

Abstract
Campylobacter hyointestinalis is a member of an emerging group of zoonotic Campylobacter spp. that are increasingly identified in both gastric and non-gastric disease in humans. Here, we discovered C. hyointestinalis in three separate classes of New Zealand ruminant livestock; cattle, sheep and deer. To investigate the relevance of these findings we performed a systematic literature review on global C. hyointestinalis epidemiology and used comparative genomics to better understand and classify members of the species. We found that C. hyointestinalis subspecies hyointestinalis has an open pangenome, with accessory gene contents involved in many essential processes such as metabolism, virulence and defence. We observed that horizontal gene transfer is likely to have played an overwhelming role in species diversification, favouring a public-goods-like mechanism of gene ‘acquisition and resampling’ over a tree-of-life-like vertical inheritance model of evolution. As a result, simplistic gene-based inferences of taxonomy by similarity are likely to be misleading. Such genomic plasticity will also mean that local evolutionary histories likely influence key species characteristics, such as host-association and virulence. This may help explain geographical differences in reported C. hyointestinalis epidemiology and limits what characteristics may be generalised, requiring further genomic studies of C. hyointestinalis in areas where it causes disease.
Introduction
Campylobacter is one of the main causative agents of human bacterial gastroenteritis worldwide1. Currently divided into 28 distinct species (http://www.bacterio.net/campylobacter.html)2,3, this bacterial genus occupies a variety of different niches, primarily the gastrointestinal tracts of many animal species where it may be either commensal or pathogenic4. The most frequently attributed sources of human campylobacteriosis are food (particularly poultry), water, and raw milk5,6. Human campylobacteriosis has symptoms including diarrhoea (often bloody), abdominal pain, nausea, and headaches7 and is most commonly caused by the species Campylobacter jejuni and Campylobacter coli8,9. However, campylobacteriosis has also been reported to be caused by so-called ‘emerging Campylobacter spp.10, including Campylobacter concisus11, Campylobacter sputorum12, Campylobacter upsaliensis12, Campylobacter ureolyticus13 and Campylobacter hyointestinalis (see below). Campylobacter spp., including the ‘emerging species’, have also been associated with other pathological presentations in humans, including bacteraemia14–18, abscesses19–21, Crohn’s disease22,23, meningitis24, neonatal infection and abortion25. Improved diagnostic technologies have revolutionised our understanding of the clinical importance of “emerging Campylobacter spp.”, while the exponentially increasing availability of genetic and genomic data means that our appreciation of each species’ characteristics and ecological associations is constantly evolving.
The taxonomic classification of Campylobacter species has historically been driven by phenotypic characterisation of bacterial isolates, with molecular characterization being only a minor component of the bacterial description26. As with all branches of taxonomy this is changing with the advent of widely available and accessible DNA sequencing technologies, but it is only very recently that the standards for description of Campylobacter species have been up-dated to a biphasic approach with genotype and phenotype descriptions both being important27. DNA-based classifications also have no universal standard, and different methodologies (eg. multi-locus sequence typing vs. whole genome sequencing) allow different resolving capacities for the distinction between strain variants as well as dealing with complex evolutionary phenomena such as recombination in different ways. Nonetheless, taxonomic updates based on DNA sequences are essential and have led to the inclusion of species previously not classified as Campylobacter28 and the exclusion and reclassification of others29.
Campylobacter hyointestinalis, originally isolated from diseased pigs30 is a member of the “emerging Campylobacter spp.” group that can also cause disease in humans. Although C. hyointestinalis was originally described as being able to grow at 43 °C31,32 it is frequently referred to as a non-thermotolerant species33. The species has two recognised subspecies; hyointestinalis and lawsonii. Molecular detection systems have been published which provide culture independent detection methods for this species34,35. The first complete genome sequences of C. hyointestinalis subspecies hyointestinalis and lawsonii were published in 201636, showing that the C. hyointestinalis genome was approximately 1.75 Mbp in length, with a GC content of approximately 34%.
Here, we provide an up to date summary of all publicly reported detections of C. hyointestinalis infection. We use genomic data generated from New Zealand isolates to examine the evolutionary processes occurring in C. hyointestinalis, and discuss how these influence our interpretation of both its characteristics and classification.
Results
Literature Survey
We conducted a systematic literature review looking for articles that reported original observations of C. hyointestinalis. A total of 75 articles were identified that satisfied the search criteria (inclusion of the text “Campylobacter hyointestinalis” at any point) from more than 1,500 articles, using a combination of different search engines for scientific literature. The summary of the data extracted from these 75 articles is provided in Supplementary Data S1.
To date, C. hyointestinalis has been reported from 30 countries across the world, and from six of the seven continents (Fig. 1A). Although the original reports of C. hyointestinalis were from pigs that displayed signs of gastric disease30, the most frequently reported source for C. hyointestinalis isolation has been the faeces of healthy cattle herds, and humans with various presentations of gastroenteritis (Fig. 1B). Ninety-three percent of all reports of C. hyointestinalis isolation have been from mammalian hosts, of which almost all were domesticated or zoo-kept animals. However, select observations of C. hyointestinalis have been made from the excretions of non-mammalian hosts, including birds, tortoises and shellfish.
Figure 1.

Summary of data from the systematic literature review carried out as part of this study. (A) Geographical distribution of publications reporting original observations of C. hyointestinalis. Countries that have reported C. hyointestinalis are displayed in blue. (B) Number of reports of C. hyointestinalis from different sources. Bars represent the number of publications that report the presence of C. hyointestinalis in each species. Purple bars represent cases where infection was in a host displaying gastric disease, blue bars represent instances where there was no disease, or the disease status of the tested host was not reported. The presented map was modified in Inkscape 0.91 (http://inkscape.org) from World with Countries – Outline by FreeVectorMaps.com. Icons made by Freepik from www.flaticon.com (https://www.flaticon.com/authors/freepik/).
Reports of C. hyointestinalis have increased slowly since its description in 1983 (Fig. 2A), suggesting that new technologies have either not been widely adopted or have not significantly improved the isolation and detection this species. The methodologies that have led to C. hyointestinalis isolation have remained relatively unchanged in this time, and culture-dependent methodologies remain the most common way of identifying C. hyointestinalis (Fig. 2B), despite the introduction of numerous different molecular techniques for the detection of Campylobacter over the same time period34,35,37. Although C. hyointestinalis is considered to be a non-thermotolerant member of the Campylobacteraeceae family33, it has been isolated most commonly under selective conditions that exploit high temperatures (>41 °C) (Fig. 2C), possibly because these are the prevailing conditions used for the identification of Campylobacter infections which are more commonly associated with the species C. jejuni and C. coli38.
Figure 2.
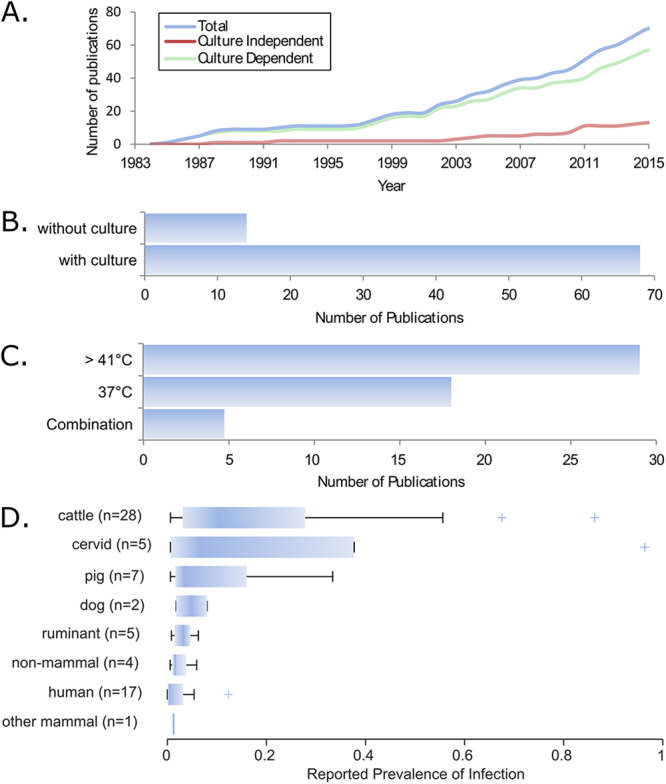
Summary of data from the literature survey carried out as part of this study. (A) Temporal trend of publications reporting original observations of C. hyointestinalis. (B) Number of publications that used culture dependent, or culture independent methods to identify C. hyointestinalis. (C) Number of publications reporting culture dependent methods with culture conditions at high (>41 °C), or low (37) temperatures for the isolation of C. hyointestinalis. (D) For population level studies, defined as those with epidemiological sampling protocols and sample sizes greater than 20 individuals, reported infection prevalence is summarised for each host species. Box plots demark 5th and 95th percentiles, interquartile ranges, median and outlying values.
From the original 75 articles, 68 datasets (including some articles describing multiple datasets) were identified reporting C. hyointestinalis with population-level sampling strategies and sample sizes greater than 20 test individuals. In these studies, the reported prevalence of infection has been greatest in cattle, cervids (deer and reindeer) and pigs (Fig. 2D).
Detection of C. hyointestinalis in New Zealand Ruminants
Campylobacter isolation was performed on faecal samples from 33 dairy, sheep and beef and deer farms across the Manawatu district of the North Island of New Zealand. In total, Campylobacter spp. were identified in 52% of ruminant faecal samples tested. C. jejuni and/or C. coli were identified in 39% of samples, whereas C. hyointestinalis was identified in 21% of samples. The use of pooled samples and a second culture condition for cattle and sheep samples is likely to have increased the detection rate of C. hyointestinalis in these samples relative to the deer population.
For deer; of 80 faecal samples taken, nine showed Campylobacter-like growth on an mCCDA plate (two stag samples, seven hind samples). Two colonies were taken from each for further examination giving a total of 22 isolates for PCR. Sixteen of the isolates and nine red deer were positive for Campylobacter, two positives from stags and seven from hinds. Two isolates from a stag tested positive for C. jejuni and all tested negative for C. coli. More extensive speciation revealed 14 isolates to be positive for C. hyointestinalis (seven hinds, one stag).
For cattle; Campylobacter was isolated from 94% of pooled faecal samples. PCR identified 57 C. jejuni and C. coli isolates, and 28 C. hyointestinalis isolates. For sheep; Campylobacter was isolated from 51% of pooled faecal samples, of which 29 were identified as C. jejuni and C. coli, and six were identified as C. hyointestinalis. Epidemiological findings are summarised in Supplementary Table S1.
From the 48 isolates identified as C. hyointestinalis, a subset of 16 was randomly selected to be characterised via whole genome sequencing. Illumina HiSeq 2 × 250 bp sequencing provided good quality draft genome assemblies for all isolates (Supplementary Table S2).
Population genetics, genomics and evolutionary analysis
Whole genome MLST analysis identified a conservative total of 255 polymorphic loci that were shared between the 58 genomes included from the non-thermophilic branch of the Campylobacteraceae. An annotated list of the 255 conserved, polymorphic loci has been provided in Supplementary Data S2. The resulting network analysis of the concatenated gene alignment from all shared loci had a tree-like structure with clade groupings that essentially matched current species-level taxonomic classifications (Fig. 3). One exception to this was C. rectus, which was predicted to have strong similarity to C. showae. Those taxa that contained distinct sub-species level taxonomic divisions included C. hyointestinalis, C. fetus and C. concisus. C. concisus currently has no formally recognised subspecies, though there are known genomospecies. Conversely, available C. fetus data originating from isolates with the subspecies classifications testudinum, fetus and venerealis formed a single distinct branch of the similarity network. C. hyointestinalis subsp. lawsonii and hyointestinalis were clearly distinguishable. All isolates obtained as part of this study belonged to the taxon C. hyointestinalis subsp. hyointestinalis.
Figure 3.
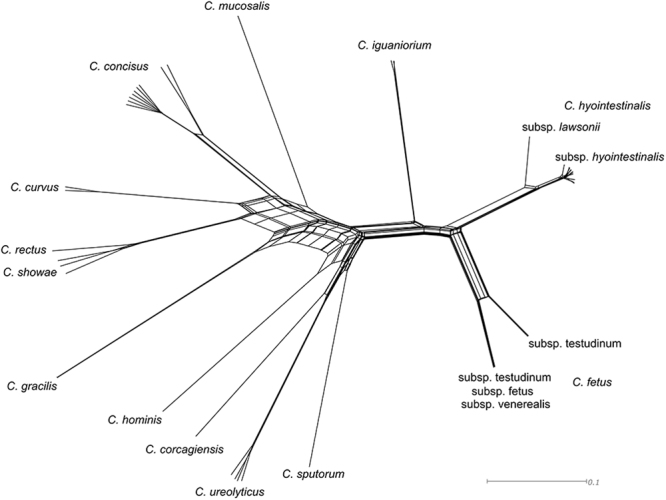
Core genome alignment network for non-thermophilic Campylobacter species. Single nucleotide polymorphism distances are calculated from 255 core genes identified by the Genome Profiler software package.
Isolates were compared to publicly deposited data from PubMLST by concatenating and aligning the appropriate seven MLST loci from the assembled genomic data. Previously unreported allelic sequences were obtained from many of the isolates, and for all loci except gltA and glyA. These were used to assign ten new sequence-type definitions (Supplementary Table S3, Supplementary Data S3). Sequence type similarity was calculated for all known C. hyointestinalis isolates using the minimum-spanning method (Fig. 4). In this analysis, most New Zealand isolates were located at terminal positions of the network, suggesting significant divergence from C. hyointestinalis variants from other geographical locations. However, p-distance analysis confirmed that all of these variants belonged to the same subspecies. Source data from the 237 available isolates and 141 different sequence types analysed showed that 94% of C. hyointestinalis subsp. lawsonii isolates had originated from swine. Contrastingly, only 6% of C. hyointestinalis subsp. hyointestinalis were isolated from swine, whereas 89% were isolated from cattle.
Figure 4.

Minimum Spanning Tree of the seven-gene MLST data from C. hyointestinalis sourced from PubMLST. Circle diameter signifies the number of reported samples in the PubMLST database, and colour depicts host species as indicated in the legend. Inset, histograms of pairwise p-distance calculations between all samples including (light red) and excluding (light blue) subspecies lawsonii from the calculations. Sequence types for which no isolate metadata were available are represented as singletons of unknown source.
We further examined the evolutionary characteristics of New Zealand C. hyointestinalis within the seven-gene MLST data by looking for signs of recombination39. By this method only 344 bp remained after stripping predicted recombinant sites from the original 3,312 bp alignment, suggesting that approximately 90% of sites within the standard MLST scheme loci may have been subject to recombination at some point (Supplementary Figure 1). Phylogenetic reconstruction from the raw data showed poor node-support, most likely as a result of this estimated high rate of recombination (data not shown). Phylogenetic reconstruction based on the recombination-stripped data had high posterior probabilities for the majority of structure-defining tree nodes, and predicted highly contrasting relationships between members of this species relative to the minimum spanning method (Supplementary Figure 2). Within data derived from our samples, the nature of this difference was most evident for sequence types N1 and N9 (all isolates from New Zealand cattle), which were erroneously assigned to a cluster with international isolates of STs 83, 67 and 6 using the minimum spanning method (Fig. 4) and predicted to have a much closer relationship to ST N5 (an isolate from New Zealand sheep) in the phylogenetic analysis (Supplementary Figure 2).
We extended our analysis of recombination rates to the whole-genome scale. Similar to regions within the seven gene MLST allele sites, recombination events were predicted to be extremely frequent between lineages, with as few as 54,000 bp (4.3%) of the total core genomic content being retained in the global clonal frame. On average, 79% ± SD 9% of genomic content on tree leaves was estimated to have been inherited vertically from its closest estimated ancestor. For isolate S1499c, shared recombination loci between genome sequences of this atypical C. hyointestinalis subspecies hyointestinalis and C. hyointestinalis subsp. lawsonii suggested that this isolate had undergone subspecies-level introgression at some point in its evolutionary history (Supplementary Figure 3).
We next examined core- and pan- genome characteristics for all C. hyointestinalis genomes available from our study and both the NCBI and ENA databases. Prokka-based annotation predicted an average of 1,828 coding sequences (±SD 80) for an average genome size of 1.77 Mbp (±SD 0.06 Mbp). No plasmids were detected when queried against the PlasmidFinder database40. OrthoMCL-based clustering executed in get_homologues estimated a core genome size of 1,271 genes (67% ± SD 3% of the total number of genes), a soft-core size (defined as orthologous genes found in all or all but one of the studied genomes) of 1,416 genes (78% ± SD 3% of the total number of genes) and a pangenome size of 3,446 genes (Fig. 5). Rarefaction analysis suggested that even when restricted to the subspecies scale, the pangenome of C. hyointestinalis could be considered as open, with a Heap’s law coefficient of 0.21 (Supplementary Figure S4). We quantified the rates of gene acquisition and loss along the predicted recombination-stripped phylogeny, using the Count software package and the pan-genome presence/absence matrix generated by get_homologues. Our analysis supported the theory that gene acquisition may drive microbial speciation, with major gene acquisition events being associated with the subspecies level divergence event of C. hyointesinalis subsp. lawsonii, as well as the atypical isolate S1499c (Fig. 6). Conversely, gene loss events were predicted to occur at more evenly-spaced intervals along the C. hyointestinalis evolutionary history. Interestingly, recombination/mutation rates also varied greatly between different C. hyointestinalis lineages (Fig. 7) and higher rates of recombination showed no correlation to higher rates of gene gain, suggesting that homologous and non-homologous recombination frequencies are determined by independent mechanisms.
Figure 5.

Pangenome structure of Campylobacter hyointestinalis. The dendrogram (left) depicts the optimal hierarchical clustering of the orthologous gene group presence/absence matrix generated, expressed in terms of λ′ heterogeneity as described in72. The heatmap (centre) depicts pairwise pangenome intersect and union sizes for all genome combinations. Numbers of unique genes associated with each genome are indicated by bars (top). Numbers of gene clusters associated with the depicted genome groups are shown as a stacked icicle plot where the intersect size (number of shared genes) in each set determines the height of each box (bottom). The figure was generated using Pedersen’s HierarchicalSets package for R72.
Figure 6.

Predicted gene gain, gene loss and recombination/mutation rates with the predicted evolutionary history of C. hyointestinalis. (a) Recombination-stripped core-gene based phylogeny, generated using BEAST2. (b) The same phylogeny as in (a), represented as two mirrored cladograms. Branch thickness denotes predicted gene gain (left) and gene loss (right) events as calculated using the Count software package74, and expressed as a percentage of the total genomic content. Branch colour denotes the log value of the predicted recombination to mutation ratio for each corresponding branch. Reference genome sequences not generated as part of this study are referred to as REF1 (GCA000705275), REF2 (GCA001643955), REF3 (GCA900116585) and LAW (C. hyointestinalis subsp. lawsonii, GCA001643975).
Figure 7.

Tanglegram depicting the relationship between the predicted evolutionary histories of C. hyointestinalis based on core gene phylogenies (left) and hierarchical clustering of the pangenome presence/absence matrix (right). Correspondences were used to designate clade groups, which are linked in the tanglegram using individual colours per clade.
The atypical isolate S1499c provided the most extreme example of genomic plasticity in this dataset, with unique predicted coding genes that accounted for more than 10% of its total gene content. Examination of the contig-aligned distribution of the inherited gene content in this isolate showed that large genomic islands had been incorporated into its genome on at least one occasion and read-mapping and coverage analysis confirmed this was unlikely to be an artefact of genomic assembly (data not shown). Isolates were grouped into descriptive clade categories based on their predicted phylogenetic and pangenomic relationships (Fig. 7), and the functions of the characteristic genes from each clade (genes that were both conserved and unique) were derived (Fig. 8). For atypical isolate S1499c, the majority of these inherited genes had unknown function. Of those genes with predicted functions, a large proportion was associated with metabolic processes and, intriguingly, many genes had functions linked to the exchange of genetic material.
Figure 8.

Accessory genome composition of the genomic clades of C. hyointestinalis. Clades are defined based on the congruence of the pangenome and phylogenetic analyses, as presented in Fig. 7. Gene groupings obtained by the use of get_homologues were used to define core and accessory genome components. Genes that are identified as unique to, and conserved within each defined clade are enumerated with respect to their functional category, as assigned using Eggnog-mapper71.
Characterisation of the predicted functions of clade-associated accessory genes showed that 25–40% of acquired genes had metabolic functions, and that gene acquisitions affecting cell membrane composition and signal transduction pathways occur frequently (Fig. 8). Additionally, multiple lineages had acquired genes that were associated with phage defence mechanisms such as restriction enzyme mediated, or CRISPR-mediated defence pathways.
To further explore the phage defence mechanisms employed by C. hyointestinalis, we characterised CRISPR elements and arrays from all available C. hyointestinalis genomes (Supplementary Table S4). All C. hyointestinalis subsp. hyointestinalis genomes contained components of at least one type I CRISPR-mediated defence system, whereas subspecies lawsonii had no CRISPR elements. Several lineages contained multiple copies of cas1, cas2 and cas9 genes. The number of CRISPR spacer sequences detectable in C. hyointestinalis genomes ranged from four to more than 100. Comparing the number of CRISPR arrays and the average spacer count per array to other Campylobacter genomes, we noted that the presence of CRISPR spacers is highly variable between Campylobacter species, and that C. hyointestinalis has both higher numbers of CRISPR array loci per genome and more individual spacer sequences per locus than most other species (Supplementary Figure 5).
Cytolethal distending toxin (CDT) is one of the main Campylobacter-associated protein toxins that has been linked to cytotoxicity41, and novel variants of CDT proteins have previously been reported in C. hyointestinalis42. We therefore compared gene similarity between all subunits of CDT identified within our genomic data to all publicly deposited gene sequences available in NCBI. We observed three distinct sub-categories for each of CDT subunits A, B and C (Supplementary Figure 6), and pseudogene variants caused by frame-shift mutations in subunits A and B (Supplementary Table 5).
Discussion
In summary, we have shown that C. hyointestinalis subspecies hyointestinalis may be commonly isolated from livestock (sheep, cattle and deer) in New Zealand. Extended literature analysis shows that this is a common, global picture but that relatively little data exist to help us understand the epidemiology of this microbial species. Current data suggest that cattle, and possibly deer, are the main reservoir hosts of C. hyointestinalis subsp. hyointestinalis whereas C. hyointestinalis subsp. lawsonii is more commonly associated with pigs (also see ref.36). Thus, C. hyointestinalis infection in humans could be an occupational or environmental hazard in rural areas, or associated with contaminated food production. For example, two articles identified raw milk consumption as a likely source of infection43,44. Geographical context and regional farming practices are also likely to influence C. hyointestinalis distribution and prevalence. C. hyointestinalis infection has frequently been reported in association with human gastrointestinal illness (see Supplementary Data S1). Subspecies-level classification of human-infecting C. hyointestinalis is absent from all these published observations. Data from PubMLST suggests that human infection is most frequently attributed to subspecies hyointestinalis, however this data is mainly derived from higher income countries.
Of particular concern to human health, studies from South Korea45,46 have identified C. hyointestinalis in cases of non-Helicobacter pylori gastrointestinal adenocarcinoma. Virulence factors secreted by H. pylori during chronic infection of the gastric mucosa are a known risk factor for carcinogenesis47. In our data, all C. hyointestinalis genomes possessed the known H. pylori-associated virulence factor vacuolating cytotoxin A (vacA)48, but lacked other known virulence factors including both cytotoxin-associated gene A (cagA) and oipA. Further studies in areas of high human C. hyointestinalis infection incidence would be required to determine whether equivalent pathologies are associated with C. hyointestinalis. Cytolethal distending toxin is the most studied proteinaceous element known to be associated with cytotoxicity41. C. hyointestinalis isolates have previously been demonstrated to secrete multiple variants of CDT49. Our genomic analysis shows that many C. hyointestinalis variants carry multiple gene paralogues of CDT subunits A and C, and that gene truncation of specific subunit types is common, likely resulting in on/off CDT variant expression as a result of genomic frame shifts. We also observe that such virulence-related genes are commonly lost and acquired as part of the organism’s accessory pangenome, suggesting that these proteins’ roles in virulence are often mediated in response to changing environmental stimuli and selection pressures. Further studies of the mechanisms of virulence in C. hyointestinalis will be required to elucidate its role in both gastric and non-gastric disease.
Our analyses show an extremely high level of plasticity between C. hyointestinalis genomes. Single nucleotide polymorphism distances between isolates of the same sub-species commonly fell in the range of 10,000 to 20,000 SNPs, but only 10% of these were predicted to fall in genomic regions with no history of recombination. Mutation and recombination are both estimated to have been significant driving forces in the diversification of this species. Our analyses suggest that recombination/mutation rates are highly lineage specific, and that recombination may be more frequent between closely related lineages (see Fig. 7). The significant role of recombination in C. hyointestinalis evolution (and other Campylobacter, or indeed all other prokaryotic species) has direct implications for our ability to classify variants using simplistic phylogenetic techniques that rely on the validity of the tree of life hypothesis. Here, we noted that even within standard typing loci (MLST), recombination histories have significantly influenced ancestry to the extent that, when recombination is not accounted for, ancestral node support is dramatically diminished. Extending this to the scale of the whole genome, we estimate that less than 5% of genomic content can be considered to be part of the organism’s clonal frame, and thus that the tree of life hypothesis can be considered invalid for the majority of species-level or sub-species level comparative applications. As has been noted elsewhere50, this highlights the importance of techniques that accommodate recombination histories39, or the preference for network-based51 or ring-based52 analyses for comparative classifications of bacteria over tree-based methods.
Furthermore, although approximately 78% of the gene content of C. hyointestinalis can be considered as conserved (core genes), a large amount of the variation between subspecies variants can be attributed to components of the accessory genome. Although total genome size is relatively stable, even at subspecies level the C. hyointestinalis genome can be considered as open (Heap’s law coefficient = 0.21). Thus gene gain and loss are frequent in C. hyointestinalis evolution, but the temporally sparse nature of available data means that quantification of the timescale of these events is not currently possible. Studies of other microbial organisms have suggested that speciation may be promoted by large scale gene acquisition events followed by differential gene loss53, and our analyses suggest that this may also be the case within C. hyointestinalis lineages. Pangenome analysis demonstrated common variation in genes associated with metabolic processes, cell surface antigen presentation, DNA exchange processes and defence mechanisms. This is likely to reflect genomic adaptation to varying environmental selection pressures in dynamic niches. In such a situation, C. hyointestinalis pangenomes may undergo adaptive change54, acquiring genes as public goods55 from communities of organisms cohabiting each niche. Genes conferring a survival advantage will be subject to positive selection, and may even displace others via adaptation to the changing requirements for survival. In the case of genes like the CDT subunits (see previously), such dynamic selection will likely influence virulence characteristics, and thus disease associations will depend greatly on a strain’s natural history, even between members of the same species or subspecies. Quantification of gene exchange in such organisms is essential to correct interpretations for classification based on similarity between lineages: mosaic genomes that result from continuously sampled genes as public goods may result in misleading tree-like evolutionary patterns55.
Overall, due to its ubiquitous nature and low suspected virulence, C. hyointestinalis is likely to remain a background source of infection in both humans and animals. However, further epidemiological and molecular investigations are warranted, particularly in developing countries where less information is available. Like most ‘emerging Campylobacter spp.’, C. hyointestinalis is not uniformly associated with human disease. Whether this is due to organism-associated traits or diagnostic bias remains to be explored on a worldwide basis. Frequent homologous and non-homologous recombination adds considerable complexity to the challenges of taxonomic classification and source attribution. This limits what can be generalised about the important traits of C. hyointestinalis such as its virulence, host-preference and metabolic capacity. Natural histories will play an important role in determining the genomic composition of each variant of C. hyointestinalis and thus studies of local variants are required to address local issues in both epidemiology and disease.
Materials and Methods
Literature survey
An initial list of publications for systematic review was compiled from the search results of both PubMed and Google Scholar, searching for any article in which the phrase “Campylobacter hyointestinalis” appeared at any position in the text. This search returned more than 1500 published research articles and reports. Further search inquiries were performed for any apparently relevant publications that were cited by articles that appeared in the original list. Articles were manually curated and assessed for their relevance. Only articles that reported original observations of C. hyointestinalis, and employed robust strategies to identify this particular species, were included in the final literature review (summarised in Supplementary Data 1).
Microbiology
Samples were collected from subprojects in our ongoingCampylobacter surveillance programme. In total, six hundred and sixty ruminant faecal samples were collected on 33 farms across the Manawatu district between 2008 and 2016, after pooling this represented a total of 225 independent samples (see Supplementary Table S1). Of these, 16 were dairy farms, seven were sheep farms, nine were sheep and beef farms and one was a red deer (Cervus elaphus) farm. Isolations from deer were performed in order to examine potential human disease sources, and therefore culture conditions were selected to mirror those used in human Campylobacter isolation. Sheep and cattle samples were collected as part of a wider, ongoing project which uses alternative culture conditions to assess the bacterial diversity of infections in ruminant livestock. Swabs were taken from fresh faeces on the ground. For deer, swab tips were inoculated into 3 ml Bolton Broth (Lab M, Bury, United Kingdom) and incubated in a microaerobic (85% nitrogen, 5% oxygen, 10% carbon dioxide) incubator (Don Whitley, Shipley, England) for 48 hours at 42 °C. Modified Cefoperazone Charcoal Deoxycholate Agar (mCCDA) plates (Fort Richard, Auckland, New Zealand) were inoculated with the broth and incubated microaerobically for 48 hours at 42 °C. For cattle and sheep, duplicate pools of four swabs were inoculated into i) 20 ml Bolton Broth and incubated as for the deer swabs and ii) 20 ml CAT Broth (Oxoid, Hampshire, England) and incubated in a hydrogen-enriched microaerobic (82% nitrogen, 3% oxygen, 10% carbon dioxide, 5% hydrogen) incubator (Don Whitley, Shipley, England) for 48 hours at 37 °C. mCCDA plates were inoculated with Bolton broth and incubated microaerobically for 48 hours at 42 °C. CAT plates (Fort Richard, Auckland, New Zealand) were inoculated with CAT broth and incubated in a hydrogen-enriched microaerobic incubator for 48 hours at 37 °C. Where growth was observed, two discrete colonies were removed and grown on separate Colombia horse blood agar plates (Fort Richard, Auckland, New Zealand) for 48 hours in the same atmosphere and temperature as the original plate.
Molecular Biology
Crude DNA extracts from colonies resembling Campylobacter were made and Campylobacter genus PCR performed using the primers of Linton56 as described by Bojanić et al.57. Positive isolates were speciated by PCR using the primers of Stucki58 for C. jejuni, those of Denis59 for C. coli and those of Linton56 for C. fetus, C. hyointestinalis, C. lari and C. upsaliensis, as described in Bojanić et al.57. Purified genomic DNA was extracted using a QIAamp Mini kit (Qiagen, Hilden, Germany) following the manufacturer’s instructions with minor modifications. Genomic DNA was prepared for sequencing using the Nextera XT library kit (Illumina, San Diego, USA). Libraries were sequenced on an Illumina HiSeq using 2 × 250 bp paired end sequencing by the Massey Genome Service (Palmerston North, New Zealand), part of New Zealand Genomics Ltd.
Bioinformatics, population genetics and evolutionary analysis
Illumina reads were trimmed using a combination of in-house software and Trimmomatic60, assembled using SPAdes version 3.9.061 and annotated using Prokka version 1.1162 as part of the Nullarbor pipeline (https://github.com/tseemann/nullarbor), which was also used to extract assembly statistics which are presented in Supplementary Table S2. Assembled draft genomes were deposited to NCBI with accession numbers NIQE00000000 – NIQT00000000.
Genus-level whole genome MLST was performed using the Genome Profiler software package63 under default settings and the complete genome of C. hyointestinalis LM926036 as a reference. Network diagrams were computed from the core genome alignment output of Genome Profiler using Splitstree version 4.14.464.
Seven-gene MLST alignments and isolate data were obtained for all isolates recorded in PubMLST65. Minimum spanning analysis was generated from MLST alignments using the PopArt software package (http://popart.otago.ac.nz), pairwise p-distances were calculated using a Jukes-Cantor substitution model in the MATLAB R2016b Statistics and the Machine Learning Toolbox. New seven-gene allele sequences were verified by PCR amplification66 and Sanger sequencing (Supplementary Data S3).
Sites of recombination and recombination to mutation ratios were estimated using Gubbins39. Phylogenetic analysis of recombination stripped alignments was performed in BEAST267 with an MCMC chain length of 100,000,000 using an HKY substitution model accounting for invariant sites, a strict molecular clock and a Yule population size model. Convergence of the phylogenetic Bayesian inference was verified in Tracer v1.6 (http://beast.bio.ed.ac.uk/Tracer), with all estimated parameters returning estimated sample sizes values greater than 200 after a 10% burn-in.
Whole genome comparison and the assignment of clusters of orthologous genes for genomes belonging to the C. hyointestinalis species was performed using get_homologues68 and the OrthoMCL clustering algorithm69. Prior to get_homologues execution, all reference genome datasets acquired from public data repositories were reannotated using Prokka version 1.1162 in order to ensure similar annotation schemes between comparisons. Protein functional groups were assigned to accessory genes using HMMER3 (http://hmmer.janelia.org/) to query BactNOG, the bacterial subset of the EggNOG4.5 database70 through the online EggNOG-mapper tool71. Pangenome composition was explored and visualised using the hierarchicalSets package in R72. Tanglegrams were generated using the dendextend package for R73. Gene gain and loss events were estimated using the Count software package74 based on the birth and death model described by Csűrös and Miklós53, and allowing variation in both the gain:loss and duplication:loss ratios.
For gene-based analysis of the cytolethal distending toxin (CDT) subunits; relevant clusters of orthologous genes were identified after functional characterisation in EggNog-mapper. Reference protein sequences were obtained from NCBI using the search terms “cytolethal distending toxin” and “organism contains ‘Campylobacter’”. Sequences were manually curated, aligned using ClustalW, and UPGMA tree representations of gene similarity were generated in the Geneious software package (Biomatters v.10.0.8). CRISPR spacer regions were detected using the CRISPRfinder75 software package, and repeats were enumerated per speices using custom-written scripts in MATLAB R2016b.
Accession numbers
Assembled draft genomes were deposited to NCBI with accession numbers NIQE00000000–NIQT00000000.
Electronic supplementary material
Acknowledgements
Cattle and sheep sampling in this study was made possible through funding from New Zealand’s Ministry of Primary Industries, grant code 17509. We would like to thank all Manawatu farmers who participated in this study, including the Massey University Deer unit. We would also like to thank Richard Fong and Pani Vijayan from the Massey Genome Service for their assistance with DNA sequencing as part of this project.
Author Contributions
D.A.W. and A.C.M. designed the study. D.A.W., A.J.O’D., A.F., L.E.R., R.N.A., H.M., P.J.B., N.P.F. and A.C.M. conducted the study. D.A.W. performed the analyses. D.A.W., N.P.F. and A.C.M. wrote the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-20889-x.
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Kaakoush NO, Castano-Rodriguez N, Mitchell HM, Man SM. Global Epidemiology of Campylobacter Infection. Clin Microbiol Rev. 2015;28:687–720. doi: 10.1128/CMR.00006-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caceres A, Munoz I, Iraola G, Diaz-Viraque F, Collado L. Campylobacter ornithocola sp. nov., a novel member of the Campylobacter lari group isolated from wild bird faecal samples. Int J Syst Evol Microbiol. 2017;67:1643–1649. doi: 10.1099/ijsem.0.001822. [DOI] [PubMed] [Google Scholar]
- 3.Gilbert MJ, et al. Campylobacter pinnipediorum sp. nov., isolated from pinnipeds, comprising Campylobacter pinnipediorum subsp. pinnipediorum subsp. nov. and Campylobacter pinnipediorum subsp. caledonicus subsp. nov. Int J Syst Evol Microbiol. 2017;67:1961–1968. doi: 10.1099/ijsem.0.001894. [DOI] [PubMed] [Google Scholar]
- 4.Iraola G, et al. Genomic evidence for the emergence and evolution of pathogenicity and niche preferences in the genus Campylobacter. Genome Biol Evol. 2014;6:2392–2405. doi: 10.1093/gbe/evu195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mullner P, et al. Assigning the source of human campylobacteriosis in New Zealand: a comparative genetic and epidemiological approach. Infect Genet Evol. 2009;9:1311–1319. doi: 10.1016/j.meegid.2009.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Friedman, C. R., Neimann, J., Wegener, H. C. & Tauxe, R. V. In Campylobacter Vol. II/6 121–138 (ASM International, 2000).
- 7.Skirrow MB. Campylobacter enteritis: a “new” disease. Br Med J. 1977;2:9–11. doi: 10.1136/bmj.2.6078.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gurtler M, Alter T, Kasimir S, Fehlhaber K. The importance of Campylobacter coli in human campylobacteriosis: prevalence and genetic characterization. Epidemiol Infect. 2005;133:1081–1087. doi: 10.1017/S0950268805004164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Karmali MA, Penner JL, Fleming PC, Williams A, Hennessy JN. The serotype and biotype distribution of clinical isolates of Campylobacter jejuni and Campylobacter coli over a three-year period. J Infect Dis. 1983;147:243–246. doi: 10.1093/infdis/147.2.243. [DOI] [PubMed] [Google Scholar]
- 10.Man SM. The clinical importance of emerging Campylobacter species. Nat Rev Gastroenterol Hepatol. 2011;8:669–685. doi: 10.1038/nrgastro.2011.191. [DOI] [PubMed] [Google Scholar]
- 11.Kalischuk LD, Inglis GD. Comparative genotypic and pathogenic examination of Campylobacter concisus isolates from diarrheic and non-diarrheic humans. BMC Microbiol. 2011;11:53. doi: 10.1186/1471-2180-11-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindblom GB, Sjogren E, Hansson-Westerberg J, Kaijser B. Campylobacter upsaliensis, C. sputorum sputorum and C. concisus as common causes of diarrhoea in Swedish children. Scand J Infect Dis. 1995;27:187–188. doi: 10.3109/00365549509019006. [DOI] [PubMed] [Google Scholar]
- 13.O’Donovan D, Corcoran GD, Lucey B, Sleator RD. Campylobacter ureolyticus. Virulence. 2014;5:498–506. doi: 10.4161/viru.28776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pacanowski J, et al. Campylobacter bacteremia: clinical features and factors associated with fatal outcome. Clin Infect Dis. 2008;47:790–796. doi: 10.1086/591530. [DOI] [PubMed] [Google Scholar]
- 15.Chua K, et al. Campylobacter insulaenigrae causing septicaemia and enteritis. J Med Microbiol. 2007;56:1565–1567. doi: 10.1099/jmm.0.47366-0. [DOI] [PubMed] [Google Scholar]
- 16.Martinot M, et al. Campylobacter lari bacteremia. Clin Microbiol Infect. 2001;7:96–97. doi: 10.1046/j.1469-0691.2001.00212.x. [DOI] [PubMed] [Google Scholar]
- 17.Kweon OJ, et al. First Case Report of Campylobacter volucris Bacteremia in an Immunocompromised Patient. J Clin Microbiol. 2015;53:1976–1978. doi: 10.1128/JCM.00442-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tee W, Mijch A. Campylobacter jejuni bacteremia in human immunodeficiency virus (HIV)-infected and non-HIV-infected patients: comparison of clinical features and review. Clin Infect Dis. 1998;26:91–96. doi: 10.1086/516263. [DOI] [PubMed] [Google Scholar]
- 19.Han XY, Tarrand JJ, Rice DC. Oral Campylobacter species involved in extraoral abscess: a report of three cases. J Clin Microbiol. 2005;43:2513–2515. doi: 10.1128/JCM.43.5.2513-2515.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wetsch NM, et al. Campylobacter curvus-associated hepatic abscesses: a case report. J Clin Microbiol. 2006;44:1909–1911. doi: 10.1128/JCM.44.5.1909-1911.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Siqueira JF, Jr., Rocas I. A 16S rDNA-based nested PCR protocol to detect Campylobacter gracilis in oral infections. Pesqui Odontol Bras. 2003;17:142–146. doi: 10.1590/S1517-74912003000200008. [DOI] [PubMed] [Google Scholar]
- 22.Man SM, et al. Campylobacter concisus and other Campylobacter species in children with newly diagnosed Crohn’s disease. Inflamm Bowel Dis. 2010;16:1008–1016. doi: 10.1002/ibd.21157. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, et al. Detection and isolation of Campylobacter species other than C. jejuni from children with Crohn’s disease. J Clin Microbiol. 2009;47:453–455. doi: 10.1128/JCM.01949-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morooka T, et al. Nosocomial meningitis due to Campylobacter fetus subsp. fetus in a neonatal intensive care unit. Eur J Pediatr. 1988;148:89–90. doi: 10.1007/BF00441825. [DOI] [PubMed] [Google Scholar]
- 25.Gurgan T, Diker KS. Abortion associated with Campylobacter upsaliensis. J Clin Microbiol. 1994;32:3093–3094. doi: 10.1128/jcm.32.12.3093-3094.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ursing JB, Lior H, Owen RJ. Proposal of minimal standards for describing new species of the family Campylobacteraceae. Int J Syst Bacteriol. 1994;44:842–845. doi: 10.1099/00207713-44-4-842. [DOI] [PubMed] [Google Scholar]
- 27.On, S. L. W., Miller, W. G., Houf, K., Fox, J. G. & Vandamme, P. Minimal standards for describing new species belonging to the families Campylobacteraceae and Helicobacteraceae: Campylobacter, Arcobacter, Helicobacter and Wolinella spp. Int J Syst Evol Microbiol, 10.1099/ijsem.0.002255 (2017). [DOI] [PMC free article] [PubMed]
- 28.Vandamme P, Debruyne L, De Brandt E, Falsen E. Reclassification of Bacteroides ureolyticus as Campylobacter ureolyticus comb. nov., and emended description of the genus Campylobacter. Int J Syst Evol Microbiol. 2010;60:2016–2022. doi: 10.1099/ijs.0.017152-0. [DOI] [PubMed] [Google Scholar]
- 29.GOODWIN CS, et al. Transfer of Campylobacter pylori and Campylobacter mustelae to Helicobacter gen. nov. as Helicobacter pylori comb. nov. and Helicobacter mustelae comb. nov., Respectively. International Journal of Systematic and Evolutionary Microbiology. 1989;39:397–405. [Google Scholar]
- 30.Gebhart CJ, Ward GE, Chang K, Kurtz HJ. Campylobacter hyointestinalis (new species) isolated from swine with lesions of proliferative ileitis. Am J Vet Res. 1983;44:361–367. [PubMed] [Google Scholar]
- 31.Gebhart CJ, Edmonds P, Ward GE, Kurtz HJ, Brenner DJ. “Campylobacter hyointestinalis” sp. nov.: a new species of Campylobacter found in the intestines of pigs and other animals. J Clin Microbiol. 1985;21:715–720. doi: 10.1128/jcm.21.5.715-720.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.On SL, Bloch B, Holmes B, Hoste B, Vandamme P. Campylobacter hyointestinalis subsp. lawsonii subsp. nov., isolated from the porcine stomach, and an emended description of Campylobacter hyointestinalis. Int J Syst Bacteriol. 1995;45:767–774. doi: 10.1099/00207713-45-4-767. [DOI] [PubMed] [Google Scholar]
- 33.Huang H, et al. Development of a monoclonal antibody-based colony blot immunoassay for detection of thermotolerant Campylobacter species. J Microbiol Methods. 2016;130:76–82. doi: 10.1016/j.mimet.2016.08.015. [DOI] [PubMed] [Google Scholar]
- 34.Lawson AJ, Shafi MS, Pathak K, Stanley J. Detection of campylobacter in gastroenteritis: comparison of direct PCR assay of faecal samples with selective culture. Epidemiol Infect. 1998;121:547–553. doi: 10.1017/S0950268898001630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Metherell LA, Logan JM, Stanley J. PCR-enzyme-linked immunosorbent assay for detection and identification of Campylobacter species: application to isolates and stool samples. J Clin Microbiol. 1999;37:433–435. doi: 10.1128/jcm.37.2.433-435.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller, W. G., Yee, E. & Chapman, M. H. Complete Genome Sequences of Campylobacter hyointestinalis subsp. hyointestinalis Strain LMG 9260 and C. hyointestinalis subsp. lawsonii Strain LMG 15993. Genome Announc4, 10.1128/genomeA.00665-16 (2016). [DOI] [PMC free article] [PubMed]
- 37.Rinttila T, Kassinen A, Malinen E, Krogius L, Palva A. Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR. J Appl Microbiol. 2004;97:1166–1177. doi: 10.1111/j.1365-2672.2004.02409.x. [DOI] [PubMed] [Google Scholar]
- 38.Brown PE, et al. Frequency and spatial distribution of environmental Campylobacter spp. Appl Environ Microbiol. 2004;70:6501–6511. doi: 10.1128/AEM.70.11.6501-6511.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Croucher NJ, et al. Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 2015;43:e15. doi: 10.1093/nar/gku1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carattoli A, et al. In silico detection and typing of plasmids using Plasmid Finder and plasmid multilocus sequence typing. Antimicrob Agents Chemother. 2014;58:3895–3903. doi: 10.1128/AAC.02412-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson WM, Lior H. A new heat-labile cytolethal distending toxin (CLDT) produced by Campylobacter spp. Microb Pathog. 1988;4:115–126. doi: 10.1016/0882-4010(88)90053-8. [DOI] [PubMed] [Google Scholar]
- 42.Samosornsuk W, et al. A new variant of cytolethal distending toxin in a clinical isolate of Campylobacter hyointestinalis. J Med Microbiol. 2015;64:1124–1134. doi: 10.1099/jmm.0.000145. [DOI] [PubMed] [Google Scholar]
- 43.Salama SM, Tabor H, Richter M, Taylor DE. Pulsed-field gel electrophoresis for epidemiologic studies of Campylobacter hyointestinalis isolates. J Clin Microbiol. 1992;30:1982–1984. doi: 10.1128/jcm.30.8.1982-1984.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Serraino A, et al. Presence of Campylobacter and Arcobacter species in in-line milk filters of farms authorized to produce and sell raw milk and of a water buffalo dairy farm in Italy. J Dairy Sci. 2013;96:2801–2807. doi: 10.3168/jds.2012-6249. [DOI] [PubMed] [Google Scholar]
- 45.Seo TH, et al. The origin of non-H. pylori-related positive Giemsa staining in human gastric biopsy specimens: A prospective study. Dig Liver Dis. 2011;43:23–27. doi: 10.1016/j.dld.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 46.Han HS, Lee KY, Lim SD, Kim WS, Hwang TS. Molecular identification of Helicobacter DNA in human gastric adenocarcinoma tissues using Helicobacter species-specific 16S rRNA PCR amplification and pyrosequencing analysis. Oncol Lett. 2010;1:555–558. doi: 10.3892/ol_00000098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Franco AT, et al. Regulation of gastric carcinogenesis by Helicobacter pylori virulence factors. Cancer Res. 2008;68:379–387. doi: 10.1158/0008-5472.CAN-07-0824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jones KR, Whitmire JM, Merrell DS. A Tale of Two Toxins: Helicobacter Pylori CagA and VacA Modulate Host Pathways that Impact Disease. Front Microbiol. 2010;1:115. doi: 10.3389/fmicb.2010.00115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamei K, et al. Campylobacter hyointestinalis Isolated from Pigs Produces Multiple Variants of Biologically Active Cytolethal Distending Toxin. Infect Immun. 2015;83:4304–4313. doi: 10.1128/IAI.00997-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Puigbo P, Wolf YI, Koonin EV. Genome-wide comparative analysis of phylogenetic trees: the prokaryotic forest of life. Methods Mol Biol. 2012;856:53–79. doi: 10.1007/978-1-61779-585-5_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Puigbo P, Wolf YI, Koonin EV. The tree and net components of prokaryote evolution. Genome Biol Evol. 2010;2:745–756. doi: 10.1093/gbe/evq062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lake JA, et al. Rings Reconcile Genotypic and Phenotypic Evolution within the Proteobacteria. Genome Biol Evol. 2016;8:578. doi: 10.1093/gbe/evw030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Csuros M, Miklos I. Streamlining and large ancestral genomes in Archaea inferred with a phylogenetic birth-and-death model. Mol Biol Evol. 2009;26:2087–2095. doi: 10.1093/molbev/msp123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McInerney JO, McNally A, O’Connell MJ. Why prokaryotes have pangenomes. Nat Microbiol. 2017;2:17040. doi: 10.1038/nmicrobiol.2017.40. [DOI] [PubMed] [Google Scholar]
- 55.McInerney JO, Pisani D, Bapteste E, O’Connell MJ. The Public Goods Hypothesis for the evolution of life on Earth. Biol Direct. 2011;6:41. doi: 10.1186/1745-6150-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Linton D, Owen RJ, Stanley J. Rapid identification by PCR of the genus Campylobacter and of five Campylobacter species enteropathogenic for man and animals. Res Microbiol. 1996;147:707–718. doi: 10.1016/S0923-2508(97)85118-2. [DOI] [PubMed] [Google Scholar]
- 57.Bojanic K, et al. Variation in the limit-of-detection of the ProSpecT Campylobacter microplate enzyme immunoassay in stools spiked with emerging Campylobacter species. J Microbiol Methods. 2016;127:236–241. doi: 10.1016/j.mimet.2016.06.016. [DOI] [PubMed] [Google Scholar]
- 58.Stucki U, Frey J, Nicolet J, Burnens AP. Identification of Campylobacter jejuni on the basis of a species-specific gene that encodes a membrane protein. J Clin Microbiol. 1995;33:855–859. doi: 10.1128/jcm.33.4.855-859.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Denis M, Refregier-Petton J, Laisney MJ, Ermel G, Salvat G. Campylobacter contamination in French chicken production from farm to consumers. Use of a PCR assay for detection and identification of Campylobacter jejuni and Camp. coli. J Appl Microbiol. 2001;91:255–267. doi: 10.1046/j.1365-2672.2001.01380.x. [DOI] [PubMed] [Google Scholar]
- 60.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bankevich A, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 63.Zhang J, Halkilahti J, Hanninen ML, Rossi M. Refinement of whole-genome multilocus sequence typing analysis by addressing gene paralogy. J Clin Microbiol. 2015;53:1765–1767. doi: 10.1128/JCM.00051-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23:254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 65.Jolley KA. & Maiden, M. C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics. 2010;11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Miller WG, et al. Multilocus sequence typing methods for the emerging Campylobacter Species C. hyointestinalis, C. lanienae, C. sputorum, C. concisus, and C. curvus. Front Cell Infect Microbiol. 2012;2:45. doi: 10.3389/fcimb.2012.00045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bouckaert R, et al. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput Biol. 2014;10:e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Contreras-Moreira B, Vinuesa P. GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl Environ Microbiol. 2013;79:7696–7701. doi: 10.1128/AEM.02411-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li L, Stoeckert CJ, Jr, Roos DS. OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003;13:2178–2189. doi: 10.1101/gr.1224503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huerta-Cepas J, et al. eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016;44:D286–293. doi: 10.1093/nar/gkv1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol10.1093/molbev/msx148 (2017). [DOI] [PMC free article] [PubMed]
- 72.Pedersen TL. Hierarchical sets: analyzing pangenome structure through scalable set visualizations. Bioinformatics. 2017;33:1604–1612. doi: 10.1093/bioinformatics/btx057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Galili T. dendextend: an R package for visualizing, adjusting and comparing trees of hierarchical clustering. Bioinformatics. 2015;31:3718–3720. doi: 10.1093/bioinformatics/btv428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Csuros M. Count: evolutionary analysis of phylogenetic profiles with parsimony and likelihood. Bioinformatics. 2010;26:1910–1912. doi: 10.1093/bioinformatics/btq315. [DOI] [PubMed] [Google Scholar]
- 75.Grissa I, Vergnaud G, Pourcel C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007;35:W52–57. doi: 10.1093/nar/gkm360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.