

Abstract
Appearance of alveolar protein-rich edema is an early event in the development of acute respiratory distress syndrome (ARDS). Alveolar edema in ARDS results from a significant increase in the permeability of the alveolar epithelial barrier, and represents one of the main factors that contribute to the hypoxemia in these patients. Damage of the alveolar epithelium is considered a major mechanism responsible for the increased pulmonary permeability, which results in edema fluid containing high concentrations of extravasated macromolecules in the alveoli. The breakdown of the alveolar-epithelial barrier is a consequence of multiple factors that include dysregulated inflammation, intense leukocyte infiltration, activation of pro-coagulant processes, cell death and mechanical stretch. The disruption of tight junction (TJ) complexes at the lateral contact of epithelial cells, the loss of contact between epithelial cells and extracellular matrix (ECM), and relevant changes in the communication between epithelial and immune cells, are deleterious alterations that mediate the disruption of the alveolar epithelial barrier and thereby the formation of lung edema in ARDS.
Keywords: Lung injury, pulmonary edema, alveolar epithelial barrier, mechanisms, tight junctions (TJs)
Introduction
Acute respiratory distress syndrome (ARDS) refers to the development of bilateral pulmonary infiltrates and hypoxemia secondary to intense and diffuse alveolar damage (DAD) (Figure 1). Sepsis, pneumonia, smoke inhalation syndrome, aspiration of gastric contents, major trauma, multiple blood product transfusions or mechanical ventilation with high tidal volume, are among the varied injurious stimuli that can cause ARDS (1). In patients with ARDS, the alveoli present an intense inflammatory response with leukocyte infiltration, activation of pro-coagulant processes, and damage of epithelial and endothelial cells that lead to the breakdown of the alveolar-epithelial barrier and, consequently, to the formation of alveolar protein-rich edema (Figure 2). Such pulmonary edema is a major factor for hypoxemia and one of the earliest events that define ARDS.
Figure 1.

Characteristic radiological and histopathological findings in patients with acute respiratory distress syndrome (ARDS). (A) Chest X-ray shows diffuse and bilateral infiltrates in a patient that fulfills criteria of ARDS; (B) representative lung tissue sections obtained in autopsies from critically-ill patients without ARDS (control group) or in patients with a clinical diagnosis of ARDS showing the anatomopathological diagnosis of diffuse alveolar damage (DAD). Hematoxylin-eosin staining shows DAD characterized by leukocyte infiltrates, increased thickness of the alveolar wall, endothelial cell damage, loss of alveolar epithelial cells with deposition of hyaline membranes on the denudated basement membrane (arrow), flooding of airspaces by protein-rich edema fluid (arrow head), alveolar hemorrhage and vascular congestion and microthrombi. (Original magnification, 40×).
Figure 2.

Increased alveolar permeability to high molecular-weight plasma proteins in acute respiratory distress syndrome (ARDS). Representative lung tissue sections obtained in autopsies from critically-ill patients without ARDS (control group) or in patients with a clinical diagnosis of ARDS showing the anatomopathological diagnosis of diffuse alveolar damage (DAD). The images correspond to merged signals of immunofluorescence labeled IgM (pink signal, originally 488 nm wavelength), DAPI staining of nuclei (light blue signal, originally 358 nm wavelength) and light microscopy of the alveolar structure obtained by differential interference contrast (DIC). Left images show IgM (pink signal) restrained within the alveolar walls in a control lung. Right images show plasma IgM extravasation (pink signal) in alveolar airspaces of a patient with ARDS-DAD. (Original magnification, 20× and 40×).
In the normal lung, fluid and small proteins pass from the intravascular to the interstitial space mostly through small gaps between capillary endothelial cells, being returned to the systemic circulation by the lymphatics. This fluid and solutes do not enter the alveoli in normal conditions because of the tightness of the alveolar epithelium (2). In patients with acute cardiogenic dysfunction or volume overload, the alveolar edema is generated by a rapid increase in the hydrostatic pressure in the pulmonary capillaries (2) and has a low protein concentration compared to plasma (3). Resolution of this cardiogenic pulmonary edema is usually rapid, in part because the alveolar-epithelial barrier is not damaged and the mechanisms of alveolar fluid clearance (AFC) are intact. In patients with ARDS, in contrast, the alveolar edema results from the loss of the alveolar endothelial and epithelial barriers, allowing fluid and large plasma proteins to move into the interstitial tissue and to flood the alveolar airspaces (4-8) (Figure 2). The alveolar epithelial damage is a critical factor that promotes the development of increased-permeability edema in ARDS. Potential operative mechanisms of alveolar epithelial damage include cell death, the loss of adequate tight junction (TJ)-mediated cell-to-cell contact, changes in extracellular matrix (ECM) components and in their contact with epithelial cells, and changes in the communication between epithelial and immune cells. These factors can be promoted by mechanical stretch, dysregulated inflammatory responses, inappropriate activation of leukocytes and platelets, and enhanced activation of pro-coagulation signals with formation of microthrombi (9-11).
Role of the alveolar epithelium in lung edema formation
In healthy alveoli, the capillary endothelium forms a semipermeable barrier to fluid exchange, whereas the alveolar epithelium is an extremely tight barrier that restricts the passage of water, electrolytes and small hydrophilic solutes to the air spaces (12,13). During lung injury, the edema fluid accumulating in airspaces is cleared by the creation of a transepithelial osmotic gradient by active sodium transport via apical membrane epithelial Na+ channels (ENaC), causing water to move passively from the airspaces to the interstitium and thereby removing excess alveolar fluid. This electrochemical gradient for Na+ influx is maintained by the basolateral Na,K-ATPase (14). In most patients with ARDS, the AFC capability is impaired, which is associated with more prolonged acute respiratory failure and higher mortality (15). Remarkably, predominant injury of the alveolar epithelium has been described in patients who died with ARDS (16), and the degree of alveolar epithelial damage appears to determine the severity of ARDS (17-19). Extensive damage of alveolar epithelial leads to the formation of alveolar edema containing high molecular-weight serum proteins, with the consequent worsening of gas exchange and a higher likelihood of disordered repair (9,20). It has also been shown that injury of the alveolar epithelium, but not of the vascular endothelium, determines the progression to lung fibrosis in these patients (19,21). Finally, the repair of alveolar epithelium is also crucial for recovery in ARDS, since it is responsible for clearing the filtered fluid and proteins from the alveolar airspaces (15). Importantly, the permeability and the AFC function of the alveolar epithelium depend on intercellular TJ complexes that allow cell-to-cell contact, as well as on the interaction between the epithelium and the ECM.
Alveolar epithelial TJ complexes as modulators of alveolar barrier permeability
TJs are heteromeric protein complexes that laterally approximate the lipid membranes of adjacent epithelial cells (22-24). The TJs constitute a regulated diffusion barrier within the intercellular space, and render the epithelium much less permeable than the endothelial barrier (11,19). In addition to controlling paracellular transport, TJs also maintain cellular polarity, regulate a variety of intracellular signals, and control the transcellular transport across the epithelium by influencing the expression of transport proteins and channels and by establishing separate intercellular compartments. All these functions are crucial for the exchange of substances between the internal and external cellular environments in the lung (13,22,24).
Damage of TJs is a major cause of epithelial barrier breakdown during lung inflammation. Dysfunction of the TJs results in increased permeability to water and proteins and in the deterioration of the AFC capacity of the epithelium, leading to the formation and perpetuation of lung edema. Furthermore, alteration of the TJs facilitates the passage of infectious agents, exogenous toxins and endogenous products into the systemic circulation (22,24,25), therefore exposing other organs and contributing to multiorgan failure.
The TJ complexes include transmembrane proteins such as occludin, claudins, tricellulin, and other junction adhesion molecules (JAM), and intracellular adaptor proteins like cingulin and zonula occludens (ZO) that ultimately bind to actin fibers of the cytoskeleton (22,24,26). Occludin, ZO-1, and claudin-4 have been shown to be important components of TJs in the alveolar epithelium (Figure 3) (25,28,29). Occludin is required for maintaining the integrity of the alveolar epithelial barrier (30,31). Claudin-4 improves the barrier function of the pulmonary epithelial barrier by promoting AFC function (32,33). ZO-1 is a scaffold protein that serves as a link between transmembrane TJ proteins (occludin, claudin) and the actin cytoskeleton (34), being an important element that influences the structure and function of the alveolar epithelial barrier (25,35). Actin and myosin, the two main components of the anchored cytoskeleton, interact to regulate cell tension and contraction, which also influence epithelial permeability. Alterations in the expression, localization and assembly of these proteins within the TJ complexes and in their interactions with the actin fibers of the cytoskeleton result in the dysfunction of TJs with the consequent increase in paracellular permeability (22,26).
Figure 3.
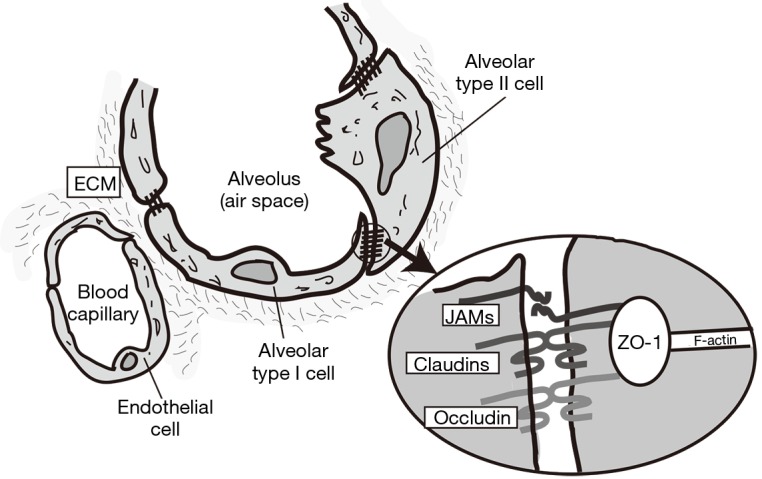
Schematic of alveolar epithelium and intercellular tight junction (TJ) structure. Squamous alveolar type I (AT-I) and cuboidal alveolar type II (AT-II) cells conform the alveolar epithelium. The tight junctions between adjacent AT-I cells are narrower than those between AT-I and AT-II cells. Occludin, claudins (cldn-3, -4 and -18) and ZOs proteins are expressed in both cells, but with different claudin expression patterns. AT-I: Cldn-18>cldn-3>cldn-4. Type II: cldn-3>cldn-4>cldn-18 (27). ECM, extracellular matrix; JAMs, junctional adhesion molecules.
The TJ complexes are dynamic and regulated structures (36). TJ assembly and disruption are regulated by several factors such as mechanical stretch (37), microbial pathogens and their products (e.g., endotoxin) (38,39), inflammatory cytokines—IL-4, IL-13, tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ) (40-44), matrix metalloproteinases (MMPs) (45), microRNAs (46), and reactive oxygen species (47-50). These stimuli activate classical signal transduction pathways involving ATP depletion (51), release of intracellular Ca2+ (52), G proteins (53), protein kinase C (PKC) (54), MAPK, PI3K (55), protein phosphatases and phosphorylation-related regulation (56-58), and small GTPases (59). Nitric oxide and peroxynitrite increase epithelial permeability by altering the expression or localization of key TJ proteins such as ZO-1 and occludin, and by promoting actin-myosin interaction and cell contraction via Rho kinase activation and MLC phosphorylation (39).
Interactions between alveolar epithelium and ECM influence alveolar barrier permeability
The ECM constitutes the three-dimensional scaffold of the alveolar epithelium and the capillary endothelium, and it is composed of the epithelial and endothelial basement membranes (BMs)—formed by type IV collagen, laminin, type V collagen and proteoglycans—and of a thin layer of interstitial connective tissue between them—formed by type I and III collagen, elastin and proteoglycans (60-64). The ECM is known to modulate cell survival, proliferation, migration and differentiation, and to have an important role in tissue morphogenesis and repair. The ECM is also crucial for the epithelial and endothelial barrier function, since it regulates cell-cell interactions and controls the trafficking of fluid and molecules in the interstitial space (60,65,66). Changes in the composition and mechanic properties of the ECM have been shown to modify the expression of TJs and the barrier function in alveolar epithelial and endothelial cells, contributing to lung edema formation (67,68). For example, alveolar epithelial type II cells cultured on laminin, the main component of the BM, exhibit higher resistance than cells on collagen-I or fibronectin (67). Changes in the ECM stiffness due to lysyl oxidase-mediated collagen crosslinking has been associated with the disruption of junctional integrity, being a potential mechanism of endotoxin-induced deterioration of pulmonary vascular permeability (68). The cell-matrix interaction is also important for the regulation of alveolar permeability, which is mainly controlled by adhesive membrane receptors called integrins that link the epithelial and endothelial cells to the BM (69-72). The structural organization and degradation of the ECM is also controlled by the proteolytic action of many proteases, including MMPs and their inhibitors (73,74). Both inflammatory and stromal cells can express MMPs, although the expression profile is both cell and stimulus specific. Recent animal and human studies have shown a role of MMP-2, -3, -7, -8 and -9, expressed by inflammatory, mesenchymal and probably epithelial cells, in the development and repair of the alveolar-capillary damage in ARDS (74-76). Although there is evidence that MMPs can alter TJ expression (45), their role in the normal lung and in the development of ARDS still needs to be elucidated.
Caveolin-1 as regulator of lung injury
Caveolin-1 is an important structural and regulatory component of caveolae, which are involved in multiple physiological processes, such as proliferation, apoptosis, cell differentiation, and regulation of membrane trafficking processes including endocytosis, exocytosis and transcytosis (77,78). Caveolin-1 has also been reported to regulate the assembly of TJ proteins (79,80). In the lung, caveolin-1 is highly expressed in epithelial cells, fibroblasts, vascular endothelial cells, smooth muscle cells, macrophages and neutrophils, and it has been involved in several pathological features of ARDS, such as epithelial and endothelial cell death, inflammation, fibrosis and alterations of the alveolar-capillary permeability in the lung (77,81). In experimental models of lung injury, the downregulation of caveolin-1 was associated with decreased expression of TJ proteins (occludin, claudin-4 and ZO-1) and increase of pulmonary epithelial permeability, whereas caveolin-1 upregulation markedly antagonized the loss of TJ proteins and the destruction of the pulmonary epithelial barrier (80,82).
Mechanisms of epithelial cell damage in ARDS
The normal alveolar epithelium is composed of type I and type II pneumocytes. Type I pneumocytes are squamous, cover 90–95% of the alveolar surface area, mediate gas exchange and barrier function, and are easily injured. They are also metabolically active, participating in host defense, alveolar remodeling and antioxidant functions. Type II pneumocytes are cuboidal cells that synthetize and release surfactant, act as a progenitor cell for both type I and type II cells, and have more proliferative capability and resistance to injury than type I cells (7). Cell death, inflammation, coagulation and mechanical stretch are considered important mechanisms that contribute to the damage of alveolar epithelial cells in the lung of patients with ARDS (9,11).
Cell death
Cell death occurs in the alveolar walls of patients with ARDS as well as of animal models of acute lung injury (ALI) induced by hyperoxia, lipopolysaccharide (LPS), bleomycin, cecal ligation and puncture, ischemia/reperfusion injury, and mechanical ventilation (83,84). In patients with ARDS, epithelial necrosis is present and can be directly caused by mechanical factors, hyperthermia, local ischemia, or bacterial products and viruses in the airspaces (9,85). In addition, epithelial cell apoptosis characterized by decreased size, nuclear DNA fragmentation and subsequent chromatin condensation has also been observed (16,86). The apoptotic changes are accompanied by activation of pro-apoptotic molecular proteins such as Bax, caspase-3, and p53 in the lung (83,87), as well as by elevated levels of caspase-cleaved cytokeratin-18, a marker for epithelial cell apoptosis, in bronchoalveolar lavage (BAL) fluid of these patients (88). Another important mechanism of alveolar epithelial injury in ARDS is the activation of the pro-apoptotic Fas/FasL pathway. This apoptotic pathway requires binding of membrane-bound or soluble FasL (sFasL) to Fas-bearing cells (86). Apoptosis of lung epithelial cells represents a potentially important mechanism contributing to the loss of alveolar epithelial cells and development of ARDS (89-91). The inhibition of apoptosis by blocking the Fas/FasL pathway or caspase activity has been shown to attenuate lung injury and protein-rich edema formation, and to prevent the lethal consequences of sepsis and ventilator induced-lung injury in animals. Importantly, these beneficial effects were accompanied by less pulmonary epithelial cell apoptosis when compared to control animals (90,91).
Although apoptosis seems to participate on lung injury, the mechanisms by which it compromises alveolar epithelial barrier function and lung edema formation have not been fully elucidated. Our group has shown that activation of Fas via intratracheal instillation of sFasL led to an increase of the alveolar capillary protein permeability, to an impairment of AFC, and to protein-rich edema formation in mouse lungs by mechanisms involving caspase-dependent apoptosis (90). However, the number of apoptotic cells identified in most models of ALI is too small to exclusively attribute the formation of lung edema to the apoptosis-mediated loss of cells. Thus, it is conceivable that the activation of apoptotic pathways also causes cellular changes that contribute to lung edema by mechanisms that do not depend on the ultimate death of epithelial cells.
Inflammation
Inflammation in the alveoli occurs early in the development of ARDS, and it is associated with changes in protein permeability and in the AFC capacity that lead to lung edema. In this setting, inflammation is characterized by marked neutrophil influx, activation of alveolar macrophages, and release of cytokines (TNF-α, TNFR, IL-1β, IL1RA, IL-6, INF-γ and G-CSF) and chemokines (IL-8, ENAP-78, MCP-1, MIP-1) into the airspaces by alveolar endothelial and epithelial cells, and by activated immune cells. IL-1β and TNF-α are biologically active cytokines in the pulmonary airspace of patients with ARDS and both seem to increase pulmonary epithelial permeability (21,62,92,93). IL-1β increases alveolar endothelial and epithelial permeability via RhoA/integrins-mediated epithelial TGF-β release, which has been shown to induce phosphorylation of adherent junction proteins and stress actin fiber formation in endothelial cells in vitro (94). IL-1β also inhibited fluid transport across the human distal lung epithelium in vitro (92). In contrast, TNF-α has shown a stimulatory effect on AFC in some animal models of ALI (pneumonia and ischemia/reperfusion injury) (95). Both effects on AFC are due to changes in the expression of the major Na+ and Cl- transporters in the lung (96). The underlying mechanisms responsible for the cytokine-induced alterations of epithelial and endothelial barriers are not totally known, but seem to involve apoptosis-dependent and apoptosis-independent mechanisms (84,97). TNF-α has been shown to disrupt TJ proteins (ZO-1, claudin 2-4-5) and β-catenin in pulmonary endothelial and epithelial cell layers (41,98-100), which can be exacerbated by interferon-gamma (IFN-γ) (101). In contrast, IFN-γ alone has been shown to enhance pulmonary epithelial barrier function and repair (102). TNF-α enhanced human pulmonary microvascular endothelial permeability and altered the actin cytoskeleton by mechanisms involving the activation of PKC, the increase of MAPK activity in a RhoA/ROCK-dependent manner, and the Rho-dependent myosin-light-chain (MLC) phosphatase inhibition (96,101,103-105). In contrast, other studies have reported that the gradual increase in permeability induced by TNF-α involved long-term reorganization of transmembrane TJ proteins—occludin and JAM-A—rather than the contractile mechanisms dependent on Rho, ROCK, and MLC Kinase (MLCK) (101,106). TNF-α, IL-1β and IL-6 can upregulate trypsin in endothelial cells, which may result in the loss of the TJ protein ZO-1 and vascular hyperpermeability via protease-activated receptor-2 (PAR-2) (107). IL-4 and IL-13 reduced the expression of ZO-1 and occludin, and diminished the repairing capacity of pulmonary epithelial cells in vitro (102). IL-1 receptor-ligand complexes increased alveolar epithelial protein permeability through activation of the tyrosine kinase receptor human epidermal growth factor receptor-2 (HER2). This HER2 activation by IL-1β required a disintegrin and metalloproteinase 17 (ADAM17)-dependent shedding of the ligand neuregulin-1 (NRG-1). Importantly, NRG-1 was detectable and elevated in pulmonary edema samples from patients with ALI, suggesting that this inflammatory signaling pathway in the lung could have diagnostic and therapeutic implications (108).
Coagulation
ARDS is characterized by the presence of intense pro-coagulant activity in the airspaces, which is triggered by vascular endothelial cell damage and increased microvascular permeability (109-111). In healthy lungs, resting endothelial cells constitute a non-thrombogenic barrier that produces anticoagulant molecules and inhibits platelet activation, thus preventing an inappropriate activation of coagulation (85). In ARDS lungs, the injury of vascular endothelial cells initiate coagulation by promoting both activation of platelets and pro-coagulant cascades and reduction of anticoagulant components and fibrinolysis, resulting in microthrombi in the pulmonary microvasculature and fibrin deposition in intra-alveolar and interstitial compartments (112,113). During the early stages of ALI/ARDS, pro-inflammatory mediators favor this pro-coagulant activity by downregulating natural anticoagulant pathways and by increasing pro-coagulant activity (109,110,114). This pro-coagulant activity is reflected by increased levels of soluble tissue factor, activated factor VII, tissue factor-dependent factor X, thrombin, fibrinopeptide A, D-dimer and fibrinogen in the alveolar airspaces. Concomitantly, there is a decrease in fibrinolytic activity, as shown by decreased levels of activated protein C (APC) and urokinase, and increased levels of fibrinolysis inhibitors such as plasminogen activator inhibitor (PAI) and α2-antiplasmin (85,109-111,114).
Several evidences indicate that pro-coagulant factors increase alveolar epithelial and endothelial barrier permeability by altering the cytoskeleton and the physical forces on cell-cell and cell-matrix interactions. Such procoagulant-induced alterations are mediated to a large extent by changes in Rac1/RhoA activity ratios, which results in the contraction of actin-myosin fibers and/or TJ proteins (115-117). Exposure of plasma components to tissue factor expressed by injured endothelial cells, macrophages, alveolar epithelial cells, or fibroblasts leads to intra-alveolar activation of coagulation and thrombin generation (109-111). Thrombin is an important pro-coagulant protein elevated in the lungs of patients with ARDS (111,118) that modifies alveolar epithelial and endothelial cell permeability by changing their contractile machinery with the formation of actin stress fibers, increasing cell contraction and stiffness, and affecting the cell-cell contact (115,119,120). Although thrombin is known to increase the endothelial barrier permeability, its effect on the alveolar epithelial barrier is still unclear. On one hand, incubation of alveolar epithelial cells with thrombin caused an elongation of ZO-1 aggregates and increased the membrane expression of ZO-1 and occludin proteins in cell-cell interface areas. Activation of Rac and Rho GTPases seemed to be involved in these effects, which were associated with enhanced epithelial cell contraction, intercellular gap formation and increased barrier permeability (115). In another study, on the contrary, thrombin induced prominent circumferential localization of actin fibers, increased MLC phosphorylation and enhanced epithelial barrier function with increased levels of the TJ proteins ZO-1 and occludin at the cell-cell interface (115,116). These differences could be explained by the degree of cell contraction and the capacity of the TJ-actin complexes to maintain the barrier function after thrombin exposure, which in turn depend on the final activation of small GTPase Rac and Rho, phosphorylation and spatial location of MLC and TJ proteins, and on the actin-myosin interaction (82). On the surface of alveolar epithelial cells, the anticoagulant protein C is activated by the thrombin-thrombomodulin complex (121) and can be inhibited by the presence of cytokines such as TNF-α, IL-1β, and IFN-γ (122). APC prevented the disruption of barrier integrity induced by thrombin in lung endothelial and alveolar epithelial cells in vitro (116). In a mouse model of Pseudomonas aeruginosa pneumonia, elevated levels of APC prevented the worsening of endothelial and alveolar epithelial protein permeability and improved AFC, effects that were mediated by the inhibition of RhoA and the activation of Rac1, and that required the endothelial protein C receptor (EPCR)/protease-activated receptor-1 (PAR-1)-dependent and sphingosine-1-phosphate (S1P) pathways (123).
Mechanical stretch
Cyclic stretch of epithelial cells during mechanical ventilation increases the release of inflammatory cytokines and induces alveolar epithelial cell death (124,125). In addition, cyclic stretch enhances protein permeability, which is associated with reduction of TJ proteins, disorganization of actin monofilaments, and elevated intracellular calcium concentrations (37). The mechanisms by which mechanical stretch alters TJ-actin complexes are not fully known. Mechanical stretch reduces the expression of occludin in the alveolar epithelium in a volume- and frequency-dependent manner by mechanisms involving PKC signaling (126), JNK activation (127) and reduction of intracellular ATP (37), and also promotes actin cytoskeletal redistribution to form peri-junctional actin rings (128). All these mechanical stretch-activated mechanisms result in an increase of epithelial barrier permeability. The stretch-mediated changes in the actin cytoskeleton of alveolar epithelial cells seem to be mediated by an early Rac1 activation that induces the phosphorylation of Akt and LIM kinase (LIMK) and decreases the phosphorylation of the actin turnover mediator cofilin (128). In addition, mechanical stretch of alveolar epithelial cells results in the production of reactive oxygen and nitrogen species—superoxide and nitric oxide—that may have a role in the dissociation of claudin-4 and claudin-7 from ZO-1 observed under these conditions (129). In accordance with these observations, reducing the intensity of mechanical stretch on epithelium by decreasing tidal volume is an important protective strategy of mechanical ventilation for patients with ALI.
Role of immune cells and their interactions on lung edema formation
In ARDS, the early activation of innate immune responses and platelets in the alveoli initiates the release of proinflammatory cytokines/chemokines and procoagulant factors, leading to the recruitment of neutrophils, lymphocytes and mononuclear phagocytes into the alveolar air space. Activated immune cells and platelets establish a paracrine communication network between the different immune, epithelial, and endothelial cells within the injured alveolus that may alter AFC and permeability, resulting in lung edema. This cell-cell interaction may be mediated by microparticle exchange that allow distant cell communication, and by intercellular gap junctions that allow communication between contiguous cells. These forms of cellular communication imply exchange of cytoplasmic constituents from the originating cell to the target cells. A wide variety of cellular molecules such as RNA, proteins and lipids can be enclosed into microparticles and be transferred to the destination cell. These molecules can also be freely secreted and serve as extracellular mediators (130-132). In pneumonia or ARDS, microparticles originated in epithelial cells, platelets, neutrophils and macrophages are found in the BAL fluid (130,133).
Microparticles contain micro-RNAs (miRNAs)—small, single-stranded noncoding RNAs—that regulate post-transcriptional gene expression and multiple cellular processes (cell proliferation, differentiation, development, survival, apoptosis, metabolism and immunity) (134-136). Pulmonary permeability can also be regulated by miRNAs. New evidences show that miRNA-155, miRNA-466d-5p and miR-466f-3p regulated lung inflammation and increased alveolar epithelial barrier permeability in experimental models of ALI (46,137,138). In particular, it has been shown that macrophage-derived miR-155 exerted these effects by promoting the expression of proinflammatory factors via SOCS-1, whereas the blockage of this miRNA prevented these changes in an endotoxin-induced ALI model in mice (137). In contrast, miRNA-147b decreased ADAM15 expression and attenuated endotoxin-induced barrier dysfunction in endothelial cells (139). Lipids such as the lysophospholipid mediator S1P are present in BAL fluid of patients with inflammatory pulmonary diseases (140-142), and are known to regulate alveolar barrier function (143). S1P is produced or secreted as an autocrine mediator into the extracellular environment, or stored within intracellular vesicles in mast cells, platelets, endothelial and epithelial cells, and regulate innate and adaptive immunity. Its expression can be up-regulated by the pro-inflammatory cytokines IL-1β and TNF-α. In the lung, there are multiple S1P receptors, which can be coupled to the small GTP-binding proteins Rac and Rho, that mediate the extracellular effects of S1P, enhancing the pulmonary endothelial barrier integrity (143,144).
Interactions between macrophages and epithelial cells
The mononuclear phagocyte system of the lung comprises resident interstitial and alveolar macrophages, dendritic cells and peripheral blood monocytes. Besides their essential host-defense functions, monocytes/macrophages have been implicated in the early alveolar epithelial damage in ALI by contributing to a detrimental immune response (137,145-149). An overly activated inflammatory response may contribute to alveolar barrier disruption by mechanisms that depend on both tissue-resident and bone marrow-derived macrophages (137,145,146,150). In injured alveoli, the recruitment of peripheral blood monocytes to the alveolar compartment is mediated by the alveolar epithelial release of chemokines such as CC-chemokine ligand 2 (CCL2) (147,151). Once recruited into the alveoli, blood monocytes acquire a lung resident macrophage phenotype and replenish the alveolar macrophage pool. Some alveolar macrophages can adhere and interact with the epithelium. Macrophage-epithelial interactions in alveoli involve Ca2+ communication through connexin 43 (Cx43)-containing alveolar gap junction channels. Increased levels of cytosolic Ca2+ modulate the expression of TJ and adhesion junction proteins, facilitating alveolar influx of immune cells across the alveolar-capillary barrier (148). Although the effect of Ca2+ dependent communication on alveolar barrier function is unknown, increased cytosolic Ca2+ indirectly can lead to mitochondrial-mediated apoptosis in epithelial cells and to the release of proinflammatory cytokines such as TNF-α in macrophages that could indirectly change the properties of the pulmonary barrier (148). In a murine model of influenza-induced pneumonia, recruited monocyte-derived macrophages contribute to alveolar epithelial cell apoptosis and alveolar barrier leakage by the release of the cytokine TNF-related apoptosis-inducing ligand (TRAIL), which seems related to autocrine interferon stimulus (149,152). Alveolar macrophages also inhibit AFC by decreasing the expression and function of Na,K-ATPase in alveolar epithelial cells via cell-cell communications, in which IFN-dependent elevation of macrophage TRAIL also seems to play a role (153,154).
Concomitant with the induction of inflammation, macrophages also initiate anti-inflammatory and tissue repair responses through diverse mechanisms, including the phenotypic conversion of macrophages from inflammatory (M1) to anti-inflammatory (M2), the induction of efferocytosis (phagocytosis of apoptotic cells) for neutrophil clearance, the secretion of cytokines such as IL-10, IL-1R antagonist (153,154), IL-4 and IL13 (155,156) and of epithelial growth factors (PDGF, FGFs, HGF, TGF-β and VEGF) (154,157,158) that contribute to pulmonary epithelial and endothelial proliferation and repair. In addition, alveolar macrophages secrete microparticles containing the anti-inflammatory proteins, SOCS1 and SOCS3 (159). Uptake of these macrophage-derived microparticles by the alveolar epithelium provides a mechanism by which activated macrophages may inhibit alveolar inflammation. In experimental models of LPS-induced lung injury, LPS stimulation enhanced the expression of miR-155 mainly in alveolar macrophages, which was thought to exacerbate alveolar permeability and lung injury via SOCS-1 down-regulation (137). In contrast, the expression of IL-1Ra by circulating monocyte-derived alveolar macrophages may have a protective effect by antagonizing IL-1β signaling and preventing the disruption and disassembly of ZO-1 in alveolar epithelial cells (94,153). Macrophage-released growth factors might also increase tightness of junctions in airway epithelial cells (154,160,161).
Neutrophils
Under certain stimuli, circulating blood neutrophils migrate from the vasculature to the airspaces in the lungs, crossing the endothelial and epithelial barriers through the paracellular spaces (and via transcellular route also in the endothelium). In the alveoli, this neutrophil transepithelial migration mainly occurs at tricellular junctions composed of two alveolar type I cells and one alveolar type II cell, in which TJ complexes form a discontinuous structure. In some circumstances, this neutrophil migration does not cause lung injury or changes in alveolar-capillary permeability (162-164). In some pathological states, however, the pulmonary influx of neutrophils into the alveolar space correlates with lung injury manifested as an increased permeability of the alveolar-capillary membrane (165). It has been proposed that these different outcomes depend on the degree of neutrophil activation and on the mechanisms that control neutrophil transmigration and barrier function.
Neutrophil paracellular transmigration involves close cell–cell contacts and highly regulated mechanisms responsible for signaling the opening and closing of the TJs without compromising barrier function (166). The mechanisms of the transepithelial migration of neutrophils are activated at sequential stages, starting by the initial adhesion to the basolateral surface, the migration through paracellular spaces and the final adhesion of the neutrophil to the apical epithelial surface. The initial adhesion of neutrophils to the basolateral surface of epithelial cells triggers intracellular signaling events within the epithelial cell such as phosphorylation of TJs and myosin light chain (MLC) (167), which in turn lead to the formation and contraction of the actomyosin ring, the opening of the TJs and a transient increase in epithelial permeability (168). This is followed by a rapid closure of the junction, in which JAM-A plays a critical role to restore the barrier function (169). Transepithelial migration depends on the interaction and activation of several surface molecules between epithelial cells and neutrophils, including the integrin associated protein CD47, the signal regulatory proteins SIRPα and SIRPβ, and the neutrophil JAM-like protein (JAML) that binds to the epithelial coxsackie and adenovirus receptor (CAR) (170). It has been suggested that neutrophil CD47 contributes to the increase in lung permeability caused by LPS on Gram-negative bacteria (171). Once translocated, neutrophils adhere to the apical epithelial surface by the adhesion molecule ICAM. At this stage, neutrophil-epithelial cell interaction results in the reestablishment of epithelial TJ complexes via adenosine-adenosine receptor binding on the apical epithelial surface (172). The neutrophil-epithelial cell interaction can also lead to tyrosine phosphorylation of TJ proteins, which is known to regulate permeability (167). The up-regulation of ICAM observed in inflamed lungs could enhance neutrophil-epithelial cell adhesiveness (167).
In pathologic conditions, an excessive and/or prolonged activation of translocating neutrophils into the airspaces can result in damage of alveolar epithelium due to several mechanisms, including: (I) release of cytotoxic substances to the extracellular environment by neutrophils, affecting neighboring and distant cells; (II) neutrophil-epithelial cell regulation of disassembly and reassembly of TJ; and (III) neutrophil-mediated mechanical force resulting in epithelial wounds. These “wounds” are thought to represent precursors of the macroscopic areas of denuded epithelium (ulcerative lesions) that characterize the DAD in ARDS. Neutrophils possess a potent antimicrobial arsenal that includes reactive oxygen species (O2− and H2O2), proteolytic enzymes (elastase and MMPs) and cationic peptides (defensins) that can be released into the extracellular environment during transepithelial migration and neutrophil–epithelial cell interactions (173). In pathological circumstances, these extracellular mediators cause a spectrum of responses in epithelial cells ranging from activation, injury and death accompanied by alteration of epithelial permeability and function (174). For example, the BAL fluid and plasma from patients with ARDS contains high levels of oxidants (likely of neutrophil origin, elastase, MMPs and defensins that correlate with the severity of lung injury and prognosis (75,175-178). Oxidants increase epithelial and endothelial permeability via disruption of TJs and redistribution of junctional proteins (176,177).
Elastase secreted by transmigrating neutrophils induces increased epithelial permeability via reorganization of the actin cytoskeleton and the intercellular junctions of epithelial cells adjacent to migrating neutrophils (178). This event also facilitates further neutrophil transmigration, resulting ultimately in the creation of circular defects (wounds) in epithelial cell monolayers in vitro (179). Elastase also cleaves BM matrix (180) and endothelium components such as E-cadherin and endothelial VE-cadherin (181).
MMPs can degrade nearly every ECM component, but their role in endothelial and epithelial homeostasis is less clear. Certain MMPs appear to play a role in modulating epithelial permeability (182), in part via proteolytic cleavage of E-cadherin and occludin, leading to TJ and adherent junction disassembly (183,184). By analogy, endothelial permeability is regulated by MMP degradation of occludin (185) and E-cadherin (186). The direct effect of MMPs on alveolar epithelial TJ proteins and permeability has not been elucidated yet.
Lastly, the cationic peptides called defensins are a major component of azurophilic granules of neutrophils and exert an antimicrobial effect against gram-positive and gram-negative bacteria, fungi and viruses via permeabilization of their cell membranes (187). As with other antimicrobial mediators, defensins induce lung injury and epithelial permeability via potential cytotoxic (188,189) and noncytotoxic (190) mechanisms.
All these effects of neutrophils on the alveolar epithelium along with the effectiveness of repair mechanisms are likely to determine the severity in macromolecular permeability and lung edema formation induced by neutrophil transmigration in ALI.
Future directions
Preventing the extravasation of fluid and proteins across the injured epithelium is important in patients at risk of developing ARDS. The degradation of protein components in the alveolar epithelial and endothelial barriers, including intercellular TJ proteins and ECM, is considered a critical process in the development of protein-rich edema in ARDS, and constitutes an attractive therapeutic target for maintaining the integrity and function of the alveolar epithelial barrier. The molecular cross-talk between immune and lung structural cell populations that leads to lung injury and pulmonary dysfunction remains incompletely elucidated. New studies are needed to improve our knowledge of cellular crosstalk in the alveoli and its role in the pathogenesis of DAD. Treatments focused on decreasing the initial epithelial injury and on accelerating repair processes of the alveolar epithelium might be of great value to improve the outcome of patients with ARDS.
Acknowledgements
Funding: This work was supported by grants (PI15/00482 to R Herrero and PI15/1942 to JA Lorente) from the Instituto de Salud Carlos III-Fondos Europeos de Desarrollo Regional (FEDER) “Una manera de hacer Europa”, Madrid, Spain.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Petty TL, Ashbaugh DG. The adult respiratory distress syndrome. Clinical features, factors influencing prognosis and principles of management. Chest 1971;60:233-9. 10.1378/chest.60.3.233 [DOI] [PubMed] [Google Scholar]
- 2.Staub NC. Pulmonary edema. Physiol Rev 1974;54:678-811. 10.1152/physrev.1974.54.3.678 [DOI] [PubMed] [Google Scholar]
- 3.Ware LB, Fremont RD, Bastarache JA, et al. Determining the aetiology of pulmonary oedema by the oedema fluid-to-plasma protein ratio. Eur Respir J 2010;35:331-7. 10.1183/09031936.00098709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ware LB, Matthay MA. Clinical practice. Acute pulmonary edema. N Engl J Med 2005;353:2788-96. 10.1056/NEJMcp052699 [DOI] [PubMed] [Google Scholar]
- 5.Fein A, Grossman RF, Jones JG, et al. The value of edema fluid protein measurement in patients with pulmonary edema. Am J Med 1979;67:32-8. 10.1016/0002-9343(79)90066-4 [DOI] [PubMed] [Google Scholar]
- 6.Carlson RW, Schaeffer RC, Carpio M, et al. Edema fluid and coagulation changes during fulminant pulmonary edema. Chest 1981;79:43-9. 10.1378/chest.79.1.43 [DOI] [PubMed] [Google Scholar]
- 7.Sibbald WJ, Cunningham DR, Chin DN. Non-cardiac or cardiac pulmonary edema? A practical approach to clinical differentiation in critically ill patients. Chest 1983;84:452-61. 10.1378/chest.84.4.452 [DOI] [PubMed] [Google Scholar]
- 8.Murray JF. Pulmonary edema: pathophysiology and diagnosis. Int J Tuberc Lung Dis 2011;15:155-60. [PubMed] [Google Scholar]
- 9.Matthay MA, Ware LB, Zimmerman GA. The acute respiratory distress syndrome. J Clin Invest 2012;122:2731-40. 10.1172/JCI60331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McVey M, Tabuchi A, Kuebler WM. Microparticles and acute lung injury. Am J Physiol Lung Cell Mol Physiol 2012;303:L364-381. 10.1152/ajplung.00354.2011 [DOI] [PubMed] [Google Scholar]
- 11.Zemans RL, Matthay MA. Bench-to-bedside review: the role of the alveolar epithelium in the resolution of pulmonary edema in acute lung injury. Crit Care 2004;8:469-77. 10.1186/cc2906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor AE, Gaar KA. Estimation of equivalent pore radii of pulmonary capillary and alveolar membranes. Am J Physiol 1970;218:1133-40. 10.1152/ajplegacy.1970.218.4.1133 [DOI] [PubMed] [Google Scholar]
- 13.Weibel ER. On the tricks alveolar epithelial cells play to make a good lung. Am J Respir Crit Care Med 2015;191:504-13. 10.1164/rccm.201409-1663OE [DOI] [PubMed] [Google Scholar]
- 14.Eaton DC, Helms MN, Koval M, et al. The contribution of epithelial sodium channels to alveolar function in health and disease. Annu Rev Physiol 2009;71:403-23. 10.1146/annurev.physiol.010908.163250 [DOI] [PubMed] [Google Scholar]
- 15.Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med 2001;163:1376-83. 10.1164/ajrccm.163.6.2004035 [DOI] [PubMed] [Google Scholar]
- 16.Bachofen M, Weibel ER. Structural alterations of lung parenchyma in the adult respiratory distress syndrome. Clin Chest Med 1982;3:35-56. [PubMed] [Google Scholar]
- 17.Matthay MA, Robriquet L, Fang X. Alveolar epithelium: role in lung fluid balance and acute lung injury. Proc Am Thorac Soc 2005;2:206-13. 10.1513/pats.200501-009AC [DOI] [PubMed] [Google Scholar]
- 18.Matthay MA, Folkesson HG, Campagna A, et al. Alveolar epithelial barrier and acute lung injury. New Horiz 1993;1:613-22. [PubMed] [Google Scholar]
- 19.Yanagi S, Tsubouchi H, Miura A, et al. Breakdown of Epithelial Barrier Integrity and Overdrive Activation of Alveolar Epithelial Cells in the Pathogenesis of Acute Respiratory Distress Syndrome and Lung Fibrosis. BioMed Res Int 2015;2015:573210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Matthay MA, Ware LB. Resolution of Alveolar Edema in Acute Respiratory Distress Syndrome. Physiology and Biology. Am J Respir Crit Care Med 2015;192:124-5. 10.1164/rccm.201505-0938ED [DOI] [PubMed] [Google Scholar]
- 21.Chambers RC, Mercer PF. Mechanisms of alveolar epithelial injury, repair, and fibrosis. Ann Am Thorac Soc 2015;12:S16-20. 10.1513/AnnalsATS.201410-448MG [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneeberger EE, Lynch RD. The tight junction: a multifunctional complex. Am J Physiol Cell Physiol 2004;286:C1213-28. 10.1152/ajpcell.00558.2003 [DOI] [PubMed] [Google Scholar]
- 23.Schneeberger EE, Karnovsky MJ. Substructure of intercellular junctions in freeze-fractured alveolar-capillary membranes of mouse lung. Circ Res 1976;38:404-11. 10.1161/01.RES.38.5.404 [DOI] [PubMed] [Google Scholar]
- 24.Denker BM, Nigam SK. Molecular structure and assembly of the tight junction. Am J Physiol 1998;274:F1-9. [DOI] [PubMed] [Google Scholar]
- 25.Wittekindt OH. Tight junctions in pulmonary epithelia during lung inflammation. Pflugers Arch 2017;469:135-47. 10.1007/s00424-016-1917-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.González-Mariscal L, Betanzos A, Nava P, et al. Tight junction proteins. Prog Biophys Mol Biol 2003;81:1-44. 10.1016/S0079-6107(02)00037-8 [DOI] [PubMed] [Google Scholar]
- 27.LaFemina MJ, Rokkam D, Chandrasena A, et al. Keratinocyte growth factor enhances barrier function without altering claudin expression in primary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 2010;299:L724-34. 10.1152/ajplung.00233.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuehn A, Kletting S, de Souza Carvalho-Wodarz C, et al. Human alveolar epithelial cells expressing tight junctions to model the air-blood barrier. ALTEX 2016;33:251-60. [DOI] [PubMed] [Google Scholar]
- 29.Ohta H, Chiba S, Ebina M, et al. Altered expression of tight junction molecules in alveolar septa in lung injury and fibrosis. Am J Physiol Lung Cell Mol Physiol 2012;302:L193-205. 10.1152/ajplung.00349.2010 [DOI] [PubMed] [Google Scholar]
- 30.Caraballo JC, Yshii C, Westphal W, et al. Ambient particulate matter affects occludin distribution and increases alveolar transepithelial electrical conductance. Respirology 2011;16:340-9. 10.1111/j.1440-1843.2010.01910.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Caraballo JC, Yshii C, Butti ML, et al. Hypoxia increases transepithelial electrical conductance and reduces occludin at the plasma membrane in alveolar epithelial cells via PKC-ζ and PP2A pathway. Am J Physiol Lung Cell Mol Physiol 2011;300:L569-78. 10.1152/ajplung.00109.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitchell LA, Overgaard CE, Ward C, et al. Differential effects of claudin-3 and claudin-4 on alveolar epithelial barrier function. Am J Physiol Lung Cell Mol Physiol 2011;301:L40-9. 10.1152/ajplung.00299.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wray C, Mao Y, Pan J, et al. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am J Physiol Lung Cell Mol Physiol 2009;297:L219-227. 10.1152/ajplung.00043.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Balda MS, Matter K. The tight junction protein ZO-1 and an interacting transcription factor regulate ErbB-2 expression. EMBO J 2000;19:2024-33. 10.1093/emboj/19.9.2024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bazzoni G, Martinez-Estrada OM, Orsenigo F, et al. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem 2000;275:20520-6. 10.1074/jbc.M905251199 [DOI] [PubMed] [Google Scholar]
- 36.Shen L. Tight junctions on the move: molecular mechanisms for epithelial barrier regulation. Ann N Y Acad Sci 2012;1258:9-18. 10.1111/j.1749-6632.2012.06613.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cavanaugh KJ, Oswari J, Margulies SS. Role of stretch on tight junction structure in alveolar epithelial cells. Am J Respir Cell Mol Biol 2001;25:584-91. 10.1165/ajrcmb.25.5.4486 [DOI] [PubMed] [Google Scholar]
- 38.Looi K, Troy NM, Garratt LW, et al. Effect of human rhinovirus infection on airway epithelium tight junction protein disassembly and transepithelial permeability. Exp Lung Res 2016:1-16. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 39.Han X, Fink MP, Uchiyama T, et al. Increased iNOS activity is essential for pulmonary epithelial tight junction dysfunction in endotoxemic mice. Am J Physiol Lung Cell Mol Physiol 2004;286:L259-67. 10.1152/ajplung.00187.2003 [DOI] [PubMed] [Google Scholar]
- 40.Coyne CB, Vanhook MK, Gambling TM, et al. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell 2002;13:3218-34. 10.1091/mbc.E02-03-0134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mazzon E, Cuzzocrea S. Role of TNF-alpha in lung tight junction alteration in mouse model of acute lung inflammation. Respir Res 2007;8:75. 10.1186/1465-9921-8-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capaldo CT, Farkas AE, Hilgarth RS, et al. Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol Biol Cell 2014;25:2710-9. 10.1091/mbc.E14-02-0773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Relova A-J, Shahana S, Makeeva N, et al. Effect of cytokines on ICAM-1 and ZO-1 expression on human airway epithelial cells. Cell Biol Int 2005;29:768-77. 10.1016/j.cellbi.2005.05.002 [DOI] [PubMed] [Google Scholar]
- 44.Patrick DM, Leone AK, Shellenberger JJ, et al. Proinflammatory cytokines tumor necrosis factor-alpha and interferon-gamma modulate epithelial barrier function in Madin-Darby canine kidney cells through mitogen activated protein kinase signaling. BMC Physiol 2006;6:2. 10.1186/1472-6793-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vermeer PD, Denker J, Estin M, et al. MMP9 modulates tight junction integrity and cell viability in human airway epithelia. Am J Physiol Lung Cell Mol Physiol 2009;296:L751-62. 10.1152/ajplung.90578.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cichon C, Sabharwal H, Rüter C, et al. MicroRNAs regulate tight junction proteins and modulate epithelial/endothelial barrier functions. Tissue Barriers 2014;2:e944446. 10.4161/21688362.2014.944446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Overgaard CE, Daugherty BL, Mitchell LA, et al. Claudins: control of barrier function and regulation in response to oxidant stress. Antioxid Redox Signal 2011;15:1179-93. 10.1089/ars.2011.3893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rao R. Oxidative stress-induced disruption of epithelial and endothelial tight junctions. Front Biosci 2008;13:7210-26. 10.2741/3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun Y, Minshall RD, Hu G. Role of claudins in oxidant-induced alveolar epithelial barrier dysfunction. Methods Mol Biol 2011;762:291-301. 10.1007/978-1-61779-185-7_21 [DOI] [PubMed] [Google Scholar]
- 50.Usatyuk PV, Parinandi NL, Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem 2006;281:35554-66. 10.1074/jbc.M607305200 [DOI] [PubMed] [Google Scholar]
- 51.Tsukamoto T, Nigam SK. Tight junction proteins form large complexes and associate with the cytoskeleton in an ATP depletion model for reversible junction assembly. J Biol Chem 1997;272:16133-9. 10.1074/jbc.272.26.16133 [DOI] [PubMed] [Google Scholar]
- 52.Stuart RO, Sun A, Panichas M, et al. Critical role for intracellular calcium in tight junction biogenesis. J Cell Physiol 1994;159:423-33. 10.1002/jcp.1041590306 [DOI] [PubMed] [Google Scholar]
- 53.Balda MS, González-Mariscal L, Contreras RG, et al. Assembly and sealing of tight junctions: possible participation of G-proteins, phospholipase C, protein kinase C and calmodulin. J Membr Biol 1991;122:193-202. 10.1007/BF01871420 [DOI] [PubMed] [Google Scholar]
- 54.Suzuki T, Elias BC, Seth A, et al. PKC eta regulates occludin phosphorylation and epithelial tight junction integrity. Proc Natl Acad Sci U S A 2009;106:61-6. 10.1073/pnas.0802741106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.González-Mariscal L, Tapia R, Chamorro D. Crosstalk of tight junction components with signaling pathways. Biochim Biophys Acta 2008;1778:729-56. 10.1016/j.bbamem.2007.08.018 [DOI] [PubMed] [Google Scholar]
- 56.Dörfel MJ, Huber O. Modulation of tight junction structure and function by kinases and phosphatases targeting occludin. J Biomed Biotechnol 2012;2012:807356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Elias BC, Suzuki T, Seth A, et al. Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions J Biol Chem 2009;284:1559-69. 10.1074/jbc.M804783200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rao R. Occludin phosphorylation in regulation of epithelial tight junctions. Ann N Y Acad Sci 2009;1165:62-8. 10.1111/j.1749-6632.2009.04054.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Citalán-Madrid AF, García-Ponce A, Vargas-Robles H, et al. Small GTPases of the Ras superfamily regulate intestinal epithelial homeostasis and barrier function via common and unique mechanisms. Tissue Barriers 2013;1:e26938. 10.4161/tisb.26938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.West JB, Mathieu-Costello O. Structure, strength, failure, and remodeling of the pulmonary blood-gas barrier. Annu Rev Physiol 1999;61:543-72. 10.1146/annurev.physiol.61.1.543 [DOI] [PubMed] [Google Scholar]
- 61.Pelosi P, Rocco PRM, Negrini D, et al. The extracellular matrix of the lung and its role in edema formation. An Acad Bras Cienc 2007;79:285-97. 10.1590/S0001-37652007000200010 [DOI] [PubMed] [Google Scholar]
- 62.Souza-Fernandes AB, Pelosi P, Rocco PRM. Bench-to-bedside review: the role of glycosaminoglycans in respiratory disease. Crit Care 2006;10:237. 10.1186/cc5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rocco PR, Negri EM, Kurtz PM, et al. Lung tissue mechanics and extracellular matrix remodeling in acute lung injury. Am J Respir Crit Care Med 2001;164:1067-71. 10.1164/ajrccm.164.6.2007062 [DOI] [PubMed] [Google Scholar]
- 64.Negrini D, Passi A, De Luca G, et al. Proteoglycan involvement during development of lesional pulmonary edema. Am J Physiol 1998;274:L203-11. [DOI] [PubMed] [Google Scholar]
- 65.Pichert A, Schlorke D, Franz S, et al. Functional aspects of the interaction between interleukin-8 and sulfated glycosaminoglycans. Biomatter 2012;2:142-8. 10.4161/biom.21316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson Z, Proudfoot AE, Handel TM. Interaction of chemokines and glycosaminoglycans: a new twist in the regulation of chemokine function with opportunities for therapeutic intervention. Cytokine Growth Factor Rev 2005;16:625-36. 10.1016/j.cytogfr.2005.04.006 [DOI] [PubMed] [Google Scholar]
- 67.Koval M, Ward C, Findley MK, et al. Extracellular matrix influences alveolar epithelial claudin expression and barrier function. Am J Respir Cell Mol Biol 2010;42:172-80. 10.1165/rcmb.2008-0270OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mammoto A, Mammoto T, Kanapathipillai M, et al. Control of lung vascular permeability and endotoxin-induced pulmonary oedema by changes in extracellular matrix mechanics. Nat Commun 2013;4:1759. 10.1038/ncomms2774 [DOI] [PubMed] [Google Scholar]
- 69.Girault A, Chebli J, Privé A, et al. Complementary roles of KCa3.1 channels and β1-integrin during alveolar epithelial repair. Respir Res 2015;16:100. 10.1186/s12931-015-0263-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sheppard D. Modulation of acute lung injury by integrins. Proc Am Thorac Soc 2012;9:126-9. 10.1513/pats.201112-052AW [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Su G, Atakilit A, Li JT, et al. Absence of integrin αvβ3 enhances vascular leak in mice by inhibiting endothelial cortical actin formation. Am J Respir Crit Care Med 2012;185:58-66. 10.1164/rccm.201108-1381OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Taooka Y, Ohe M, Chen L, et al. Increased expression levels of integrin α9β1 and CD11b on circulating neutrophils and elevated serum IL-17A in elderly aspiration pneumonia. Respiration 2013;86:367-75. 10.1159/000345390 [DOI] [PubMed] [Google Scholar]
- 73.Lanchou J, Corbel M, Tanguy M, et al. Imbalance between matrix metalloproteinases (MMP-9 and MMP-2) and tissue inhibitors of metalloproteinases (TIMP-1 and TIMP-2) in acute respiratory distress syndrome patients. Crit Care Med 2003;31:536-42. 10.1097/01.CCM.0000048626.02184.F8 [DOI] [PubMed] [Google Scholar]
- 74.Elkington PTG, Friedland JS. Matrix metalloproteinases in destructive pulmonary pathology. Thorax 2006;61:259-66. 10.1136/thx.2005.051979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davey A, McAuley DF, O’Kane CM. Matrix metalloproteinases in acute lung injury: mediators of injury and drivers of repair. Eur Respir J 2011;38:959-70. 10.1183/09031936.00032111 [DOI] [PubMed] [Google Scholar]
- 76.Hendrix AY, Kheradmand F. The Role of Matrix Metalloproteinases in Development, Repair, and Destruction of the Lungs. Prog Mol Biol Transl Sci 2017;148:1-29. 10.1016/bs.pmbts.2017.04.004 [DOI] [PubMed] [Google Scholar]
- 77.Jin Y, Lee SJ, Minshall RD, et al. Caveolin-1: a critical regulator of lung injury. Am J Physiol Lung Cell Mol Physiol 2011;300:L151-60. 10.1152/ajplung.00170.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Insel PA, Head BP, Ostrom RS, et al. Caveolae and lipid rafts: G protein-coupled receptor signaling microdomains in cardiac myocytes. Ann N Y Acad Sci 2005;1047:166-72. 10.1196/annals.1341.015 [DOI] [PubMed] [Google Scholar]
- 79.Nighot PK, Leung L, Ma TY. Chloride channel ClC-2 enhances intestinal epithelial tight junction barrier function via regulation of caveolin-1 and caveolar trafficking of occludin. Exp Cell Res 2017;352:113-22. 10.1016/j.yexcr.2017.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gao C, Li R, Huan J, et al. Caveolin-1 siRNA increases the pulmonary microvascular and alveolar epithelial permeability in rats. J Trauma 2011;70:210-9. 10.1097/TA.0b013e3181e7432d [DOI] [PubMed] [Google Scholar]
- 81.Maniatis NA, Kardara M, Hecimovich D, et al. Role of caveolin-1 expression in the pathogenesis of pulmonary edema in ventilator-induced lung injury. Pulm Circ 2012;2:452-60. 10.4103/2045-8932.105033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jin Y, Kim HP, Chi M, et al. Deletion of caveolin-1 protects against oxidative lung injury via up-regulation of heme oxygenase-1. Am J Respir Cell Mol Biol 2008;39:171-9. 10.1165/rcmb.2007-0323OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Martin TR, Nakamura M, Matute-Bello G. The role of apoptosis in acute lung injury. Crit Care Med 2003;31:S184-188. 10.1097/01.CCM.0000057841.33876.B1 [DOI] [PubMed] [Google Scholar]
- 84.Lipke AB, Matute-Bello G, Herrero R, et al. Febrile-range hyperthermia augments lipopolysaccharide-induced lung injury by a mechanism of enhanced alveolar epithelial apoptosis. J Immunol 2010;184:3801-13. 10.4049/jimmunol.0903191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang Y, Tang H. Aberrant coagulation causes a hyper-inflammatory response in severe influenza pneumonia. Cell Mol Immunol 2016;13:432-42. 10.1038/cmi.2016.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Albertine KH, Soulier MF, Wang Z, et al. Fas and fas ligand are up-regulated in pulmonary edema fluid and lung tissue of patients with acute lung injury and the acute respiratory distress syndrome. Am J Pathol 2002;161:1783-96. 10.1016/S0002-9440(10)64455-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herrero R, Kajikawa O, Matute-Bello G, et al. The biological activity of FasL in human and mouse lungs is determined by the structure of its stalk region. J Clin Invest 2011;121:1174-90. 10.1172/JCI43004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lee KS, Choi YH, Kim YS, et al. Evaluation of bronchoalveolar lavage fluid from ARDS patients with regard to apoptosis. Respir Med 2008;102:464-9. 10.1016/j.rmed.2007.10.001 [DOI] [PubMed] [Google Scholar]
- 89.Martin TR, Hagimoto N, Nakamura M, et al. Apoptosis and epithelial injury in the lungs. Proc Am Thorac Soc 2005;2:214-20. 10.1513/pats.200504-031AC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Herrero R, Tanino M, Smith LS, et al. The Fas/FasL pathway impairs the alveolar fluid clearance in mouse lungs. Am J Physiol Lung Cell Mol Physiol 2013;305:L377-88. 10.1152/ajplung.00271.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Perl M, Lomas-Neira J, Chung CS, et al. Epithelial cell apoptosis and neutrophil recruitment in acute lung injury-a unifying hypothesis? What we have learned from small interfering RNAs. Mol Med Camb Mass 2008;14:465-75. 10.2119/2008-00011.Perl [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pugin J, Ricou B, Steinberg KP, et al. Proinflammatory activity in bronchoalveolar lavage fluids from patients with ARDS, a prominent role for interleukin-1. Am J Respir Crit Care Med 1996;153:1850-6. 10.1164/ajrccm.153.6.8665045 [DOI] [PubMed] [Google Scholar]
- 93.Pugin J, Verghese G, Widmer MC, et al. The alveolar space is the site of intense inflammatory and profibrotic reactions in the early phase of acute respiratory distress syndrome. Crit Care Med 1999;27:304-12. 10.1097/00003246-199902000-00036 [DOI] [PubMed] [Google Scholar]
- 94.Ganter MT, Roux J, Miyazawa B, et al. Interleukin-1beta causes acute lung injury via alphavbeta5 and alphavbeta6 integrin-dependent mechanisms. Circ Res 2008;102:804-12. 10.1161/CIRCRESAHA.107.161067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Rezaiguia S, Garat C, Delclaux C, et al. Acute bacterial pneumonia in rats increases alveolar epithelial fluid clearance by a tumor necrosis factor-alpha-dependent mechanism. J Clin Invest 1997;99:325-35. 10.1172/JCI119161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Petrache I, Birukova A, Ramirez SI, et al. The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability. Am J Respir Cell Mol Biol 2003;28:574-81. 10.1165/rcmb.2002-0075OC [DOI] [PubMed] [Google Scholar]
- 97.Bruewer M, Luegering A, Kucharzik T, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol 2003;171:6164-72. 10.4049/jimmunol.171.11.6164 [DOI] [PubMed] [Google Scholar]
- 98.Hermanns MI, Kasper J, Dubruel P, et al. An impaired alveolar-capillary barrier in vitro: effect of proinflammatory cytokines and consequences on nanocarrier interaction. J R Soc Interface 2010;7:S41-54. 10.1098/rsif.2009.0288.focus [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li Q, Zhang Q, Wang M, et al. Interferon-gamma and tumor necrosis factor-alpha disrupt epithelial barrier function by altering lipid composition in membrane microdomains of tight junction. Clin Immunol 2008;126:67-80. 10.1016/j.clim.2007.08.017 [DOI] [PubMed] [Google Scholar]
- 100.Hardyman MA, Wilkinson E, Martin E, et al. TNF-α-mediated bronchial barrier disruption and regulation by src-family kinase activation. J Allergy Clin Immunol 2013;132:665-675.e8. 10.1016/j.jaci.2013.03.005 [DOI] [PubMed] [Google Scholar]
- 101.Al-Sadi R, Boivin M, Ma T. Mechanism of cytokine modulation of epithelial tight junction barrier. Front Biosci (Landmark Ed) 2009;14:2765-78. 10.2741/3413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ahdieh M, Vandenbos T, Youakim A. Lung epithelial barrier function and wound healing are decreased by IL-4 and IL-13 and enhanced by IFN-gamma. Am J Physiol Cell Physiol 2001;281:C2029-38. 10.1152/ajpcell.2001.281.6.C2029 [DOI] [PubMed] [Google Scholar]
- 103.Koss M, Pfeiffer GR, Wang Y, et al. Ezrin/radixin/moesin proteins are phosphorylated by TNF-alpha and modulate permeability increases in human pulmonary microvascular endothelial cells. J Immunol 2006;176:1218-27. 10.4049/jimmunol.176.2.1218 [DOI] [PubMed] [Google Scholar]
- 104.Mong PY, Petrulio C, Kaufman HL, et al. Activation of Rho kinase by TNF-alpha is required for JNK activation in human pulmonary microvascular endothelial cells. J Immunol 2008;180:550-8. 10.4049/jimmunol.180.1.550 [DOI] [PubMed] [Google Scholar]
- 105.Nwariaku FE, Rothenbach P, Liu Z, et al. Rho inhibition decreases TNF-induced endothelial MAPK activation and monolayer permeability. J Appl Physiol 2003;95:1889-95. 10.1152/japplphysiol.00225.2003 [DOI] [PubMed] [Google Scholar]
- 106.McKenzie JA, Ridley AJ. Roles of Rho/ROCK and MLCK in TNF-alpha-induced changes in endothelial morphology and permeability. J Cell Physiol 2007;213:221-8. 10.1002/jcp.21114 [DOI] [PubMed] [Google Scholar]
- 107.Wang S, Le TQ, Kurihara N, Chida J, et al. Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza. J Infect Dis 2010;202:991-1001. 10.1086/656044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Venugopal R, Galam L, Cox R, Fukumoto J, et al. Inflammasome Inhibition Suppresses Alveolar Cell Permeability Through Retention of Neuregulin-1 (NRG-1). Cell Physiol Biochem 2015;36:2012-24. 10.1159/000430169 [DOI] [PubMed] [Google Scholar]
- 109.Frantzeskaki F, Armaganidis A, Orfanos SE. Immunothrombosis in Acute Respiratory Distress Syndrome: Cross Talks between Inflammation and Coagulation. Respiration 2017;93:212-25. 10.1159/000453002 [DOI] [PubMed] [Google Scholar]
- 110.Welty-Wolf KE, Carraway MS, Ortel TL, et al. Coagulation and inflammation in acute lung injury. Thromb Haemost 2002;88:17-25. [PubMed] [Google Scholar]
- 111.Glas GJ, Van Der Sluijs KF, Schultz MJ, et al. Bronchoalveolar hemostasis in lung injury and acute respiratory distress syndrome. J Thromb Haemost 2013;11:17-25. 10.1111/jth.12047 [DOI] [PubMed] [Google Scholar]
- 112.van Hinsbergh VW. Endothelium--role in regulation of coagulation and inflammation. Semin Immunopathol 2012;34:93-106. 10.1007/s00281-011-0285-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Armstrong SM, Darwish I, Lee WL. Endothelial activation and dysfunction in the pathogenesis of influenza A virus infection. Virulence 2013;4:537-42. 10.4161/viru.25779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Laterre PF, Wittebole X, Dhainaut JF. Anticoagulant therapy in acute lung injury. Crit Care Med 2003;31:S329-36. 10.1097/01.CCM.0000057912.71499.A5 [DOI] [PubMed] [Google Scholar]
- 115.Kawkitinarong K, Linz-McGillem L, Birukov KG, et al. Differential regulation of human lung epithelial and endothelial barrier function by thrombin. Am J Respir Cell Mol Biol 2004;31:517-27. 10.1165/rcmb.2003-0432OC [DOI] [PubMed] [Google Scholar]
- 116.Puig F, Fuster G, Adda M, et al. Barrier-protective effects of activated protein C in human alveolar epithelial cells. PloS One 2013;8:e56965. 10.1371/journal.pone.0056965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vandenbroucke E, Mehta D, Minshall R, et al. Regulation of endothelial junctional permeability. Ann N Y Acad Sci 2008;1123:134-45. 10.1196/annals.1420.016 [DOI] [PubMed] [Google Scholar]
- 118.Levi M, Schultz MJ, Rijneveld AW, et al. Bronchoalveolar coagulation and fibrinolysis in endotoxemia and pneumonia. Crit Care Med 2003;31:S238-42. 10.1097/01.CCM.0000057849.53689.65 [DOI] [PubMed] [Google Scholar]
- 119.Trepat X, Grabulosa M, Buscemi L, et al. Thrombin and histamine induce stiffening of alveolar epithelial cells. J Appl Physiol 2005;98:1567-74. 10.1152/japplphysiol.00925.2004 [DOI] [PubMed] [Google Scholar]
- 120.Gavara N, Sunyer R, Roca-Cusachs P, et al. Thrombin-induced contraction in alveolar epithelial cells probed by traction microscopy. J Appl Physiol 2006;101:512-20. 10.1152/japplphysiol.00185.2006 [DOI] [PubMed] [Google Scholar]
- 121.Ware LB, Fang X, Matthay MA. Protein C and thrombomodulin in human acute lung injury. Am J Physiol Lung Cell Mol Physiol 2003;285:L514-21. 10.1152/ajplung.00442.2002 [DOI] [PubMed] [Google Scholar]
- 122.Hataji O, Taguchi O, Gabazza EC, et al. Activation of protein C pathway in the airways. Lung 2002;180:47-59. 10.1007/s004080000080 [DOI] [PubMed] [Google Scholar]
- 123.Bir N, Lafargue M, Howard M, et al. Cytoprotective-selective activated protein C attenuates Pseudomonas aeruginosa-induced lung injury in mice. Am J Respir Cell Mol Biol 2011;45:632-41. 10.1165/rcmb.2010-0397OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Vlahakis NE, Schroeder MA, Limper AH, et al. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol 1999;277:L167-73. [DOI] [PubMed] [Google Scholar]
- 125.Song MJ, Davis CI, Lawrence GG, et al. Local influence of cell viability on stretch-induced permeability of alveolar epithelial cell monolayers. Cell Mol Bioeng 2016;9:65-72. 10.1007/s12195-015-0405-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu M, Gu C, Wang Y. Upregulation of the tight junction protein occludin: effects on ventilation-induced lung injury and mechanisms of action. BMC Pulm Med 2014;14:94. 10.1186/1471-2466-14-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Cohen TS, Gray Lawrence G, Khasgiwala A, et al. MAPK activation modulates permeability of isolated rat alveolar epithelial cell monolayers following cyclic stretch. PloS One 2010;5:e10385. 10.1371/journal.pone.0010385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dipaolo BC, Davidovich N, Kazanietz MG, at al. Rac1 pathway mediates stretch response in pulmonary alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol 2013;305:L141-53. 10.1152/ajplung.00298.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Song MJ, Davidovich N, Lawrence GG, et al. Superoxide mediates tight junction complex dissociation in cyclically stretched lung slices. J Biomech 2016;49:1330-5. 10.1016/j.jbiomech.2015.10.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Buesing KL, Densmore JC, Kaul S, et al. Endothelial microparticles induce inflammation in acute lung injury. J Surg Res 2011;166:32-9. 10.1016/j.jss.2010.05.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Brown GT, McIntyre TM. Lipopolysaccharide signaling without a nucleus: kinase cascades stimulate platelet shedding of proinflammatory IL-1β-rich microparticles. J Immunol 2011;186:5489-96. 10.4049/jimmunol.1001623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Camaioni C, Gustapane M, Cialdella P, et al. Microparticles and microRNAs: new players in the complex field of coagulation. Intern Emerg Med 2013;8:291-6. 10.1007/s11739-011-0705-5 [DOI] [PubMed] [Google Scholar]
- 133.Brodsky SV, Zhang F, Nasjletti A, et al. Endothelium-derived microparticles impair endothelial function in vitro. Am J Physiol Heart Circ Physiol 2004;286:H1910-5. 10.1152/ajpheart.01172.2003 [DOI] [PubMed] [Google Scholar]
- 134.Schickel R, Boyerinas B, Park SM, et al. MicroRNAs: key players in the immune system, differentiation, tumorigenesis and cell death. Oncogene 2008;27:5959-74. 10.1038/onc.2008.274 [DOI] [PubMed] [Google Scholar]
- 135.Vaporidi K, Vergadi E, Kaniaris E, et al. Pulmonary microRNA profiling in a mouse model of ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 2012;303:L199-207. 10.1152/ajplung.00370.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sessa R, Hata A. Role of microRNAs in lung development and pulmonary diseases. Pulm Circ 2013;3:315-28. 10.4103/2045-8932.114758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Wang W, Liu Z, Su J, et al. Macrophage micro-RNA-155 promotes lipopolysaccharide-induced acute lung injury in mice and rats. Am J Physiol Lung Cell Mol Physiol 2016;311:L494-506. 10.1152/ajplung.00001.2016 [DOI] [PubMed] [Google Scholar]
- 138.Yehya N, Yerrapureddy A, Tobias J, et al. MicroRNA modulate alveolar epithelial response to cyclic stretch. BMC Genomics 2012;13:154. 10.1186/1471-2164-13-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chatterjee V, Beard RS, Reynolds JJ, et al. MicroRNA-147b regulates vascular endothelial barrier function by targeting ADAM15 expression. PloS One 2014;9:e110286. 10.1371/journal.pone.0110286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Milara J, Navarro R, Juan G, et al. Sphingosine-1-phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax 2012;67:147-56. 10.1136/thoraxjnl-2011-200026 [DOI] [PubMed] [Google Scholar]
- 141.Ghidoni R, Caretti A, Signorelli P. Role of Sphingolipids in the Pathobiology of Lung Inflammation. Mediators Inflamm 2015;2015:487508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yang Y, Uhlig S. The role of sphingolipids in respiratory disease. Ther Adv Respir Dis 2011;5:325-44. 10.1177/1753465811406772 [DOI] [PubMed] [Google Scholar]
- 143.Natarajan V, Dudek SM, Jacobson JR, et al. Sphingosine-1-phosphate, FTY720, and sphingosine-1-phosphate receptors in the pathobiology of acute lung injury. Am J Respir Cell Mol Biol 2013;49:6-17. 10.1165/rcmb.2012-0411TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Aoki M, Aoki H, Ramanathan R, et al. Sphingosine-1-Phosphate Signaling in Immune Cells and Inflammation: Roles and Therapeutic Potential. Mediators Inflamm 2016;2016:8606878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Frank JA, Wray CM, McAuley DF, et al. Alveolar macrophages contribute to alveolar barrier dysfunction in ventilator-induced lung injury. Am J Physiol Lung Cell Mol Physiol 2006;291:L1191-8. 10.1152/ajplung.00055.2006 [DOI] [PubMed] [Google Scholar]
- 146.Zareie M, McKay DM, Kovarik GG, et al. Monocyte/macrophages evoke epithelial dysfunction: indirect role of tumor necrosis factor-alpha. Am J Physiol 1998;275:C932-9. 10.1152/ajpcell.1998.275.4.C932 [DOI] [PubMed] [Google Scholar]
- 147.Herbold W, Maus R, Hahn I, et al. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect Immun 2010;78:2620-30. 10.1128/IAI.01169-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bhattacharya J, Westphalen K. Macrophage-epithelial interactions in pulmonary alveoli. Semin Immunopathol 2016;38:461-9. 10.1007/s00281-016-0569-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Högner K, Wolff T, Pleschka S, et al. Macrophage-expressed IFN-β contributes to apoptotic alveolar epithelial cell injury in severe influenza virus pneumonia. PLoS Pathog 2013;9:e1003188. 10.1371/journal.ppat.1003188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Herold S, von Wulffen W, Steinmueller M, et al. Alveolar epithelial cells direct monocyte transepithelial migration upon influenza virus infection: impact of chemokines and adhesion molecules. J Immunol 2006;177:1817-24. 10.4049/jimmunol.177.3.1817 [DOI] [PubMed] [Google Scholar]
- 151.Winter C, Taut K, Srivastava M, et al. Lung-specific overexpression of CC chemokine ligand (CCL) 2 enhances the host defense to Streptococcus pneumoniae infection in mice: role of the CCL2-CCR2 axis. J Immunol 2007;178:5828-38. 10.4049/jimmunol.178.9.5828 [DOI] [PubMed] [Google Scholar]
- 152.Herold S, Steinmueller M, von Wulffen W, et al. Lung epithelial apoptosis in influenza virus pneumonia: the role of macrophage-expressed TNF-related apoptosis-inducing ligand. J Exp Med 2008;205:3065-77. 10.1084/jem.20080201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Herold S, Tabar TS, Janssen H, et al. Exudate macrophages attenuate lung injury by the release of IL-1 receptor antagonist in gram-negative pneumonia. Am J Respir Crit Care Med 2011;183:1380-90. 10.1164/rccm.201009-1431OC [DOI] [PubMed] [Google Scholar]
- 154.Aggarwal NR, King LS, D’Alessio FR. Diverse macrophage populations mediate acute lung inflammation and resolution. Am J Physiol Lung Cell Mol Physiol 2014;306:L709-25. 10.1152/ajplung.00341.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Booth BW, Adler KB, Bonner JC, et al. Interleukin-13 induces proliferation of human airway epithelial cells in vitro via a mechanism mediated by transforming growth factor-alpha. Am J Respir Cell Mol Biol 2001;25:739-43. 10.1165/ajrcmb.25.6.4659 [DOI] [PubMed] [Google Scholar]
- 156.White SR, Martin LD, Abe MK, et al. Insulin receptor substrate-1/2 mediates IL-4-induced migration of human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2009;297:L164-73. 10.1152/ajplung.90453.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Medeiros AI, Serezani CH, Lee SP, et al. Efferocytosis impairs pulmonary macrophage and lung antibacterial function via PGE2/EP2 signaling. J Exp Med 2009;206:61-8. 10.1084/jem.20082058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Granata F, Frattini A, Loffredo S, et al. Production of vascular endothelial growth factors from human lung macrophages induced by group IIA and group X secreted phospholipases A2. J Immunol 2010;184:5232-41. 10.4049/jimmunol.0902501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Bourdonnay E, Zasłona Z, Penke LRK, et al. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. J Exp Med 2015;212:729-42. 10.1084/jem.20141675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Herold S, Mayer K, Lohmeyer J. Acute lung injury: how macrophages orchestrate resolution of inflammation and tissue repair. Front Immunol 2011;2:65. 10.3389/fimmu.2011.00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Terakado M, Gon Y, Sekiyama A, et al. The Rac1/JNK pathway is critical for EGFR-dependent barrier formation in human airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 2011;300:L56-63. 10.1152/ajplung.00159.2010 [DOI] [PubMed] [Google Scholar]
- 162.Martin TR. Neutrophils and lung injury: getting it right. J Clin Invest 2002;110:1603-5. 10.1172/JCI0217302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Martin TR, Pistorese BP, Chi EY, et al. Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest 1989;84:1609-19. 10.1172/JCI114338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wiener-Kronish JP, Albertine KH, Matthay MA. Differential responses of the endothelial and epithelial barriers of the lung in sheep to Escherichia coli endotoxin. J Clin Invest 1991;88:864-75. 10.1172/JCI115388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Worthen GS, Haslett C, Rees AJ, et al. Neutrophil-mediated pulmonary vascular injury. Synergistic effect of trace amounts of lipopolysaccharide and neutrophil stimuli on vascular permeability and neutrophil sequestration in the lung. Am Rev Respir Dis 1987;136:19-28. 10.1164/ajrccm/136.1.19 [DOI] [PubMed] [Google Scholar]
- 166.Nash S, Stafford J, Madara JL. The selective and superoxide-independent disruption of intestinal epithelial tight junctions during leukocyte transmigration. Lab Invest 1988;59:531-7. [PubMed] [Google Scholar]
- 167.Edens HA, Levi BP, Jaye DL, et al. Neutrophil transepithelial migration: evidence for sequential, contact-dependent signaling events and enhanced paracellular permeability independent of transjunctional migration. J Immunol 2002;169:476-86. 10.4049/jimmunol.169.1.476 [DOI] [PubMed] [Google Scholar]
- 168.Turner JR. “Putting the squeeze” on the tight junction: understanding cytoskeletal regulation. Semin Cell Dev Biol 2000;11:301-8. 10.1006/scdb.2000.0180 [DOI] [PubMed] [Google Scholar]
- 169.Liu Y, Nusrat A, Schnell FJ, et al. Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci 2000;113:2363-74. [DOI] [PubMed] [Google Scholar]
- 170.Zen K, Liu Y, McCall IC, et al. Neutrophil migration across tight junctions is mediated by adhesive interactions between epithelial coxsackie and adenovirus receptor and a junctional adhesion molecule-like protein on neutrophils. Mol Biol Cell 2005;16:2694-703. 10.1091/mbc.E05-01-0036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Liu Y, Bühring HJ, Zen K, et al. Signal regulatory protein (SIRPalpha), a cellular ligand for CD47, regulates neutrophil transmigration. J Biol Chem 2002;277:10028-36. 10.1074/jbc.M109720200 [DOI] [PubMed] [Google Scholar]
- 172.Lawrence DW, Comerford KM, Colgan SP. Role of VASP in reestablishment of epithelial tight junction assembly after Ca2+ switch. Am J Physiol Cell Physiol 2002;282:C1235-45. 10.1152/ajpcell.00288.2001 [DOI] [PubMed] [Google Scholar]
- 173.Dengler V, Downey GP, Tuder RM, et al. Neutrophil intercellular communication in acute lung injury. Emerging roles of microparticles and gap junctions. Am J Respir Cell Mol Biol 2013;49:1-5. 10.1165/rcmb.2012-0472TR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Okrent DG, Lichtenstein AK, Ganz T. Direct cytotoxicity of polymorphonuclear leukocyte granule proteins to human lung-derived cells and endothelial cells. Am Rev Respir Dis 1990;141:179-85. 10.1164/ajrccm/141.1.179 [DOI] [PubMed] [Google Scholar]
- 175.Quinlan GJ, Lamb NJ, Tilley R, et al. Plasma hypoxanthine levels in ARDS: implications for oxidative stress, morbidity, and mortality. Am J Respir Crit Care Med 1997;155:479-84. 10.1164/ajrccm.155.2.9032182 [DOI] [PubMed] [Google Scholar]
- 176.Rao RK, Basuroy S, Rao VU, et al. Tyrosine phosphorylation and dissociation of occludin-ZO-1 and E-cadherin-beta-catenin complexes from the cytoskeleton by oxidative stress. Biochem J 2002;368:471-81. 10.1042/bj20011804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Musch MW, Walsh-Reitz MM, Chang EB. Roles of ZO-1, occludin, and actin in oxidant-induced barrier disruption. Am J Physiol Gastrointest Liver Physiol 2006;290:G222-31. 10.1152/ajpgi.00301.2005 [DOI] [PubMed] [Google Scholar]
- 178.Ginzberg HH, Cherapanov V, Dong Q, et al. Neutrophil-mediated epithelial injury during transmigration: role of elastase. Am J Physiol Gastrointest Liver Physiol 2001;281:G705-17. 10.1152/ajpgi.2001.281.3.G705 [DOI] [PubMed] [Google Scholar]
- 179.Nusrat A, Parkos CA, Liang TW, et al. Neutrophil migration across model intestinal epithelia: monolayer disruption and subsequent events in epithelial repair. Gastroenterology 1997;113:1489-500. 10.1053/gast.1997.v113.pm9352851 [DOI] [PubMed] [Google Scholar]
- 180.Shapiro SD. Neutrophil elastase: path clearer, pathogen killer, or just pathologic? Am J Respir Cell Mol Biol 2002;26:266-8. 10.1165/ajrcmb.26.3.f233 [DOI] [PubMed] [Google Scholar]
- 181.Carden D, Xiao F, Moak C, et al. Neutrophil elastase promotes lung microvascular injury and proteolysis of endothelial cadherins. Am J Physiol 1998;275:H385-92. [DOI] [PubMed] [Google Scholar]
- 182.Castaneda FE, Walia B, Vijay-Kumar M, et al. Targeted deletion of metalloproteinase 9 attenuates experimental colitis in mice: central role of epithelial-derived MMP. Gastroenterology 2005;129:1991-2008. 10.1053/j.gastro.2005.09.017 [DOI] [PubMed] [Google Scholar]
- 183.Gorodeski GI. Estrogen decrease in tight junctional resistance involves matrix-metalloproteinase-7-mediated remodeling of occludin. Endocrinology 2007;148:218-31. 10.1210/en.2006-1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Pflugfelder SC, Farley W, Luo L, et al. Matrix metalloproteinase-9 knockout confers resistance to corneal epithelial barrier disruption in experimental dry eye. Am J Pathol 2005;166:61-71. 10.1016/S0002-9440(10)62232-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Alexander JS, Elrod JW. Extracellular matrix, junctional integrity and matrix metalloproteinase interactions in endothelial permeability regulation. J Anat 2002;200:561-74. 10.1046/j.1469-7580.2002.00057.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Navaratna D, McGuire PG, Menicucci G, et al. Proteolytic degradation of VE-cadherin alters the blood-retinal barrier in diabetes. Diabetes 2007;56:2380-7. 10.2337/db06-1694 [DOI] [PubMed] [Google Scholar]
- 187.Aarbiou J, Rabe KF, Hiemstra PS. Role of defensins in inflammatory lung disease. Ann Med 2002;34:96-101. 10.1080/07853890252953482 [DOI] [PubMed] [Google Scholar]
- 188.Aarbiou J, Tjabringa GS, Verhoosel RM, et al. Mechanisms of cell death induced by the neutrophil antimicrobial peptides alpha-defensins and LL-37. Inflamm Res 2006;55:119-27. 10.1007/s00011-005-0062-9 [DOI] [PubMed] [Google Scholar]
- 189.Van Wetering S, Mannesse-Lazeroms SP, Dijkman JH, et al. Effect of neutrophil serine proteinases and defensins on lung epithelial cells: modulation of cytotoxicity and IL-8 production. J Leukoc Biol 1997;62:217-26. 10.1002/jlb.62.2.217 [DOI] [PubMed] [Google Scholar]
- 190.Nygaard SD, Ganz T, Peterson MW. Defensins reduce the barrier integrity of a cultured epithelial monolayer without cytotoxicity. Am J Respir Cell Mol Biol 1993;8:193-200. 10.1165/ajrcmb/8.2.193 [DOI] [PubMed] [Google Scholar]