced acute lung injury is TLR-4 dependent. Am J Physiol Regul Integr Comp Physiol. 2004;287:R592–R599. doi: 10.1152/ajpregu.00412.2003. [DOI] [PubMed] [Google Scholar]
- 7.Pascual JL, Ferri LE, Seely AJ, Campisi G, Chaudhury P, Giannias B, Evans DC, Razek T, Michel RP, Christou NV. Hypertonic saline resuscitation of hemorrhagic shock diminishes neutrophil rolling and adherence to endothelium and reduces in vivo vascular leakage. Ann Surg. 2002;236:634–642. doi: 10.1097/00000658-200211000-00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rizoli SB, Kapus A, Fan J, Li YH, Marshall JC, Rotstein OD. Immunomodulatory effects of hypertonic resuscitation on the development of lung inflammation following hemorrhagic shock. J Immunol. 1998;161:6288–6296. [PubMed] [Google Scholar]
- 9.Angle N, Hoyt DB, Coimbra R, Liu F, Herdon-Remelius C, Loomis W, Junger WG. Hypertonic saline resuscitation diminishes lung injury by suppressing neutrophil activation after hemorrhagic shock. Shock. 1998;9:164–170. doi: 10.1097/00024382-199803000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Angle N, Cabello-Passini R, Hoyt DB, Loomis WH, Shreve A, Namiki S, Junger WG. Hypertonic saline infusion: can it regulate human neutrophil function? Shock. 2000;14:503–508. [PubMed] [Google Scholar]
- 11.Ciesla DJ, Moore EE, Gonzalez RJ, Biffl WL, Silliman CC. Hypertonic saline inhibits neutrophil (PMN) priming via attenuation of p38 MAPK signaling. Shock. 2000;14:265–269. doi: 10.1097/00024382-200014030-00004. discussion 269–270. [DOI] [PubMed] [Google Scholar]
- 12.Junger WG, Hoyt DB, Davis RE, Herdon-Remelius C, Namiki S, Junger H, Loomis W, Altman A. Hypertonicity regulates the function of human neu-trophils by modulating chemoattractant receptor signaling and activating mitogen-activated protein kinase p38. J Clin Invest. 1998;101:2768–2779. doi: 10.1172/JCI1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orlic T, Loomis WH, Shreve A, Namiki S, Junger WG. Hypertonicity increases cAMP in PMN and blocks oxidative burst by PKA-dependent and -independent mechanisms. Am J Physiol Cell Physiol. 2002;282:C1261–C1269. doi: 10.1152/ajpcell.00479.2001. [DOI] [PubMed] [Google Scholar]
- 14.Cuschieri J, Gourlay D, Garcia I, Jelacic S, Maier RV. Hypertonic preconditioning inhibits macrophage responsiveness to endotoxin. J Immunol. 2002;168:1389–1396. doi: 10.4049/jimmunol.168.3.1389. [DOI] [PubMed] [Google Scholar]
- 15.Shukla A, Hashiguchi N, Chen Y, Coimbra R, Hoyt DB, Junger WG. Osmotic regulation of cell function and possible clinical applications. Shock. 2004;21:391–400. doi: 10.1097/00024382-200405000-00001. [DOI] [PubMed] [Google Scholar]
- 16.Ayala A, Perrin MM, Meldrum DR, Ertel W, Chaudry IH. Hemorrhage induces an increase in serum TNF which is not associated with elevated levels of endotoxin. Cytokine. 1990;2:170–174. doi: 10.1016/1043-4666(90)90012-i. [DOI] [PubMed] [Google Scholar]
- 17.Mukhopadhyay S, Hoidal JR, Mukherjee TK. Role of TNFalpha in pulmonary pathophysiology. Respir Res. 2006;7:125. doi: 10.1186/1465-9921-7-125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Atsuta J, Sterbinsky SA, Plitt J, Schwiebert LM, Bochner BS, Schleimer RP. Phenotyping and cytokine regulation of the BEAS-2B human bronchial epithelial cell: demonstration of inducible expression of the adhesion molecules VCAM-1 and ICAM-1. Am J Respir Cell Mol Biol. 1997;17:571–582. doi: 10.1165/ajrcmb.17.5.2685. [DOI] [PubMed] [Google Scholar]
- 19.Song Y, Ao L, Raeburn CD, Calkins CM, Abraham E, Harken AH, Meng X. A low level of TNF-alpha mediates hemorrhage-induced acute lung injury via p55 TNF receptor. Am J Physiol Lung Cell Mol Physiol. 2001;281:L677–L684. doi: 10.1152/ajplung.2001.281.3.L677. [DOI] [PubMed] [Google Scholar]
- 20.Thorley AJ, Ford PA, Giembycz MA, Goldstraw P, Young A, Tetley TD. Differential regulation of cytokine release and leukocyte migration by lipopolysaccharide-stimulated primary human lung alveolar type II epithelial cells and macrophages. J Immunol. 2007;178:463–473. doi: 10.4049/jimmunol.178.1.463. [DOI] [PubMed] [Google Scholar]
- 21.Hacker H, Karin M. Regulation and function of IKK and IKK-related kinases. Sci STKE. 2006;2006:re13. doi: 10.1126/stke.3572006re13. [DOI] [PubMed] [Google Scholar]
- 22.Ciesla DJ, Moore EE, Biffl WL, Gonzalez RJ, Silliman CC. Hypertonic saline attenuation of the neutrophil cytotoxic response is reversed upon restoration of normotonicity and reestablished by repeated hypertonic challenge. Surgery. 2001;129:567–575. doi: 10.1067/msy.2001.113286. [DOI] [PubMed] [Google Scholar]
- 23.Fehrenbach A, Pufe T, Wittwer T, Nagib R, Dreyer N, Pech T, Petersen W, Fehrenbach H, Wahlers T, Richter J. Reduced vascular endothelial growth factor correlates with alveolar epithelial damage after experimental ischemia and reperfusion. J Heart Lung Transplant. 2003;22:967–978. doi: 10.1016/s1053-2498(02)01157-9. [DOI] [PubMed] [Google Scholar]
- 24.Mura M, Han B, Andrade CF, Seth R, Hwang D, Waddell TK, Keshavjee S, Liu M. The early responses of VEGF and its receptors during acute lung injury: implication of VEGF in alveolar epithelial cell survival. Crit Care. 2006;10:R130. doi: 10.1186/cc5042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ware LB, Matthay MA. The acute respiratory distress syndrome. N Engl J Med. 2000;342:1334–1349. doi: 10.1056/NEJM200005043421806. [DOI] [PubMed] [Google Scholar]
- 26.Islam A, Adamik B, Hawari F I, Ma G, Rouhani FN, Zhang J, Levine SJ. Extracellular TNFR1 release requires the calcium-dependent formation of a nucleobindin 2-ARTS-1 complex. J Biol Chem. 2006;281:6860–6873. doi: 10.1074/jbc.M509397200. [DOI] [PubMed] [Google Scholar]
- 27.Sheikh-Hamad D, Gustin MC. MAP kinases and the adaptive response to hypertonicity: functional preservation from yeast to mammals. Am J Physiol Renal Physiol. 2004;287:F1102–F1110. doi: 10.1152/ajprenal.00225.2004. [DOI] [PubMed] [Google Scholar]
- 28.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 29.Holden NS, Catley MC, Cambridge LM, Barnes PJ, Newton R. ICAM-1 expression is highly NF-kappaB–dependent in A549 cells. No role for ERK and p38 MAPK. Eur J Biochem. 2004;271:785–791. doi: 10.1111/j.1432-1033.2004.03982.x. [DOI] [PubMed] [Google Scholar]
- 30.Skerrett SJ, Liggitt HD, Hajjar AM, Ernst RK, Miller SI, Wilson CB. Respiratory epithelial cells regulate lung inflammation in response to inhaled endotoxin. Am J Physiol Lung Cell Mol Physiol. 2004;287:L143–L152. doi: 10.1152/ajplung.00030.2004. [DOI] [PubMed] [Google Scholar]
- 31.Sadikot RT, Han W, Everhart MB, Zoia O, Peebles RS, Jansen ED, Yull FE, Christman JW, Blackwell TS. Selective I kappa B kinase expression in airway epithelium generates neutrophilic lung inflammation. J Immunol. 2003;170:1091–1098. doi: 10.4049/jimmunol.170.2.1091. [DOI] [PubMed] [Google Scholar]
- 32.Poynter ME, Irvin CG, Janssen-Heininger YM. A prominent role for airway epithelial NF-kappa B activation in lipopolysaccharide-induced airway inflammation. J Immunol. 2003;170:6257–6265. doi: 10.4049/jimmunol.170.12.6257. [DOI] [PubMed] [Google Scholar]
- 33.Staudenmayer KL, Maier RV, Jelacic S, Bulger EM. Hypertonic saline modulates innate immunity in a model of systemic inflammation. Shock. 2005;23:459–463. doi: 10.1097/01.shk.0000160523.37106.33. [DOI] [PubMed] [Google Scholar]
- 34.Simon RH, DeHart PD, Todd RF., 3rd Neutrophil-induced injury of rat pulmonary alveolar epithelial cells. J Clin Invest. 1986;78:1375–1386. doi: 10.1172/JCI112724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beck-Schimmer B, Madjdpour C, Kneller S, Ziegler U, Pasch T, Wuthrich RP, Ward PA, Schimmer RC. Role of alveolar epithelial ICAM-1 in lipopolysaccharide-induced lung inflammation. Eur Respir J. 2002;19:1142–1150. doi: 10.1183/09031936.02.00236602. [DOI] [PubMed] [Google Scholar]
- 36.Beck-Schimmer B, Schimmer RC, Schmal H, Flory CM, Friedl HP, Pasch T, Ward P. Characterization of rat lung ICAM-1. Inflamm Res. 1998;47:308–315. doi: 10.1007/s000110050334. [DOI] [PubMed] [Google Scholar]
- 37.Schmal H, Czermak BJ, Lentsch AB, Bless NM, Beck-Schimmer B, Friedl HP, Ward PA. Soluble ICAM-1 activates lung macrophages and enhances lung injury. J Immunol. 1998;161:3685–3693. [PubMed] [Google Scholar]
- 38.Powers KA, Woo J, Khadaroo RG, Papia G, Kapus A, Rotstein OD. Hypertonic resuscitation of hemorrhagic shock upregulates the anti-inflammatory response by alveolar macrophages. Surgery. 2003;134:312–318. doi: 10.1067/msy.2003.246. [DOI] [PubMed] [Google Scholar]
- 39.Donaldson SH, Bennett WD, Zeman KL, Knowles MR, Tarran R, Boucher RC. Mucus clearance and lung function in cystic fibrosis with hypertonic saline. N Engl J Med. 2006;354:241–250. doi: 10.1056/NEJMoa043891. [DOI] [PubMed] [Google Scholar]
- 40.Elkins MR, Robinson M, Rose BR, Harbour C, Moriarty CP, Marks GB, Belousova EG, Xuan W, Bye PT. A controlled trial of long-term inhaled hypertonic saline in patients with cystic fibrosis. N Engl J Med. 2006;354:229–240. doi: 10.1056/NEJMoa043900. [DOI] [PubMed] [Google Scholar]