

Abstract
Increasing evidence suggests that cytokines and chemokines play crucial roles in chronic itch. In the present study, we evaluated the roles of tumor necrosis factor-alpha (TNF-α) and its receptors TNF receptor subtype-1 (TNFR1) and TNFR2 in acute and chronic itch in mice. Compared to wild-type (WT) mice, TNFR1-knockout (TNFR1-KO) and TNFR1/R2 double-KO (DKO), but not TNFR2-KO mice, exhibited reduced acute itch induced by compound 48/80 and chloroquine (CQ). Application of the TNF-synthesis inhibitor thalidomide and the TNF-α antagonist etanercept dose-dependently suppressed acute itch. Intradermal injection of TNF-α was not sufficient to evoke scratching, but potentiated itch induced by compound 48/80, but not CQ. In addition, compound 48/80 induced TNF-α mRNA expression in the skin, while CQ induced its expression in the dorsal root ganglia (DRG) and spinal cord. Furthermore, chronic itch induced by dry skin was reduced by administration of thalidomide and etanercept and in TNFR1/R2 DKO mice. Dry skin induced TNF-α expression in the skin, DRG, and spinal cord and TNFR1 expression only in the spinal cord. Thus, our findings suggest that TNF-α/TNFR1 signaling is required for the full expression of acute and chronic itch via peripheral and central mechanisms, and targeting TNFR1 may be beneficial for chronic itch treatment.
Keywords: Itch, Tumor necrosis factor, Tumor necrosis factor receptor, Spinal cord, Central sensitization
Introduction
Itch (pruritus) is an unpleasant sensation that elicits the desire or reflex to scratch [1, 2]. Acute itch protects against potentially harmful irritations, while chronic itch can greatly reduce the quality of life [3]. Chronic itch is often associated with dermatological diseases [4, 5], as well as hepatic [6, 7], metabolic [8], and renal diseases [9, 10]. Because of the great variety of pathophysiological conditions underlying chronic itch, typical allergenic and anti-inflammatory treatments are not always effective. For example, antihistamines are prescribed for allergic itch, but other chronic itch conditions are resistant to this treatment. Thus, histamine-independent (non-histaminergic) mechanisms are critically involved in the pathogenesis of chronic itch. However, the molecular mechanisms underlying non-histaminergic itch remain unclear.
Recent studies have shown that itch shares many neural mediators and signaling pathways with pain [11–13]. Accordingly, many of the receptor systems known to modulate pain have also been examined for their effects on itch [13, 14]. It is well documented that cytokines and chemokines play key roles in driving neuroinflammation and the development of chronic pain [15–19]. Emerging evidence suggests that cytokines and chemokines also serve as key itch mediators [20]. For example, cytokines such as interleukin-31 (IL-31) [21, 22] and thymic stromal lymphopoietin [5] contribute to atopic dermatitis-induced itch via direct activation of primary sensory neurons in dorsal root ganglia (DRG) through transient receptor potential cation channel subfamily V member 1 (TRPV1) and/or the TRP cation channel member A1 (TRPA1). In addition, up-regulation of C-X-C motif chemokine 10 mediates allergic itch via the activation of C-X-C motif chemokine receptor 3 on a subset of DRG neurons in mice [23]. Although other cytokines such as IL-2, IL-4, IL-6, IL-8, and IL-13 have also been implicated in chronic itch [20, 24], direct evidence is currently limited.
Tumor necrosis factor alpha (TNF-α) is one of the best known pro-inflammatory cytokines and is produced by mast cells, macrophages, fibroblasts, endothelial cells, Schwann cells, microglia, and astrocytes [25, 26]. It is known to act on two distinct receptors, TNF receptor 1 (TNFR1) and TNFR2 [27]. It has been documented that TNF-α plays a key role in the pathogenesis of chronic pain, including that due to inflammation [28, 29], neuropathy [26, 30], and cancer [31, 32]. Peripheral administration of TNF-α can excite or sensitize primary afferent fibers to mechanical and heat stimulation in mice, possibly due to the up-regulation of TRPV1 [33, 34] and/or p38-dependent modulation of tetrodotoxin-resistant Na+ channels in primary afferent neurons [35, 36]. Local application of the TNF-α antagonist etanercept has also been shown to partially block mechanical hyperalgesia in a cancer pain model [31, 32, 37]. Our previous work demonstrated that TNF-α evokes a significant increase in the frequency of spontaneous excitatory postsynaptic currents in spinal lamina II neurons through the activation of both TNFR1 and TNFR2 [28]. TNF-α also increases N-methyl-D-aspartate (NMDA) currents in spinal lamina II neurons by acting on TNFR1 [28]. We also demonstrated that TNFR1 and TNFR2 play distinct roles in regulating different phases of inflammatory pain in mice [28]. In a soft-tissue cancer model, TNFR2-knockout (KO) mice display delayed onset of thermal hyperalgesia [32]. In bone-cancer pain, both TNFR1 and TNFR2 are required for the development of mechanical hyperalgesia in mice [31]. Together, these data suggest that TNF-α plays a central role in regulating synaptic plasticity in the spinal cord and chronic pain via TNFR1 and/or TNFR2. However, the roles of TNF-α in the regulation of itch sensation are unclear. The present study was designed to test the hypothesis that peripheral and spinal TNF-α/TNFR signaling play important roles in the processing of acute and chronic itch in mice.
Materials and Methods
Animals
Male CD1 mice (8–10 weeks) were purchased from the Shanghai SLAC Laboratory Animal Co. (Shanghai, China). C57BL/6 wild-type mice and mice deficient in TNFR1 (TNFR1-KO), TNFR2 (TNFR2-KO), or TNFR1 and 2 (TNFR1/R2 DKO) were obtained from the Jackson Laboratory (Bar Harbor, ME). All animals were maintained under a 12 h light/dark cycle with food and water available ad libitum at standard room temperature (22 ± 2 °C) and humidity (60%–80%). All animal procedures were performed according to the guidelines of the International Association for the Study of Pain and were approved by the Ethics Committee for the Use of Experimental Animals in Soochow University. Experiments were performed in a blinded manner between 09:00 and 16:00 in a sound-attenuated room.
Mouse Model of Acute Itch
As previously reported, mice were shaved at the nape of the neck (15 × 10 mm2) one day before intradermal (i.d.) injection. Mice were placed in small plastic chambers (10 × 10 × 12.5 cm3) on an elevated metal mesh floor and allowed 30 min for habituation before behavioral testing. Compound 48/80 (100 μg; Sigma-Aldich, St. Louis, MO) or chloroquine (CQ; 200 μg; Sigma) was injected i.d. in the nape at a volume of 50 μL under brief anesthesia with isoflurane. Immediately after the injection, mice were returned to their chambers and recorded for 30 min (Sony HDR-CX610, Shanghai, China). The video was re-played offline and scratching behavior was quantified in a blinded manner. As we postulated that multiple injections of thalidomide would have a cumulative inhibitory effect on TNF synthesis, thalidomide (50 mg/kg, i.p.; Sigma) was injected twice (16 h and 30 min before compound 48/80 and chloroquine injection). Intrathecal (i.t.) injection of etanercept (Enbrel®; 0.1, 1, and 10 μg; Pfizer, New York, NY) or i.d. injection of etanercept (1 and 10 μg) was performed 20 min before injection of compound 48/80 and CQ. Thalidomide was dissolved in 10% dimethylsulfoxide and etanercept in sterile saline.
Mouse Model of Chronic Itch Induced by Dry Skin
As previously described, the nape was shaved with electric clippers and depilatory paste 3 days prior to treatment. Dry skin was induced under isoflurane anesthesia by treatment with a 1:1 mixture of acetone and diethyl ether for 15 s followed by clean water for 10 s (AEW), twice a day (10:00 and 17:00) for 5 days. Control animals were treated with water only. The drug-treatment group also received thalidomide (50 mg/kg; i.p.) or etanercept (10 μg; i.t., 10 μg; i.d., or 1 mg/kg; i.p.) on day 6 before testing scratching behavior. Spontaneous scratching behavior was video-recorded for 1 h and quantified in a blinded manner.
Alloknesis Assay
Alloknesis after acute itch and under chronic itch conditions was evaluated as in a previous report [43]. Briefly, 30 min after injection of compound 48/80, a von Frey filament (0.7 mN) was applied to the affected skin. The number of scratch bouts directed to the site after mechanical stimulation was counted. The alloknesis (touch-evoked itch) score was determined by calculating the total number of scratches elicited by ten mechanical stimuli. In the dry-skin model, the von Frey filament was applied at the border of the treatment area after AEW treatment for 5 days to elicit scratching and the alloknesis score was calculated.
Real-Time Quantitative RT-PCR Analysis
We collected the treated skin, C3–C4 DRGs, and C3–C4 spinal cord 30 min after compound 48/80 or CQ injection, or after 1, 3, or 5 days of AEW treatment. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s guidelines. One microgram of total RNA was reverse-transcribed for each sample using a RevertAid First Strand cDNA Synthesis Kit according to the manufacturer’s protocol (Thermo Fisher Scientific, Waltham, MA). Real-time PCR analyses were performed using the SYBR Green master mix (Roche, Basle, Switzerland) and an Opticon real-time PCR detection system (ABI 7500, Life Technologies, Thermo Fisher Scientific, Darmstadt, Germany). Specific fragments were amplified with the following primers: TNFα: 5′- GTTCTCTTCAAGGGACAAGGCTG-3′; 5′-TCCTGGTATGAGATAGCAAATCGG-3′; TNFR1: 5′-TGAGTGCGTCCCTTGCAGCC-3′; 5′-AACC AGGGGCAACAGCACCG-3′; TNFR2: 5′-GTCATGGCGGAGGCCCAAGG-3′; 5′-GCGCTGGCTTGGGAAGAGCA-3′; CX3CR1: 5′-CCAGAGCCGTCAGACTCATC-3′; 5′-CTGTCTCCGTCACACTGAGG-3′; GAPDH: 5′-GAAGGTCGGTGTGAACGGAT-3′; 5′-AATCTCCACTTTGCCACTGC-3′. The targeted gene expression level was normalized to GAPDH.
Western Blotting
After AEW treatment for 5 days, on day 6, mice were terminally anesthetized with isoflurane and transcardially perfused with PBS and the treated skin, C3–C4 DRGs, and C3–C4 spinal cord were rapidly removed and homogenized in lysis buffer containing a cocktail of phosphatase inhibitors and protease inhibitors for total protein extraction assays as in our previous report [43]. The protein concentrations were measured with the Pierce BCA Protein Assay (Thermo), and equal amounts of protein (25 μg) were loaded onto each lane and separated in 10% SDS-PAGE. After transfer, the blots were blocked with 5% non-fat milk in Tris-buffered saline at room temperature for 1 h and PVDF membranes were incubated overnight at 4 °C with primary polyclonal anti-TNF-α (rabbit, 1:1000; Abcam, Cambridge, MA). For loading control, the blots were probed with α-tubulin antibody (mouse, 1:1000; Vazyme, Nanjing, China). The blots were washed and incubated with horseradish peroxidase-conjugated goat anti-rabbit or goat anti-mouse IgG secondary antibody (1:2000; Vazyme). Protein bands were visualized using an enhanced chemiluminescence detection kit (Pierce) and the band densities were analyzed using the Molecular Imager ChemiDoc XRS + System (Bio-Rad). Data from 4–5 mice were used for statistical analysis.
Statistical Analysis
All data are expressed as mean ± SEM. Statistical analyses were carried out using Graphpad Prism version 6.02 (Graphpad software, La Jolla, CA). Unpaired Student’s t-test was used for two-group comparison. One-way ANOVA with the Bonferroni post-hoc test was used for multiple comparisons. Two-way repeated-measured ANOVA was also used to analyze data with multiple time points. P < 0.05 was considered statistically significant.
Results
TNFR1, but not TNFR2, is Required for Both Histaminergic and Non-histaminergic Itch in Mice
First, we used the TNFR1-KO, TNFR2-KO, and TNFR1/R2-DKO mice to explore the potential role of TNF-α/TNFR signaling in acute itch, which is traditionally divided into histaminergic and non-histaminergic itch in humans and rodents [1, 38]. Consistent with previous reports [39, 40], i.d. injection of compound 48/80, which induces mast cell degranulation and histamine release [41, 42], induced histaminergic itch (Fig. 1). The compound 48/80-induced scratching was significantly reduced in TNFR1-KO and TNFR1/R2 DKO mice (P < 0.05; Figure 1A, B), but not in TNFR2-KO mice. CQ is an anti-malarial drug that induces non-histaminergic itch via activation of Mas-related G protein-coupled receptor A3 (MrgprA3) and TRPA1 in primary sensory neurons in mice [38, 49]. Similarly, CQ-induced scratching was also lower in TNFR1-KO and TNFR1/R2 DKO mice (P < 0.05; Fig. 1C, D), but not in TNFR2-KO mice. Thus, these results suggested that TNFR1 is required for both histaminergic and non-histaminergic itch in mice.
Fig. 1.

TNFR1, but not TNFR2, is required for both histaminergic and non-histaminergic itch in mice. A Time course of acute histaminergic itch induced by compound 48/80 (48/80) in wild-type (WT), TNFR1-KO, TNFR2-KO, and TNFR1/R2 DKO mice. B Total scratching bouts within 30 min induced by 48/80 in all four groups of mice. C Time course of acute non-histaminergic itch induced by chloroquine (CQ) in WT, TNFR1-KO, TNFR2-KO, and TNFR1/R2 DKO mice. D Total scratching bouts within 30 min induced by CQ in all four groups of mice. Data are presented as mean ± SEM ( P < 0.05 vs WT group one-way ANOVA followed by Bonferroni post hoc test; n = 5–7 mice per group).
P < 0.05 vs WT group one-way ANOVA followed by Bonferroni post hoc test; n = 5–7 mice per group).
Thalidomide and Etanercept Inhibit both Histaminergic and Non-histaminergic Itch in Mice
We subsequently investigated whether inhibition of TNF-α synthesis and anti-TNF-α treatment reduce acute itch in mice. Systemic administration of thalidomide dose-dependently suppressed the compound 48/80-induced histaminergic itch (P < 0.05; Fig. 2A, B). Similarly, i.t. (1 and 10 μg) and i.d. (10 μg) administration of etanercept both significantly attenuated the compound 48/80-induced itch (both P < 0.05; Fig. 2C–F). Meanwhile, systemic administration of thalidomide dose-dependently suppressed the CQ-induced non-histaminergic itch (P < 0.05; Fig. 3A, B). And i.t. injection of etanercept (10 μg) attenuated CQ-induced itch (P < 0.05; Fig. 3C, D). In contrast, i.d. administration of etanercept (1 and 10 μg) had little effect on CQ-induced non-histaminergic itch (P > 0.05; Fig. 3E, F). Together, these results suggested that inhibition of TNF-α synthesis and treatment with a TNF-α antagonist reduce both histaminergic and non-histaminergic itch in mice in a dose-dependent manner, although the effect of etanercept on CQ-induced non-histaminergic itch was limited.
Fig. 2.

Inhibitory effects of thalidomide and etanercept on acute histaminergic itch in mice. A–B Time course (A) and total scratching bouts within 30 min (B) of compound 48/80-induced itch in vehicle- and thalidomide-treated mice. C–D Time course (C) and total scratching bouts within 30 min (D) of compound 48/80-induced itch after i.t. injection of vehicle or etanercept. E–F Time course (E) and total scratching bouts within 30 min (F) of compound 48/80-induced itch after i.d. injection of vehicle or etanercept. Data are presented as mean ± SEM (
P < 0.05 vs vehicle group, one-way ANOVA followed by Bonferroni post hoc test; n = 6–8 mice per group).
Fig. 3.

Inhibitory effects of thalidomide or etanercept on acute non-histaminergic itch in mice. A–B Time course (A) and total scratching bouts within 30 min (B) of CQ-induced itch in vehicle- and thalidomide-treated mice. C–D Time course (C) and total scratching bouts within 30 min (D) of CQ-induced itch after i.t. injection of vehicle or etanercept. E–F Time course (E) and total scratching bouts within 30 min (F) of CQ-induced itch after i.d. injection of vehicle or etanercept. Data are presented as mean ± SEM (
P < 0.05 vs vehicle group, one-way ANOVA followed by Bonferroni post hoc test; n = 6–8 mice per group).
Peripheral and Central TNF-α Play Distinct Roles in Acute Histaminergic and Non-Histaminergic Itch in Mice
The i.d. injection of TNF-α was not sufficient to evoke scratching (P > 0.05; Fig. 4A). However, i.d. injection of TNF-α potentiated the compound 48/80-induced scratching (P < 0.05; Fig. 4B), but had no effect on CQ-induced acute itch (P > 0.05; Fig. 4B). We further examined the expression changes of TNF-α after application of compound 48/80 or CQ using q-PCR. The results showed that compound 48/80 up-regulated TNF-α expression in the skin (P < 0.05; Fig. 4C), but not in the DRG and spinal cord. In contrast, CQ up-regulated TNF-α expression in the DRG and spinal cord (P < 0.05; Fig. 4D), but not in the skin. Thus, TNF-α in the skin is mainly involved in compound 48/80-induced acute itch, while TNF-α in the DRG and spinal cord is involved in both compound 48/80- and CQ-induced acute itch in mice.
Fig. 4.

Peripheral and central TNF-α play distinct roles in compound 48/80- and CQ-induced acute itch in mice. A I.d. injection of TNF-α (0.2–20 ng) was not able to elicit scratching (P > 0.05; one-way ANOVA followed by Bonferroni post hoc test; n = 6-8 mice per group). B I.d. injection of TNF-α (20 ng) enhanced compound 48/80- but not CQ-induced scratching (*P < 0.05 vs saline control group; Student’s t test; n = 6-8 mice per group). C Quantitative real-time PCR (q-PCR) analysis showed compound 48/80-induced up-regulation of TNF-α expression in the skin, but not in the DRG or spinal cord. D Q-PCR analysis showed CQ-induced up-regulation of TNF-α expression in the DRG and spinal cord, but not in the skin. Data are presented as mean ± SEM (
P < 0.05, 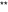 P < 0.01 vs saline control group; Student’s t test; n = 3–4 mice per group).
P < 0.01 vs saline control group; Student’s t test; n = 3–4 mice per group).
TNF-α/TNFR Signaling is Required for Dry Skin-Induced Chronic Itch and Alloknesis in Mice
To further investigate the role of TNF-α/TNFR in chronic itch, we subsequently determined the effects of thalidomide or etanercept on dry skin-induced chronic itch [43]. We found that AEW-treated mice developed spontaneous scratching behavior (Fig. 5A) and alloknesis (Fig. 5B). I.t. injection of TNF-α was not able to evoke scratching in control and AEW-treated mice (Fig. 5C), suggesting that TNF-α in the spinal cord is not sufficient to induce itch. Systemic injection of thalidomide (50 mg/kg; i.p.) suppressed the AEW-induced chronic itch (P < 0.05; Fig. 5D). I.p. (1 mg/kg) or i.t. (10 μg) injection of etanercept suppressed AEW-induced chronic itch (P < 0.05; Fig. 5E, F), but i.d. injection of etanercept (10 μg) did not have this effect (P > 0.05; Fig. 5G). The AEW-induced chronic itch was also lower in TNFR1/R2 DKO mice (P < 0.05; Fig. 5H). In addition, thalidomide (50 mg/kg; i.p.) and etanercept (10 μg; i.t.) both suppressed the AEW-induced alloknesis (P < 0.05; Fig. 5I, J). Finally, the compound 48/80-induced alloknesis was suppressed by i.t. injection of etanercept (10 μg; P < 0.05; Fig. 5K). Thus, the results indicated that TNF-α/TNFR signaling is required for the full development of dry skin-induced chronic itch and alloknesis in mice.
Fig. 5.

TNF-α contributes to dry skin-induced chronic itch and alloknesis in mice. A–B Time courses of spontaneous scratching (A) and alloknesis (B) induced by AEW treatment for 5 days. C I.t. injection of TNF-α was not sufficient to evoke scratching in control (Ctrl) and AEW-treated mice. D Systemic administration of thalidomide (50 mg/kg; i.p.) suppressed spontaneous itch induced by AEW treatment. E–F I.p. injection of etanercept (E; 1 mg/kg) and i.t. injection of etanercept (F; 10 μg) suppressed spontaneous itch induced by AEW treatment. G I.d. injection of etanercept (10 μg) did not suppress spontaneous itch induced by AEW treatment. H Spontaneous itch induced by AEW treatment was significantly lower in TNFR1/R2 DKO mice. I I.p. injection of thalidomide (50 mg/kg) suppressed AEW treatment-induced alloknesis. J I.t. injection of etanercept (10 μg) suppressed AEW treatment-induced alloknesis. K I.t. injection of etanercept (10 μg) suppressed compound 48/80-induced alloknesis. Data are presented as mean ± SEM (
P < 0.05, 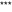 P < 0.001 vs corresponding group, Student’s t test; n = 6–8 mice per group).
P < 0.001 vs corresponding group, Student’s t test; n = 6–8 mice per group).
Effects of AEW-Induced Dry Skin on the Expression of TNF-α and TNFRs in the Skin, DRG, and Spinal Cord in Mice
To further explore the possible regulation of TNF-α/TNFR signaling under chronic itch conditions, we determined the expression changes of TNF-α, TNFR1, and TNFR2 in the skin, DRG, and spinal cord after AEW treatment in mice using Western blotting and q-PCR analysis. Western blotting data showed dry skin-induced up-regulation of TNF-α expression in the skin, DRG, and spinal cord (P < 0.05; Fig. 6A, B). Q-PCR analysis also confirmed the increased mRNA expression of TNF-α expression in the skin, DRG, and spinal cord (P < 0.05; Fig. 6C). We further examined the time-course of TNF-α expression in the spinal cord after AEW treatment and found that its expression started to increase after 1 day of AEW treatment and persisted for at least 5 days (Fig. 6D). We also investigated whether AEW treatment induced the activation of microglia, a major source of TNF-α in the central nervous system. The results showed that CX3C chemokine receptor 1 (CX3CR1), a molecular marker of microglia in the central nervous system, was transiently up-regulated after 1 day of AEW treatment (P < 0.05; Fig. 6E). Finally, we found that the expression of TNFR1 was up-regulated in the spinal cord, but not in the skin and DRG, following AEW treatment (Fig. 6F). In contrast, the expression of TNFR2 in the skin, DRG, or spinal cord was not affected by AEW treatment (Fig. 6F). Thus, up-regulation of TNF-α/TNFR1 signaling may contribute to the development of chronic itch.
Fig. 6.

Dry skin induces up-regulation of TNF-α/TNFR1 signaling in mice. A–B Western blots (A) and analysis (B) showing that AEW induced up-regulation of TNF-α at the protein level in the skin, DRG, and spinal cord. C Q-PCR analysis confirmed the AEW-induced up-regulation of TNF-α expression at the mRNA level in the skin, DRG, and spinal cord. D–E Time course of the up-regulation of TNF-α mRNA (D) and CX3CR1 mRNA (E) in the spinal cord induced by AEW treatment. F Q-PCR analysis showing that the mRNA expression of TNFR1, but not TNFR2, was up-regulated in the spinal cord, but not in the skin and DRG. We collected all tissues for Western blotting or q-PCR after AEW treatment for 5 days (
P < 0.05,
P < 0.01,
P < 0.001 vs corresponding control group; Student’s t test; n = 3–4 mice per group).
Discussion
Itch is a major somatic sensation along with touch, pain, and temperature sensations [1, 44]. Acute itch serves as a self-protective mechanism [45], while chronic itch represents a clinical problem, commonly accompanying skin, systemic, and metabolic disorders. Although antihistamines are often used to treat allergic itch [46], they are often ineffective for most kinds of chronic itch, suggesting histamine-independent mechanisms are involved. Histamine, mainly released from mast cells [47] and keratinocytes [48], binds H1 and H4 receptors to activate phospholipase C beta 3 and TRPV1 on free nerve terminals in skin to elicit histaminergic itch. In contrast, multiple mechanisms underlie non-histaminergic itch. Many efforts have been made to reveal the molecular and cellular bases of this type of itch. MrgprA3 and MrgprC11 coupling with TRPA1 have been demonstrated to mediate non-histaminergic itch induced by CQ and the endogenous neuropeptide BAM8-22, respectively [38, 49, 50]. Our recent studies revealed that the activation of Toll-like receptors (TLRs), such as TLR7 and TLR3, induces non-histaminergic itch, possibly via direct coupling to TRPA1 in primary sensory neurons in mice [44]. In addition, oxidative stress induces non-histaminergic itch through direct activation of TRPA1, and antioxidants strongly attenuate acute and chronic itch in mice [40, 51]. Recent studies have also provided new insights into the ionic mechanisms of acute itch, such as TRPV4 regulation of histamine- and serotonin-induced itch [52, 53], Cav3.2 regulation of H2S-induced itch [54], and acid-sensing ion channel 3 regulation of CQ-induced itch [55]. Increasing evidence supports a critical role of cytokines and chemokines in the pathogenesis of chronic itch [20]. Unfortunately, there is no report on the role of TNF-α, one of the best known pro-inflammatory cytokines, in acute and chronic itch.
In the present study, we aimed to investigate the role of TNF-α/TNFR1 signaling in acute and chronic itch. For acute itch, we used the mouse histaminergic itch model induced by compound 48/80 [56], and the non-histaminergic itch model induced by CQ [49]. We first demonstrated that TNFR1, but not TNFR2, is required for acute histaminergic and non-histaminergic itch using KO mice. The TNF-synthesis inhibitor thalidomide and the TNF-α antagonist etanercept dose-dependently suppressed acute itch. In addition, compound 48/80 induced TNF-α mRNA expression in the skin, while CQ induced its expression in the DRG and spinal cord. Thalidomide and etanercept also attenuated dry skin-induced chronic itch and alloknesis. Dry skin induced the up-regulation of TNF-α expression in the skin, DRG, and spinal cord and TNFR1 only in the spinal cord. Thus, our findings suggested that TNF-α/TNFR1 signaling is required for the full expression of acute and chronic itch, possibly via distinct peripheral and central mechanisms.
TNF-α is produced by different cell types in the skin, such as mast cells, macrophages, and fibroblasts [25, 26]. As TNFR1 is expressed by DRG neurons [57], we hypothesized that TNF released by peripheral immune cells or skin-resident cells sensitizes primary afferent nerve fibers to exogenous or endogenous pruritogens. We found that i.t. application of the TNF-α antagonist etanercept attenuated compound 48/80- and CQ-induced acute itch. In contrast, i.d. injection of etanercept only suppressed the compound 48/80- but not CQ-induced acute itch. Accordingly, TNF-α potentiated compound 48/80- but not CQ-induced acute itch, although i.d. injection of TNF-α was not sufficient to induce scratching. Thus, these data suggest that TNF-α in the skin is predominantly involved in compound 48/80-induced itch, while TNF-α in the spinal cord contributes to both compound 48/80- and CQ-induced acute itch in mice. Furthermore, q-PCR analysis showed that compound 48/80 induced up-regulation of TNF-α in the skin, while CQ-induced up-regulation of TNF-α in the DRG and spinal cord. We postulated that the acute inhibitory effects of thalidomide and etanercept on acute itch may be attributed to their inhibition of the synthesis or release of TNF, and the up-regulation of TNF mRNA may be involved in alloknesis, which developed later.
What is the role of TNF-α/TNFR signaling in chronic itch? We found that thalidomide or etanercept significantly attenuated dry skin-induced chronic itch and alloknesis in mice. In addition, these phenomena were lower in TNFR1/R2 DKO mice. Finally, we demonstrated that TNF-α was up-regulated in the skin, DRG, and spinal cord and TNFR1 was up-regulated in the spinal cord under dry skin-induced chronic itch conditions. Recent studies have emphasized that microglia play an important role in the development of chronic itch in mice. For example, previous work showed that the acute scratching induced by pruritogens is accompanied by activation of microglial cells in the spinal cord [58]. Microglia are also activated in the spinal cord under chronic itch condition, such as atopic dermatitis [59, 60]. Given that microglia are considered to be the main source of TNF-α in the central nervous system [61], we further showed that CX3CR1 (a microglial marker) was transiently up-regulated in the spinal cord of dry-skin mice, suggesting that microglia are transiently activated in these mice. It has been clearly demonstrated that glutamate and natriuretic precursor peptide B (NppB) released from primary pruriceptive neurons activate secondary spinal cord natriuretic peptide receptor A (NPRA)-positive neurons, which use gastrin-releasing peptide (GRP) to activate GRP receptor (GRPR)-expressing excitatory interneurons in the superficial laminae of the dorsal horn to transmit itch signals [42, 62, 63]. Our previous study showed that TNF-α increases the NMDA currents in spinal lamina II neurons, and this increase is abolished in TNFR1-KO mice, but retained in TNFR2-KO mice [28]. This result provided an important clue that the facilitating effects of TNF-α on glutamate neurotransmission may participate in the central sensitization for chronic itch. However, the effects of TNF-α on NppB/NPRA or GRP/GRPR signaling in the spinal cord remain unclear. Together, the results suggest that TNF-α, possibly derived from microglia, contributes to the development of chronic itch, perhaps by the activation of TNFR1 in the spinal cord.
In summary, we have demonstrated a critical role of TNF-α/TNFR1 signaling in acute and chronic itch by distinct peripheral and central mechanisms. Given that the TNF antagonist etanercept is already available for treating inflammation-related diseases, such as arthritis, anti-TNF-α therapy may be a new option for the treatment of devastating chronic itch.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31371179 and 81300968), and from the Natural Science Foundation of Jiangsu Province, China (BK20140372), the Scientific Funding from Jiangsu Province, China (2015-JY-029), the Second Affiliated Hospital of Soochow University Preponderant Clinic Discipline Group Project Funding (XKQ2015007) and a Project funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Jiangsu Province, China.
Footnotes
Xiuhua Miao and Ya Huang have contributed equally to this work.
References
- 1.Green D, Dong X. The cell biology of acute itch. J Cell Biol. 2016;213:155–161. doi: 10.1083/jcb.201603042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.LaMotte RH, Dong X, Ringkamp M. Sensory neurons and circuits mediating itch. Nat Rev Neurosci. 2014;15:19–31. doi: 10.1038/nrn3641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yosipovitch G, Bernhard JD. Clinical practice. Chronic pruritus. N Engl J Med. 2013;368:1625–1634. doi: 10.1056/NEJMcp1208814. [DOI] [PubMed] [Google Scholar]
- 4.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–1494. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 5.Wilson SR, The L, Batia LM, Beattie K, Katibah GE, McClain SP, et al. The epithelial cell-derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell. 2013;155:285–295. doi: 10.1016/j.cell.2013.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kremer AE, Oude Elferink RP, Beuers U. Pathophysiology and current management of pruritus in liver disease. Gastroenterol Clin Biol. 2011;35:89–97. doi: 10.1016/j.clinre.2010.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Kremer AE, Martens JJ, Kulik W, Rueff F, Kuiper EM, van Buuren HR, et al. Lysophosphatidic acid is a potential mediator of cholestatic pruritus. Gastroenterology. 1018;2010(139):1008–1018. doi: 10.1053/j.gastro.2010.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Yamaoka H, Sasaki H, Yamasaki H, Ogawa K, Ohta T, Furuta H, et al. Truncal pruritus of unknown origin may be a symptom of diabetic polyneuropathy. Diabetes Care. 2010;33:150–155. doi: 10.2337/dc09-0632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mettang T, Kremer AE. Uremic pruritus. Kidney Int. 2015;87:685–691. doi: 10.1038/ki.2013.454. [DOI] [PubMed] [Google Scholar]
- 10.Berger TG, Steinhoff M. Pruritus and renal failure. Semin Cutan Med Surg. 2011;30:99–100. doi: 10.1016/j.sder.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhand A, Aminoff MJ. The neurology of itch. Brain. 2014;137:313–322. doi: 10.1093/brain/awt158. [DOI] [PubMed] [Google Scholar]
- 12.Stander S, Schmelz M. Chronic itch and pain–similarities and differences. Eur J Pain. 2006;10:473–478. doi: 10.1016/j.ejpain.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 13.Liu T, Ji RR. New insights into the mechanisms of itch: are pain and itch controlled by distinct mechanisms? Pflugers Arch. 2013;465:1671–1685. doi: 10.1007/s00424-013-1284-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luo J, Feng J, Liu S, Walters ET, Hu H. Molecular and cellular mechanisms that initiate pain and itch. Cell Mol Life Sci. 2015;72:3201–3223. doi: 10.1007/s00018-015-1904-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ji RR, Xu ZZ, Gao YJ. Emerging targets in neuroinflammation-driven chronic pain. Nat Rev Drug Discov. 2014;13:533–548. doi: 10.1038/nrd4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. 2013;154:2185–2197. doi: 10.1016/j.pain.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao YJ, Ji RR. Chemokines, neuronal-glial interactions, and central processing of neuropathic pain. Pharmacol Ther. 2010;126:56–68. doi: 10.1016/j.pharmthera.2010.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ji RR, Berta T, Nedergaard M. Glia and pain: is chronic pain a gliopathy? Pain. 2013;154(Suppl 1):S10–S28. doi: 10.1016/j.pain.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354:572–577. doi: 10.1126/science.aaf8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Storan ER, O’Gorman SM, McDonald ID, Steinhoff M. Role of cytokines and chemokines in itch. Handb Exp Pharmacol. 2015;226:163–176. doi: 10.1007/978-3-662-44605-8_9. [DOI] [PubMed] [Google Scholar]
- 21.Cevikbas F, Wang X, Akiyama T, Kempkes C, Savinko T, Antal A, et al. A sensory neuron-expressed IL-31 receptor mediates T helper cell-dependent itch: Involvement of TRPV1 and TRPA1. J Allergy Clin Immunol. 2014;133:448–460. doi: 10.1016/j.jaci.2013.10.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dillon SR, Sprecher C, Hammond A, Bilsborough J, Rosenfeld-Franklin M, Presnell SR, et al. Interleukin 31, a cytokine produced by activated T cells, induces dermatitis in mice. Nat Immunol. 2004;5:752–760. doi: 10.1038/ni1084. [DOI] [PubMed] [Google Scholar]
- 23.Qu L, Fu K, Yang J, Shimada SG, LaMotte RH. CXCR3 chemokine receptor signaling mediates itch in experimental allergic contact dermatitis. Pain. 2015;156:1737–1746. doi: 10.1097/j.pain.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oh MH, Oh SY, Lu J, Lou H, Myers AC, Zhu Z, et al. TRPA1-dependent pruritus in IL-13-induced chronic atopic dermatitis. J Immunol. 2013;191:5371–5382. doi: 10.4049/jimmunol.1300300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McDermott MF. TNF and TNFR biology in health and disease. Cell Mol Biol (Noisy-le-grand) 2001, 47: 619–635. [PubMed]
- 26.Leung L, Cahill CM. TNF-alpha and neuropathic pain–a review. J Neuroinflammation. 2010;7:27. doi: 10.1186/1742-2094-7-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hehlgans T, Mannel DN. The TNF-TNF receptor system. Biol Chem. 2002;383:1581–1585. doi: 10.1515/BC.2002.178. [DOI] [PubMed] [Google Scholar]
- 28.Zhang L, Berta T, Xu ZZ, Liu T, Park JY, Ji RR. TNF-alpha contributes to spinal cord synaptic plasticity and inflammatory pain: distinct role of TNF receptor subtypes 1 and 2. Pain. 2011;152:419–427. doi: 10.1016/j.pain.2010.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Junger H, Sorkin LS. Nociceptive and inflammatory effects of subcutaneous TNFalpha. Pain. 2000;85:145–151. doi: 10.1016/S0304-3959(99)00262-6. [DOI] [PubMed] [Google Scholar]
- 30.Vogel C, Stallforth S, Sommer C. Altered pain behavior and regeneration after nerve injury in TNF receptor deficient mice. J Peripher Nerv Syst. 2006;11:294–303. doi: 10.1111/j.1529-8027.2006.00101.x. [DOI] [PubMed] [Google Scholar]
- 31.Geis C, Graulich M, Wissmann A, Hagenacker T, Thomale J, Sommer C, et al. Evoked pain behavior and spinal glia activation is dependent on tumor necrosis factor receptor 1 and 2 in a mouse model of bone cancer pain. Neuroscience. 2010;169:463–474. doi: 10.1016/j.neuroscience.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 32.Constantin CE, Mair N, Sailer CA, Andratsch M, Xu ZZ, Blumer MJ, et al. Endogenous tumor necrosis factor alpha (TNFalpha) requires TNF receptor type 2 to generate heat hyperalgesia in a mouse cancer model. J Neurosci. 2008;28:5072–5081. doi: 10.1523/JNEUROSCI.4476-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rozas P, Lazcano P, Pina R, Cho A, Terse A, Pertusa M, et al. Targeted overexpression of tumor necrosis factor-alpha increases cyclin-dependent kinase 5 activity and TRPV1-dependent Ca2+ influx in trigeminal neurons. Pain. 2016;157:1346–1362. doi: 10.1097/j.pain.0000000000000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Russell FA, Fernandes ES, Courade JP, Keeble JE, Brain SD. Tumour necrosis factor alpha mediates transient receptor potential vanilloid 1-dependent bilateral thermal hyperalgesia with distinct peripheral roles of interleukin-1beta, protein kinase C and cyclooxygenase-2 signalling. Pain. 2009;142:264–274. doi: 10.1016/j.pain.2009.01.021. [DOI] [PubMed] [Google Scholar]
- 35.Jin X. Gereau RWt. Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J Neurosci. 2006;26:246–255. doi: 10.1523/JNEUROSCI.3858-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gudes S, Barkai O, Caspi Y, Katz B, Lev S, Binshtok AM. The role of slow and persistent TTX-resistant sodium currents in acute tumor necrosis factor-alpha-mediated increase in nociceptors excitability. J Neurophysiol. 2015;113:601–619. doi: 10.1152/jn.00652.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson SR, Gerhold KA, Bifolck-Fisher A, Liu Q, Patel KN, Dong X, et al. TRPA1 is required for histamine-independent, Mas-related G protein-coupled receptor-mediated itch. Nat Neurosci. 2011;14:595–602. doi: 10.1038/nn.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu T, Xu ZZ, Park CK, Berta T, Ji RR. Toll-like receptor 7 mediates pruritus. Nat Neurosci. 2010;13:1460–1462. doi: 10.1038/nn.2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu T, Ji RR. Oxidative stress induces itch via activation of transient receptor potential subtype ankyrin 1 in mice. Neurosci Bull. 2012;28:145–154. doi: 10.1007/s12264-012-1207-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schlosburg JE, Boger DL, Cravatt BF, Lichtman AH. Endocannabinoid modulation of scratching response in an acute allergenic model: a new prospective neural therapeutic target for pruritus. J Pharmacol Exp Ther. 2009;329:314–323. doi: 10.1124/jpet.108.150136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gao Y, Fang X, Sun H, Wang Y, Yao LJ, Li JP, et al. Toll-like receptor 4-mediated myeloid differentiation factor 88-dependent signaling pathway is activated by cerebral ischemia-reperfusion in hippocampal CA1 region in mice. Biol Pharm Bull. 2009;32:1665–1671. doi: 10.1248/bpb.32.1665. [DOI] [PubMed] [Google Scholar]
- 43.Liu T, Han Q, Chen G, Huang Y, Zhao LX, Berta T, et al. Toll-like receptor 4 contributes to chronic itch, alloknesis, and spinal astrocyte activation in male mice. Pain. 2016;157:806–817. doi: 10.1097/j.pain.0000000000000439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu T, Gao YJ, Ji RR. Emerging role of Toll-like receptors in the control of pain and itch. Neurosci Bull. 2012;28:131–144. doi: 10.1007/s12264-012-1219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baral P, Mills K, Pinho-Ribeiro FA, Chiu IM. Pain and itch: beneficial or harmful to antimicrobial defense? Cell Host Microbe. 2016;19:755–759. doi: 10.1016/j.chom.2016.05.010. [DOI] [PubMed] [Google Scholar]
- 46.Shim WS, Oh U. Histamine-induced itch and its relationship with pain. Mol Pain. 2008;4:29. doi: 10.1186/1744-8069-4-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McNeil B, Dong X. Peripheral mechanisms of itch. Neurosci Bull. 2012;28:100–110. doi: 10.1007/s12264-012-1202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shimizu K, Andoh T, Yoshihisa Y, Shimizu T. Histamine released from epidermal keratinocytes plays a role in alpha-melanocyte-stimulating hormone-induced itching in mice. Am J Pathol. 2015;185:3003–3010. doi: 10.1016/j.ajpath.2015.07.015. [DOI] [PubMed] [Google Scholar]
- 49.Liu Q, Tang Z, Surdenikova L, Kim S, Patel KN, Kim A, et al. Sensory neuron-specific GPCR Mrgprs are itch receptors mediating chloroquine-induced pruritus. Cell. 2009;139:1353–1365. doi: 10.1016/j.cell.2009.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sikand P, Dong X, LaMotte RH. BAM8-22 peptide produces itch and nociceptive sensations in humans independent of histamine release. J Neurosci. 2011;31:7563–7567. doi: 10.1523/JNEUROSCI.1192-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou FM, Cheng RX, Wang S, Huang Y, Gao YJ, Zhou Y, et al. Antioxidants attenuate acute and chronic itch: peripheral and central mechanisms of oxidative stress in pruritus. Neurosci Bull. 2016 doi: 10.1007/s12264-016-0076-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Y, Fang Q, Wang Z, Zhang JY, MacLeod AS, Hall RP, et al. Transient receptor potential vanilloid 4 ion channel functions as a pruriceptor in epidermal keratinocytes to evoke histaminergic itch. J Biol Chem. 2016;291:10252–10262. doi: 10.1074/jbc.M116.716464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Akiyama T, Ivanov M, Nagamine M, Davoodi A, Carstens MI, Ikoma A, et al. Involvement of TRPV4 in serotonin-evoked scratching. J Invest Dermatol. 2016;136:154–160. doi: 10.1038/JID.2015.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang XL, Tian B, Huang Y, Peng XY, Chen LH, Li JC, et al. Hydrogen sulfide-induced itch requires activation of Cav3.2 T-type calcium channel in mice. Sci Rep 2015, 5: 16768. [DOI] [PMC free article] [PubMed]
- 55.Lei Z, Sami Shaikh A, Zheng W, Yu X, Yu J, Li J. Non-proton ligand sensing domain of acid sensing ion channel 3 is required for itch sensation. J Neurochem. 2016;139:1093–1101. doi: 10.1111/jnc.13869. [DOI] [PubMed] [Google Scholar]
- 56.Chatterjea D, Paredes L, Martinov T, Balsells E, Allen J, Sykes A, et al. TNF-alpha neutralizing antibody blocks thermal sensitivity induced by compound 48/80-provoked mast cell degranulation. F1000Res 2013, 2: 178. [DOI] [PMC free article] [PubMed]
- 57.Wheeler MA, Heffner DL, Kim S, Espy SM, Spano AJ, Cleland CL, et al. TNF-alpha/TNFR1 signaling is required for the development and function of primary nociceptors. Neuron. 2014;82:587–602. doi: 10.1016/j.neuron.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang Y, Dun SL, Chen YH, Luo JJ, Cowan A, Dun NJ. Scratching activates microglia in the mouse spinal cord. J Neurosci Res. 2015;93:466–474. doi: 10.1002/jnr.23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang Y, Yan J, Hu R, Sun Y, Ma Y, Chen Z, et al. Microglia are involved in pruritus induced by DNFB via the CX3CR1/p38 MAPK pathway. Cell Physiol Biochem. 2015;35:1023–1033. doi: 10.1159/000373929. [DOI] [PubMed] [Google Scholar]
- 60.Torigoe K, Tominaga M, Ko KC, Takahashi N, Matsuda H, Hayashi R, et al. Intrathecal minocycline suppresses itch-related behavior and improves dermatitis in a mouse model of atopic dermatitis. J Invest Dermatol. 2016;136:879–881. doi: 10.1016/j.jid.2015.12.037. [DOI] [PubMed] [Google Scholar]
- 61.Berta T, Park CK, Xu ZZ, Xie RG, Liu T, Lu N, et al. Extracellular caspase-6 drives murine inflammatory pain via microglial TNF-alpha secretion. J Clin Invest. 2014;124:1173–1186. doi: 10.1172/JCI72230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sun YG, Chen ZF. A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature. 2007;448:700–703. doi: 10.1038/nature06029. [DOI] [PubMed] [Google Scholar]
- 63.Mishra SK, Hoon MA. The cells and circuitry for itch responses in mice. Science. 2013;340:968–971. doi: 10.1126/science.1233765. [DOI] [PMC free article] [PubMed] [Google Scholar]