

Abstract
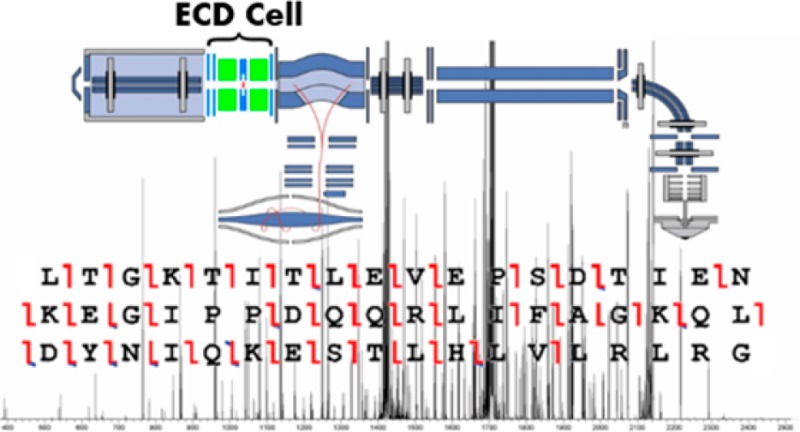
As the application of mass spectrometry intensifies in scope and diversity, the need for advanced instrumentation addressing a wide variety of analytical needs also increases. To this end, many modern, top-end mass spectrometers are designed or modified to include a wider range of fragmentation technologies, for example, ECD, ETD, EThcD, and UVPD. Still, the majority of instrument platforms are limited to more conventional methods, such as CID and HCD. While these latter methods have performed well, the less conventional fragmentation methods have been shown to lead to increased information in many applications including middle-down proteomics, top-down proteomics, glycoproteomics, and disulfide bond mapping. We describe the modification of the popular Q Exactive Orbitrap mass spectrometer to extend its fragmentation capabilities to include ECD. We show that this modification allows ≥85% matched ion intensity to originate from ECD fragment ion types as well as provides high sequence coverage (≥60%) of intact proteins and high fragment identification rates with ∼70% of ion signals matched. Finally, the ECD implementation promotes selective disulfide bond dissociation, facilitating the identification of disulfide-linked peptide conjugates. Collectively, this modification extends the capabilities of the Q Exactive Orbitrap mass spectrometer to a range of new applications.
Keywords: electron capture dissociation, middle-down, top-down, bottom-up, Orbitrap, Q Exactive, disulfide, protein, mass spectrometry, post-translational modifications
Introduction
The identity, abundance, structure, and degree of modification of proteins within a cell regulate the biological processes that occur. Because of the importance of understanding these protein characteristics, the field of proteomics aims to analyze the entirety of the proteoforms present within a system. Mass spectrometry (MS) has evolved to become a key analytical technique in the field of proteomics owing to its sensitivity, speed, and selectivity.1 In a standard MS proteomics experiment, the proteome of interest is isolated, digested by an enzyme, typically trypsin, and then the resulting peptides are separated with liquid chromatography (LC), fragmented, and mass-analyzed with tandem MS (MS/MS) using collisional activation. Then, the protein of origin is identified through database searching, matching experimental with theoretical fragmentation spectra.2,3 Collectively, this workflow is termed bottom-up proteomics. While bottom-up proteomics excels in identifying proteins, it also has limitations. Information about post-translational modifications (PTMs), residue point mutations, disulfide bond linkages, and protein microheterogeneity can be easily lost. To address these challenges, other methodologies have been developed. By analyzing longer peptide segments, which can be generated by enzymes other than trypsin, or analyzing intact proteins through techniques termed middle-down and top-down proteomics, respectively, more in-depth information about the protein’s structure, function, and biological state, particularly concerning the influence of multiple PTMs, can be ascertained.4−7 However, while the separation, fragmentation, and database searching of tryptic peptides have been thoroughly optimized, there is still a need to optimize the workflows for these emerging applications.
Bottom-up proteomics has so far relied heavily on the use of collisional activation, such as collisional-induced dissociation (CID) and higher-energy collisional induced dissociation (HCD), as these perform very well for tryptic peptides. When CID and HCD are applied in middle-down or top-down proteomic workflows, these techniques become limited in extent of backbone cleavage, termed sequence coverage, and PTM retention, hampering the identification and localization of modifications and mutations.8,9 To improve upon these issues, alternative fragmentation methods have been developed.10−12 Of note are electron-based fragmentation methods, for example, electron-capture dissociation (ECD) and electron-transfer dissociation (ETD).
ECD operates by passing a multiply charged precursor ion through a confined low-energy electron beam. As the precursor ion moves through this beam, an electron capture event can occur, giving rise to charge reduction and subsequent covalent bond cleavage. The predominant bonds cleaved are the backbone N–Cα bond, yielding c and z ions, with the exception of N-terminal proline cleavage, due to the proline’s cyclic nature.10,13,14 Unlike CID, the backbone fragmentation process is more stochastic, giving rise to higher sequence coverage.15 In addition to these protein backbone cleavages, ECD has been demonstrated to preferentially cleave disulfide bonds.16 Disulfide bonds present an analytical challenge both in top-down proteomics and in mapping of the disulfide bonds, as CID and HCD do not cleave the disulfide bond without special functionalization of the disulfide bond,17 thus hampering the fragmentation and identification of the linked conjugated proteins/peptides. Collectively, these attributes of ECD make it advantageous for inclusion in instrument platforms to extend the instrument’s capabilities.
For some time, ECD has been available on FT-ICR instruments.18 Additionally, in recent years two new approaches on hybrid Q-ToF instruments as well as ECD implementations on ion trap instruments have been reported.19−22 Furthermore, an electromagnetostatic cell, which uses a magnetic field to confine low-energy electrons in a flight path of the analyte ion, facilitating ECD fragmentation without the need for long reaction times or ultrahigh vacuum has been reported.23−26 Herein, we describe a novel modification of one of the most widely used proteomic instrument platforms, the Q Exactive Orbitrap mass spectrometer, to enable ECD fragmentation through the use of a newly designed electromagnetostatic ECD cell, utilizing a double-pass design to increase fragmentation efficiency and sequence coverage of analyte ions, while also allowing for a combination of ECD and HCD. We use standard peptides and proteins to demonstrate successful ECD fragmentation, with data showing high sequence coverage for peptides and proteins and ≥85% of matched fragments coming from c/z-type fragment ions as well as the applicability of the modification to a range of applications including top-down identification and disulfide bond mapping.
Instrumentation and Methods
ECD Cell: Design and Principles of Operation
The cell, shown in Figure 1, is housed in an aluminum sleeve that fits on the front end of the HCD-cell and connects to the exit lens from the C-trap. Internally the ECD cell is composed of seven electrostatic lenses (E1 through E7) with an internal diameter of 3.0 mm. The width of each electrode is listed in Supplemental Table 1. E4 houses the filament, a single loop of W4 rhenium wire (Scientific Instrument Services, Ringoes, NJ), which is heated through variable current regulation and is biased with an independent electrostatic potential. Two samarium–cobalt (Sm2Co17) ring magnets, which are magnetized through the thickness of the ring and have an operational temperature of up to 350 °C (Grade SM2435, Chino Magnetism, Fairfield, NJ), are on either side of E4 with opposing polarities facing each other to establish a “bottle” configuration of the magnetic field (Supplemental Figure 1). During ECD operation, the filament current is increased to start the emission of electrons from the filament surface. The magnetic field confines these free electrons in the radial dimension, while a potential barrier that is applied to both E1 and E6 confines the electrons in the axial dimension. ECD occurs when a precursor ion is moved through this confined electron beam (ca. electron energy <1 eV).
Figure 1.
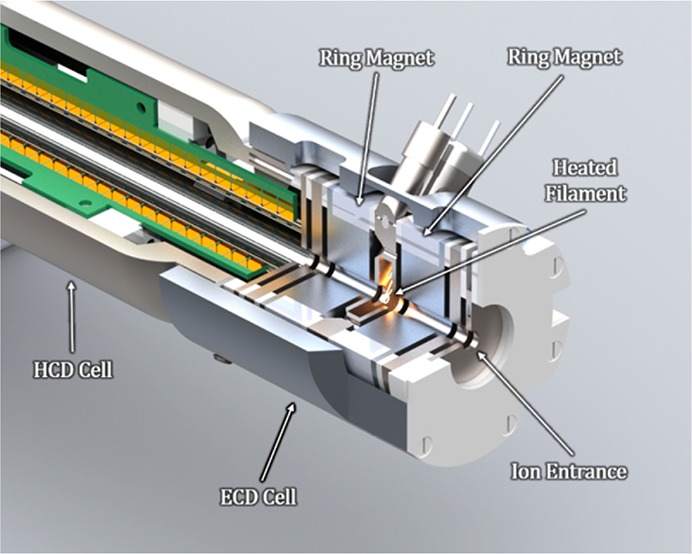
Schematic of the ECD cell attached to the front of the HCD-cell. The ECD cell consists of seven electrostatic electrodes and two ring magents. Electrons are generated by a heated rhenium filament at the center of the cell.
It is of interest to consider the choice to implement ECD over ETD, which has been developed on many commercial and research instrument platforms. ECD and ETD result in similar fragmentation patterns and offer many similar analytical advantages; however, the implementations of the two techniques are different. With ECD, as described above, a precursor cation captures an electron to initiate the first steps of fragmentation; however, the use of electrons does not allow for trapping of the cation and electron together, and thus the overlap between cations and electrons is often very short, leading to low fragmentation efficiencies. In contrast, ETD uses an anionic transfer reagent, often a small molecule such as fluoranthene, which can be trapped concurrently with the precursor cation, allowing for better overlap between the two species. To perform ETD, the instrument must include an ion trap capable of trapping both cations and anions simultaneously, as shown by the implementation of ETD on several linear ion trap orbitrap mass spectrometers. Here we aimed to increase the fragmentation capabilities of the Q Exactive mass spectrometer through the least invasive means possible in a way that can be implemented without needing to access proprietary internal programming of commercial instrument platforms. Owing to the base hardware design of the Q Exactive Orbitrap mass spectrometer, implementation of ETD would require major modification to the ion timing workflow coding within the embedded PC of the instrument and major modifications to hardware design itself: Both of which would preclude the majority of researchers from being able to implement such modifications on their own machine and workflows. Instead, the design of the ECD cell described herein does not require major hardware modifications and can operate autonomously from the internal programming of the mass spectrometer. Moreover, because of the similarity in analytical metrics of the two fragmentation techniques, the inclusion of ECD is more amenable to and will have a greater impact on this instrument platform.
Sample Preparation, Data Acquisition, and Data Analysis
Solutions of substance P (SP), myoglobin, and ubiquitin (Sigma-Aldrich, Zwijndrecht, Netherlands) were prepared to concentrations of 2 μM using 99.9% water and 0.1% formic acid. Aliquots of each solution were loaded into a gold-coated glass capillary emitter and biased +1.2 to 1.35 kV with respect to heated inlet of the mass spectrometer. ECD fragmentation mass spectra were collected on a Q Exactive Orbitrap mass spectrometer modified with the inclusion of an ECD cell. The terminal electrode of the ECD cell was electrically coupled to the C-trap exit lens. The electrostatic potentials applied to other lenses as well as the current applied to the filament were manually tuned to optimize transmission and fragmentation efficiency. The filaments used were an uncoated rhenium wire (ECD of SP) or one coated with yttrium oxide (ECD of intact proteins). Mass spectra of intact proteins were collected with 50 μscans at 140 000 mass resolution and an ion injection time of 100 ms. Mass spectra of peptides were collected with 10 μscans at 35 000 mass resolution and an ion injection time of 50 ms. A typical peptide fragmentation spectra is obtainable in <30 s, while protein ECD spectra typically require longer (∼1 min) acquisitions to obtain good signal-to-noise fragmentation spectra. The injection energy into the HCD-cell was tuned to minimize collisional activation during ECD-only fragmentation and set to 25 normalized collision energy (NCE) during ECD-HCD fragmentation. Methods for ECD of phosphorylated peptides are presented in the Supporting Information. For HCD fragmentation of ubiquitin and myoglobin, the instrument was unmodified; that is, the ECD cell modification was removed, and the NCE was scanned from 15 to 25. To address the specificity of ECD toward disulfide bond dissociation, a nonreduced tryptic digest of human serum albumin (HSA) (Sigma-Aldrich, Zwijndrecht, Netherlands) was prepared to generate disulfide-linked peptide species and analyzed with LC–MS/MS. The ECD spectra were collected with 5 μscans at 17 500 mass resolution. Further details regarding the method of digestion and LC–MS/MS workflow can be found in the Supporting Information.
For tandem spectra of SP, fragments were manually analyzed for b, c, y, and z ion types. For intact protein analyses, extracted mass spectra were deconvoluted using Auto Xtract in Protein Deconvolution 4.0 (Thermo Fisher Scientific, Bremen, Germany). Deconvolution settings were S/N Threshold: 1, Fit Factor: 80%, and Remainder Threshold: 25%. Deconvoluted results were searched using ProSight Lite.27 Ion types matched were b, y, c, and z with a 10 ppm mass error limit.
Results and Discussion
Performance and Advantages of ECD Cell Design
The ECD fragmentation spectrum of SP [M + 2H]2+ ion (Figure 2a) demonstrates the advantages of the design of the modification. The spectrum consists of a full range of c ions, with only the prohibited N-terminal proline cleavages missing, and one z ion, confirming that the ECD cell is well functioning and capable of selective ECD fragmentation (matched fragments are listed in Supplemental Table 2). Of interest is the charge state of the precursor. While both ECD and ETD have demonstrated their utility to numerous applications in the proteomic field, one inherent limitation is the charge-state dependence; that is, as the charge state is reduced, the fragmentation efficiency also reduces.28 Thus [M+2H]2+ ions present a challenging case for electron fragmentation methods; however, the ECD cell design takes steps to address this limitation. The placement of the device between the HCD-cell and the C-trap allows for analytes to undergo two ECD fragmentation events, one upon going into the HCD-cell from the C-trap and the second when being ejected from the HCD-cell for mass analysis. Thus even for the lowest observed charge state of SP, the ECD fragmentation spectrum produces 80% sequence coverage with >90% of the matched ion intensity arising from ECD type fragment ions (Figure 2a, inset). Moreover, ECD analysis of phosphorylated peptide ions demonstrates that the device is capable of maintaining the labile modification and provides site localization information (Supplemental Figures 2–5).
Figure 2.

ECD and EChcD spectra of substance P. The ECD (a) spectrum shows ≥90% fragment intensity from c/z ion types. EChcD (b) fragmentation shows a combination of both b/y and c/z ion types. Insets show the fragment ion intensity percentage. Annotations are predominate b, c, y, and z ions. b/y ions are shown as blue brackets and c/z ions are shown as red brackets. Typical peptide fragmentation spectra are obtainable in <30 s of data acquisition.
Other approaches to increase fragmentation efficiency include the use of supplemental activation. Of note, Frese et al. created a hybrid technique termed EThcD whereby the ion of interest is first subject to ETD fragmentation and then the remaining precursor is fragmented with HCD, allowing for the benefits of both techniques to be present in the resulting fragmentation spectrum.29 Analogous to this, the ECD cell modification can be operated in a dual mode with both ECD and HCD, newly termed here as EChcD. During EChcD, the precursor is subjected to the first round of ECD fragmentation; however, the injection energy into the HCD-cell is increased to a 25 NCE, fragmenting the remaining precursor with HCD fragmentation. The EChcD fragmentation spectrum for the [M+2H]2+ precursor of SP is shown in Figure 2b. Similar to EThcD, the spectrum shows a combination of both ECD and HCD fragment types (Figure 2b, inset). All of the c ions present in the ECD spectrum are retained, while there are an additional nine b ions and two y ions present, giving full sequence coverage with multiple b/c pairings, termed “golden pairs”, of the peptide ion. (Matched fragment ions are listed in Supplemental Table 3.) These data indicate that the ECD cell adds a high degree of functionality, that is, the ability to do HCD, ECD, EChcD, to the mass spectrometer and allows for enhanced workflows to be utilized.
Application to Top-Down Proteomics
Fragmentation of longer peptides (middle-down) or intact proteins (top-down) via HCD or CID is largely hampered by the selective cleavage of labile bonds that can lead to poor sequence coverage for peptides or proteins with molecular weights >10 kDa.9 To address this, the application of electron-based fragmentation methods, surface-induced dissociation, and photodissociation have been used, and the modification of commercial instrumentation platforms to include these fragmentation types is an active field of study.9,30 To explore the utility of the ECD modification to the field of top-down proteomics, the analysis of two midsized model proteins, ubiquitin and myoglobin, was performed.
The ECD fragmentation spectrum of ubiquitin (Figure 3a) shows a high specificity for the formation of c/z ion types with 85% of the total fragment composition originating from these ions (Supplemental Table 4). The same specificity is shown for the fragmentation of myoglobin (Figure 3b, Supplemental Table 5), with 93% fragment composition coming from ECD fragment types. Furthermore, these ECD spectra exhibit high sequence coverage, 80 and 60% for ubiquitin and myoglobin, respectively. To benchmark the ECD fragmentation performance versus the unmodified instrument, HCD fragmentation was performed at a range of NCE values for both protein ions. HCD of ubiquitin shows that as NCE is increased from 15 to 25 the sequence coverage also increases and reaches a maximum that is similar to that obtained with ECD (Table 1). For myoglobin, HCD performs substantially worse than ECD, and the opposite trend is shown with maximal sequence coverage, 45%, occurring at 15 NCE and dropping to 18% at 25 NCE. These data demonstrate that the ECD modification outperforms the unmodified instrument for top-down analysis of myoglobin as well as highlight an apparent limitation of HCD for top-down proteomic experiments; that is, HCD must perform well for a wide range of proteins to be effective in these experiments.
Figure 3.

ECD fragmentation spectra of ubiquitin [M+8H]8+ (a) and Myoglobin [M+20H]20+ (b). Below each spectrum are the corresponding deconvoluted matched fragment ions, as obtained from Prosight Lite. Red brackets are matched c/z ions, while blue brackets are matched b/y ions. Data acquisition was performed for ∼1 min.
Table 1. Collision Energy (NCE), Sequence Coverage, and Percentage of Explained Fragments for the HCD and ECD Fragmentation of Ubiquitin and Myoglobin.
| analyte | NCE | % sequence coverage | % fragments explained |
|---|---|---|---|
| ubiquitin | 15 | 53 | 25 |
| 18 | 67 | 23 | |
| 20 | 69 | 20 | |
| 22 | 75 | 14 | |
| 25 | 81 | 11 | |
| ECD | 80 | 69 | |
| myoglobin | 15 | 45 | 23 |
| 18 | 48 | 13 | |
| 20 | 43 | 10 | |
| 22 | 28 | 5 | |
| 25 | 18 | 3 | |
| ECD | 60 | 74 |
During to- and middle-down experiments, proteins of varying identity, molecular weight, chemical composition, and degree of modification elute from an LC column and will be subjected to fragmentation in the mass spectrometer. To elucidate full information from this experiment, the fragmentation technique must perform well for a wide range of proteins. The top-down data show that HCD fragmentation needs to be optimized for each individual protein; for example, NCE 15 produces the highest sequence coverage for myoglobin, while NCE 25 is required for ubiquitin. In contrast, ECD produces high sequence coverage for both proteins without optimizing parameters for each individual analyte and thus will be more amenable to a range of unknown proteins.
In addition to the need of a versatile fragmentation method, a successful top-down proteomic experiment also requires the elucidation of protein identity from a fragmentation mass spectrum by searching against the full proteome database and this search returning the correct protein identification. To this end, the quality and percentage of explained fragments present in the spectrum becomes increasingly important as it limits search space and reduces the chances of falsely assigning an unknown protein.31 At 15 NCE, the fragmentation spectrum of ubiquitin resulted in the highest percentage of matched fragments with 25%, but as the collision energy increased to 25 NCE, the number of explained fragments decreased to 11%. Similarly, the fragmentation of myoglobin occurred whereby an NCE of 15 generated 23% explainable fragments and decreased to 3% at 25 NCE. In contrast, ECD spectra resulted in 69% of explained fragments for ubiquitin and 60% for myoglobin. These data indicate that ECD is a more specific fragmentation technique with regard to ion types produced, which when applied to an unknown protein sample will aid in correctly identifying the analyte and limit the search space.
Application of ECD for Disulfide Mapping
A potential interesting field of application of the ECD modification is the use in disulfide mapping. Because disulfides play a critical role in the high-order structure of proteins, postcolumn, online disulfide reduction is gaining a lot of focus in recent years as it facilitates the identification of disulfide-bonded species and their constituting peptides within a protein sequence.32−34 ECD has previously been shown to dissociate many disulfide bonds more rapidly than any other bonds.16 To explore this bond specificity in disulfide mapping, the disulfide-bonded species from a nonreduced tryptic digestion of human serum albumin (HSA) were subjected to ECD fragmentation. As HSA contains 17 disulfide bonds and 8 occurrences of two adjacent cysteine residues, proteolytic digestion of HSA will inherently produce multichain species connected by two or more disulfide bonds. Figure 4 shows two examples of disulfide-linked peptides from HSA, each consisting of three peptides linked by two disulfide bonds. With the ECD modification, selective disulfide dissociation was observed, as the most intense fragment ions were the free disulfide-cleaved peptides (Figure 4). This observation of free reduced peptides allows for easy identification of which peptides constitute the intact disulfide-bonded precursor. In addition to the free peptides, partially reduced peptide fragments were also observed in the spectra. These partially reduced species are the result of selective cleavage of only one of the two disulfide bonds. If a peptide involved in disulfide bonding contains more than a single cysteine residue, for example, T21 and T75 peptides, then these partially reduced species have the potential to unravel the exact cysteine pairings down to the residue level by further fragmentation of these species.33 However, depending on the purpose of disulfide mapping in specific studies, confirmation of expected peptides to be disulfide-bonded after enzymatic digestion is often sufficient. For this, the ECD modification on a Q Exactive Orbitrap shows a promising potential.
Figure 4.

ECD spectra of two disulfide linked tripeptide conjugates from a HSA digest. For both, the spectra demonstrate that the disulfide bonds are selectively cleaved and release both free peptides and partially reduced peptide fragments. Numberings are according to an in silico tryptic digestion of HSA. Data were obtained with single MS scans.
Conclusions
An electromagnetostatic ECD cell modification was designed for the benchtop Q Exactive Orbitrap mass spectrometer and characterized. Data presented herein demonstrate that the modification adds ECD fragmentation capabilities and produces >85% signal intensity for ECD fragment ion types, that is, c and z ions, while being able to provide high sequence coverage for both peptides and proteins. The ECD cell is even able to produce this high sequence coverage for the [M+2H]2+ ion of SP without the need for supplemental activation owing to its two-stage ECD fragmentation design. In addition, HCD functionality of the instrument is maintained and allows for the combination of ECD with HCD, termed here EChcD, providing complementary fragmentation information similar to that of EThcD. The utility to workflows such as top-down proteomics and disulfide bond mapping is also demonstrated. For top-down experiments, the modification shows high specificity in fragments generated as the number of match fragments is >60% in ECD as compared with <25% with HCD, which will allow for more precise identification of unknown proteins during full proteome experiments.
Finally, selective disulfide bond cleavage is shown by the full and partial reduction of peptides linked through multiple disulfide bonds. The full reduction of these species allows for the identification of linked peptides through their intact mass, while the partially reduced species could potentially be used to elucidate the specific residues linked for peptides containing more than one cysteine. Collectively, the inclusion of the ECD cell modification expands the utility of the Q Exactive to workflows that were previously unavailable on this platform, increasing its versatility for modern proteomic research.
Acknowledgments
K.L.F. and A.J.R.H. are funded by the large-scale proteomics facility Proteins@Work (Project 184.032.201) embedded in The Netherlands Proteomics Centre and supported by The Netherlands Organization for Scientific Research (NWO). They acknowledge additional support through the European Union Horizon 2020 program FET-OPEN project MSmed, Project 686547. C.N.C. was supported by The Danish Agency for Science, Technology and Innovation and the Novo Nordisk STAR program. The Novo Nordisk Foundation Center for Protein Research (CPR) is supported financially by the Novo Nordisk Foundation (grant agreement NNF14CC0001). This work was also supported by an NIH SBIR (R44 GM122131-01 awarded to V.G.V.).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jproteome.7b00622.
Methods for LC–MS, ECD of phosphopeptides, electron trajectory simulations, and fragmentation tables. Supplemental Figure 1: Magnetic vector potentials. Supplemental Figure 2: ECD fragmentation spectrum of a singly phosphorylated peptide ion. Supplemental Figure 3: ECD fragmentation spectrum of a peptide with a phosphorylation and acetylation modification. Supplemental Figure 4: ECD fragmentation spectrum of a singly phosphorylated peptide ion. Supplemental Figure 5: ECD fragmentation spectrum of a singly phosphorylated peptide ion. Supplemental Table 1: Width of each electrode in the ECD cell. Supplemental Table 2: Matched fragments for ECD of [M+2H]2+ substance P. Supplemental Table 3: Matched fragment ions for EChcD of [M+2H]2+ substance P. Supplemental Table 4: Matched fragment ions for ECD of [M+8H]8+ ubiquitin Supplemental Table 5: Matched fragments for ECD of [M+20H]20+ myoglobin. (PDF)
The authors declare the following competing financial interest(s): V.G.V., Y.V.V., N.I.L., and J.S.B. are employees of eMSion, the company that aims to commercialize the ECD cell designed and described here.
Supplementary Material
References
- Aebersold R.; Mann M. Mass spectrometry-based proteomics. Nature 2003, 422 (6928), 198–207. 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Cox J.; Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26 (12), 1367–1372. 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J. C.; Creasy D. M.; Cottrell J. S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20 (18), 3551–3567. . [DOI] [PubMed] [Google Scholar]
- McLafferty F. W.; Breuker K.; Jin M.; Han X. M.; Infusini G.; Jiang H.; Kong X. L.; Begley T. P. Top-down MS, a powerful complement to the high capabilities of proteolysis proteomics. FEBS J. 2007, 274 (24), 6256–6268. 10.1111/j.1742-4658.2007.06147.x. [DOI] [PubMed] [Google Scholar]
- Cristobal A.; Marino F.; Post H.; van den Toorn H. W. P.; Mohammed S.; Heck A. J. R. Toward an Optimized Workflow for Middle-Down Proteomics. Anal. Chem. 2017, 89 (6), 3318–3325. 10.1021/acs.analchem.6b03756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiatsiani L.; Heck A. J. R. Proteomics beyond trypsin. FEBS J. 2015, 282 (14), 2612–2626. 10.1111/febs.13287. [DOI] [PubMed] [Google Scholar]
- Lossl P.; Brunner A. M.; Liu F.; Leney A. C.; Yamashita M.; Scheltema R. A.; Heck A. J. R. Deciphering the Interplay among Multisite Phosphorylation, Interaction Dynamics, and Conformational Transitions in a Tripartite Protein System. ACS Cent. Sci. 2016, 2 (7), 445–455. 10.1021/acscentsci.6b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raspopov S. A.; El-Faramawy A.; Thomson B. A.; Siu K. W. M. Infrared Multiphoton Dissociation in Quadrupole Time-of-Flight Mass Spectrometry: Top-Down Characterization of Proteins. Anal. Chem. 2006, 78 (13), 4572–4577. 10.1021/ac052248i. [DOI] [PubMed] [Google Scholar]
- Shaw J. B.; Li W.; Holden D. D.; Zhang Y.; Griep-Raming J.; Fellers R. T.; Early B. P.; Thomas P. M.; Kelleher N. L.; Brodbelt J. S. Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J. Am. Chem. Soc. 2013, 135 (34), 12646–12651. 10.1021/ja4029654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubarev R. A.; Kelleher N. L.; McLafferty F. W. Electron capture dissociation of multiply charged protein cations. A nonergodic process. J. Am. Chem. Soc. 1998, 120 (13), 3265–3266. 10.1021/ja973478k. [DOI] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (26), 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodbelt J. S. Photodissociation mass spectrometry: new tools for characterization of biological molecules. Chem. Soc. Rev. 2014, 43 (8), 2757–2783. 10.1039/C3CS60444F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syrstad E. A.; Turecček F. Toward a general mechanism of electron capture dissociation. J. Am. Soc. Mass Spectrom. 2005, 16 (2), 208–224. 10.1016/j.jasms.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Horn D. M.; Fridriksson E. K.; Kelleher N. L.; Kruger N. A.; Lewis M. A.; Carpenter B. K.; McLafferty F. W. Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal. Chem. 2000, 72 (3), 563–573. 10.1021/ac990811p. [DOI] [PubMed] [Google Scholar]
- Cooper H. J.; Hakansson K.; Marshall A. G. The role of electron capture dissociation in biomolecular analysis. Mass Spectrom. Rev. 2005, 24 (2), 201–222. 10.1002/mas.20014. [DOI] [PubMed] [Google Scholar]
- Zubarev R. A.; Kruger N. A.; Fridriksson E. K.; Lewis M. A.; Horn D. M.; Carpenter B. K.; McLafferty F. W. Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity. J. Am. Chem. Soc. 1999, 121 (12), 2857–2862. 10.1021/ja981948k. [DOI] [Google Scholar]
- Gunawardena H. P.; O’Hair R. A. J.; McLuckey S. A. Selective Disulfide Bond Cleavage in Gold(I) Cationized Polypeptide Ions Formed via Gas-Phase Ion/Ion Cation Switching. J. Proteome Res. 2006, 5 (9), 2087–2092. 10.1021/pr0602794. [DOI] [PubMed] [Google Scholar]
- Qi Y. L.; Volmer D. A. Electron-based Fragmentation Methods in Mass Spectrometry: An Overview. Mass Spectrom. Rev. 2017, 36 (1), 4–15. 10.1002/mas.21482. [DOI] [PubMed] [Google Scholar]
- Baba T.; Campbell J. L.; Le Blanc J. C. Y.; Hager J. W.; Thomson B. A. Electron Capture Dissociation in a Branched Radio-Frequency Ion Trap. Anal. Chem. 2015, 87 (1), 785–792. 10.1021/ac503773y. [DOI] [PubMed] [Google Scholar]
- Robb D. B.; Brown J. M.; Morris M.; Blades M. W. Tandem Mass Spectrometry Using the Atmospheric Pressure Electron Capture Dissociation Ion Source. Anal. Chem. 2014, 86 (9), 4439–4446. 10.1021/ac5002959. [DOI] [PubMed] [Google Scholar]
- Ding L.; Brancia F. L. Electron Capture Dissociation in a Digital Ion Trap Mass Spectrometer. Anal. Chem. 2006, 78 (6), 1995–2000. 10.1021/ac0519007. [DOI] [PubMed] [Google Scholar]
- Silivra O. A.; Kjeldsen F.; Ivonin I. A.; Zubarev R. A. Electron capture dissociation of polypeptides in a three-dimensional quadrupole ion trap: Implementation and first results. J. Am. Soc. Mass Spectrom. 2005, 16 (1), 22–27. 10.1016/j.jasms.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Voinov V. G.; Deinzer M. L.; Barofsky D. F. Electron capture dissociation in a linear radiofrequency-free magnetic cell. Rapid Commun. Mass Spectrom. 2008, 22 (19), 3087–3088. 10.1002/rcm.3709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Kapp E. A.; Lothian A.; Roberts A. M.; Vasil’ev Y. V.; Boughton B. A.; Barnham K. J.; Kok W. M.; Hutton C. A.; Masters C. L.; Bush A. I.; Beckman J. S.; Dey S. G.; Roberts B. R. Characterization and Identification of Dityrosine Cross-Linked Peptides Using Tandem Mass Spectrometry. Anal. Chem. 2017, 89 (11), 6136–6145. 10.1021/acs.analchem.7b00941. [DOI] [PubMed] [Google Scholar]
- Voinov V. G.; Hoffman P. D.; Bennett S. E.; Beckman J. S.; Barofsky D. F. Electron Capture Dissociation of Sodium-Adducted Peptides on a Modified Quadrupole/Time-of-Flight Mass Spectrometer. J. Am. Soc. Mass Spectrom. 2015, 26 (12), 2096–2104. 10.1007/s13361-015-1230-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voinov V. G.; Bennett S. E.; Beckman J. S.; Barofsky D. F. ECD of Tyrosine Phosphorylation in a Triple Quadrupole Mass Spectrometer with a Radio-Frequency-Free Electromagnetostatic Cell. J. Am. Soc. Mass Spectrom. 2014, 25 (10), 1730–1738. 10.1007/s13361-014-0956-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellers R. T.; Greer J. B.; Early B. P.; Yu X.; LeDuc R. D.; Kelleher N. L.; Thomas P. M. ProSight Lite: Graphical software to analyze top-down mass spectrometry data. Proteomics 2015, 15 (7), 1235–1238. 10.1002/pmic.201400313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavarone A. T.; Paech K.; Williams E. R. Effects of charge state and cationizing agent on the electron capture dissociation of a peptide. Anal. Chem. 2004, 76 (8), 2231–2238. 10.1021/ac035431p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J. R.; Mohammed S. Toward Full Peptide Sequence Coverage by Dual Fragmentation Combining Electron-Transfer and Higher-Energy Collision Dissociation Tandem Mass Spectrometry. Anal. Chem. 2012, 84 (22), 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Blackwell A. E.; Dodds E. D.; Bandarian V.; Wysocki V. H. Revealing the Quaternary Structure of a Heterogeneous Noncovalent Protein Complex through Surface-Induced Dissociation. Anal. Chem. 2011, 83 (8), 2862–2865. 10.1021/ac200452b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii A. I. A survey of computational methods and error rate estimation procedures for peptide and protein identification in shotgun proteomics. J. Proteomics 2010, 73 (11), 2092–2123. 10.1016/j.jprot.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Lei Q. P.; Washabaugh M. Characterization of IgG2 Disulfide Bonds with LC/MS/MS and Postcolumn Online Reduction. Anal. Chem. 2016, 88 (10), 5080–5087. 10.1021/acs.analchem.5b04368. [DOI] [PubMed] [Google Scholar]
- Cramer C. N.; Kelstrup C. D.; Olsen J. V.; Haselmann K. F.; Nielsen P. K. Complete Mapping of Complex Disulfide Patterns with Closely-Spaced Cysteines by In-Source Reduction and Data-Dependent Mass Spectrometry. Anal. Chem. 2017, 89 (11), 5949–5957. 10.1021/acs.analchem.7b00424. [DOI] [PubMed] [Google Scholar]
- Cramer C. N.; Haselmann K. F.; Olsen J. V.; Nielsen P. K. Disulfide Linkage Characterization of Disulfide Bond-Containing Proteins and Peptides by Reducing Electrochemistry and Mass Spectrometry. Anal. Chem. 2016, 88 (3), 1585–1592. 10.1021/acs.analchem.5b03148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.