

Abstract
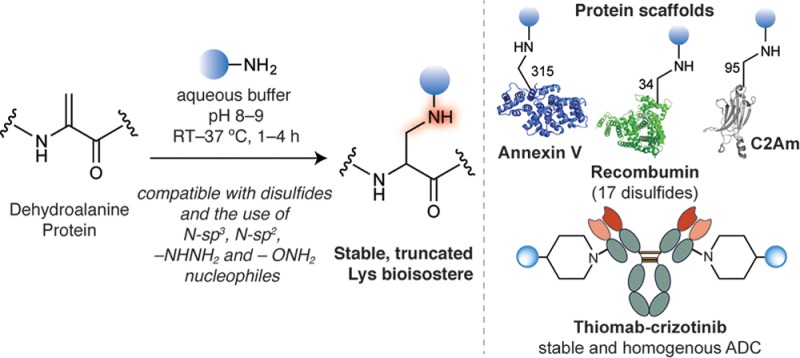
Chemical modification of proteins is essential for a variety of important diagnostic and therapeutic applications. Many strategies developed to date lack chemo- and regioselectivity as well as result in non-native linkages that may suffer from instability in vivo and adversely affect the protein’s structure and function. We describe here the reaction of N-nucleophiles with the amino acid dehydroalanine (Dha) in a protein context. When Dha is chemically installed in proteins, the addition of a wide-range N-nucleophiles enables the rapid formation of amine linkages (secondary and tertiary) in a chemoselective manner under mild, biocompatible conditions. These new linkages are stable at a wide range of pH values (pH 2.8 to 12.8), under reducing conditions (biological thiols such as glutathione) and in human plasma. This method is demonstrated for three proteins and is shown to be fully compatible with disulfide bridges, as evidenced by the selective modification of recombinant albumin that displays 17 structurally relevant disulfides. The practicability and utility of our approach is further demonstrated by the construction of a chemically modified C2A domain of Synaptotagmin-I protein that retains its ability to preferentially bind to apoptotic cells at a level comparable to the native protein. Importantly, the method was useful for building a homogeneous antibody-drug conjugate with a precise drug-to-antibody ratio of 2. The kinase inhibitor crizotinib was directly conjugated to Dha through its piperidine motif, and its antibody-mediated intracellular delivery results in 10-fold improvement of its cancer cell-killing efficacy. The simplicity and exquisite site-selectivity of the aza-Michael ligation described herein allows the construction of stable secondary and tertiary amine-linked protein conjugates without affecting the structure and function of biologically relevant proteins.
Introduction
For a variety of important diagnostic and therapeutic applications, there is considerable interest in the covalent modification of proteins.1−6 This field has grown to include a diverse range of reactions that modify a variety of different proteins with unique functions. The modification of such proteins enables the synthesis of protein conjugates suitable for the study of post-translational modifications,3 the imaging of biological processes6 and the construction of protein conjugates for targeted therapeutics, such as antibody-drug conjugates (ADCs).7,8
Despite the great interest in the field of covalent chemical modification of proteins over the past decade, many of the documented methods lack site-selectivity within the chemically complex protein environment. For this reason, there is a pressing need for the development of reactions that modify defined amino acids within a protein’s structure. For many applications, particularly for the development of therapeutics, introducing these modifications selectively at particular sites is of paramount importance. In one example, it has been demonstrated previously that a heterogeneously labeled ADC is significantly less efficacious than a homogeneous, but otherwise identical, conjugate.9 For this application as well as many others, the development of a site-selective conjugation methodology is essential.
One particularly powerful approach to achieve site-selective chemical modification of proteins utilizes engineered noncanonical amino acids.2 These amino acids can be incorporated into a protein’s structure using triplet amber codon suppression10,11 or auxotrophic strains of bacteria.12 The utilization of these techniques to include unnatural functionalities, such as alkynes, azides, ketones, alkenes or tetrazines into a protein has greatly accelerated the expansion of site-selective protein modification.2,5 Although reactions that target such functional groups are highly useful and often show a high degree of chemoselectivity, even in living systems,2 the installation of noncanonical amino acids requires a high degree of protein engineering that can lead to low expression levels, which can be detrimental for many diagnostic and therapeutic applications. A complementary approach exploits the unique reactivity of the N-terminal position of a given protein, for instance, via imidazolidinone formation using 2-pyridinecarboxaldehyde.13 While N-terminal modification methods are attractive because they do not require protein engineering, they are limited to the N-terminal position as the site of attachment. More recently, the introduction of a specific amino acid sequence that enhances the reactivity of a particular amino acid side chain, for example cysteine (Cys), has enabled site-selective protein modification even if other cysteine residues were present in the protein sequence.14 However, this method requires extensive engineering of the native protein sequence for efficient bioconjugation.
The most commonly used approach for the site-selective modification of therapeutic proteins remains the Michael addition of the sulfhydryl side-chain of Cys residues on the protein’s surface with maleimide reagents.15 For example, a recently Food and Drug Administration approved ADC, brentuximab vedotin, used maleimide chemistry to conjugate a cleavable linker bearing a cytotoxic drug to genetically engineered Cys residues on the surface of an antibody against CD-30, a marker used to target Hodgkin’s Lymphoma.16 A potential drawback of this approach is that the thioether succinimide linkage that results from this reaction can rapidly undergo retro-Michael addition under physiological conditions, resulting in early release of the cargo from the carrier antibody leading to off-target toxicity.17 In a complementary approach to direct Cys-based conjugation methods, Cys has been utilized as a precursor to chemically install the amino acid dehydroalanine (Dha) on proteins.18,19 Its unique electrophilic character, when compared with the natural nucleophilic residues, enabled the development of a robust and reliable method to form stable thioether-linkages between the protein and modification (e.g., post-translational modifications (PTMs) such as phosphorylation, acetylation, methylation or glycosylation) through the Michael addition of suitable thiol reagents.20,21 Although this methodology has found many applications in protein modification, the small molecule sulfur nucleophiles used may interfere with existing disulfide bonds present in the protein, especially in cases where these are solvent exposed, forming mixed disulfides that may disrupt the protein native structure and function.
On the basis of these considerations, we postulated that the development of a method for the chemical site-selective modification of Dha—accessed through ready chemical conversion from Cys—that is disulfide compatible, not limited to terminal positions and affords a native chemical linkage between protein and the desired modification would find great utility for the modification of proteins that are already available through large-scale production and that have been engineered with additional Cys residues. These include, for example, several recombinant proteins that are used as biomarkers for disease imaging or proteins that serve as vehicles for drug-delivery purposes such as albumins or antibodies.7,22
Here, we report a simple and robust methodology for protein site-selective bioconjugation based on the addition of N-nucleophiles to engineered internal Dha residues in a protein context. This reaction, which we refer to as aza-Michael ligation, proceeds in a chemoselective fashion under mild conditions (25 to 37 °C in buffered aqueous solution at pH 8.0) and results in a natural secondary amine linkage (truncated Lys analogue) that is stable to degradation in biological conditions. To date, generation of a secondary amine bond on a protein has been limited to reductive amination strategies that requires either the use of highly reactive reducing agents such as NaBH4 or NaBH3CN,23 or the use of an iridium catalyst, [Cp*Ir(4,4′-dimethoxy-2,2′-bipyridine)(H2O)]SO4, in the presence of formate ions.24 Additionally, acrylamide-containing noncanonical amino acids have been genetically encoded on proteins and reacted with N-nucleophiles in a proximity-driven reaction.25 We demonstrate the potential of the aza-Michael ligation method for three different proteins that contain naturally occurring or genetically engineered surface-exposed Cys residues. We also show the high degree of disulfide compatibility of this novel method, whose simplicity and robustness is likely to find broad applicability for the modification of proteins used in diagnostic and therapeutic applications.
Results and Discussion
Amine Additions to Dha on Amino Acids
Our design of a conjugation method to site-selectively install amine linked modifications on a protein was inspired by the potential reactivity of Dha toward N-nucleophiles (Figure 1a). There are very few examples of the reaction of Dha with N-nucleophiles on peptides,26−28 and to an even lesser extent on proteins—in a single example, Dha has been used as a precursor for the installation of an isomer of histidine through the reaction with imidazole.29−32
Figure 1.
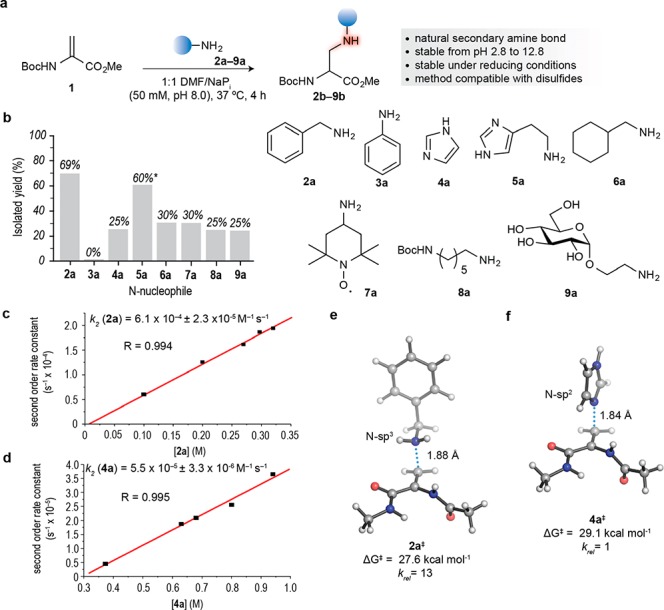
Reaction between a protected Dha amino acid derivative and amine nucleophiles. (a) Reaction of Boc-Dha methyl ester 1 with N-nucleophiles. (b) Graphical representation of the isolated yields of the reaction of 1 and N-nucleophiles 2a–9a. General conditions for amine addition to Dha: Boc-Dha methyl ester 1 (1 equiv) and N-nucleophile 2a–9a (1.5 equiv) in a 1:1 mixture of DMF/sodium phosphate buffer (50 mM, pH 8.0) at 37 °C for 4 h. All yields were calculated after SiO2 flash column chromatography purification with the exception of the addition of 5a to 1 for which conversion using the crude mixture is indicated* (1.7:1 N-sp3/N-sp2 ratio). (c,d) Experimental determination of the second order rate constant for the addition of benzylamine 2a and imidazole 4a to Boc-Dha methyl ester 1, respectively. (e,f) Transition structures and associated activation free energies (ΔG‡) at 25 °C and relative reaction rates (krel) calculated with PCM(water)/M06-2X/6-311+g(2d,p) for the aza-Michael ligation of model dehydro amino acid Ac-Dha-NHMe with benzylamine 2a and imidazole 4a, respectively. NaPi, sodium phosphate buffer; DMF, dimethylformamide; Boc, tert-butyloxycarbonyl.
We first investigated the potential of Dha as an aza-Michael acceptor within a small molecule context. This was performed by examining the reaction between a Boc-Dha methyl ester 1 and a variety of small molecule nitrogen nucleophiles 2a–9a with different N-hybridizations, which are representative of common motifs found in drug fragments, spectroscopic probes, linkers and PTMs (Figure 1b). These reactions were carried out in aqueous conditions, specifically in a 1:1 mixture of DMF and 50 mM sodium phosphate buffer at near neutral pH 8.0 and 37 °C. These reaction conditions fulfill the essential requirements for a potentially useful protein modification reaction.
Chemical yields were calculated after SiO2 flash chromatography purification for each reaction and ranged between 25–30%, with the exception of benzylamine 2a, for which the reaction proceeded with 69% yield (Figure 1b). Poorly nucleophilic amines, such as aniline 3a, proved to be unreactive under the conditions tested. We also evaluated the chemoselectivity (N-sp3 vs N-sp2) of the aza-Michael addition by reacting Boc-Dha methyl ester 1 with histamine 5a, which has two possible reactive nitrogen atoms. Under the conditions tested, 60% conversion in a 1.7:1 sp3/sp2 ratio was observed, suggesting predominant addition of the primary amine to Dha 1 (Figure S1). Next, we studied the diastereoselectivity of the addition reaction by reacting dipeptide Boc-Ala-Dha methyl ester S1 with 2a. As expected, this afforded the corresponding aza-Michael ligation product S2 in 40% yield as a 1:1 mixture of diastereomers (Figure S2). Using the product of benzylamine 2a addition to the amino acid Dha 1, we also evaluated the stability of the newly formed secondary amine linkage under a variety of pH conditions. We found that the secondary amine linkage was stable from pH 2.8 to pH 12.8, as confirmed by 1H NMR. Peaks corresponding to the alkene group of Dha or products resulting from peptide cleavage/BnNH2 release were not observed in these spectra, indicating that there was no detectable degradation of the product under this range of pH conditions (Figure S3). These data show that although the secondary amine may be protonated in slightly acidic biological environments, it should remain stable, an important feature for diagnostic and therapeutic applications of protein conjugates.
Kinetic Measurements and Theoretical Calculations
Building upon these results, we then examined the kinetics of the reaction between Dha 1 and two N-nucleophiles, specifically, benzylamine 2a and imidazole 4a, by calculating second order rate constants. By monitoring the reaction using 1H NMR, we determined the second order rate constants (k2) to be 6.1 × 10–4 and 5.6 × 10–5 M–1 s–1, respectively (Figure 1c and 1d). In the case of benzylamine 2a this value is of the same magnitude as the second order rate constants of common protein modification reactions, such as the oxime and Staudinger ligations.2 The superior reactivity of benzylamine 2a compared to imidazole 4a in the aza-Michael reaction, especially in the protein context (vide infra), was examined computationally using abbreviated models for the dehydro amino acid (Figure 1e and 1f, Figure S4, Table S1). Using the M06-2X33 DFT functional, which has been proved to give accurate results for Michael addition reactions,34 the experimental reactivity trends were reproduced, revealing the higher nucleophilic character of the N-sp3 in 2a compared to the reactive N-sp2 in 4a. The calculated relative reaction rates at 25 °C for benzylamine 2a (krel = 13) and imidazole 4a (krel = 1) correlate well with the experimental values (krel = 11 and 1 for 2a and 4a, respectively) when the dehydro amino acid is in the same local environment and peptide backbone conformation (Figure 1e and 1f). Once the ability of the aza-Michael addition to Dha derivative 1 was demonstrated with various amines, we expanded our investigation to proteins with chemically engineered, internal Dha residues on their surface.
Benzylamine Addition to Dha-Tagged Proteins
With these encouraging small-molecule results in hand, we next examined the reaction with a model protein to evaluate its potential as a novel protein ligation methodology for the construction of chemically defined secondary amine linked protein conjugates. To this aim, we selected a single cysteine mutant of the C2A domain of Synaptotagmin-I (C2Am).35 We selected this protein due to its diagnostic relevance as an apoptosis imaging agent, as well as the accessibility of the engineered Cys residue on the protein surface. We successfully generated Dha from the engineered Cys at position 95 on the C2Am protein by the previously reported bis-alkylation-elimination method.19 To achieve this we treated C2Am-Cys95 with α,α′-dibromo-adipyl(bis)amide 10 (92.4 mM) and complete conversion to C2Am-Dha95 was observed after 3 h at room temperature and 1 h at 37 °C (Figure S10). Chemical mutation of Cys to Dha proceeds with minimal perturbation of the global structure of the protein as evidenced by Circular Dichroism analysis (CD) (SI). Having obtained successfully the Dha residue at position 95 of C2Am, we began our study with the addition of benzylamine 2a (11 mM), the amine that achieved the highest Dha conversion in the small molecule experiments (Figure 2a). We obtained complete conversion after 3 h at room temperature in sodium phosphate buffer pH 8.0, as confirmed by liquid-chromatography mass-spectrometry (LC–MS) analysis (Figure 2b; see Figure S5 for a typical conjugation analysis by LC–MS). The presence of the expected secondary benzylamine linkage at position 95 was further confirmed by tryptic digest and MS/MS analysis (Figure 2e).
Figure 2.
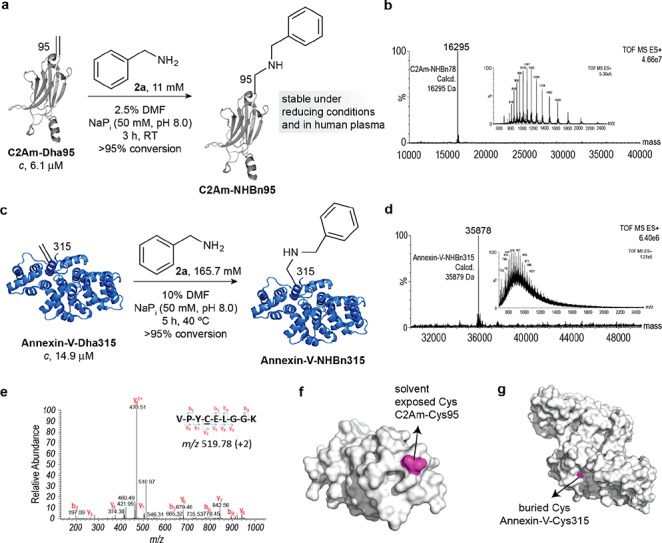
Chemical site-selective Dha modification with benzylamine on proteins. (a) Addition of 2a to the engineered Dha residue at position 95 of the C2Am. (b) ESI–MS spectrum of C2A-NHBn. (c) Benzylamine 2a addition to the engineered Dha at position 315 on the surface of Annexin-V. (d) ESI–MS spectrum of Annexin-V-NHBn. (e) MS/MS spectrum of the m/z 519.78 doubly charged ion of the tryptic peptide VPYCELGGK, containing the −NHBn– modification at the original Cys95 residue. The fragment ions generated are consistent with the mass of the modification. (f,g) Surface representation of C2Am and Annexin-V, respectively, showing in purple the Cys residue that is converted into Dha. While in C2Am the reactive Cys is exposed to the solvent, in Annexin-V this residue is found in a more buried position.
Next, we sought to broaden the scope of this reaction to other proteins. Thus, we investigated the addition of benzylamine 2a to an engineered Dha residue in Annexin-V (Figure 2c). Annexin-V is another apoptosis imaging agent, functionally similar to C2Am in that it binds to the phosphatidylserine (PS) externalised to the outer leaflet of the plasma membrane of cells undergoing apoptosis.36 It contains a single free Cys that is positioned in a more hindered and challenging position when compared to that on C2Am. We synthesized Dha from this single Cys by incubating Annexin-V-Cys315 (27.8 μM) with α,α′-dibromo-adipyl(bis)amide 10 (14 mM) for 4 h at 37 °C (Figure S15). We observed full addition of benzylamine 2a (165.7 mM) to Annexin-V-Dha315 (14.9 μM) after 5 h at 40 °C, as shown by LC–MS (Figure 2d). Although these conditions are harsher when compared to the addition of 2a to C2Am-Dha95, this is unsurprising due to the buried nature of the free Cys of Annexin-V, as clearly observed in a surface representation of the modification sites on both proteins (Figure 2f and 2g).
Scope of N-Nucleophile Additions to Dha-Tagged Proteins
Having demonstrated that it is possible to generate a secondary amine bond at the internal position of Dha-tagged proteins, we explored the scope of the aza-Michael addition ligation to Dha on C2Am-Dha95 by exhaustively testing a wide range of different N-nucleophile reagents, as shown in Figure 3a. Similar results to those obtained with benzylamine 2a were obtained in the addition of cyclohexylmethylamine 6a with complete addition observed after 3 h at room temperature. A complete addition was also observed with 4-hydroxybenzylamine 11, 4-(aminomethyl)benzoic acid 12 and 4-(aminomethyl)cyclohexanecarboxylic acid 15 after 24 h at 37 °C. Interestingly, full conversion was also observed after 24 h at 37 °C with histamine 5a, an important neurotransmitter and immunomodulatory small molecule.37 The addition of 4-aminobutyl β-d-galactopyranoside 22, as an example of protein glycosylation via direct attachment of sugar units, also proceeded with complete conversion. Interestingly, we found that the secondary amine piperidine 23 (example of N-sp3 heterocyclic motif found in many biologically active drugs–see Table S4) adds efficiently to Dha to form a tertiary amine-linked conjugate (Figure S39). Unlike other amines, complete conversion was achieved with the more nucleophilic 23 using only 300 equiv. Of note, we also observed full conversion by addition of “softer” nucleophilic species such as hydroxylamine 24 and hydrazine 25 as shown by LC–MS (Figure S41 and S42). The modification site for the hydrazine addition product was also confirmed by peptide mapping and MS/MS analysis (Figure S58). These results expand the scope of N-nucleophiles that may be successfully reacted with Dha beyond simple amine handles. These reactions also compare favorably in terms of mildness and operational simplicity with standard reductive amination-like protocols using hydroxylamines/hydrazines with aldehyde/ketone handles. Among the other amines tested, imidazole 4a (example of N-sp2 heterocyclic motif found in many biologically active amines), N-Boc-1,6-hexadiamine 8a (aliphatic linker), 4-bromobenzylamine 13 (handle for C–C cross-couplings), phenylethylamine 14 (an important neuromodulator) showed the greatest conversions–up to 90% after 16–48 h at 37 °C. All other amines surveyed showed conversions ranging between 40%–60% upon incubation at 37 °C for 24–48 h, with the aqueous solubility of each amine nucleophile being a key factor in determining its reactivity. Yet, the lower conversion could be mitigated by the addition of variable amounts of DMF as a cosolvent (SI).
Figure 3.
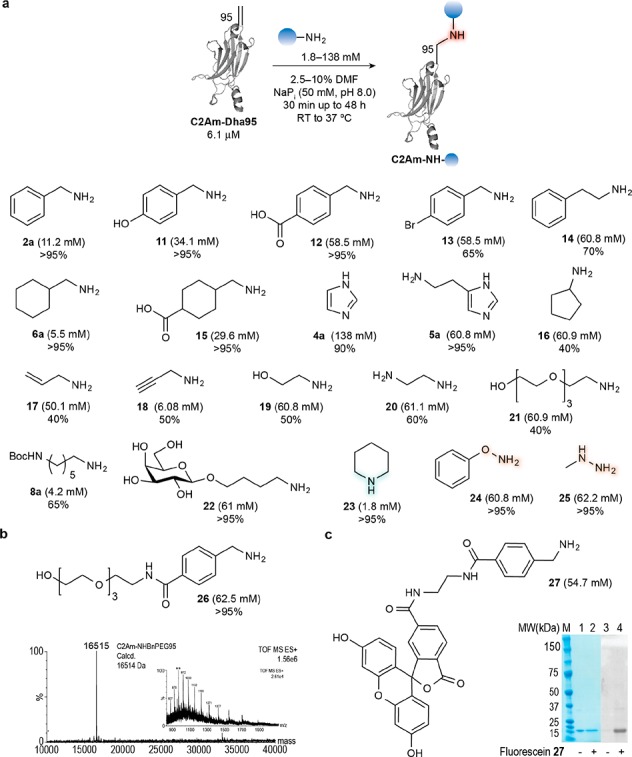
Site-selective modification of C2Am-Dha95 with a range of amines. (a) The reaction of C2Am-Cys95 with a wide variety of N-nucleophiles. The conversions listed here are the maximum conversions that could be obtained by reacting the different N-nucleophiles with C2Am-Dha95. Further experimental details on each reaction may be found in Supporting Information. (b) The reaction of compound 26 with C2Am-Dha95 went to completion as detected by ESI–MS shown here. (c) Treatment of C2Am-Dha95 with compound 27 afforded a fluorescent protein conjugate, as detected by SDS-PAGE gel shown here. Lanes 1 and 2, Coomassie staining. Lanes 3 and 4, fluorescence.
Protein Labeling Using Aminomethylbenzoic Acid Derivatives
To explore the versatility of aza-Michael ligation on proteins, particularly toward PEGylation and fluorescent labeling,5,38 we investigated the addition of tetraethylene glycols 21 and 26 and fluorescein derivative 27 to C2Am-Dha95. p-Aminomethylbenzoic derivatives 26 and 27 were readily synthesized from accessible p-aminomethylbenzoic acid 12. The basic structure of this compound is composed of a primary amine for conjugate addition to Dha and a carboxylic acid for functionalization with a variety of different molecules and proved to be one of the best p-substituted benzylamine performers with C2Am-Dha95 (see SI for synthesis details). Preliminary ligations with 21 afforded the corresponding PEGylation product in 40% conversion (Figure 3a). In contrast, treatment of 26 (62.5 mM) with C2Am (6.1 μM) for 48 h at 37 °C gave full conversion to the corresponding PEGylated protein, as shown by LC–MS (Figure 3b). Similarly, when C2Am-Dha95 was reacted with the fluorescein derivative 27, a fluorescence conjugate was formed as evidenced by fluorescent SDS-PAGE gel (Figure 3c). This result shows that through the use of the appropriate primary amine handle, this reaction may be generalized to connect a variety of important synthetic compounds to biomolecules.
Aza-Michael Ligation Is Disulfide Compatible
To verify the disulfide compatibility of this new amine addition methodology, we first took advantage of the ready dimer formation of C2Am through a disulfide bond between the engineered free Cys residue at position 95. After leaving C2Am-Cys95 open to the atmosphere for 1 h at 37 °C, we observed complete conversion to the oxidized 32 kDa dimer from the original 16 kDa protein (Figure S45). We then treated this protein dimer with a model thiol, β-mercaptoethanol 28, and benzylamine 2a to compare the compatibility of these nucleophiles with disulfide bonds (Figure 4a). We were encouraged that we observed maintenance of the dimer, and thus preservation of the disulfide bond integrity, when the C2Am-Cys95 dimer was treated with 2a (56.6 mM – the same amine concentration required to afford complete conversion on Dha) for 2 h at room temperature, as shown by LC–MS analysis (Figure 4b). Instead, when this dimer was treated with β-mercaptoethanol 28 (22.5 mM) under the same conditions, 2 h at room temperature, complete reduction of the disulfide bond was readily achieved with no significant dimer observed in the mass spectrum (Figure 4c).
Figure 4.

Addition of N-nucleophiles to Dha on proteins is compatible with disulfides. (a) Comparative experiment where the readily formed disulfide dimer of C2Am-Cys95 was exposed to a S-nucleophile, β-mercaptoethanol 28, and to an N-nucleophile, benzylamine 2a. (b) ESI–MS spectrum of the reaction of disulfide dimer of C2Am-Cys95 with benzylamine shows the unreacted dimer of C2Am-Cys95 indicating that benzylamine does not cross-react with the disulfide. (c) ESI–MS spectrum of the reaction of disulfide dimer of C2Am-Cys95 with β-mercaptoethanol 28 shows rapid disulfide reduction and formation of the monomer C2Am-Cys95. (d) Conversion of the free Cys34 of Recombumin to Dha and addition of benzylamine 2a. (e) ESI–MS spectrum of the product of the reaction of Recombumin-Cys34 with α,α′-dibromo-adipyl(bis)amide 10. (f) Reaction of Recombumin-Dha34 with benzylamine 2a results in a homogeneous conjugate Recombumin-NHBn34, as shown in the ESI–MS spectrum. (g) Surface representation of Recombumin showing the Cys residue at site 34 (in pink) that is first converted into Dha and then used as a handle for aza-Michael ligation. A number of disulfides bridges that are solvent exposed are shown in yellow.
To further demonstrate the chemoselectivity of the reaction, we next evaluated amine addition to a particularly relevant protein, albumin. Human serum albumin is the most abundant protein found in plasma. It is comprised of 585 amino acids and contains 17 structural disulfides plus one free Cys residue at position 34. Plasma derived human albumin has been used in the clinic for decades to expand plasma volume in order to counter severe blood loss as well as in the formulation of active pharmaceutical ingredients.39 In the interest of having a well-defined single pure protein for these therapeutic applications, recombinant human albumin has become available and gained wide acceptance as a bespoke human albumin for formulation, drug delivery and imaging applications.22 Thus, we investigated the addition of benzylamine 2a to the single engineered Dha residue in a recombinant human albumin—Recombumin (Albumedix Ltd.) (Figure 4d). We first confirmed the reactivity of the free Cys34 residue by reaction with Ellman’s reagent, which resulted in full conversion to the corresponding disulfide adduct (Figure S53), showing the thiol of Cys34 to be in its reduced and reactive form. Dha was then installed into albumin by reaction of the single Cys residue at position 34 with α,α′-dibromo-adipyl(bis)amide reagent 10 (22.5 mM). However, although we found monoalkylation of the single Cys to proceed rapidly to completion in 1.5 h at 37 °C, elimination proved to be more difficult. Bis-alkylation and elimination to yield the desired Dha residue were ultimately completed through the addition of 3 M guanidinium hydrochloride to slightly destabilize the protein structure and by temporarily raising the pH to 12.40 Under these conditions, the monoalkylated Cys34 in albumin (Figure S54) yielded the Dha residue with full conversion (Figure 4e). The reluctance of the elimination reaction to occur could be a reflection of the distinct local environment of the groove where Cys34 is found.41 This residue was then reacted with benzylamine 2a to yield the desired conjugate (Figure 4f) as shown by ESI–MS analysis. Importantly, we conclude that the new method for installing secondary amine bonds on proteins via aza-Michael ligation with amine nucleophiles is fully compatible with disulfide bonds, even in cases where these disulfides are solvent exposed (Figure 4g).
Protein Conjugates Are Stable to Biological Thiols
Upon obtaining the benzylamine conjugated C2Am protein, C2Am-NHBn, we aimed to verify its potential value of the secondary amine linkage. First, we sought out to demonstrate the linker’s stability in human plasma. Linker stability is an essential requirement for the development of useful protein conjugates for therapeutic and diagnostic applications since systemic, nontargeted release of the conjugated payload would result in undesired side-toxicity and nonspecific imaging, respectively. Importantly, no detectable degradation of the benzylamine conjugate was observed after incubation with human plasma for 24 h at 37 °C, demonstrating the conjugate’s stability under these conditions (Figures S48 and S49). Complete stability in human plasma was also observed for the conjugate formed after the reaction of C2Am with piperidine 23 that features a tertiary amine bond (Figure S40). Additionally, when C2Am-NHBn was incubated under reducing conditions (1 mM glutathione for 24 h at 37 °C), the conjugate remained intact, further demonstrating its stability in biologically relevant conditions (Figures S50 and S51).
Protein Conjugates Retain Biological Function
Albumin has an extended serum half-life of approximately 3 weeks due to its size and neonatal Fc receptor (FcRn) mediated recycling that prevents intracellular degradation.42 Albumin binds FcRn in a noncooperative and strictly pH dependent manner, with strong binding at pH 6.0 that becomes progressively weaker approaching physiological pH.43 Using Surface Plasma Resonance (SPR), we confirmed that the conjugate Recombumin-NHBn34 binds to FcRn in a reversible and pH dependent manner (Figure 5a, Table S2). While the rate of association was slower than the unmodified Recombumin-Cys34 control, the binding event was comparable (Figure 5a, Table S2). This result highlights the utility of our method to provide functional protein conjugates regardless of exposure and reactivity of the target Cys.
Figure 5.
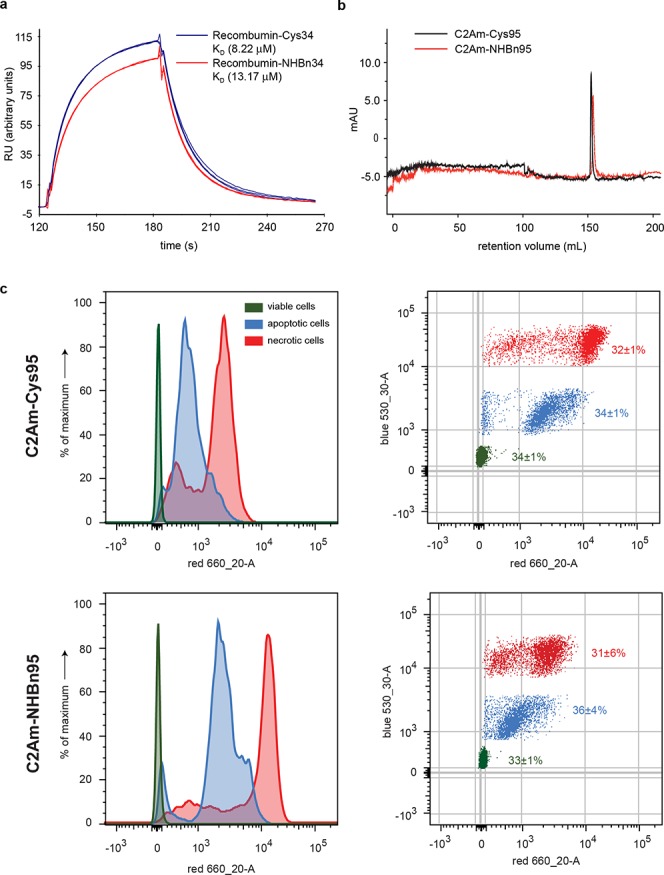
Assessment of the biological activity of Recombumin and C2Am protein after chemical modification. (a) Biacore SPR analysis of Recombumin-Cys34, blue, and Recombumin-NHBn34, red, n = 3 replicates. (b) Fast protein liquid chromatography analysis utilizing a HiPrep S FF (GE Healthcare)–affinity column comparing C2Am-Cys, black, and C2Am-NHBn, red. (c) Flow cytometry plots obtained by fluorescence activated cell sorting (FACS) of C2Am-Cys (top) and C2Am-NHBn (bottom) labeling viable (green), apoptotic (blue) and necrotic (red) EL4 cells. Cell populations (%) for C2Am-Cys95; C2Am-NHBn95:33 ± 1%; 34 ± 1% (viable), 36 ± 4%; 34 ± 1% (apoptotic), 31 ± 6%; 32 ± 1% (necrotic). Apoptotic/viable MFI ratios (C2Am-Cys95; C2Am-NHBn95): 35 ± 1%; 87 ± 11%. Necrotic/viable MFI ratios (C2Am-Cys95; C2Am-NHBn95): 98 ± 4%; 411 ± 32%. MFI-median fluorescence intensity at 660 nm. Data is mean ± s.d., n = 3 replicates, 2 independent experiments.
C2Am has been validated in vivo for detecting apoptotic tissue, using a variety of methods, including magnetic resonance imaging (MRI)44 and single photon emission computed tomography (SPECT).45 C2Am specifically labels apoptotic cells by binding to phosphatidylserine (PS) available on their cell membrane. Therefore, we verified that C2Am modified through a secondary amine linkage retained its natural high affinity for PS and the ability to detect apoptotic cells in vitro. Using affinity chromatography, we verified that the conjugate C2Am-NHBn shows similar binding to PS as the original unaltered C2Am protein (Figure 5b). Additionally, using flow cytometry, we proved that C2Am-NHBn retained its ability to preferentially bind to apoptotic cells over viable cells, at a level comparable to the unmodified protein (Figure 5c). With these results, we surmised that the benzylamine adduct retained the original biological function of C2Am even upon modification, highlighting the potential of our conjugation method for the construction of functional modified proteins.
Construction of Homogeneous Antibody–Drug Conjugates
Tumour specific antibody-mediated delivery of cytotoxic molecules holds great promise for cancer therapy.5,8,16 To further demonstrate the utility of our method, we sought to build a homogeneous ADC through aza-Michael ligation at Dha. We chose a Thiomab antibody targeting Her2 receptor. This antibody has been engineered so that it possesses an additional Cys residue at position 205 in each light-chain (Thiomab LC-V205C).9 Most ADCs developed to date utilize DNA alkylating agents or tubulin polymerization inhibitors as payloads. These highly potent cytotoxic motifs are usually released upon cleavage of a conditionally labile linker between the antibody and the payload. While ADCs are finding significant clinical use for cancer treatment, there are increasing reports of side-effects and toxicity associated with ADC therapy.46 Such off-target toxicity may result, for instance, from premature drug release in the blood and/or the intrinsic cytotoxicity of the payloads. Considering these issues, we chose to develop a strategy based on our aza-Michael addition to Dha to incorporate novel payloads with differentiated mechanisms of action into antibodies. Our strategy relies on the formation of a fully stable ADC where the drug is directly conjugated to the antibody through an amine linkage. Upon internalization, cellular processing and degradation in the endosome and lysosome, the drug is released intracellularly.
We searched for terminal piperidine motifs that could serve as amine handle for direct aza-Michael site-selective antibody conjugation in approved/investigational anticancer drugs. The search pooled several kinase inhibitors (Table S4) from which we selected crizotinib 30 as a suitable drug to test our hypothesis. Crizotinib is a known inhibitor of the MET, ALK and ROS1 kinases47 that has been approved for the treatment of ALK-rearranged nonsmall-cell lung carcinoma (NSCLC). It has also been recently described to promote T cell interactions with monocytes, as well as with cancer cells, through inhibition of the receptor tyrosine kinase MSTR1 and subsequent up-regulation of the expression of major histocompatibility complex molecules.48 Chemically, the piperidine motif was found to be highly reactive and to form a stable tertiary amine linkage upon aza-Michael addition to Dha. Crizotinib also features a 2-amino pyridine motif that we found to be highly unreactive even when used in large excess (Figure S71). This is expected since piperidine is a much better nucleophile than anilines in the Mayr’s scale.
We started by treating Thiomab LC-V205C with tris(2-carboxyethyl)phosphine (TCEP) followed by dehydroascorbic acid mediated disulfide reoxidation. This procedure is required to ensure that the engineered Cys is in its free form and readily available for chemical manipulation.9 Using typical Dha bis-alkylation/elimination procedure with 10 on Thiomab LC-V205C proved unsuccessful under several conditions (large excess, high temperature—up to 40 °C, mild denaturing conditions and high pH 10). In all cases not even the alkylation product could be detected using LC–MS. Instead methyl 2,5 dibromo-pentanoate2929 proved to be highly reactive with Thiomab LC-V205C and enabled the formation of Thiomab-Dha at 37 °C. Of note, we found that removal of the excess reagent using size exclusion chromotography after 2 h at pH 8.0 followed by buffer exchange to pH 10.0 and further shaking for an additional 22 h period to be necessary to facilitate elimination to Dha (Figure 6a). Analysis by LC–MS showed the presence of two Dha residues per light-chain while no modifications were observed in the heavy-chain (Figure 6b). This data suggests that the inter heavy-chain disulfides were successfully reoxidized while the one between the Cys on the light-chain and the hinge Cys on the heavy-chain was not. Control experiments with thiol-specific Ellman’s reagent also showed two modifications within the light-chain of Thiomab LC-V205C supporting the presence of two reactive Cys residues (Figure S65). Nevertheless, Thiomab-Dha was tested for its binding affinity to Her2 using biolayer interferometry and we found that the binding capability of Thiomab-Dha remained comparable to the nonmodified antibody (Figure 6d). Next, we tested the reactivity of Thiomab-Dha toward piperidine. Using a small excess of 23 (3 mM, 100 equiv), complete conversion to the tertiary-amine modified antibody was observed after 2 h at 37 °C (Figure 6e). Two modifications per light-chain were detected by LC–MS while no modifications were found on the heavy-chain further highlighting the compatibility of the aza-Michael addition approach with disulfides. When the same procedure was applied using Crizotinib, a chemically defined ADC was obtained bearing only one drug molecule per light-chain (Figure 6e). This may be rationalized on the basis of steric hindrance—the engineered Cys is within proximity of the Cys that is usually present as a disulfide with the hinge Cys of the heavy-chain. In addition, solvent accessible surface area (SASA) calculated through 100 ns molecular dynamic simulations in explicit water for Thiomab (PDB id: 5d6c) indicated a greater SASA value for the 205 residue in comparison to the inner 194 residue. This data suggests that Dha at position 205 is likely the one that undergoes aza-Michael addition with 30 (Figure S70). This homogeneous ADC bearing a defined drug-to-antibody ratio (DAR) of 2 featured a tertiary amine bond that is fully stable in human plasma (Figures S40 and S68) and showed an important, 10-fold improvement in cell-killing activity when compared to the free drug as assessed by CellTiter-Blue assay in SKBR3 breast cancer cells that overexpress Her2 (Figures 6f,g). Naked Thiomab showed no effect on cell viability (control) (Figure S73). Furthermore, we confirmed that the modified antibody retained its specificity and capacity to bind to the Her2 antigen in SKBR3 cells as demonstrated by Flow Cytometry analysis (Figure 6h). With these experiments, we provide evidence of the potential of this site-selective conjugation method for the construction of stable and homogeneous ADCs with a defined DAR. In addition, this work highlights the potential of directly conjugating drugs that are outside the traditional choices for ADC construction to increase their cancer cell killing efficiency by antibody-mediated intracellular delivery. Moreover, with the advent of ADCs for treating infectious diseases49 our approach may have broader applicability beyond cancer therapy.
Figure 6.

Construction of a stable and chemically defined ADC through direct, aza-Michael conjugation of crizotinib to Dha-tagged Thiomab and its biological evaluation. (a) Reaction scheme for the conversion of Thiomab LC-V205C to Thiomab-Dha using a bis-alkylation/elimination procedure. (b) ESI–MS spectra of the light- and heavy-chains of Thiomab LC-V205C after reduction and reoxidation protocol. (c) ESI–MS spectra of the light- and heavy-chains of the reaction of Thiomab LC-V205C with methyl 2,5-dibromopentanoate 29 shows the formation of two Dha residues per light-chain. (d) KD constants derived from BLI experiments for Thiomab and Thiomab-Dha. For the BLI curves and fitting curves obtained for Thiomab and Thiomab-30 with Her2 receptor see the SI. (e) Reaction scheme for the reaction of Thiomab-Dha with either piperidine 23 or crizotinib 30. ESI-MS spectra of the light-chain shows the addition of two piperidine molecules and the addition of one crizotinib 30, respectively. (f) SKBR3 cells viability after treatment with Thiomab-30 for 24 h. See SI for data with crizotinib and naked Thiomab (control). Results are shown as percentage of control (medium + vehicle – PBS) and correspond to 3 biological replicates (mean ± s.d.). (g) IC50 of Thiomab-30 in SKBR3 cells. (h) Thiomab-30 binding affinity to SKBR3 cells measured by flow cytometry.
Conclusion
We have described a thorough evaluation of N-nucleophile additions to internal Dha residues as a chemoselective and biocompatible protein site-selective modification methodology for the construction of homogeneous protein conjugates. We first demonstrated its potential as a protein modification reaction on a small molecule level both in terms of general reactivity and kinetics. The second rate order constants were similar to the rates of widely used protein modification reactions including the Staudinger ligation50 and the oxime reaction between noncanonical ketone amino acids and aminooxy reagents, which are used currently for the assembly of ADCs that are under evaluation in the clinic.51 We then conducted a thorough evaluation of the addition of various amine nucleophiles to the Dha engineered from single Cys residue in the site-directed mutant of C2Am. From this study, we demonstrated that some amines, in particular benzylamine, cyclohexylamine and piperidine as well as their derivatives, and histamine, are reactive toward the Dha residue at near neutral pH. Hydroxylamines and hydrazines also reacted efficiently with Dha, further expanding the scope of N-nucleophiles that can be used to chemically site-selectively modify Dha on proteins. In addition, we noticed that the aza-Michael addition is not (or is poorly) diastereoselective. While it is very challenging to calculate the diastereomeric ratio on intact proteins, it is well documented that the ratio is dependent on the amino acid sequence adjacent to the site of modification, at least in peptide models.52 However, for most diagnostic and therapeutic applications racemization of the α-carbon of Cys will not be detrimental. Even synthetic histones either bearing thioether-linked PTM mimics that were produced through thiol-Michael addition at Dha20 or bearing PTMs installed through carbon free radical chemistry at Dha53,54 were found to be fully functional despite the likely generation of a diastereomeric mixture. The same was observed for the installation of mimics of phosphorylation on protein kinase p38α and on a single single-domain antibody cAb-Lys3.21,55 However, and in one case, only the natural diasterioisomer of a γ-thialysine mimic of Lys165 in the enzyme N-acetylneuraminic acid lyase (NAL) was able to refold correctly and retain enzymatic activity.56 Therefore, the lack of stereoselectivity of amine addition to Dha should be of little relevance to most applications. However, in those applications where stereoselectivity is needed, modulation of the local amino acid environment could be investigated as a means to promote stereoselective Dha additions.
The utility of the novel conjugation procedure was demonstrated by extending the reaction to other proteins of biological interest, namely Annexin-V as well as a recombinant human albumin used clinically. Albumin provides an example of a protein that although it displays 35 Cys, with 34 of those involved in 17 disulfide bridges, some of which are solvent exposed, it can successfully be modified using the reported postexpression chemical conversion of Cys to Dha followed by aza-Michael ligation. Following modification, the Recombumin-NHBn34 retained reversible pH dependent receptor binding. We also confirmed that modified C2Am is stable under reducing conditions and in human plasma, while retaining the protein’s native biological function. Importantly, the scope of the method could be expanded to build an ADC with a precise DAR of 2 through direct site-selective conjugation of the piperidine motif present in the anticancer drug crizotinib. Its antibody-mediated intracellular delivery resulted in a 10-fold improvement in cancer cell killing activity.
Taken together, we have explored the potential of amine additions to Dha as a successful, disulfide compatible protein modification reaction for site-selective introduction of secondary and tertiary amine linked modifications into proteins. This is demonstrated by the application of this reaction to three structurally distinct proteins, making it a ligation of potential importance for the construction of homogeneously labeled proteins for imaging (C2Am and Annexin-V) and therapeutic (Recombumin and Trastuzumab) applications. Considering the simple setup of this bioconjugation method and the use of easily derivatized amine reagents, we anticipate that this method will become a much-used tool for the chemical site-selective modification of proteins with diagnostic and therapeutic relevance.
Experimental Section
Dha Formation on Proteins
To an aliquot of purified, reduced protein, a freshly prepared solution of α,α′-dibromo-adipyl(bis)amide 10 in DMF was added and the resulting mixture was vortexed for 30 s and then was shaken at room temperature and/or warmed to 37 °C. The reaction progress was monitored by LC–MS with samples taken after defined time points by aliquoting 2.5 μL of the reaction mixture and diluting it with 8 μL of 50 mM sodium phosphate buffer at pH 8.0. Ten μL of this diluted sample was injected on the LC–MS. Full conversion to the expected Dha product was observed after several hours, with the time to completion depending on the protein. Small molecules were removed from the reaction mixture by loading the sample onto a Zeba Spin Desalting Column previously equilibrated with 50 mM sodium phosphate buffer at pH 8.0. The sample was eluted via centrifugation (2 min, 1500g). The protein solution was then flash frozen with liquid nitrogen and stored at −20 °C.
Aza-Michael Ligation on Proteins
An aliquot of Dha-modified protein in 50 mM sodium phosphate buffer at pH 8.0 was thawed. A nitrogen nucleophile was added at room temperature (reaction concentration ranging from 5–138 mM) and the resulting mixture vortexed for 30 s. Of note, the final pH of the reaction mixture may vary between 8 to 9 depending on the amine nucleophile used. The reaction progress was monitored by LC–MS with time points taken at defined time points ranging from 15 min to 48 h. Time points were taken by aliquoting 5 μL of the reaction mixture and diluting it with 6 μL of 50 mM sodium phosphate buffer at pH 8.0. Ten μL of this diluted sample was injected into the LC–MS.
Surface Plasmon Resonance Analysis of Recombumin-Cys34 and Recombumin-NHBn34
SPR experiments were performed using a Biacore 3000 instrument (GE Healthcare). Flow cells of CM5 sensor chips were coupled with soluble human FcRn (1505 RU) using amine coupling chemistry as described in the protocol provided by the manufacturer (GE Healthcare). The coupling was performed by injecting 10 μg/mL of the protein in 10 mM sodium acetate pH 4.5 (GE healthcare). Phosphate buffer (25 mM Na-acetate, 25 mM NaH2PO4, 150 mM NaCl, 0.01% T-20, pH 5.5) was used as running buffer and dilution buffer. Regeneration of the surfaces were performed using injections of HBS-EP buffer (0.01 M HEPES, 0.15 M NaCl, 3 mM EDTA, 0.005% surfactant P20) at pH 7.4 (GE Healthcare). Post immobilization, the chip was left to stabilize with a constant flow (5 μL/min) of running buffer. The chip surface was conditioned by injecting 3× injections of running buffer followed by 3× injections of regeneration buffer. Surfaces were checked for activity with an unmodified Recombumin-Cys34 albumin control. For determination of binding kinetics, serial dilutions of Recombumin-Cys34 and Recombumin-NHBn34 (10–0 μM) were injected over immobilized receptor at a constant flow rate (30 μL/min) at 25 °C. In all experiments, data were zero adjusted and the reference cell subtracted. Data evaluations were performed using BIAevaluation 4.1 software (GE Healthcare).
Flow Cytometry of Cell Death Using C2Am-Cys95 or C2Am-NHBn95
Murine lymphoma (EL4) cells (ATCC, Piscataway, NJ) were propagated in RPMI 1640 media (Sigma) supplemented with 10% fetal calf serum (FCS) and 2 mM l-glutamine (Sigma). Cell number and viability were monitored using the trypan blue dye exclusion assay on a Vi-cell system (Beckman Coulter, Brea, CA). EL4 cell death was induced by addition of 10 μM etoposide (Teva, Leeds, UK) for 18 h at 37 °C. In preparation for flow cytometry, cells (10 million) were then pelleted (600 g, 4 °C, 4 min), washed in ice-cold HBS+ buffer (HBS, 2 mM CaCl2, 1% FCS), and resuspended in 100 μL of the same buffer containing either C2Am-Cys95 or C2Am-NHBn95 (2 μM), in combination with 50 nM of the necrosis probe Sytox green (Life Technologies, Grand Island, NY) for 15 min at 37 °C, with orbital shaking (300 r.p.m.). The resulting cell suspension was washed twice with cold HBS+ buffer, kept briefly on ice, and analyzed in a LSRII cytometer (BD Biosciences, Rockville, MD), equipped with 488 and 630 nm lasers and counting 20 000 cells per event.
Dha Formation on Thiomab LC-V205C
To a 100 μL aliquot of reduced/reoxidized Thiomab (10.0 μM, 1.0 nmol), a freshly prepared solution of methyl 2,5-dibromopentanoate (1.0 μL of 50.0 mM solution, 0.05 μmol) was added and the resulting mixture was vortexed for 30 s and then was shaken at 37 °C. The reaction progress was monitored by LC–MS. After 2 h, small molecules were removed from the reaction mixture by a buffer exchange column Viva 500 (10 kDa). The sample was eluted via centrifugation (5 min, 1500g) using sodium phosphate buffer (50 mM, pH 10.0) to dilute the sample. The reaction mixture was further shaken at 37 °C for 22 h. After this time, small molecules were removed from the reaction mixture by loading the sample onto a Zeba Spin desalting column previously equilibrated with sodium phosphate buffer (50 mM, pH 8.0). The sample was eluted via centrifugation (2 min, 1500g). Samples for LC–MS analysis were prepared by aliquoting 5 μL of the reaction mixture followed by dilution with 5 μL of sodium phosphate buffer (50 mM, pH 8.0). Ten μL of this diluted sample was injected on the LC–MS. Complete conversion to a single product with a mass corresponding to the formation of two Dha in the light-chain was observed after 24 h (calculated mass light-chain bearing two Dha, 23373, observed mass, 23376). The heavy-chain remained intact after Dha formation.
Antigen Binding Properties of Thiomab-Dha
Biotinylation of antibodies. Nonmodified Thiomab and Thiomab-Dha were conjugated to a biotin linker (Biotin-(PEG)4-N-hydroxysuccinimide, Thermofisher Scientific) in order to carry out Biolayer Interferometry (BLI) experiments using Streptavidin (SA) Biosensors. A solution of EZ-Link NHS-(PEG)4-Biotin (20 μL, 200 mM in PBS, pH 7.4) was added to the corresponding antibody (20 μL, 20 mM in PBS, pH 7.4) and was shacked at room temperature for 30 min. The crude reaction mixture was buffer exchanged (3×) with PBS pH 7.4 to remove the excess of biotin-linker, obtaining a biotin-to-antibody ratio of ∼1 to 2 (determined using the Pierce Biotin Quantitation Kit, ThermoFisher Scientific). Biolayer interferometry. Binding assays were performed on an Octet Red Instrument (fortéBIO). Ligand immobilization, binding reactions, regeneration and neutralizations were conducted in wells of black polypropylene 96-well microplates. Thiomab and Thiomab-Dha were immobilized on Streptavidin (SA) Biosensors in PBS pH 7.4 with 0.1% BSA and 0.02% tween at 30 °C. Binding analysis were carried out at 25 °C, 1,000 r.p.m. in PBS pH 7.4 with 0.1% BSA and 0.02% tween. Association time was 600 s, followed by 2,200 s of dissociation, using different concentrations (200, 66.6, 22.2, 7.4, and 2.47 nM) of ErbB2/Her2 Recombinant Protein Antigen to obtain the association curve. Glycine pH 2.0 was used as a regeneration buffer. Data were analyzed using Data Analysis (fortéBIO), with Savitzky-Golay filtering. Binding was fitted to a 2:1 Heterogeneous ligand model, steady state analysis was performed to obtain the binding kinetics constants (KD).
Aza-Michael Addition of Crizotinib 30 to Thiomab-Dha
A 40 μL aliquot of Thiomab-Dha (10 μM, 399 pmol) in 50 mM sodium phosphate buffer at pH 8.0 was thawed. Crizotinib 30 (0.54 μL of a 222 mM solution in DMF) was added at 37 °C and the resulting mixture vortexed for 30 s. The reaction progress was monitored by LC–MS. Small molecules were removed from the reaction mixture by loading the sample onto a Zeba Spin Desalting Column previously equilibrated with sodium phosphate buffer (50 mM, pH 8.0). The sample was eluted via centrifugation (2 min, 1500g). When the reaction was scaled up for in vitro studies, this procedure was repeated 3 times to optimize the efficiency of the method. These columns are described to have at least 95% retention (removal) of salts and other small molecules (<1000 MW). Samples for LC–MS analysis were prepared by aliquoting 5 μL of the reaction mixture and diluting it with 5 μL of sodium phosphate buffer (50 mM, pH 8.0). Ten μL of this diluted sample was injected on the LC–MS. Complete conversion to Thiomab-30 was observed after 24 h (calculated mass light-chain, 23822, observed mass, 23826). The heavy-chain remained intact after aza-Michael addition of 30 to Thiomab-Dha.
Cell Viability Assay
10 000 cells/well were seeded in 96 well-plates and were treated with crizotinib, Thiomab or Thiomab-30 24 h after seeding, to allow the cells to stabilize. The cells were incubated with several concentrations of crizotinib (0.5, 1, 2.5, 5, 10, 15, 25, 50, 75, 100 μM), Thiomab (0.5, 1, 2.5, 5, 8 μM) and Thiomab-30 (0.5, 1, 2.5, 5, 8 μM) for 24 h. After this incubation period, the culture medium was removed and the cells were incubated with CellTiter-Blue (Promega) for 90 min at 37 °C. Cell viability was evaluated by measuring the Emission Intensity in RFUs, relative fluorescent units, with an Infinite M200 plate reader. IC50s were calculated using GraphPad Prism5 software.
Binding Affinity Determined by Flow Cytometry Analysis
The binding affinity of the antibody Thiomab-30 was determined by Flow Cytometry analysis. For this purpose, SKBR3 cells (with high expression of Her2 receptor) and Hek293T cells (with low expression of Her2 receptor) were plated in 96 well plates (100 000 cells per well) and incubated with 10 μL of 1 μM Thiomab-30 at room temperature. After 1 h of incubation, cells were washed with medium and were incubated with 50 μL/well of Goat anti-Human IgG (H+L) cross-adsorbed secondary antibody (10 μg/mL, Alexa Fluor 647, Thermo Scientific), for 1 h. After this incubation period, cells were washed by adding 100 μL of 10% FBS in PBS pH 7.4 and centrifuged for 5 min at 400g.The supernatant was then removed, the cells were resuspended in 400 μL of 10% FBS in PBS pH 7.4 and transferred to flow cytometry tubes. Acquisition was performed using a BD LSR Fortessa set up with a 640 nm laser and a 670/14 nm band-pass filter (combination used for APC detection). Data analysis was performed with FlowJo (version 6.3.4, FlowJo) software. Data represents mean ± s.d. of 3 biological replicates and only single-cell events are shown.
Acknowledgments
We thank the Gates-Cambridge Trust Foundation (scholarship to A.M.F.), Xunta de Galicia (Postdoctoral Fellowship to M.J.M.), Campus Iberus and MINECO (“Programa de ayudas para la movilidad del personal docente e investigador” and CTQ2015-67727-R both to F.C.), the European Commission (Marie Curie CIG to G.J.L.B., Marie Curie IEF to O.B., Marie-Skłodowska-Curie IF to T.R. and the Marie Curie ITN project ProteinConjugates to G.J.L.B. and P.A.), MINECO (CTQ2015-70524-R and RYC2013-14706 grants to G.J.O.), FCT Portugal (FCT Investigator to G.J.L.B. and Ph.D. studentship to A.G.), and the EPSRC for financial support. BIFI (Memento cluster) and UR (Beronia cluster) are acknowledged for computer support, Dr. Mike Deery and Ms. Julie Howard for help with mass spectrometry analysis. We also thank Dr. Justin Chalker for useful discussions and Genentech, Inc. for providing purified 4D5 LC-V205C THIOMAB. G.J.L.B. is a Royal Society University Research Fellow and holds an ERC Starting Grant (TagIt).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10702.
Detailed synthetic procedures and methods, characterization of all protein conjugates, and additional supporting figures (PDF)
Author Contributions
# A.M.F., M.J.M., O.B., F.C., and A.G. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Stephanopoulos N.; Francis M. B. Nat. Chem. Biol. 2011, 7, 876–884. 10.1038/nchembio.720. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. Chem. Rev. 2014, 114, 4764–4806. 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- Spicer C. D.; Davis B. G. Nat. Commun. 2014, 5, 4740. 10.1038/ncomms5740. [DOI] [PubMed] [Google Scholar]
- Boutureira O.; Bernardes G. J. L. Chem. Rev. 2015, 115, 2174–2195. 10.1021/cr500399p. [DOI] [PubMed] [Google Scholar]
- Krall N.; da Cruz F. P.; Boutureira O.; Bernardes G. J. L. Nat. Chem. 2016, 8, 103–113. 10.1038/nchem.2393. [DOI] [PubMed] [Google Scholar]
- Xue L.; Karpenko I. A.; Hiblot J.; Johnsson K. Nat. Chem. Biol. 2015, 11, 917–923. 10.1038/nchembio.1959. [DOI] [PubMed] [Google Scholar]
- Chari R. V. J.; Miller M. L.; Widdison W. C. Angew. Chem., Int. Ed. 2014, 53, 3796–3827. 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- Chudasama V.; Maruani A.; Caddick S. Nat. Chem. 2016, 8, 114–119. 10.1038/nchem.2415. [DOI] [PubMed] [Google Scholar]
- Junutula J. R.; Raab H.; Clark S.; Bhakta S.; Leipold D. D.; Weir S.; Chen Y.; Simpson M.; Tsai S. P.; Dennis M. S.; Lu Y.; Meng Y. G.; Ng C.; Yang J.; Lee C. C.; Duenas E.; Gorrell J.; Katta V.; Kim A.; McDorman K.; Flagella K.; Venook R.; Ross S.; Spencer S. D.; Lee Wong W.; Lowman H. B.; Vandlen R.; Sliwkowski M. X.; Scheller R. H.; Polakis P.; Mallet W. Nat. Biotechnol. 2008, 26, 925–932. 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- Xie J.; Schultz P. G. Nat. Rev. Mol. Cell Biol. 2006, 7, 775–782. 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- Davis L.; Chin J. W. Nat. Rev. Mol. Cell Biol. 2012, 13, 168–182. 10.1038/nrm3286. [DOI] [PubMed] [Google Scholar]
- Johnson J. A.; Lu Y. Y.; Van Deventer J. A.; Tirrell D. A. Curr. Opin. Chem. Biol. 2010, 14, 774–780. 10.1016/j.cbpa.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald J. I.; Munch H. K.; Moore T.; Francis M. B. Nat. Chem. Biol. 2015, 11, 326–331. 10.1038/nchembio.1792. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Welborn M.; Zhu T.; Yang N. J.; Santos M. S.; Van Voorhis T.; Pentelute B. L. Nat. Chem. 2016, 8, 120–128. 10.1038/nchem.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalker J. M.; Bernardes G. J. L.; Lin Y. A.; Davis B. G. Chem. - Asian J. 2009, 4, 630–640. 10.1002/asia.200800427. [DOI] [PubMed] [Google Scholar]
- Senter P. D.; Sievers E. L. Nat. Biotechnol. 2012, 30, 631–637. 10.1038/nbt.2289. [DOI] [PubMed] [Google Scholar]
- Lyon R. P.; Setter J. R.; Bovee T. D.; Doronina S. O.; Hunter J. H.; Anderson M. E.; Balasubramanian C. L.; Duniho S. M.; Leiske C. I.; Li F.; Senter P. D. Nat. Biotechnol. 2014, 32, 1059–1062. 10.1038/nbt.2968. [DOI] [PubMed] [Google Scholar]
- Bernardes G. J. L.; Chalker J. M.; Errey J. C.; Davis B. G. J. Am. Chem. Soc. 2008, 130, 5052–5053. 10.1021/ja800800p. [DOI] [PubMed] [Google Scholar]
- Chalker J. M.; Gunnoo S. B.; Boutureira O.; Gerstberger S. C.; Fernandez-Gonzalez M.; Bernardes G. J. L.; Griffin L.; Hailu H.; Schofield C. J.; Davis B. G. Chem. Sci. 2011, 2, 1666–1676. 10.1039/c1sc00185j. [DOI] [Google Scholar]
- Chalker J. M.; Lercher L.; Rose N. R.; Schofield C. J.; Davis B. G. Angew. Chem., Int. Ed. 2012, 51, 1835–1839. 10.1002/anie.201106432. [DOI] [PubMed] [Google Scholar]
- Chooi K. P.; Galan S. R. G.; Raj R.; McCullagh J.; Mohammed S.; Jones L. H.; Davis B. G. J. Am. Chem. Soc. 2014, 136, 1698–1701. 10.1021/ja4095318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleep D. Expert Opin. Drug Delivery 2015, 12, 793–812. 10.1517/17425247.2015.993313. [DOI] [PubMed] [Google Scholar]
- Baslé E.; Joubert N.; Pucheault M. Chem. Biol. 2010, 17, 213–227. 10.1016/j.chembiol.2010.02.008. [DOI] [PubMed] [Google Scholar]
- McFarland J. M.; Francis M. B. J. Am. Chem. Soc. 2005, 127, 13490–13491. 10.1021/ja054686c. [DOI] [PubMed] [Google Scholar]
- Furman J. L.; Kang M.; Choi S.; Cao Y.; Wold E. D.; Sun S. B.; Smider V. V.; Schultz P. G.; Kim C. H. J. Am. Chem. Soc. 2014, 136, 8411–8417. 10.1021/ja502851h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naidu B. N.; Li W.; Sorenson M. E.; Connolly T. P.; Wichtowski J. A.; Zhang Y.; Kim O. K.; Matiskella J. D.; Lam K. S.; Bronson J. J.; Ueda Y. Tetrahedron Lett. 2004, 45, 1059–1063. 10.1016/j.tetlet.2003.11.081. [DOI] [Google Scholar]
- Ferreira P. M. T.; Maia H. L. S.; Monteiro L. S.; Sacramento J.; Sebastiao J. J. Chem. Soc., Perkin Trans. 2000, 1, 3317–3324. 10.1039/b003353g. [DOI] [Google Scholar]
- Naidu B. N.; Sorenson M. E.; Connolly T. P.; Ueda Y. J. Org. Chem. 2003, 68, 10098–10102. 10.1021/jo034762z. [DOI] [PubMed] [Google Scholar]
- Dadová J.; Isenegger P. G.; Wu K.-J.; Errey J. C.; Bernardes G. J. L.; Chalker J. M.; Raich L.; Rovira C.; Davis B. G. ACS Cent. Sci. 2017, 3, 1168. 10.1021/acscentsci.7b00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardes G. J. L.; Chalker J. M.; Davis B. G.. PCT/GB2009/000194, 2011.
- Chalker J. M. D.Phil. Thesis, University of Oxford, 2012. [Google Scholar]
- Bernardes G. J. L. D.Phil. Thesis, University of Oxford, 2008. [Google Scholar]
- Zhao Y.; Truhlar D. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Smith J. M.; Jami Alahmadi Y.; Rowley C. N. J. Chem. Theory Comput. 2013, 9, 4860–4865. 10.1021/ct400773k. [DOI] [PubMed] [Google Scholar]
- Alam I. S.; Neves A. A.; Witney T. H.; Boren J.; Brindle K. M. Bioconjugate Chem. 2010, 21, 884–891. 10.1021/bc9004415. [DOI] [PubMed] [Google Scholar]
- Logue S. E.; Elgendy M.; Martin S. J. Nat. Protoc. 2009, 4, 1383–1395. 10.1038/nprot.2009.143. [DOI] [PubMed] [Google Scholar]
- Haas H.; Panula P. Nat. Rev. Neurosci. 2003, 4, 121–130. 10.1038/nrn1034. [DOI] [PubMed] [Google Scholar]
- Pelegri-O’Day E. M.; Lin E.-W.; Maynard H. D. J. Am. Chem. Soc. 2014, 136, 14323–14332. 10.1021/ja504390x. [DOI] [PubMed] [Google Scholar]
- Kratz F.; Elsadek B. J. Controlled Release 2012, 161, 429–445. 10.1016/j.jconrel.2011.11.028. [DOI] [PubMed] [Google Scholar]
- Meuzelaar H.; Panman M. R.; Woutersen S. Angew. Chem., Int. Ed. 2015, 54, 15255–15259. 10.1002/anie.201508601. [DOI] [PubMed] [Google Scholar]
- Stewart A. J.; Blindauer C. A.; Berezenko S.; Sleep D.; Tooth D.; Sadler P. J. FEBS J. 2005, 272, 353–362. 10.1111/j.1742-4658.2004.04474.x. [DOI] [PubMed] [Google Scholar]
- Andersen J. T.; Dalhus B.; Viuff D.; Thue Ravn B.; Gunnarsen K. S.; Plumridge A.; Bunting K.; Antunes F.; Williamson R.; Athwal S.; Allan E.; Evans L.; Bjørås M.; Kjærulff S.; Sleep D.; Sandlie I.; Cameron J. J. Biol. Chem. 2014, 289, 13492–13502. 10.1074/jbc.M114.549832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. T.; Dee Qian J.; Sandlie I. Eur. J. Immunol. 2006, 36, 3044–3051. 10.1002/eji.200636556. [DOI] [PubMed] [Google Scholar]
- Krishnan A. S.; Neves A. A.; de Backer M. M.; Hu D.-E.; Davletov B.; Kettunen M. I.; Brindle K. M. Radiology 2008, 246, 854–862. 10.1148/radiol.2463070471. [DOI] [PubMed] [Google Scholar]
- Neves A. A.; Xie B.; Fawcett S.; Alam I. S.; Witney T. H.; de Backer M. M.; Summers J.; Hughes W.; McGuire S.; Soloviev D.; Miller J.; Howat W. J.; Hu D. E.; Rodrigues T. B.; Lewis D. Y.; Brindle K. M. J. Nucl. Med. 2017, 58, 881–887. 10.2967/jnumed.116.183004. [DOI] [PubMed] [Google Scholar]
- Donaghy H. mAbs 2016, 8, 659–671. 10.1080/19420862.2016.1156829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. J.; Tran-Dubé M.; Shen H.; Nambu M.; Kung P.-P.; Pairish M.; Jia L.; Meng J.; Funk L.; Botrous I.; McTigue M.; Grodsky N.; Ryan K.; Padrique E.; Alton G.; Timofeevski S.; Yamazaki S.; Li Q.; Zou H.; Christensen J.; Mroczkowski B.; Bender S.; Kania R. S.; Edwards M. P. J. Med. Chem. 2011, 54, 6342–6363. 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- Vladimer G. I.; Snijder B.; Krall N.; Bigenzahn J. W.; Huber K. V. M.; Lardeau C.-H.; Sanjiv K.; Ringler A.; Berglund U. W.; Sabler M.; de la Fuente O. L.; Knobl P.; Kubicek S.; Helleday T.; Jager U.; Superti-Furga G. Nat. Chem. Biol. 2017, 13, 681–690. 10.1038/nchembio.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehar S. M.; Pillow T.; Xu M.; Staben L.; Kajihara K. K.; Vandlen R.; DePalatis L.; Raab H.; Hazenbos W. L.; Hiroshi Morisaki J.; Kim J.; Park S.; Darwish M.; Lee B.-C.; Hernandez H.; Loyet K. M.; Lupardus P.; Fong R.; Yan D.; Chalouni C.; Luis E.; Khalfin Y.; Plise E.; Cheong J.; Lyssikatos J. P.; Strandh M.; Koefoed K.; Andersen P. S.; Flygare J. A.; Wah Tan M.; Brown E. J.; Mariathasan S. Nature 2015, 527, 323–328. 10.1038/nature16057. [DOI] [PubMed] [Google Scholar]
- Lang K.; Chin J. W. ACS Chem. Biol. 2014, 9, 16–20. 10.1021/cb4009292. [DOI] [PubMed] [Google Scholar]
- Tian F.; Lu Y.; Manibusan A.; Sellers A.; Tran H.; Sun Y.; Phuong T.; Barnett R.; Hehli B.; Song F.; DeGuzman M. J.; Ensari S.; Pinkstaff J. K.; Sullivan L. M.; Biroc S. L.; Cho H.; Schultz P. G.; DiJoseph J.; Dougher M.; Ma D.; Dushin R.; Leal M.; Tchistiakova L.; Feyfant E.; Gerber H.-P.; Sapra P. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 1766–1771. 10.1073/pnas.1321237111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt U.; Öhler E. Angew. Chem., Int. Ed. Engl. 1976, 15, 42–42. 10.1002/anie.197600421. [DOI] [PubMed] [Google Scholar]
- Wright T. H.; Bower B. J.; Chalker J. M.; Bernardes G. J. L.; Wiewiora R.; Ng W.-L.; Raj R.; Faulkner S.; Vallée M. R. J.; Phanumartwiwath A.; Coleman O. D.; Thézénas M.-L.; Khan M.; Galan S. R. G.; Lercher L.; Schombs M. W.; Gerstberger S.; Palm-Espling M. E.; Baldwin A. J.; Kessler B. M.; Claridge T. D. W.; Mohammed S.; Davis B. G. Science 2016, 354, 597. 10.1126/science.aag1465. [DOI] [PubMed] [Google Scholar]
- Yang A.; Ha S.; Ahn J.; Kim R.; Kim S.; Lee Y.; Kim J.; Söll D.; Lee H.-Y.; Park H.-S. Science 2016, 354, 623–626. 10.1126/science.aah4428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnoo S. B.; Finney H. M.; Baker T. S.; Lawson A. D.; Anthony D. C.; Davis B. G. Nat. Commun. 2014, 5, 4388. 10.1038/ncomms5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Timms N.; Windle C. L.; Polyakova A.; Ault J. R.; Trinh C. H.; Pearson A. R.; Nelson A.; Berry A. ChemBioChem 2013, 14, 474–481. 10.1002/cbic.201200714. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.