

Abstract
Background
Common wheat is a typical allohexaploid species (AABBDD) derived from the interspecific crossing between allotetraploid wheat (AABB) and Aegilops tauschii (DD). Wide variation in grain size and shape observed among Aegilops tauschii can be retained in synthetic allohexaploid wheats, but the underlying mechanism remains enigmatic. Here, the natural and resynthesized allohexaploid wheats with near-identical AB genomes and different D genomes (TAA10 and XX329) were employed for analysis.
Results
Significant differences in grain size and weight between TAA10 and XX329 were observed at the early stages of development, which could be mainly attributed to the higher growth rates of the pericarp and endosperm cells in XX329 compared to TAA10. Furthermore, comparative transcriptome analysis identified that 8891 of 69,711 unigenes (12.75%) were differentially expressed between grains at 6 days after pollination (DAP) of TAA10 and XX329, including 5314 up-regulated and 3577 down-regulated genes in XX329 compared to TAA10. The MapMan functional annotation and enrichment analysis revealed that the differentially expressed genes were significantly enriched in categories of cell wall, carbohydrate and hormone metabolism. Notably, consistent with the up-regulation of sucrose synthase genes in resynthesized relative to natural allohexaploid wheat, the resynthesized allohexaploid wheat accumulated much higher contents of glucose and fructose in 6-DAP grains than those of the natural allohexaploid wheat.
Conclusions
These data indicated that the genetic variation of the D genome induced drastic alterations of gene expression in grains of the natural and resynthesized allohexaploid wheats, which may contribute to the observed differences in grain size and weight.
Electronic supplementary material
The online version of this article (10.1186/s12870-018-1248-y) contains supplementary material, which is available to authorized users.
Keywords: Allohexaploid wheat, D genome, Grain size and weight, Gene expression
Background
Wheat is the leading food crop produced, consumed, and traded worldwide today. To meet the demand for burgeoning human population, we need wheat cultivars with higher yield potential [1]. The grain yield of wheat is mainly determined by the number of grains per m2 and grain weight. Modern breeding has greatly improved wheat yield by increasing grains per m2 due to the utilization of dwarfing genes (Rht) in 1960s and 1970s [2]. Analysis of 1800 cultivars exhibited that thousand grain weight (TGW) increased from a mean 31.5 g in the 1940s to 44.64 g in the 2000s, with an average 2.19 g increase in each decade, indicating that TGW is an important target for the improvement of wheat yield potential [3]. In the past two decades, the successful application of quantitative-genetic methodology has facilitated the identification of numerous QTL for TGW on all 21 wheat chromosomes [4–8]. Recently, several genes associated with wheat grain size and weight were isolated by using a homology-based approach, such as TaCwi, TaGW2, TaTGW6, TaGS1a, TaGASR7-A1 and TaCYP78A3 [9–15], which enhanced our knowledge about grain size and weight determination in wheat.
Grain development is an important and complex process in the wheat life cycle and directly affects the grain weight, which can be divided into five main phases: fertilization, “coenocytic” endosperm, cellularization and early grain filling, maximum grain filling, and desiccation [16]. At the level of gene expression, growing studies exhibited that the large changes in transcript/protein abundance were associated with distinct phases of wheat grain development. For example, novel distinct spatial gene expression patterns during wheat caryopsis development have been revealed by using a novel high-throughput mRNA in situ hybridization [17]. Distinct co-expression clusters reflecting the spatiotemporal progression during wheat endosperm development were identified by the cell type-specific expression analysis [18]. Although these studies provided a number of candidate genes important for grain growth and development, the molecular mechanisms involved in the determination of grain weight and size are still poorly understood.
Common wheat is a natural allohexaploid with A, B and D genomes contributed by Triticum urartu, a close relative of Aegilops speltoides and Aegilops tauschii (syn. Ae. squarrosa, Tritcum tauschii), respectively [19]. However, only a few Aegilops tauschii’s intraspecific lineages contributed to the evolution of common wheat, which resulted in limited D-genome variation represented in hexaploid bread wheat [20]. Thus, to increase the D-genome diversity in bread wheat, Aegilops tauschii has been used to introgress various traits of economic importance into bread wheat [21–24]. Specifically, wide variation in grain size and shape observed among Ae. tauschii accessions is retained in synthetic allohexaploid wheat lines [25–27]. Moreover, several studies have been carried out to identify beneficial QTL for grain weight from the diploid D donor of common wheat [28, 29]. For instance, a major QTL QGw.caas-3D controlling higher grain weight was identified on chromosome 3D of synthetic hexaploid wheat Am3 [29]. In 1964, an allotetraploid wheat (ETW) containing the AABB component was extracted from the allohexaploid bread wheat (TAA10). Afterwards, a resynthesized allohexaploid wheat (XX329) was produced by crossing ETW and the Ae. tauschii subsp. strangulate (TQ18). Interestingly, the grain size and weight of the resynthesized allohexaploid wheat (XX329) are much higher than that of TAA10 [30]. Since the AABB genomes of XX329 should be very similar to that of the donor TAA10, the observed variation in grain size and weight between these two genotypes may be mainly attributed to the differences of D genome, but the underlying molecular basis is still poorly understood.
The wheat caryopsis is a complex tissue in which maternal and endosperm tissues follow distinct but coordinated developmental programs [17]. During the initial phase of grain growth, maternal tissues (the pericarp of the grains) undergo a remarkable expansion as they are the main component of grain at this time. Thereby, grain size and weight determination are driven by pericarp growth [31]. Here, to dissect the underlying molecular basis for the variation of grain size and weight between natural and resynthesized allohexaploid wheats (TAA10 and XX329), the dynamic grain size and weight of these two genotypes were investigated, and grains at 6 DAP were selected for comparative transcriptome analysis. The results revealed that the variation in grain size could be attributed to the difference in pericarp expansion between TAA10 and XX329. Notably, the enrichment of differentially expressed genes involved in carbohydrate and cell wall metabolism may play important roles in the observed differences between TAA10 and XX329 in terms of grain size and weight.
Methods
Plant materials
The natural allohexaploid wheat TAA10 and the resynthesized allohexaploid wheat XX329 were grown in three biological replicates (5 rows/replicate) in Shangzhuang, Beijing (E116°, N40°) in the autumn of 2015. Grains were well distributed in rows that were 1.5 m long and 0.3 m apart with a sowing rate at 20 grains per row.
Grain weight and volume measurements
Developing grains of TAA10 and XX329 for grain weight and volume measurements were collected at 2, 4, 6, 8, 10, 15, 20, 25, 30 and 35 days after pollination (DAP) with three biological replicates. In each replicate for each developmental stage, five spikes were sampled and the grains in eight central spikelets of each spike were used to measure grain fresh weight, dry weight and volume. The dry weight of grains was recorded after oven drying at 105 °C for 15 min and 65 °C for 48 h. The grain volume was determined using the “water displacement” principle, which was measured as the volume change of 95% alcohol after placing the grains in the measuring tube.
Cytological observations and measurements
For cytological observations, the grains of TAA10 and XX329 at 2, 4, 6, 8, 10 and 15 DAP were collected with three biological replicates. Each replicate contained the grains in eight central spikelets of one spike. Grains from different samples were fixed in 50% (v/v) ethanol, 5% (v/v) acetic acid, and 3.7% (w/v) formaldehyde over 12 h at 4 °C, followed by dehydration and embedding in paraffin. Cross sections (3 μm) from the middle part of grains were cut using a Leica Ultracut rotary microtome and stained with Periodic acid Schiff (PAS). Photographs were taken with Pannoramic MIDI (3DHISTECH, Ltd., Hungary).
Cross sections of three grains for each developmental stage (2, 4, 6, 8, 10 and 15 DAP) were used to measure cell area. On each cross section, the cell area of the pericarp was measured by selecting three regions within five-row cells from the epicarp inwards, and the cell area of the endosperm was measured using three randomly selected regions of the endosperm. In each of the measured regions, between 20 and 40 cells were measured with CaseViewer 2.0 (3DHISTECH, Ltd., Hungary).
Sugar content analysis
For sugar content analysis, the grains at 6 DAP of TAA10 and XX329 with three biological replicates were sampled, immediately frozen in liquid nitrogen and stored at − 80 °C.Each replicate contained three spikes and the grains in eight central spikelets of each spike were sampled. Sugar analysis was performed using ten grains in each sample, which were weighed and prepared for measurement. Soluble sugars of grain samples were extracted and dissolved in water, and the contents of sucrose, D-glucose and D-fructose in the aqueous extracts were enzymatically determined using a kit from R-Biopharm (Darmstadt, Germany) according to the manual (https://food.r-biopharm.com/wp-content/uploads/sites/2/2014/04/Roche_IFU_Sucrose-Glucose-fructose_EN_10716260035_2014-01.pdf). Statistical analysis of the difference in the sugar content between TAA10 and XX329 was performed using Student’s t-test.
RNA extraction and transcriptome sequencing
Total RNA was extracted from grains at 6 DAP of TAA10 and XX329 with three biological replicates using TRIzol reagent (Invitrogen), according to the manual. RNA concentration was measured using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc., USA) and RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc., USA). Non-stranded paired-end sequencing libraries with an average insert size of 400 bp were prepared with TruSeq RNA Sample Preparation Kit v2 (Illumina, USA) and sequenced using HiSeq 2000 (Illumina, USA) based on the manufacturer’s instructions. Raw data obtained from Illumina sequencing were processed and filtered using the Illumina pipeline (http://www.illumina.com) to generate FastQ files. Low quality reads were filtered with the following parameters: removing the reads containing adaptor sequences, more than 3% ambiguous bases (noted as N) and 50% low quality bases (Phred quality score Q < 30). Finally, approximately 6 Gb high-quality 125-bp paired-end reads were generated from each library. The RNA-Seq reads used for this study were deposited at the National Center for Biotechnology Information Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) under the accession number SRP117710.
Alignment of RNA-Seq reads and expression analysis
The high-quality paired-end RNA-Seq reads from each library were aligned to the wheat reference genome (version TGACv1, http://plants.ensembl.org/Triticum_aestivum/) [32] using Hisat2 with parameters “--phred33 --max-intronlen 7000 -k 10 -t -p 4 --no-unal --ignore-quals --rdg 3,1 --rfg 3,1 --score-min L,0,-0.19”, and alignments with no more than one mismatch were retained [33]. Then, multiply mapped reads were filtered out by customized Perl script, and reads uniquely mapped to the annotated wheat reference genes were counted by HTSeq-count [34]. The uniquely mapped reads counts were used in the following FPKM (Fragments per Kilobase per Million mapped reads) calculation and differential expression analysis which were performed by edgeR package in R software [35]. Wheat reference genes with more than 2-fold changes and false discovery rate (FDR, Benjamini and Hochberg’s method)-adjusted P-value less than 0.05 were considered as differentially expressed genes.
MapMan analysis
Wheat transcripts were annotated using the MapMan Mercator tool (http://mapman.gabipd.org /web/guest/mercator). The functional category analysis of differentially expressed genes was performed by MapMan version 3.6.0 [36]. Significantly overrepresented MapMan functional categories were determined by Fisher’s Exact Test (P-value < 0.05) and enrichment fold ≥1.5 compared with the whole genome background.
Quantitative reverse transcription PCR (qRT-PCR) analysis
Total RNA (2 μg) from each sample was used to generate complementary DNA (cDNA) templates using HiScript Q RT SuperMix for qPCR (Vazyme Biotech, Ltd., China), according to the manufacturer’s instructions. QRT-PCR was performed using a SYBR Green PCR Master Mix (Vazyme Biotech, Ltd., China) on the CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories, Inc., USA). The PCR conditions consisted of an initial step at 95 °C for 3 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 15 s and 72 °C for 30 s. Specific primer pairs for qRT-PCR analysis are listed in Table S6. For each sample, the PCR analysis was independently repeated in triplicate, and the quantification of gene expression was carried out by the relative quantification method (2-ΔΔCT method) with β-actin as an endogenous control [37].
Results
Dynamic comparison of developing grains between natural and resynthesized allohexaploid wheats
To explore the underlying mechanism for the variation of grain size and weight between natural and resynthesized allohexaploid wheats (TAA10 and XX329), we firstly measured the grain volume of these two genotypes at 2, 4, 6, 8, 10, 15, 20 and 35 DAP. For each genotype, grain volume rapidly increased during the early stages of development and reached their maximal volume at about 10 DAP, which was consistent with previous studies [16]. When comparing these two genotypes, the grain volume of XX329 was significantly higher than that of TAA10 from 4 DAP and acquired the largest difference at 10 DAP. After that, the superiority of XX329 was maintained until all grains matured (Fig. 1b). Next, we measured the fresh and dry grain weight of TAA10 and XX329 at different stages of development. The fresh grain weight of TAA10 and XX329 consecutively increased from 2 DAP and acquired their maximal fresh weight at about 25 DAP, thereafter decreased until the grains matured (Fig. 1c). Different from the fresh grain weight, the dry grain weight of each genotype continued to increase until grain maturation at about 35 DAP (Fig. 1d). Notably, XX329 exhibited significantly higher fresh and dry grain weight than that of TAA10 at each stages of grain development, and the difference in dry grain weight between XX329 and TAA10 reached the maximum at about 35 DAP when all grains matured (Fig. 1c-d).
Fig. 1.
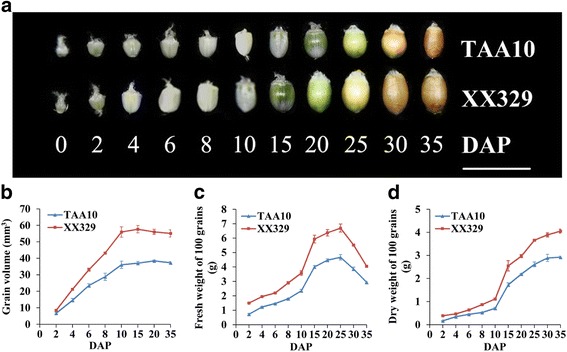
Dynamic comparison of grains at different developmental stages between the natural allohexaploid wheat TAA10 and the resynthesized allohexaploid wheat XX329. a Grain morphology at 0, 2, 4, 6, 8, 10, 15, 20, 25, 30 and 35 DAP of TAA10 and XX329. The scale bar represents 1 cm. b Grain volume at 2, 4, 6, 8, 10, 15, 20 and 35 DAP of TAA10 and XX329. The values are shown as means ± SE. c-d 100-grain fresh weight and dry weight at 2, 4, 6, 8, 10, 15, 20, 25, 30 and 35 DAP of TAA10 and XX329. The values are shown as means ± SE
The development of maternal grain tissue is mediated by regulated cell expansion and coordinated with endosperm growth [38]. Thus, we decided to measure and compare the cell size of the pericarp and endosperm in TAA10 and XX329 during the early stages of grain development (2, 4, 6, 8, 10 and 15 DAP) by cytological observation. The pericarp cell area of each genotype increased by 2 fold from 2 to 15 DAP and the growth rate of pericarp cells at 2-8 DAP was faster than that at 8-15 DAP. Moreover, the pericarp cells of XX329 exhibited faster growth rate and greater cell area than those of TAA10 during the early stages of grain development (Fig. 2i-p; Fig. 3a). Correspondingly, the growth of the endosperm cells kept pace with that of the pericarp cells. The endosperm cell size of each genotype increased by 3-4 folds from 6 to 15 DAP. Moreover, XX329 exhibited much greater endosperm cell area than that of TAA10 from 8 to 15 DAP (Fig. 2q-v; Fig. 3b). Notably, we found that the cellularization of the endosperm was complete and the starch granules were initially formed in quantity in the endosperm cells by 6 DAP (Fig. 2). Taken together, grains at 6 DAP were selected for further comparative transcriptome analysis.
Fig. 2.
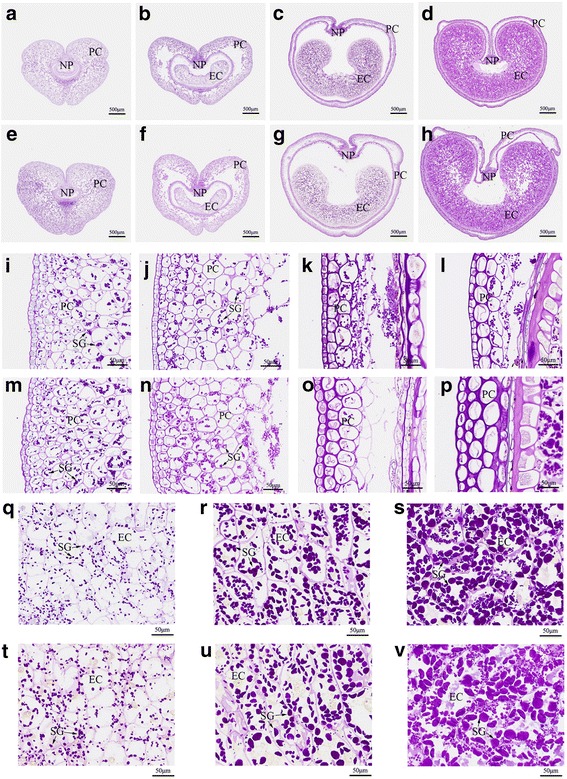
Cytological observations by cross-sections of grains at different developmental stages between the natural allohexaploid wheat TAA10 and the resynthesized allohexaploid wheat XX329. a-d Cross-sections of grains at 2 (a), 6 (b), 10 (c) and 15 (d) DAP in TAA10.The scale bars represent 500 μm. e-h Cross-sections of grains at 2 (e), 6 (f), 10 (g) and 15 (h) DAP in XX329. The scale bars represent 500 μm. i-l The pericarp cells of grains at 2 (i), 6 (j), 10 (k) and 15 (l) DAP in TAA10. The scale bars represent 50 μm. m-p The pericarp cells of grains at 2 (m), 6 (n), 10 (o) and 15 (p) DAP in XX329. The scale bars represent 50 μm. q-s The endosperm cells of grains at 6 (q), 10 (r) and 15 (s) DAP in TAA10. The scale bars represent 50 μm. t-v The endosperm cells of grains at 6 (t), 10 (u) and 15 (v) DAP in XX329. The scale bars represent 50 μm. NP, nucellar projection; PC, pericarp cell; EC, endosperm cell; SG, starch granule
Fig. 3.
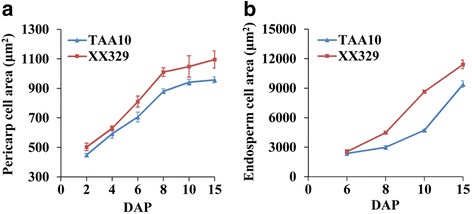
Cell area of the pericarp and endosperm during the early stages of grain development between the natural allohexaploid wheat TAA10 and the resynthesized allohexaploid wheat XX329. a Cell area of the pericarp at 2, 4, 6, 8, 10 and 15 DAP of TAA10 and XX329. The values are shown as means ± SE. b Cell area of the endosperm at 6, 8, 10 and 15 DAP of TAA10 and XX329. The values are shown as means ± SE
Comparative transcriptome profile of 6-DAP grains between natural and resynthesized allohexaploid wheats
To dissect the potential molecular basis for the difference in grain size between natural and resynthesized allohexaploid wheats, we performed RNA sequencing of 6-DAP grains of TAA10 and XX329 with three biological replicates. After removal of low-quality sequencing reads, a total of approximately 162.2 million 125 bp paired-end reads were generated, with an average of 27.0 million filtered reads for each library. About 65.4%-74.5% of the high-quality reads were uniquely mapped to wheat reference genome sequence. The ratios of uniquely mapped reads for TAA10 and XX329 on the A, B and D genomes of allohexaploid wheat ranged from 21.5 to 23.7%, indicating that these reads were equally distributed among the three subgenomes (Fig. 4a).
Fig. 4.
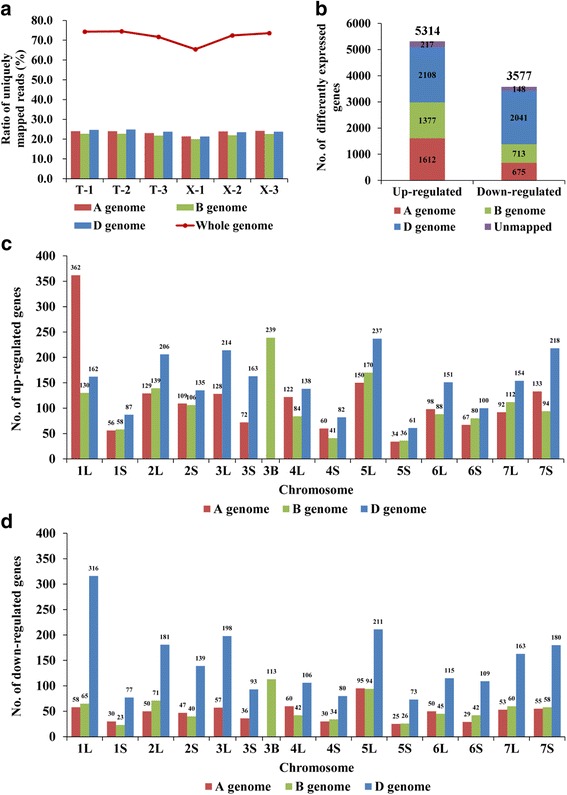
Transcriptome sequencing reads mapping and differentially expressed analysis. a The ratio of the uniquely mapped reads in each biological replicate of TAA10 and XX329. b The numbers of up-regulated and down-regulated genes on the A, B and D genomes in the resynthesized allohexaploid wheat XX329 relative to the natural allohexaploid wheat TAA10. (c-d) The numbers of up-regulated (c) and down-regulated genes (d) in XX329 relative to TAA10 on the long and short arms of different chromosomes. There is no separation for long and short arms on the chromosome 3B
To identify the differentially expressed genes between 6-DAP grains of the natural allohexaploid TAA10 and resynthesized allohexaploid wheat XX329, we performed the expression analysis using the uniquely mapped reads for each library. The correlation coefficients of different biological replicates ranged from 0.972 to 0.986 (Additional file 1: Figure S1). In total, 69,711 TGACv1 reference genes were expressed (FPKM ≥0.5 and Reads Counts ≥10) among the two genotypes and the number in TAA10 and XX329 was 69,162 and 69,195, respectively. Of 69,711 genes, 8891 (12.75%) were differentially expressed genes between TAA10 and XX329 (fold change ≥2 and false discovery rate (FDR) adjusted P < 0.05) (Additional file 2: Table S1). Compared to TAA10, the number of up-regulated genes in XX329 (5314) was much higher than that of down-regulated genes (3577) (Fig. 4b). To validate the RNA-Seq results, qRT-PCR was performed for 9 randomly selected differentially expressed genes. As shown in Fig. 5, the fold changes in gene expression determined by qRT-PCR were consistent with the changes of normalized expression level (FPKM) determined by RNA-Seq. The detailed information for these genes was shown in Additional file 3: Table S6.
Fig. 5.
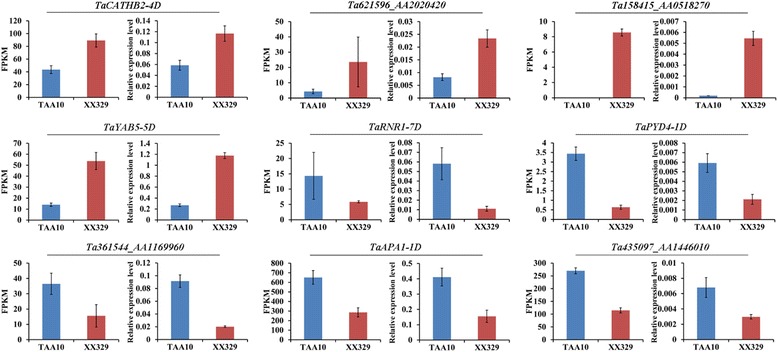
Verification of 9 differentially expressed genes by qRT-PCR. The values are shown as means ± SD
According to the wheat genome annotation (version TGACv1, http://plants.ensembl.org/Triticum_aestivum/), 8526 (95.89%) differentially expressed genes could be assigned to chromosomes, and the numbers on the A, B and D genomes were 2287, 2090 and 4149, respectively (Fig. 4b). Notably, the number of down-regulated genes in XX329 relative to TAA10 on each chromosome of the D genome was higher than that on each corresponding chromosome of the A and B genomes. In addition, we found that down-regulated genes were enriched on chromosome 1DL, whereas up-regulated genes were overrepresented on chromosome 1AL (Fig. 4c-d).
Functional annotation and categorization of the differentially expressed genes
The MapMan analysis was performed to analyze the functional annotation and categorization of the differentially expressed genes. In total, 7039 of 8891 differentially expressed genes between TAA10 and XX329 could be assigned to the MapMan categories, of which 2022 were “not assigned” to a functional category. The number of genes were assigned to the functional category “RNA” was highest (842), followed by “protein” (815), “misc.” (811) and “signaling” (397) (Table 1; Additional file 4: Table S2). Furthermore, the functional enrichment analysis of differentially expressed genes revealed that 6 primary functional categories were significantly overrepresented, including “misc.”, “cell wall”, “secondary metabolism” “major CHO (carbohydrate) metabolism”, “hormone metabolism” and “gluconeogenese/ glyoxylate cycle” (Table 1). In addition, enriched functional subcategories in each overrepresented category were further investigated. The results exhibited the numbers of enriched secondary functional categories in “misc.”, “cell wall”, “secondary metabolism”, “major CHO (carbohydrate) metabolism”, “hormone metabolism” and “gluconeogenese/ glyoxylate cycle” were 10, 4, 4, 2, 2 and 1, respectively (Additional file 5: Table S3). Although the precise roles of these differentially expressed genes are unknown, the alteration of genes may contribute to the observed variation in grain size between TAA10 and XX329.
Table 1.
Numbers of differentially expressed genes and enrichment significance in 35 MapMan primary functional categories
| Primary functional category | No. of differentially expressed genes | P-value |
|---|---|---|
| 1 PS | 31 | 0.992 |
| 2 major CHO (carbohydrate) metabolism | 73 | 7.92E-11 |
| 3 minor CHO metabolism | 37 | 0.232 |
| 4 glycolysis | 8 | 0.858 |
| 5 fermentation | 7 | 0.403 |
| 6 gluconeogenese/glyoxylate cycle | 6 | 0.020 |
| 7 OPP | 3 | 0.950 |
| 8 TCA / org. Transformation | 10 | 0.993 |
| 9 mitochondrial electron transport / ATP synthesis | 11 | 0.999 |
| 10 cell wall | 201 | 1.54E-14 |
| 11 lipid metabolism | 86 | 0.922 |
| 12 N-metabolism | 5 | 0.773 |
| 13 amino acid metabolism | 72 | 0.407 |
| 14 S-assimilation | 1 | 0.871 |
| 15 metal handling | 11 | 0.889 |
| 16 secondary metabolism | 221 | 1.60E-12 |
| 17 hormone metabolism | 226 | 3.66E-06 |
| 18 Co-factor and vitamine metabolism | 2 | 0.999 |
| 19 tetrapyrrole synthesis | 1 | 0.999 |
| 20 stress | 394 | 0.256 |
| 21 redox | 28 | 0.999 |
| 22 polyamine metabolism | 3 | 0.752 |
| 23 nucleotide metabolism | 28 | 0.957 |
| 24 Biodegradation of Xenobiotics | 27 | 0.052 |
| 25 C1-metabolism | 5 | 0.831 |
| 26 misc | 811 | 1.96E-29 |
| 27 RNA | 842 | 0.647 |
| 28 DNA | 158 | 0.999 |
| 29 protein | 815 | 0.999 |
| 30 signalling | 397 | 0.999 |
| 32 μ RNA, natural antisense etc. | 0 | 0.962 |
| 31 cell | 179 | 0.999 |
| 33 development | 222 | 0.893 |
| 34 transport | 303 | 0.960 |
| 35 not assigned | 2022 | 0.004 |
Carbohydrate metabolism is defined as the various biochemical processes responsible for the formation, breakdown and interconversion of carbohydrates. In wheat developing grains, sucrose metabolism is important for providing material and energy source to the biosynthesis of cell wall and starch. Our MapMan analysis revealed that differentially expressed genes involved in sucrose degradation exhibited significantly enrichment, including sucrose synthase (SUS) and invertase genes. Eight of 10 differentially expressed genes encoding sucrose synthase were up-regulated in XX329 relative to TAA10. Of 29 differentially expressed genes encoding cell wall (13), vacuolar (15) and neutral (1) invertases, the number of up-regulated genes in XX329 relative to TAA10 was 7, 7 and 0, respectively (Table 2; Fig. 6a). Consistently, XX329 accumulated much higher contents of sucrose, D-glucose and D-fructose than TAA10 (Fig. 6c-e). In addition, our MapMan analysis revealed that 21 differentially expressed genes were enriched in the starch biosynthesis pathway, which encoded four key enzymes, that is starch branching enzyme (SBE), ADP-glucose pyrophosphorylase (AGPase), starch synthases (SS) and starch debranching enzyme (DBE). All differentially expressed genes encoding the AGPase plastidial large subunit (4) and DBE (2) including Isoamylase 1 (1) and Isoamylase 2 (1) were significantly up-regulated in XX329 compared to TAA10. Of 10 differentially expressed SBE genes, 9 SBE2 genes were up-regulated, whereas the other one SBE1 gene was down-regulated in XX329 relative to TAA10. Three of 5 differentially expressed SS genes were up-regulated in XX329 compared to TAA10 (Table 2; Fig. 6b). To further validate the accuracy of RNA-seq results, qRT-PCR was performed for 5 randomly selected differently expressed genes involved in carbohydrate metabolism, among which the expression changes of 4 genes were consistent with the RNA-seq results (Additional file 6: Figure S2).
Table 2.
The differentially expressed genes involved in sucrose degradation and starch synthesis pathways
| Functional category | Gene IDa | Description | Log2 fold changeb | FDR-adjusted P-value |
|---|---|---|---|---|
| Major CHO metabolism. Degradation. sucrose. Invertases | TRIAE_CS42_4AL_TGACv1_288169_AA0939320 | Vacuolar invertase | 1.42 | 7.74E-06 |
| TRIAE_CS42_4AL_TGACv1_288169_AA0939350 | Vacuolar invertase | 2.27 | 5.23E-04 | |
| TRIAE_CS42_4DL_TGACv1_342608_AA1117890 | Cell wall invertase | 1.56 | 2.30E-02 | |
| TRIAE_CS42_5BL_TGACv1_404129_AA1285640 | Cell wall invertase | 6.21 | 4.38E-21 | |
| TRIAE_CS42_5DL_TGACv1_433511_AA1415210 | Cell wall invertase | 2.29 | 8.19E-06 | |
| TRIAE_CS42_6AS_TGACv1_487805_AA1573040 | Cell wall invertase | 1.81 | 1.56E-09 | |
| TRIAE_CS42_6BS_TGACv1_514515_AA1660840 | Cell wall invertase | 1.24 | 2.21E-09 | |
| TRIAE_CS42_7AS_TGACv1_569119_AA1807690 | Vacuolar invertase | 2.38 | 5.75E-21 | |
| TRIAE_CS42_7AS_TGACv1_569629_AA1820610 | Vacuolar invertase | 1.49 | 1.16E-02 | |
| TRIAE_CS42_7AS_TGACv1_571086_AA1844330 | Vacuolar invertase | 1.26 | 3.85E-04 | |
| TRIAE_CS42_7DS_TGACv1_622623_AA2042790 | Vacuolar invertase | 1.42 | 9.07E-06 | |
| TRIAE_CS42_7DS_TGACv1_622976_AA2048120 | Vacuolar invertase | 3.26 | 4.97E-28 | |
| TRIAE_CS42_U_TGACv1_645154_AA2143350 | Cell wall invertase | 1.05 | 1.71E-03 | |
| TRIAE_CS42_U_TGACv1_645154_AA2143370 | Cell wall invertase | 1.03 | 2.48E-07 | |
| TRIAE_CS42_1AL_TGACv1_002147_AA0039110 | Cell wall invertase | −3.03 | 1.42E-07 | |
| TRIAE_CS42_1BL_TGACv1_030243_AA0083360 | Cell wall invertase | −3.29 | 5.86E-07 | |
| TRIAE_CS42_1DL_TGACv1_061368_AA0193220 | Cell wall invertase | −2.93 | 5.66E-05 | |
| TRIAE_CS42_2AL_TGACv1_093126_AA0272730 | Cell wall invertase | −1.95 | 1.27E-04 | |
| TRIAE_CS42_2BL_TGACv1_130262_AA0407610 | Cell wall invertase | −3.38 | 6.12E-09 | |
| TRIAE_CS42_2DL_TGACv1_159903_AA0544110 | Cell wall invertase | −1.14 | 4.71E-04 | |
| TRIAE_CS42_4AL_TGACv1_288169_AA0939300 | Vacuolar invertase | −4.23 | 6.36E-07 | |
| TRIAE_CS42_6AS_TGACv1_486383_AA1560850 | Vacuolar invertase | −3.18 | 4.03E-06 | |
| TRIAE_CS42_6BS_TGACv1_513852_AA1650710 | Vacuolar invertase | −1.89 | 4.46E-02 | |
| TRIAE_CS42_6BS_TGACv1_515140_AA1667640 | Vacuolar invertase | −3.17 | 9.25E-05 | |
| TRIAE_CS42_6DS_TGACv1_542882_AA1732150 | Vacuolar invertase | −2.92 | 3.76E-07 | |
| TRIAE_CS42_7AS_TGACv1_570692_AA1839380 | Vacuolar invertase | −2.64 | 2.06E-05 | |
| TRIAE_CS42_7DS_TGACv1_621614_AA2021150 | Vacuolar invertase | −1.54 | 8.16E-03 | |
| TRIAE_CS42_7DS_TGACv1_622753_AA2044760 | Vacuolar invertase | −4.78 | 1.20E-19 | |
| TRIAE_CS42_U_TGACv1_642336_AA2115990 | Neutral invertase | −1.11 | 8.73E-03 | |
| Major CHO metabolism. Degradation. sucrose. Susy | TRIAE_CS42_1AL_TGACv1_002916_AA0046300 | Sucrose synthase | 4.20 | 1.42E-09 |
| TRIAE_CS42_2AS_TGACv1_112101_AA0330380 | Sucrose synthase | 2.25 | 2.76E-16 | |
| TRIAE_CS42_2BS_TGACv1_145971_AA0451230 | Sucrose synthase | 1.85 | 1.34E-11 | |
| TRIAE_CS42_2DL_TGACv1_159703_AA0541730 | Sucrose synthase | 1.89 | 5.80E-05 | |
| TRIAE_CS42_2DS_TGACv1_177457_AA0577630 | Sucrose synthase | 2.71 | 9.79E-17 | |
| TRIAE_CS42_4AL_TGACv1_288793_AA0958320 | Sucrose synthase | 1.30 | 1.73E-05 | |
| TRIAE_CS42_7AS_TGACv1_569135_AA1808380 | Sucrose synthase | 2.60 | 1.96E-16 | |
| TRIAE_CS42_7DS_TGACv1_622658_AA2043420 | Sucrose synthase | 2.94 | 1.09E-24 | |
| TRIAE_CS42_6DL_TGACv1_527850_AA1709240 | Sucrose synthase | −1.74 | 3.14E-07 | |
| TRIAE_CS42_7DL_TGACv1_604951_AA2003310 | Sucrose synthase | −1.96 | 3.18E-09 | |
| Major CHO metabolism. Synthesis. starch. AGPase | TRIAE_CS42_1AL_TGACv1_000939_AA0022300 | AGPase plastidial large subunit | 9.51 | 9.06E-110 |
| TRIAE_CS42_1BL_TGACv1_031712_AA0119450 | AGPase plastidial large subunit | 1.26 | 9.23E-05 | |
| TRIAE_CS42_7AS_TGACv1_569682_AA1821750 | AGPase plastidial large subunit | 1.29 | 2.65E-06 | |
| TRIAE_CS42_7DS_TGACv1_622617_AA2042690 | AGPase plastidial large subunit | 1.52 | 6.37E-08 | |
| Major CHO metabolism. Synthesis. starch. Debranching | TRIAE_CS42_1DL_TGACv1_061778_AA0203340 | Isoamylase 2 | 1.15 | 3.90E-07 |
| TRIAE_CS42_7DS_TGACv1_622495_AA2040790 | Isoamylase 1 | 1.07 | 6.13E-04 | |
| Major CHO metabolism. Synthesis. starch. Starch branching | TRIAE_CS42_2AL_TGACv1_095785_AA0314520 | Starch branching enzyme 2 | 2.85 | 4.50E-10 |
| TRIAE_CS42_2BL_TGACv1_132845_AA0440100 | Starch branching enzyme 2 | 1.43 | 3.85E-05 | |
| TRIAE_CS42_2DL_TGACv1_158200_AA0512220 | Starch branching enzyme 2 | 1.87 | 1.96E-09 | |
| TRIAE_CS42_7AL_TGACv1_556597_AA1766610 | Starch branching enzyme 2 | 2.83 | 1.68E-14 | |
| TRIAE_CS42_7AL_TGACv1_556597_AA1766630 | Starch branching enzyme 2 | 2.97 | 3.57E-14 | |
| TRIAE_CS42_7BL_TGACv1_577598_AA1879190 | Starch branching enzyme 2 | 4.12 | 3.24E-14 | |
| TRIAE_CS42_7BL_TGACv1_580343_AA1913520 | Starch branching enzyme 2 | 2.28 | 6.22E-08 | |
| TRIAE_CS42_7DL_TGACv1_603128_AA1976400 | Starch branching enzyme 2 | 2.77 | 2.09E-05 | |
| TRIAE_CS42_7DL_TGACv1_603128_AA1976410 | Starch branching enzyme 2 | 4.79 | 9.97E-23 | |
| TRIAE_CS42_7AL_TGACv1_556924_AA1773500 | Starch branching enzyme 1 | −1.06 | 2.63E-02 | |
| Major CHO metabolism. Synthesis. starch.starch synthase | TRIAE_CS42_1AS_TGACv1_020151_AA0075220 | Starch synthase 3 | 1.77 | 3.39E-12 |
| TRIAE_CS42_1BL_TGACv1_030553_AA0093880 | Starch synthase | 1.93 | 1.18E-10 | |
| TRIAE_CS42_1BS_TGACv1_050983_AA0177050 | Starch synthase 3 | 1.07 | 2.87E-05 | |
| TRIAE_CS42_2BL_TGACv1_131141_AA0423210 | Starch synthase 3 | −1.09 | 9.71E-04 | |
| TRIAE_CS42_2DL_TGACv1_157939_AA0502660 | Starch synthase | −1.02 | 1.31E-03 |
aGenes for qRT-PCR analysis are shown in bold
bFold change indicates that gene expression change in XX329 compared to TAA10
Fig. 6.
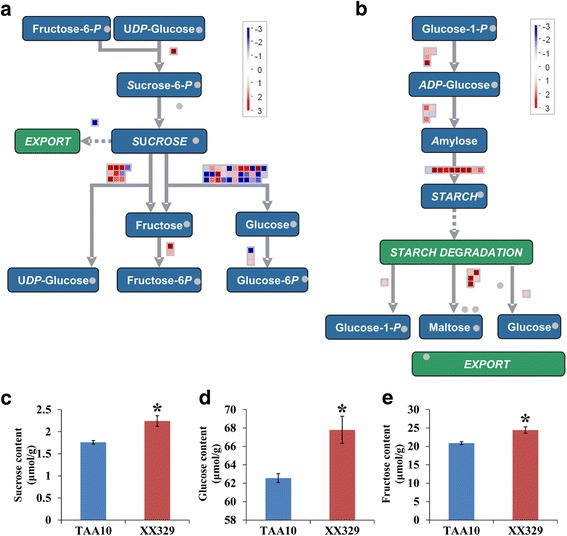
MapMan pathways in sucrose-starch metabolism and sugar content analysis. a-b Differentially expressed genes between TAA10 and XX329 in sucrose degradation (a) and starch synthesis (b) pathways. Individual genes are represented by small squares. The colour of squares represents the value of log2 fold change. Red represents up-regulation and blue represents down-regulation in XX329 relative to TAA10. c-e Sucrose (c), glucose (d) and fructose (e) contents in grains at 6 DAP of TAA10 and XX329. The values are shown as means ± SE. * indicates the significance at the 0.05 level (Student’s t test)
Grain growth during early development involved in the cell expansion of the pericarp required a mass of cell wall biosynthesis. Interestingly, we found that 201 differentially expressed genes were enriched in the primary functional category “cell wall”, among which 4 functional subcategories exhibited significant overrepresentation, including “cellulose synthesis”, “degradation”, “modification” and “cell wall proteins” (Additional file 5: Table S3). For the subcategory of cellulose synthesis, 26 differentially expressed genes were significantly overrepresented, mainly including cellulose synthase and COBRA genes (12 and 8), and the number of up-regulated genes encoding cellulose synthase and COBRA in XX329 relative to TAA10 was 5 and 6, respectively. Moreover, we identified 57 differentially expressed genes involved in cell wall degradation, including 22 up-regulated and 35 down-regulated genes, such as cellulase and pectinase genes. Notably, of differentially expressed genes encoding expansin (26) and Xyloglucan transglucosylase/hydrolase (XTH) (31) associated with cell wall modification, the number of up-regulated genes in XX329 relative to TAA10 was 16 and 18, respectively. In addition, of 24 differentially expressed genes enriched in the functional subcategory “cell wall proteins”, all five genes encoding arabinogalactan protein (AGP) were up-regulated in XX329 compared to TAA10, whereas only 7 of 15 genes encoding Leucine-rich repeat (LRR) family protein were up-regulated (Additional file 7: Table S4).
Based on the MapMan analysis, we found that 226 differentially expressed genes were enriched in the primary functional category “hormone metabolism”, including auxin, abscisic acid, ethylene and gibberellins metabolism (Additional file 5: Table S3).. Of 10 differentially expressed genes involved in auxin metabolism, 8 genes encoded the IAA-leucine-resistant (ILR) proteins regulating auxin homeostasis were up-regulated in XX329 compared to TAA10. Moreover, 7 genes involved in signal transduction of abscisic acid, such as ABI3 and FUS3, were also significantly up-regulated in XX329 compared to TAA10. Notably, we identified that a large number of differentially expressed genes (108) were associated with the ethylene metabolism, which were involved in the ethylene biosynthesis, degradation and signal transduction. In addition, the expression of 9 GA20ox genes involved in gibberellins biosynthesis were significantly altered, among which 6 were up-regulated and 3 were down-regulated in XX329 relative to TAA10 (Additional file 8: Table S5).
Discussion
Genetic variation of D genome induced drastic changes of gene expression in allohexaploid wheats
Aegilops tauschii, the D-genome progenitor of Triticum aestivum, encompasses extensive variation for various traits of potential economic importance, including yield, biotic and abiotic stress tolerance, quality and nutrition [21–24, 39, 40]. Until now, most studies focused on understanding the genetic and morphological diversity of Ae. tauschii germplasm and their functions under a allohexaploid genetic background, but little is known about their effects on gene expression [41–43]. The natural and resynthesized allohexaploid wheats with near-identical A and B genomes could provide a good example for investigating the effects of different D genomes on gene expression during allopolyploid wheat formation and evolution [44]. Recently, it was reported that the genetic variability of the progenitor of the D genome (Ae. tauschii) does not lead to significant changes in gene expression in leaves of resynthesized allohexaploid wheats, and similar features were observed by direct comparison between natural and resynthesized allohexaploid wheats [30]. In the present study, we found that 8891 genes were differentially expressed between 6-DAP grains of natural and resynthesized allohexaploid wheats with near-identical AB genomes, which is much higher than that in leaves of previous studies [30], suggesting that the genetic variation of the D genome induced drastic alterations of gene expression in grains of allohexaploid wheats. Interestingly, the number of down-regulated genes on the D genome of resynthesized relative to natural allohexaploid wheat (2041) is much higher than that on A and B genomes (675 and 713), which may be due to modifications of D genome to the genomic shocks that occurred during and after allopolyploidization [45]. It should to be noted that the number of up-regulated genes on the AB genome in resynthesized relative to natural allohexaploid wheat (2989) was significantly higher than that of down-regulated genes (1388), suggesting that the D genome of Aegilops tauschii mainly contributed to positive effects on gene expression of AB genomes by inter-genomic interactions in resynthesized allohexaploid wheats.
Accumulated data revealed that the repression of one homologous gene is compensated by the activation of the others in allopolyploid wheat [44, 46, 47]. For example, a recent study of leaf gene expression in bread wheat identified many small regions of genome dominance (transcripts of homoeologues from one genome were more abundant than the others) and many larger regions of genome repression (transcripts of homoeologues from one genome were less abundant than the others) [47]. Interestingly, we found that the genomic region on chromosome 1DL enriched down-regulated genes in resynthesized relative to the natural allohexaploid wheat. In contrast, up-regulated genes in resynthesized allohexaploid wheat were overrepresented on the corresponding homoeologous region of chromosome 1AL. Taken together, these data provide further evidence for the dominance/repression of gene expression on specific genomic regions in resynthesized allohexaploid wheat, which may play an important role in homoeologous genome divergence and contribute to genomic asymmetry. However, this feature is not observed on chromosome 1BL, which is needed for further investigation.
Alteration of sucrose metabolism was essential for the formation of large grains in resynthesized allohexaploid wheats
Grain development is highly dependent on the metabolic utilization of sucrose, which can provide carbon for the synthesis of cell wall polysaccharides and starch [16, 48]. In this study, we found that the expression level of 26 genes involved in sucrose degradation in 6-DAP grains of the resynthesized allohexaploid wheat was significantly higher than those of the natural allohexaploid wheat. Sucrose synthase is widely believed to be the main route of entry of carbon from sucrose into cellular metabolism in plants, which catalyzed the reaction of sucrose to UDP-glucose and fructose [49]. In wheat, genetic studies exhibited that sucrose synthase 1 and 2 genes (TaSus1 and TaSus2) are associated with TGW [50]. Interestingly, 8 of 10 (80%) genes encoding sucrose synthase were up-regulated in resynthesized relative to natural allohexaploid wheat. On the other hand, sucrose is also a signal molecule in regulating gene expression and normal grain development in diverse cell types [51, 52]. In general, sucrose favors differentiation and maturation, whereas hexoses favor cell division and expansion [53, 54]. Consistent with this notion, the resynthesized allohexaploid wheat showed higher contents of glucose and fructose in grains at 6 DAP than those of the natural allohexaploid wheat. Collectively, these data indicated that the up-regulation of sucrose synthase genes in resynthesized allohexaploid wheat could accelerate the process of cell growth compared to natural allohexaploid wheat, which plays important roles in the differences in grain size and weight between the natural and resynthesized allohexaploid wheats.
Rate of cell expansion contributed to the variation in grain size between resynthesized and natural allohexaploid wheats
Grain morphogenesis is dependent on the regulation of cell division and expansion [55]. Cell proliferation is almost completed by 2 DAP in the pericarp. Later, filial grain organs grow and expand rapidly within the maternal pericarp. Thereby, the growth of the endosperm must be coordinated with the maternal pericarp development, which collectively affects the grain development [38]. In the present study, we found that grains of natural and resynthesized allohexaploid wheats reach their maximal volume at about 10 DAP, whereas the grain size of the resynthesized allohexaploid wheat is significantly larger than that of the natural allohexaploid wheat. In accordance with the difference of grain size, the cell areas and growth rates of the pericarp and endosperm in the resynthesized allohexaploid wheat are higher than those in the natural allohexaploid wheat during the early stages of grain development. Therefore, the observed variation in grain size between natural and resynthesized allohexaploid wheats could be traced back to the different growth rate of the filial tissues.
Cell growth requires the cell wall to be irreversibly stretched through a wall loosening process, followed by the deposition of new wall material [56]. An important observation of this study is that the differentially expressed genes between natural and resynthesized allohexaploid wheats were significantly enriched in cell wall metabolism, which was well correlated with the difference in grain size between natural and resynthesized allohexaploid wheats at the critical period of early grain expansion (6 DAP). For example, expansins are the only proteins to directly induce cell wall extension by disrupting the hydrogen bonds between cellulose microfibrils and crosslinking glycans and permitting turgor-driven cell wall extension [57, 58]. In wheat, expansins are shown directly to induce cell wall extension, and expression of expansin genes is associated with grain size [59]. Interestingly, 16 of 26 (61.5%) differentially expressed expansins genes were up-regulated in resynthesized relative to natural allohexaploid wheat. Genes of the COBRA family are involved in various types of cell expansion and cell wall biosynthesis [60]. Of 8 differentially expressed COBRA-like genes, 6 (75%) were up-regulated in resynthesized relative to natural allohexaploid wheat. In expanding plant cells, the cellulose–xyloglucan network is considered the main load-bearing network that controls the extent of cell expansion [61]. XTH genes are important wall-loosening genes whose function in wall remodelling is to cleave xyloglucan to incorporate new molecules and catalyse linkage between xyloglucan and cellulose [62, 63]. We found that 18 of 31 (58.1%) genes encoding XTH proteins were up-regulated in resynthesized relative to natural allohexaploid wheat. However, different from the expression patterns of XTH genes, the majority of differentially expressed cellulose synthase genes (7/12, 58.3%) were down-regulated in resynthesized relative to natural allohexaploid wheat. Taken together, we proposed that these differentially expressed genes may contribute to the variation in grain size and weight between resynthesized and natural allohexaploid wheats.
Comparison of differentially expressed genes with previously known QTL and candidate genes controlling wheat grain size and weight
Recently, we identified three QTL regions associated with grain size and shape on chromosomes 2DS, 2DL and 7DS, respectively, using the F2 and F2:3 populations derived from the natural allohexaploid wheat TAA10 and resynthesized allohexaploid wheat XX329 [64]. In the present study, comparative transcriptome analysis at 6-DAP grains between TAA10 and XX329 were performed, which may provide useful information for discovering the candidate genes underpinning phenotypic variation [65]. Comparative analysis revealed that the numbers of differentially expressed genes located within the QTL regions on chromosomes 2DS, 2DL and 7DS were 43, 78 and 48, respectively (Additional file 9: Table S7). Notably, one up-regulated SBE2 gene (TRIAE_CS42_2DL_TGACv1_158200_AA0512220) in resynthesized relative to natural allohexaploid wheat was located within the QTL region on chromosome 2DL. In addition, one up-regulated GATL (Galacturonosyltransferase-Like) gene (TRIAE_CS42_7DS_TGACv1_ 622210_AA2035360) in resynthesized relative to natural allohexaploid wheat within the QTL region on chromosome 7DL was involved in cell wall biosynthesis [66]. Collectively, these genes are possible candidates for the detected QTL controlling grain size and shape, and detailed studies would be necessary to evaluate the relationship of QTL to differentially expressed genes identified in the present study.
Up to date, some wheat genes controlling grain size and weight were successfully isolated by the homology-based approach, including TaCwi-A1, TaGW2, TaTGW6, TaGS1a, TaGASR7-A1, TaCYP78A3, TaCWI, TaCKX6-D1 and TaSAP1-A1 [9–15, 67–69]. However, none of these genes were differentially expressed between 6-DAP grains of the natural allohexaploid TAA10 and resynthesized allohexaploid wheat XX329 (Additional file 9: Table S7). In addition, although a large number of up-regulated genes (362) in resynthesized relative to the natural allohexaploid wheat were overrepresented in the genomic region on chromosome 1AL, to the best of our knowledge, no known QTL controlling grain size and weight matched to the genomic region on chromosome 1AL that enriched differentially expressed genes, which deserve further investigation.
Conclusions
Significant differences in grain size and weight at the early stages of development were observed between the natural and resynthesized allohexaploid wheats (TAA10 and XX329), which could be mainly attributed to the higher growth rates of the pericarp and endosperm cells in XX329 compared to TAA10. Comparative transcriptome analysis indicated that the differentially expressed genes were significantly enriched in the functional categories of cell wall, carbohydrate and hormone metabolism, which may play important roles in the observed difference between TAA10 and XX329 in terms of grain size and weight. Notably, consistent with the up-regulation of sucrose synthase genes in resynthesized relative to natural allohexaploid wheat, the resynthesized allohexaploid wheat showed higher contents of glucose and fructose in grains at 6 DAP than those of the natural allohexaploid wheat, indicating that alteration of sucrose metabolism was essential for the formation of large grains in the resynthesized allohexaploid wheat.
Additional files
Correlation coefficients among the three biological replicates of TAA10 and XX329 by Pearson correlation analysis. (XLSX 2028 kb)
Detailed information of the 8891 differentially expressed genes between the natural and resynthesized allohexaploid wheats (TAA10 and XX329). (XLSX 473 kb)
Detailed information of genes used for qRT-PCR analysis. (XLSX 69 kb)
The MapMan functional categories of the differentially expressed genes between the natural and resynthesized allohexaploid wheats (TAA10 and XX329). (XLSX 27 kb)
The MapMan enrichment analysis of the differentially expressed genes between the natural and resynthesized allohexaploid wheats (TAA10 and XX329). (XLSX 22 kb)
Verification of 5 differentially expressed genes involved in carbohydrate metabolism by qRT-PCR. (XLSX 12 kb)
The differentially expressed genes in the enriched MapMan categories of cell wall metabolism. (XLSX 33 kb)
The differentially expressed genes in the enriched MapMan categories of hormone metabolism. (DOCX 156 kb)
Detailed information of genes associated with known QTL and candidate genes controlling wheat grain size and weight. (DOCX 143 kb)
Acknowledgements
We thank Professor Bao Liu (Northeast Normal University, Changchun 130024, China) for providing seeds of TAA10 and XX329.
Funding
This work was financially supported by the National Key Research and Development Program of China (Grant No. 2016YFD0100801), the Major Program of the National Natural Science Foundation of China (Grant No. 31290212), the National Natural Science Foundation of China (Grant No. 91435204) and the National Natural Science Foundation of China (Grant No. 31601305).
Availability of data and materials
All relevant data are available within the manuscript. The RNA-Seq reads used for this study were deposited at the National Center for Biotechnology Information Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) under the accession number SRP117710.
Abbreviations
- AGPase
ADP-glucose pyrophosphorylase
- DAP
Days after pollination
- DBE
Starch debranching enzyme
- FDR
False discovery rate
- FPKM
Fragments per kilobase per Million reads
- ILR
IAA-leucine-resistant
- PCR
Polymerase chain reaction
- QRT-PCR
Quantitative reverse transcription PCR
- SBE
Starch branching enzyme
- SS
Starch synthase
- SUS
Sucrose synthase
- TGW
Thousand grain weight
- XTH
Xyloglucan transglucosylase/hydrolase
Authors’ contributions
ZN and QS conceived the project; LY, HX, XZ, AZ and FL carried out experiments; ZL, MX, HP and YY analyzed the data; LY and ZN wrote the manuscript. All authors have read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12870-018-1248-y) contains supplementary material, which is available to authorized users.
Contributor Information
Lei Yan, Email: yanlei19369@163.com.
Zhenshan Liu, Email: shanzhenliu@163.com.
Huanwen Xu, Email: xhwnefu@163.com.
Xiaoping Zhang, Email: xiaopingzhang@163.com.
Aiju Zhao, Email: zhaoaiju81@126.com.
Fei Liang, Email: feiliangsh@163.com.
Mingming Xin, Email: mingmingxin@cau.edu.cn.
Huiru Peng, Email: penghuiru@cau.edu.cn.
Yingyin Yao, Email: yingyin@cau.edu.cn.
Qixin Sun, Email: qxsun@cau.edu.cn.
Zhongfu Ni, Email: nizf@cau.edu.cn.
References
- 1.Smith R. Increasing wheat yield requires genetics, management: Southwest Farm Press. http:// southwestfarmpress.com/wheat/increasing-wheat-yieldrequires-genetics-management. Accessed 6 Feb 2014.
- 2.Wilhelm EP, Howells RM, Alkaff N, Jia J, Baker C, Leveringtonwaite MA, et al. Genetic characterization and mapping of the Rht-1 homoeologs and flanking sequences in wheat. Theor Appl Genet. 2013;126(5):1321–1336. doi: 10.1007/s00122-013-2055-3. [DOI] [PubMed] [Google Scholar]
- 3.Wang LF, Ge HM, Hao CY, Dong YS, Zhang XY. Identifying loci influencing 1,000-kernel weight in wheat by microsatellite screening for evidence of selection during breeding. PLoS One. 2012;7(2):e29432. doi: 10.1371/journal.pone.0029432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell KG, Bergman CJ, Gualberto DG, Anderson JA, Giroux MJ, Hareland G, et al. Quantitative trait loci associated with kernel traits in a soft × hard wheat cross. Crop Sci. 1999;39:1184–1195. doi: 10.2135/cropsci1999.0011183X003900040039x. [DOI] [Google Scholar]
- 5.Varshney RK, Prasad M, Roy JK, Kumar N, Singh H, Dhaliwal HS, et al. Identification of eight chromosomes and one microsatellite marker on 1AS associated with QTL for grain weight in bread wheat. Theor Appl Genet. 2000;100:1290–1295. doi: 10.1007/s001220051437. [DOI] [Google Scholar]
- 6.Groos C, Robert N, Bervas E, Charmet G. Genetic analysis of grain proteincontent, grain yield and thousand-kernel weight in bread wheat. Theor Appl Genet. 2003;106:1032–1040. doi: 10.1007/s00122-002-1111-1. [DOI] [PubMed] [Google Scholar]
- 7.Kumar N, Kulwal PL, Gaur A, Tyagi AK, Khurana JP, Khurana P, et al. QTL analysis for grain weight in common wheat. Euphytica. 2006;151:135–144. doi: 10.1007/s10681-006-9133-4. [DOI] [Google Scholar]
- 8.Farré A, Sayers L, Leverington-Waite M, Goram R, Orford S, Wingen L, et al. Application of a library of near isogenic lines to understand context dependent expression of QTL for grain yield and adaptive traits in bread wheat. BMC Plant Biol. 2016;16:1–13. doi: 10.1186/s12870-016-0849-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma DY, Yan J, He ZH, Wu L, Xia XC. Characterization of a cell wall invertase gene TaCwi-A1 on common wheat chromosome 2A and development of functional markers. Mol Breed. 2012;29:43–52. doi: 10.1007/s11032-010-9524-z. [DOI] [Google Scholar]
- 10.Su ZQ, Hao CY, Wang LF, Dong YC, Zhang XY. Identification and development of a functional marker of TaGW2 associated with grain weight in bread wheat (Triticum aestivum L.) Theor Appl Genet. 2011;122:211–223. doi: 10.1007/s00122-010-1437-z. [DOI] [PubMed] [Google Scholar]
- 11.Hu MJ, Zhang HP, Cao JJ, Zhu XF, Wang SX, et al. Characterization of an IAA-glucose hydrolase gene TaTGW6 associated with grain weight in common wheat (Triticum aestivum L.) Mol Breed. 2016;36:1–11. doi: 10.1007/s11032-015-0425-z. [DOI] [Google Scholar]
- 12.Hanif M, Gao FM, Liu JD, Wen WE, Zhang YJ, et al. TaTGW6-A1, an ortholog of rice TGW6, is associated with grain weight and yield in bread wheat. Mol Breed. 2016;36:1. doi: 10.1007/s11032-015-0425-z. [DOI] [Google Scholar]
- 13.Guo Y, Sun JJ, Zhang GZ, Wang YY, Kong FM, et al. Haplotype, molecular marker and phenotype effects associated with mineral nutrient and grain size traits of TaGS1 a in wheat. Field Crops Res. 2013;154:119–125. doi: 10.1016/j.fcr.2013.07.012. [DOI] [Google Scholar]
- 14.Ling HQ, Zhao SC, Liu DC, Wang JY, Sun H, et al. Draft genome of the wheat A-genome progenitor Triticum urartu. Nature. 2013;496:87–90. doi: 10.1038/nature11997. [DOI] [PubMed] [Google Scholar]
- 15.Ma M, Wang Q, Li ZJ, Cheng HH, Li ZJ, et al. Expression of TaCYP78A3, a gene encoding cytochrome P450 CYP78A3 protein in wheat (Triticum aestivum L.), affects seed size. Plant J. 2015;83:312–325. doi: 10.1111/tpj.12896. [DOI] [PubMed] [Google Scholar]
- 16.Saulnier L, Guillon F, Chateigner-Boutin AL. Cell wall deposition and metabolism in wheat grain. J Cereal Sci. 2012;56(1):91–108. doi: 10.1016/j.jcs.2012.02.010. [DOI] [Google Scholar]
- 17.Drea S, Leader D, Arnold B, Shaw P, Dolan L, Doonan J. Systematic spatial analysis of gene expression during wheat caryopsis development. Plant Cell. 2005;17(8):2172–2185. doi: 10.1105/tpc.105.034058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pfeifer M, Kugler KG, Sandve SR, Zhan B, Rudi H, Hvidsten TR, et al. Genome interplay in the grain transcriptome of hexaploid bread wheat. Science. 2014;345(6194):1250091. doi: 10.1126/science.1250091. [DOI] [PubMed] [Google Scholar]
- 19.Petersen G, Seberg O, Yde M, Berthelsen K. Phylogenetic relationships of Triticum and Aegilops and evidence for the origin of the a, B, and D genomes of common wheat (Triticum aestivum) Mol Phylogenet Evol. 2006;39(1):70–82. doi: 10.1016/j.ympev.2006.01.023. [DOI] [PubMed] [Google Scholar]
- 20.Cox TS. Deepening the wheat gene pool. J Crop Prod. 1997;1(1):1–25. doi: 10.1300/J144v01n01_01. [DOI] [Google Scholar]
- 21.Dvorak J, Luo M, Yang Z, Zhang H. The structure of the Aegilops tauschii genepool and the evolution of hexaploid wheat. Theor Appl Genet. 1998;97:657–670. doi: 10.1007/s001220050942. [DOI] [Google Scholar]
- 22.Reynolds M, Dreccer F, Trethowan R. Drought-adaptive traits derived from wheat wild relatives and landraces. J Exp Bot. 2007;58:177–186. doi: 10.1093/jxb/erl250. [DOI] [PubMed] [Google Scholar]
- 23.Mujeeb-Kazi A, Gul A, Ahmad I, Farooq M, Rizwan S, Bux H, et al. Aegilops tauschii, as a spot blotch (Cochliobolus sativus) resistance source for bread wheat improvement. Pak J Bot. 2007;39:1207–1216. [Google Scholar]
- 24.Miranda LM, Murphy JP, Marshall D, Cowger C, Leath S. Chromosomal location of a novel Aegilops tauschii derived powdery mildew resistance gene introgressed into common wheat (Triticum aestivum L.) Theor Appl Genet. 2007;114(8):1451–1456. doi: 10.1007/s00122-007-0530-4. [DOI] [PubMed] [Google Scholar]
- 25.Okamoto Y, Nguyen AT, Yoshioka M, Iehisa JC, Takumi S. Identification of quantitative trait loci controlling grain size and shape in the D genome of synthetic hexaploid wheat lines. Breed Sci. 2013;63:423–429. doi: 10.1270/jsbbs.63.423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu M, Chen G, Zhang L, Liu Y, Liu D, Wang J, et al. QTL mapping for important agronomic traits in synthetic hexaploid wheat derived from Aegiliops tauschii ssp. tauschii. J Integr Agr. 2014;13:1835–1844. doi: 10.1016/S2095-3119(13)60655-3. [DOI] [Google Scholar]
- 27.Rasheed A, Xia X, Ogbonnaya F, Mahmood T, Zhang Z, Mujeeb-Kazi A, et al. Genome-wide association for grain morphology in synthetic hexaploid wheats using digital imaging analysis. BMC Plant Biol. 2014;14(1):128. doi: 10.1186/1471-2229-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Röder MS, Huang XQ, Börner A. Fine mapping of the region on wheat chromosome 7D controlling grain weight. Funct Integr Genomics. 2008;8:79–86. doi: 10.1007/s10142-007-0053-8. [DOI] [PubMed] [Google Scholar]
- 29.Liao X, Wang J, Zhou R, Ren Z, Jia J. Mining favorable alleles of QTLs conferring thousand-grain weight from synthetic wheat. Acta Agron Sin. 2008;34:1877–1884. doi: 10.1016/S1875-2780(09)60012-2. [DOI] [Google Scholar]
- 30.Zhang H, Zhu B, Qi B, Gou X, Dong Y, Xu C, et al. Evolution of the BBAA component of bread wheat during its history at the allohexaploid level. Plant Cell. 2014;26:2761–2776. doi: 10.1105/tpc.114.128439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brinton J, Simmonds J, Minter F, Leveringtonwaite M, Snape J, Uauy C. Increased pericarp cell length underlies a major quantitative trait locus for grain weight in hexaploid wheat. New Phytol. 2017;215(3):1026–1038. doi: 10.1111/nph.14624. [DOI] [PubMed] [Google Scholar]
- 32.Clavijo BJ, Venturini L, Schudoma C, Accinelli GG, Kaithakottil G, Wright J, et al. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations. Genome Res. 2017;27(5):885. doi: 10.1101/gr.217117.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 2015;12(4):357–360. doi: 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anders S, Pyl PT, Huber W. HTSeq--a python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Usadel B, Nagel A, Thimm O, Redestig H, Blaesing OE, Palacios-Rojas N, et al. Extension of the visualization tool mapman to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 2005;138(3):1195–1204. doi: 10.1104/pp.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 38.Radchuk V, Weier D, Radchuk R, Weschke W, Weber H. Development of maternal seed tissue in barley is mediated by regulated cell expansion and cell disintegration and coordinated with endosperm growth. J Exp Bot. 2011;62(3):1217. doi: 10.1093/jxb/erq348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu SS, Khan K, Klindworth DL, Nygard G. Evaluation and characterization of high-molecular weight 1D glutenin subunits from Aegilops tauschii in synthetic hexaploid wheats. J Cereal Sci. 2010;52(2):333–336. doi: 10.1016/j.jcs.2010.05.004. [DOI] [Google Scholar]
- 40.Wang K, An XL, Pan LP, Dong K, Gao LY, Wang SL, et al. Molecular characterization of HMW-GS 1Dx3(t) and 1Dx4(t) genes from Aegilops tauschii and their potential value for wheat quality improvement. Hereditas. 2012;149(1):41. doi: 10.1111/j.1601-5223.2011.02215.x. [DOI] [PubMed] [Google Scholar]
- 41.Lage J, Warburton ML, Crossa J, Skovmand B, Andersen SB. Assessment of genetic diversity in synthetic hexaploid wheats and their Triticum dicoccum and Aegilops tauschii parents using AFLPs and agronomic traits. Euphytica. 2003;134(3):305–317. doi: 10.1023/B:EUPH.0000004953.85283.f4. [DOI] [Google Scholar]
- 42.Jones H, Gosman N, Horsnell R, Rose GA, Everest LA, Bentley AR, et al. Strategy for exploiting exotic germplasm using genetic, morphological, and environmental diversity: the Aegilops tauschii, coss. Example. Theor Appl Genet. 2013;126(7):1793–1808. doi: 10.1007/s00122-013-2093-x. [DOI] [PubMed] [Google Scholar]
- 43.Mansouri S, Mehrabi AA, Kahrizi D. Evaluation of genetic diversity of Aegilops tauschii, accessions using morphological characters. J Crop Sci Biotechnol. 2013;16(3):197–200. doi: 10.1007/s12892-013-0017-6. [DOI] [Google Scholar]
- 44.Akhunova AR, Matniyazov RT, Liang H, Akhunov ED. Homoeolog-specific transcriptional bias in allopolyploid wheat. BMC Genomics. 2010;11(1):505. doi: 10.1186/1471-2164-11-505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ozkan H, Levy AA, Feldman M. Allopolyploidy-induced rapid genome evolution in the wheat (Aegilops–triticum) group. Plant Cell. 2001;13(8):1735–1747. doi: 10.1105/tpc.13.8.1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shitsukawa N, Tahira C, Kassai K, et al. Genetic and epigenetic alteration among three homoeologous genes of a class E MADS box gene in hexaploid wheat. Plant Cell. 2007;19(6):1723–1737. doi: 10.1105/tpc.107.051813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Harper AL, Martin T, He Z, Leah C, Alison F, Simon G, et al. Genome distribution of differential homoeologue contributions to leaf gene expression in bread wheat. Plant Biotechnol J. 2016;14(5):1207–1214. doi: 10.1111/pbi.12486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sreenivasulu N, Borisjuk L, Junker BH, Mock HP, Rolletschek H, Seiffert U, et al. Barley grain development toward an integrative view. Int Rev Cell Mol Biol. 2010;281:49. doi: 10.1016/S1937-6448(10)81002-0. [DOI] [PubMed] [Google Scholar]
- 49.Barratt DHP, Derbyshire P, Findlay K, Pike M, Wellner N, Lunn J, et al. Normal growth of arabidopsis requires cytosolic invertase but not sucrose synthase. Proc Natl Acad Sci U S A. 2009;106(31):13124. doi: 10.1073/pnas.0900689106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hou J, Zhang XY. Global selection on sucrose synthase haplotypes during a century of wheat breeding. Plant Physiol. 2014;164(4):1918. doi: 10.1104/pp.113.232454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koch KE. Carbohydrate-modulated gene expression in plants. Annu Rev Plant Physiol Plant Mol Biol. 1996;47(1):509–540. doi: 10.1146/annurev.arplant.47.1.509. [DOI] [PubMed] [Google Scholar]
- 52.Smeekens S, Rook F. Sugar sensing and sugar-mediated signal transduction in plants. Plant Physiol. 1997;115(1):7. doi: 10.1104/pp.115.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wobus U, Weber H. Sugars as signal molecules in plant seed development. Biol Chem. 1999;380(7-8):937–944. doi: 10.1515/BC.1999.116. [DOI] [PubMed] [Google Scholar]
- 54.Weschke W, Panitz R, Gubatz S, Wang Q, Radchuk R, Weber H, et al. The role of invertases and hexose transporters in controlling sugar ratios in maternal and filial tissues of barley caryopses during early development. Plant J. 2003;33(2):395–411. doi: 10.1046/j.1365-313X.2003.01633.x. [DOI] [PubMed] [Google Scholar]
- 55.Dai X, You C, Chen G, Li X, Zhang Q, Wu C. OsBC1L4 encodes a COBRA-like protein that affects cellulose synthesis in rice. Plant Mol Biol. 2011;75(4-5):333–345. doi: 10.1007/s11103-011-9730-z. [DOI] [PubMed] [Google Scholar]
- 56.Cosgrove DJ. Growth of the plant cell wall. Nat Rev Mol Cell Biol. 2005;6(11):850–861. doi: 10.1038/nrm1746. [DOI] [PubMed] [Google Scholar]
- 57.McQueen-Mason SJ, Durachko DM, Cosgrove DJ. Two endogenous proteins that induce cell wall extension in plants. Plant Cell. 1992;4:1425–1433. doi: 10.1105/tpc.4.11.1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McQueen-Mason SJ, Cosgrove DJ. Expansin mode of action on cell wall (analysis of wall hydrolysis, stress relaxation and binding) Plant Physiol. 1995;107:87–100. doi: 10.1104/pp.107.1.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carolina LX, Ricardo R, Gomez LD, Jaime H, Adolfo I, Mcqueen-Mason SJ, et al. Expansins expression is associated with grain size dynamics in wheat (Triticum aestivum L.) J Exp Bot. 2010;61(4):1147–1157. doi: 10.1093/jxb/erp380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ben-Tov D, Abraham Y, Stav S, Thompson K, Loraine A, Elbaum R, et al. COBRA-LIKE2, a member of the Glycosylphosphatidylinositol-anchored COBRA-LIKE family, plays a role in cellulose deposition in Arabidopsis seed coat mucilage Secretory cells. Plant Physiol. 2015;167(3):711–724. doi: 10.1104/pp.114.240671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pauly M, Albersheim P, Darvill A, York WS. Molecular domains of the cellulose/xyloglucan network in the cell walls of higher plants. Plant J. 1999;20(6):629–639. doi: 10.1046/j.1365-313X.1999.00630.x. [DOI] [PubMed] [Google Scholar]
- 62.Fry SC, Smith RC, Renwick KF, Martin DJ, Hodge SK, Matthews KJ. Xyloglucan endotransglycosylase, a new wall-loosening enzyme-activity from plants. Biochem J. 1992;282:821–828. doi: 10.1042/bj2820821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nishitani K, Tominaga R. Endoxyloglucan transferase, a novel class of glycosyltransferase that catalyzes transfer of a segment of xyloglucan molecule to another xyloglucan molecule. J Biol Chem. 1992;267:21058–21064. [PubMed] [Google Scholar]
- 64.Yan L, Liang F, Xu H, Zhang X, Zhai H, Sun Q, et al. Identification of QTL for grain size and shape on the D genome of natural and synthetic allohexaploid wheats with near-identical AABB genomes. Front Plant Sci. 2017;8:1705. doi: 10.3389/fpls.2017.01705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Habib A, Powell JJ, Stiller J, Liu M, Shabala S, Zhou M, et al. A multiple near isogenic line (multi-NIL) RNA-seq approach to identify candidate genes underpinning QTL. Theor Appl Genet. 2017(3):1–12. [DOI] [PubMed]
- 66.Kong Y, Zhou G, Yin Y, Xu Y, Pattathil S, Hahn MG. Molecular analysis of a family of Arabidopsis genes related to galacturonosyltransferases. Plant Physiol. 2011;155(4):1791. doi: 10.1104/pp.110.163220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Jiang Y, Jiang Q, Hao C, Hou J, Wang L, Zhang H, et al. A yield-associated gene TaCWI, in wheat: its function, selection and evolution in global breeding revealed by haplotype analysis. Theor Appl Genet. 2015;128(1):131. doi: 10.1007/s00122-014-2417-5. [DOI] [PubMed] [Google Scholar]
- 68.Zhang L, Zhao YL, Gao LF, Zhao GY, Zhou RH, Zhang BS, et al. TaCKX6-D1, the ortholog of rice OsCKX2, is associated with grain weight in hexaploid wheat. New Phytol. 2012;195(3):574. doi: 10.1111/j.1469-8137.2012.04194.x. [DOI] [PubMed] [Google Scholar]
- 69.Chang J, Zhang J, Mao X, Li A, Jia J, Jing R. Polymorphism of TaSAP1-A1, and its association with agronomic traits in wheat. Planta. 2013;237(6):1495–1508. doi: 10.1007/s00425-013-1860-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Correlation coefficients among the three biological replicates of TAA10 and XX329 by Pearson correlation analysis. (XLSX 2028 kb)
Detailed information of the 8891 differentially expressed genes between the natural and resynthesized allohexaploid wheats (TAA10 and XX329). (XLSX 473 kb)
Detailed information of genes used for qRT-PCR analysis. (XLSX 69 kb)
The MapMan functional categories of the differentially expressed genes between the natural and resynthesized allohexaploid wheats (TAA10 and XX329). (XLSX 27 kb)
The MapMan enrichment analysis of the differentially expressed genes between the natural and resynthesized allohexaploid wheats (TAA10 and XX329). (XLSX 22 kb)
Verification of 5 differentially expressed genes involved in carbohydrate metabolism by qRT-PCR. (XLSX 12 kb)
The differentially expressed genes in the enriched MapMan categories of cell wall metabolism. (XLSX 33 kb)
The differentially expressed genes in the enriched MapMan categories of hormone metabolism. (DOCX 156 kb)
Detailed information of genes associated with known QTL and candidate genes controlling wheat grain size and weight. (DOCX 143 kb)
Data Availability Statement
All relevant data are available within the manuscript. The RNA-Seq reads used for this study were deposited at the National Center for Biotechnology Information Short Read Archive (http://www.ncbi.nlm.nih.gov/sra/) under the accession number SRP117710.