

Abstract
Data transparency has been an important aspect of medical research as it helps in enabling evidence-based decisions in medicine which leads to foster trust among the patients and research community alike. Currently, it is one of the key talking points owing to a number of initiatives taken by the pharmaceutical organizations, regulatory bodies, and the other decision enablers of the industry. Thanks to this, there are a number of ways by which a single piece of datum is available through multiple access points, namely, clinical trial disclosures (CTDs), clinical study reports (CSRs), plain language summaries, and scientific publications including abstracts, posters, and manuscripts, to name a few. This may pose a burden of documentation on the pharmaceutical organizations, demanding downsizing of medical writing documents. Since CTDs, CSRs, and other regulatory document are more or less template driven; there may not be much scope to interfere with their structure and submission timings. Scientific publications, on the other hand, provide the flexibility of presenting the clinical data that is typically not dependent on a particular format and timelines. The present paper discusses how the upcoming data transparency initiatives could affect the publication practices across the pharmaceutical industry and what could pharmaceutical companies do to get the maximum benefit out of the data transparency initiatives.
Keywords: Authorship, data transparency, independent clinical data analysis, publication planning, publications
WHAT IS DATA TRANSPARENCY AND WHY IT MATTERS?
Evidence-based medical practice relies heavily on available data in the public domain so that informed health-care decisions can be made. Bringing in data from clinical trials within reach of clinicians, regulators, and external stakeholders alike enhances the applicability of the clinical trial data.
Data sharing help clinical investigators to weigh and/or substantiate the clinical question and share thought processes, potentiating scientific research, strengthening collaborations between multiple stakeholders, enhancing integrity and trust in clinical research among the general public, patients, and physicians.
Efforts, thus have been made to provide an easy access of data with a promise that it is coming from an authentic source, for example, Food and Drug Administration (FDA) modernization act of 1997,[1] which led to the inception of ClinicalTrials. Gov in 2000 and the World Health Organization (WHO) resolution of 2005,[2] which led to the establishment of the WHO international clinical trials registry platform, bringing transparency in data reported on the public registries.
The instigating factor for promoting data transparency are the instances of fabrication or concealment of clinical data, wherein withholding vital information led to serious repercussions. In one of the incidences, a pharmaceutical organization was under scrutiny for being non-transparent with respect to efficacy and safety of oseltamivir, an antiviral medication, indicated for the treatment of influenza. The adverse events of the medication were downplayed while exaggerating the efficacy.[3] The drug was approved for use in haste as authorities like the WHO and the European Medicines Agency (EMA) did not scrutinize the trial results before approving them.[4] In addition, majority of the data were unpublished over a decade post approval.[5] On the contrary, published data suggested that oseltamivir could induce sudden deterioration of health leading to death, which were consistent with animal toxicity studies.[6] Moreover, unpublished trial records and journal publications indicated that a number of adverse events mentioned in the clinical study reports (CSRs) were either omitted or hidden, indicating a reporting bias, putting patients at a great risk.[7]
In another case, there were attempts to conceal the cardiovascular risks of an antidiabetic drug rosiglitazone.[8,9] Post its launch in 1999, more than 100,000 cases of cardiovascular events such as heart attack and stroke including death were reported. In an article published in the New England Journal of Medicine, it was noted that rosiglitazone increases the risk of heart attack by 43%.[10] The drug was later banned in the European Union (EU) in 2010, while FDA allowed the drug to stay with the addition of black box warning. The drug manufacturer took a $2.4 billion penalty to cover the litigation charges in 2010.[11,12] However, after continued monitoring of the drug, the FDA lifted the Risk Evaluation and Mitigation Strategy in December 2015.[13]
These incidences highlight the importance and need of data transparency among the stakeholders of the clinical trial data to prevent such irreparable damages.
DATA TRANSPARENCY INITIATIVES: THE STORY SO FAR
The past one decade has seen a lot of promises and implementation in promoting transparency in clinical research. The practice of bringing transparency in the form of disclosures was first started with the registration of clinical trials in ClinicalTrials. Gov which primarily included interventional trials in 2000.[1]
To give an impetus to the transparency initiatives, the International Committee of Medical Journal Editors (ICMJE), initiated prospective registration of trials as a condition of publication in its member journals.[14] Similar thoughts were echoed by “All trials”[15] and endorsed by the WHO to publish results from every trial. Of late, the EMA developed its program to publish clinical data as their flagship policy of 2016.[16] The idea was to avoid duplication of studies, foster trust and confidence in the public, and help researchers to assess clinical data.[16] Recently, the ICMJE has mandated sharing of clinical data including the de-identified individual patient data (IPD) with the external investigators, as an ethical obligation for publication in its member journals from July 2018.[17] The ICMJE apprised that the authors propose a plan of data sharing, including where the data will be stored and how its access will be given to others.
Although there have been many initiatives and rulings toward data transparency, the pharmaceutical organizations are juggling with huge tasks of adapting to these requirements. In a published article, it was observed that out of 318 trials for 15 drugs in 2012, only 57% were registered in clinicaltrials.gov.[18] Furthermore, only 20% of the trials reported the results. Interestingly, 56% of the trials were published in medical journals,[18] suggesting a huge gap between the trial results disclosed and the ones that were being published as journal articles [Figure 1] which could be the case of the selective reporting, wherein only studies with positive outcomes were published.
Figure 1.

Percentage of registered, reported and published clinical trials. From http://bioethicsinternational.org/good-pharma-scorecard-overview/ethics-transparency/
On a brighter note, major pharmaceutical companies such as GlaxoSmithKline (GSK) and Janssen (Johnson & Johnson [J & J]) have become sensitive towards data transparency, which has become a driving factor for other pharmaceutical organizations. In a bid to assess adherence to baseline transparency requirements, it was observed that both GSK and J & J were 100% compliant, inferred by the Good Pharma Scorecard index (outcome-driven and performance-based assessment tool).[19] Other pharmaceutical organizations are also recognizing the importance of data transparency and catching up with the current practices and transparency standards, setting up robust mechanisms and policies pertaining to data disclosure in the public domain.
DATA TRANSPARENCY IN THE CURRENT SCENARIO: IS THIS TOO MUCH?
A close look at the data transparency initiatives suggests that pharmaceutical companies have their tasks in hand at each step of drug development process [Figure 2]. Before clinical trial begins, companies disclose their protocol to trial registers. During the trial, documents such as interim results, randomization records, and site monitoring reports have to be shared for public and participants. When a trial ends, the results are disclosed on the trial registers. Apart from these, there are regulatory documents that include, Investigator Brochures, CSR, reports such as drug safety update reports, summary of clinical efficacy, summary of clinical safety, to name a few, required for the submission process.
Figure 2.

Document types to be prepared during various stages of clinical trials
Plain language summaries (PLS) are a recent addition to the family of clinical documents. The need for PLS documents ascended as most of the data that were being disseminated by the pharmaceutical companies were either intended for regulatory submissions or the academicians or researchers. Trial participants, in general, who do not have the medical/science background, were not be able to comprehend the results of the clinical trials. Thus, there were demands doing round to communicate and disseminate the results in a plain and simple language that even a layperson could understand. In 2014, EU parliament passed a resolution (Regulation (EU) No 536/2014 (2014)) stating that pharmaceutical companies should provide the clinical trial results in a language that is understandable to a layperson, within the defined timelines.[20]
Making PLS mandatory will further ensure that pharmaceutical companies will have to bring out every positive/negative outcome result, a step towards making clinical data more transparent and easily accessible to the general public.
However, having so much documentation for the same data (off course from different perspectives) may cause a problem of too many for the pharmaceutical companies. So much so, some of the documents could lose their relevance or may demand teething changes in their structure. This may particularly happen to the publications documents. If we carefully look at a manuscript structure based on clinical trial, it essentially consists of four sections, namely, introduction, methods, results, and discussion. Of these four sections, introduction and discussion are very subjective, where the author builds up the context and discusses the clinical data results, respectively. In general, these two sections are of more use to a researcher building the thought process. Methods and results section consists of procedures, practices and clinical data that may or may not be a repetition to an already disclosed information as regulatory documents. In the time to come, we may see pharmaceutical companies, in a bid to lower documentation practice, press for linking the data from the randomized clinical trials with the manuscripts, publishing just the discussion section, which may lead to the end of the publications the way we have been seeing. However, it would be interesting to watch how the publication houses will respond to this change. For review articles, meta-analysis and abstracts, it is highly unlikely that there could be a significant impact.
TRANSPARENCY AND PUBLICATIONS
Publications are under high scrutiny now considering these transmit the data to a wide audience, and therefore, it is important to understand the possible impact of data transparency initiatives on publications.
Although one may disagree, the sole purpose of publications, be it an abstract, poster, or manuscript, is to create and generate interest among the academicians, researchers, and physicians alike, in a particular area so that these people may contribute in the form of new ideas, new initiatives, and policies that will benefit the end user, i.e., the patients. For example, publications in the form of reviews and meta-analysis help in validating or rejecting a clinical question by systematically analyzing the existing literature and help in establishing practice guidelines in a particular therapeutic area. On the other hand, post hoc analysis that includes the patient level data from an already conducted clinical trial helps in unearthing patterns, pathophysiology/genetics, diagnostic markers or other factors that may impact health, not evident in the parent trial conducted with a different endpoint(s).
Publications act as a beacon that spreads the light of knowledge far in the sea of medical science. With clear messaging and wide reach, pharmaceutical organizations are dependent largely on the publications (as publication planning), to present ideas for new drugs in the launching phase and prospective innovations, etc.
However, in the hindsight, it seems that data transparency have impacted publication practices in one or the other way [Figure 3]. This is because publications are marred with certain practices such as selective publication, publication bias and ghostwriting that leads to distortion of medical literature.
Figure 3.
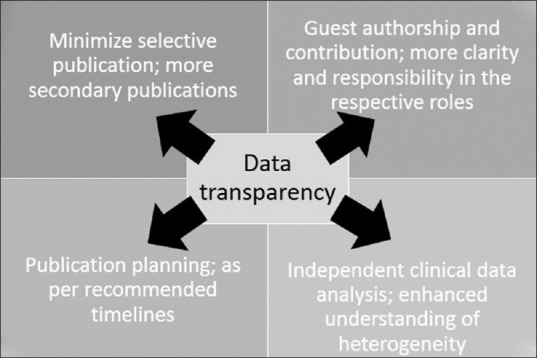
Data transparency and its impact on publications
MINIMIZE SELECTIVE PUBLICATIONS, MORE SECONDARY PUBLICATIONS
Selective publications pose a threat to medical research in general. This is because most of the interventional studies are sponsored by the industry and no company would prefer publishing negative data under their name. Thus, outcomes pertaining to adverse events, lack of efficacy, or any undesired event may not be conveyed across.
In principle, posting of clinical trial data on the public sites and the strict implementation of this policy over the years has ensured published data either in the form of abstract/poster or manuscripts, could be traced back to its source trial. Largely, transparency initiatives will ensure that the practice of selective publication will be abolished or to an extent will be reduced, as the pharmaceutical organizations will be bound to publish each trial disclosed on the public registries. Furthermore, it will also encourage publications of secondary endpoints since the clinical data are already present in the public domain and the physicians or the key stakeholders who are following the clinical data will be interested in those publications.
GUEST AUTHORSHIP AND CONTRIBUTIONS
Guest authorship refers to the practice in publications wherein the pharmaceutical organizations invite key opinion leaders (KOLs) or independent investigators to become an author in an attempt to provide an academic flavor to their publications. However, these KOLs generally do not have access to the trial data and hence may not be able to independently analyze and interpret it. With the ICMJE announcement that the clinical data are to be shared with all the investigators and authors,[14] there is a great opportunity for the pharmaceutical organizations to streamline their process of selecting authors for the publications. Further, sharing of trial data with the authors will empower them to clearly interpret the data and its implications in the clinical setting, and they will act more responsibly while reviewing the documents. From being “Guest” authors, the KOLs/external authors will become stakeholders of the data and the publication alike. This will also impact the disclosure statement related to funding and conflict of interest as they will not be merely endorsing a particular drug/molecule but they will have to be more accountable with it.
PUBLICATION PLANNING: AS PER RECOMMENDED TIMELINES
Essentially, publication planning is a powerful process wherein the pharmaceutical companies publish their trial results in the form of abstract, opinion-based papers, etc., strategically disseminating medical knowledge during the drug approval and marketing phase.
As a thumb rule, the number of publications should peak around the time that coincides or is near to the launch of the product that may or may not depend on the completion date for a particular trial. On the other hand, the International Federations of Pharmaceutical Manufacturers and Associations (IFPMA) code of practices recommend that manuscripts should be submitted to the medical journals within 12 months (or 18 months at the latest) of trial completion.[21] Furthermore, the recent Good Publication Practices (GPP) 3 update suggests that before publishing the clinical data, the pharmaceutical companies will be required to publish their background data including new techniques and methods.[22] This will ensure that data dissemination will be prompt data dissemination, bringing in more transparency in the practices followed in the industry. This recommendation will further ensure that the drug approval and launch process will hasten up as publications play a vital role in educating the regulators and KOLs alike and will help them to make their decision.
As far as the question of creating the noise during the launch of the drug is concerned, the pharmaceutical companies can still publish their primary results first, preceded by the secondary data that may include further analysis of data (like post hoc analysis) or review articles, steered by the encouraging primary results. Most importantly, as disclosure of clinical protocol ensures that each trial is in public domain before its commencement, implementation of the IFPMA code of practices/recommendations along with the GPP 3 guidelines will further ensure that even the negative results are being published. However, pharmaceutical companies might not prefer to present the negative results as full publication; these may still find their way as “letter to editor,” newsletters, short summaries. Further, pharmaceutical companies might consider to publish these results in the form of post hoc analysis to explore treatment in a subgroup population. This may open new avenues for research particularly for other investigators to explore.
INDEPENDENT CLINICAL DATA ANALYSIS
Recently, there have been requests for sharing de-identified IPD from clinical trials. This may include demographics, baseline characteristics, and other clinical laboratory results. GSK is one of the few organizations that have the provision of sharing its redacted patient-level data with researchers meeting certain criteria.[23] Initiatives like these will help in validating key findings along with bringing in public a large amount unpublished data in the form of meta-analysis and pooled analysis. Although meta-analysis and pooled analysis are still being published, independent data analysis will help in analyzing rare outcomes from the data that was not in the picture from a long time. Furthermore, this will help in understanding the heterogeneity among the studies included in the meta-analysis and pooled analysis, including the heterogeneity in the trial population, procedures, outcome measures and intervention effects.
CONCLUSION
So far, we have seen that the pharmaceutical organizations and the regulators are becoming sensitive towards the data transparency, although it may take a few years for the 100% implementation of the existing transparency measures. Considering the changing landscape wherein we are bound to see an enhanced convergence of healthcare and technology, high research and development expenses and rising expectations for a return of investment, will further give a push to data sharing practices. With transparency in data sharing practices benefitting publication industry in multiple ways, as shown above, the benefits will become more evident in terms of winning public and regulator's trust, faster approvals and ease to capture the commercial markets.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.U.S. Food and Drug. Food and Drug Administration Modernization Act (FDAMA) of 1997. [Last accessed on 2017 Jul 13]. Available from: https://www.fda.gov/RegulatoryInformation/LawsEnforcedbyFDA/SignificantAmendmentstotheFDCAct/FDAMA/default.htm .
- 2.International Clinical Trials Registry Platform (ICTRP) WHO Statement on Public Disclosure of Clinical Trial Results. [Last accessed on 2017 Jul 13]. Available from: http://www.who.int/ictrp/results/reporting/en/
- 3.Jack A. Tamiflu: “A nice little earner”. Br Med J. 2014;348:g2524. doi: 10.1136/bmj.g2524. [DOI] [PubMed] [Google Scholar]
- 4.The Guardian. What the Tamiflu Saga tells us about drug Trials and Big Pharma. [Last accessed on 2017 Jul 13]. Available from: https://www.theguardian.com/business/2014/apr/10/tamiflu-saga-drug-trials-big-pharma .
- 5.Tamiflu Campaign. Tamiflu Data: Who Saw What When. [Last accessed on 2017 Jul 13]. Available from: http://www.bmj.com/tamiflu .
- 6.Ward P, Small I, Smith J, Suter P, Dutkowski R. Oseltamivir (Tamiflu) and its potential for use in the event of an influenza pandemic. J Antimicrob Chemother. 2005;55(Suppl 1):i5–i21. doi: 10.1093/jac/dki018. [DOI] [PubMed] [Google Scholar]
- 7.Tamiflu Review Shows Desperate Need for Full Clinical Trials Data Transparency. Health Action International. [Last accessed on 2017 Jul 13]. Available from: http://www.haiweb.org/tamiflu-review-shows-desperate-need-for-full-clinical-trials-data-transparency/
- 8.Drazen JM, Wood AJ. Don't mess with the DSMB. N Engl J Med. 2010;363:477–8. doi: 10.1056/NEJMe1007445. [DOI] [PubMed] [Google Scholar]
- 9.Bracken MB. Rosiglitazone and cardiovascular risk. N Engl J Med. 2007;357:937–8. doi: 10.1056/NEJMc071602. [DOI] [PubMed] [Google Scholar]
- 10.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–71. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 11.Independent. GSK's Avandia Banned in Europe on Heart Worries. [Last accessed on 2017 Jul 13]. Available from: http://www.independent.co.uk/news/business/news/gsks-avandia-banned-in-europe-on-heart-worries-2088080.html .
- 12.NPR. Glaxo to Take $2. Glaxo to Take $24 Billion Charge to Cover Avandia Cases, Other Legal Woes. [Last accessed on 2017 Jul 13]. Available from: http://www.npr.org/sections/health-shots/2010/07/15/128534637/glaxo-takes-2-4-billion-charge-to-cover-avandia-cases .
- 13.FDA Drug Safety Communication: FDA Eliminates the Risk Evaluation and Mitigation Strategy (REMS) for Rosiglitazone.Containing Diabetes Medicines. [Last accessed on 2017 Jul 13]. Available from: https://www.fda.gov/downloads/Drugs/DrugSafety/UCM477575.pdf .
- 14.International Committee of Medical Journal Editors. Clinical Trials Registration. [Last accessed on 2017 Jul 13]. Available from: http://www.icmje.org/about-icmje/faqs/clinical-trials-registration/
- 15.All Trials. All Trials Registered|All Results Reported. [Last accessed on 2017 Jul 13]. Available from: http://www.alltrial.snet/
- 16.European Medicines Agency. Clinical Data Publication. [Last accessed on 2017 Jul 13]. Available from: http://www.ema.europa.eu/ema/curl=pages/special_topics/general/general_content_000555.jsp .
- 17.Taichman DB, Sahni P, Pinborg A, Peiperl L, Laine C, James A, et al. Data sharing statements for clinical trials: A requirement of the International Committee of Medical Journal Editors. Bull World Health Organ. 2017;95:482–3. doi: 10.2471/BLT.17.196733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miller JE, Korn D, Ross JS. Clinical trial registration, reporting, publication and FDAAA compliance: A cross-sectional analysis and ranking of new drugs approved by the FDA in 2012. BMJ Open. 2015;5:e009758. doi: 10.1136/bmjopen-2015-009758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bioethics International. Transparency Ranking of Drugs Approved By the FDA in 2012. [Last accessed on 2017 Jul 13]. Available from: http://www.bioethicsinternational.org/good-pharma-scorecard-overview/ethics-transparency/
- 20.Regulation (eu) no 536/2014 of the European Parliament and of the Council of 16 April 2014 on Clinical Trials on Medicinal Products for Human Use, and Repealing Directive 2001/20/EC. [Last accessed on 2017 Jul 13]. Available from: http://www.ec.europa.eu/health/sites/health/files/files/eudralex/vol-1/reg_2014_536/reg_2014_536_en.pdf .
- 21.International Federation of Pharmaceutical Manufacturers & Associations; European Federation of Pharmaceutical Industries and Associations; Japan Pharmaceutical Manufacturers Association; Pharmaceutical Research and Manufacturers of America. Joint Position on the Publication of Clinical Trial Results in the Scientific Literature. 2010. Jun 10, [Last accessed on 2017 Jul 13]. Available from: https://www.ifpma.org/resource-centre/new-industry-position-requires-submission-for-journal-publication-of-all-phase-iii-clinical-trials/
- 22.Battisti WP, Wager E, Baltzer L, Bridges D, Cairns A, Carswell CI, et al. Good publication practice for communicating company-sponsored medical research: GPP3. Ann Intern Med. 2015;163:461–4. doi: 10.7326/M15-0288. [DOI] [PubMed] [Google Scholar]
- 23.Cressey D. Drug-company data vaults to be opened. Nature. 2013;495:419–20. doi: 10.1038/495419a. [DOI] [PubMed] [Google Scholar]