

Abstract
BACKGROUND
Loss-of-function variants in the angiopoietin-like 3 gene (ANGPTL3) have been associated with decreased plasma levels of triglycerides, low-density lipoprotein (LDL) cholesterol, and high-density lipoprotein (HDL) cholesterol. It is not known whether such variants or therapeutic antagonism of ANGPTL3 are associated with a reduced risk of atherosclerotic cardiovascular disease.
METHODS
We sequenced the exons of ANGPTL3 in 58,335 participants in the DiscovEHR human genetics study. We performed tests of association for loss-of-function variants in ANGPTL3 with lipid levels and with coronary artery disease in 13,102 case patients and 40,430 controls from the DiscovEHR study, with follow-up studies involving 23,317 case patients and 107,166 controls from four population studies. We also tested the effects of a human monoclonal antibody, evinacumab, against Angptl3 in dyslipidemic mice and against ANGPTL3 in healthy human volunteers with elevated levels of triglycerides or LDL cholesterol.
RESULTS
In the DiscovEHR study, participants with heterozygous loss-of-function variants in ANGPTL3 had significantly lower serum levels of triglycerides, HDL cholesterol, and LDL cholesterol than participants without these variants. Loss-of-function variants were found in 0.33% of case patients with coronary artery disease and in 0.45% of controls (adjusted odds ratio, 0.59; 95% confidence interval, 0.41 to 0.85; P = 0.004). These results were confirmed in the follow-up studies. In dyslipidemic mice, inhibition of Angptl3 with evinacumab resulted in a greater decrease in atherosclerotic lesion area and necrotic content than a control antibody. In humans, evinacumab caused a dose-dependent placebo-adjusted reduction in fasting triglyceride levels of up to 76% and LDL cholesterol levels of up to 23%.
CONCLUSIONS
Genetic and therapeutic antagonism of ANGPTL3 in humans and of Angptl3 in mice was associated with decreased levels of all three major lipid fractions and decreased odds of atherosclerotic cardiovascular disease. (Funded by Regeneron Pharmaceuticals and others; ClinicalTrials.gov number, NCT01749878.)
Angiopoietin-like proteins (ANGPTLs) have been established as important regulators of lipoprotein metabolism and have thus emerged as attractive targets for modulation of lipid levels and cardiovascular disease risk. Loss-of-function variants in ANGPTL4, a negative regulator of lipoprotein lipase (LPL) activity, are associated with decreased triglyceride levels, elevated high-density lipoprotein (HDL) cholesterol levels, and a reduced risk of coronary heart disease in humans.1–5 In addition, loss-of-function variants in LPL have been shown to increase the risk of coronary artery disease, and gain-of-function variants have been shown to decrease the risk.6–8
ANGPTL3 is an endogenous inhibitor of LPL that is related to ANGPTL4. Rare loss-of-function variants in ANGPTL3 have been shown to be associated with decreased triglyceride levels as well as decreased low-density lipoprotein (LDL) cholesterol and HDL cholesterol levels in family and general population studies of humans.2,9–19 Deletion of Angptl3 was also reported to reduce the development of atherosclerosis in Apoe-deficient mice.20
In this study, we examined the relationship between ANGPTL3 loss-of-function variants and coronary artery disease in a large number of participants sampled from a large U.S. health care system, with follow-up studies in four population cohorts. We also evaluated the effects of pharmacologic antagonism of ANGPTL3 with a human monoclonal antibody on lipid metabolism and atherosclerosis in a mouse model of atherosclerosis and on lipid levels in human volunteers.
METHODS
STUDY DESIGN AND OVERSIGHT
The studies were designed and data analyses were performed by authors who were employees of the sponsor, Regeneron Pharmaceuticals, and of the Regeneron Genetics Center, with the involvement of two of the academic authors. Authors who are employees of the sponsor wrote the first draft of the manuscript, made the decision to submit the manuscript for publication, and vouch for the accuracy and completeness of the data and all analyses. The DiscovEHR study, other studies involving humans, and studies in animals were funded by Regeneron Pharmaceuticals; other funding sources are listed at the end of the article. The studies were approved by the Geisinger Health System institutional review board and each participating site. All participants in the studies involving humans gave informed written consent.
HUMAN GENETICS COHORTS
We conducted human genetics studies using DNA samples and data from five cohorts. These included the first 58,335 adult enrollees of European ancestry who provided consent to participate in the MyCode Community Health Initiative21 of the Geisinger Health System (DiscovEHR study); a total of 107,888 persons from the Copenhagen City Heart Study, the Copenhagen General Population Study, and the Copenhagen Ischemic Heart Disease Study22,23; 7549 persons from the University of Pennsylvania Medicine BioBank (Penn); 5988 persons from the Duke Catheterization Genetics cohort (Duke)24; and 9058 Taiwanese Chinese persons from the Taiwan Metabochip (TAICHI) consortium.25,26 Full study descriptions are provided in the Methods section and Tables S1 and S2 in the Supplementary Appendix (available with the full text of this article at NEJM.org).
ASCERTAINMENT OF ANGPTL3 LOSS-OF-FUNCTION VARIANTS AND ANGPTL3 CONCENTRATION
In the DiscovEHR study, Penn, Duke, and TAICHI, ascertainment of ANGPTL3 loss-of-function variants was performed by whole-exome sequencing at the Regeneron Genetics Center, as described in the Supplementary Appendix. In the Copenhagen population studies, participants were directly genotyped for the p.Ser17Ter, p.Asn121fs, p.Asn147fs, and c.495+6T→C variants with the use of the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) and TaqMan-based assays or with the use of an allele-specific polymerase-chain-reaction system (KASPer, LGC Genomics). Quantification of serum ANGPTL3 concentration was performed as described in the Supplementary Appendix.
STUDIES IN ANIMALS
APOE*3Leiden.CETP mice, an established model for hyperlipidemia with all features of mixed or familial dysbetalipoproteinemia and atherosclerosis,27 were used for the evaluation of the effects of a fully human anti-ANGPTL3 monoclonal antibody, evinacumab (REGN1500), on atherosclerosis.28–30 (For antibody description and full experimental details, see the Supplementary Appendix.) A total of 50 female transgenic mice31 on a Western-type diet were matched to one control group (20 mice) and one evinacumab-treated group (30 mice) on the basis of body weight and plasma levels of total cholesterol and triglycerides and were treated with evinacumab or isotype-matched antibody with irrelevant specificity (control antibody) by weekly subcutaneous injections at a dose of 25 mg per kilogram of body weight for 13 weeks. Mice in which a mouse antihuman antibody response developed were excluded (see the Supplementary Appendix). At 13 weeks, the mice were euthanized and atherosclerosis development in the aortic root was measured (15 mice per group), as described in the Supplementary Appendix. The study was conducted at TNO Metabolic Health Research (the Netherlands). All experiments involving animals were approved by the Institutional Animal Care and Use Committee of the Netherlands Organization for Applied Scientific Research.
EVINACUMAB CLINICAL TRIAL
A phase 1, first-in-human, randomized, placebo-controlled, double-blind, ascending single-dose clinical trial was performed to assess the safety, side-effect profile, and pharmacodynamics of REGN1500 (evinacumab) administered subcutaneously or intravenously to participants with varying degrees of dyslipidemia (see the Supplementary Appendix). The trial protocol is available at NEJM.org. The results for Group A of this trial are described in this report; Group A enrolled healthy men and women 18 to 65 years of age with a fasting triglyceride level of 150 to 450 mg per deciliter (1.7 to 5.1 mmol per liter) or an LDL cholesterol level of 100 mg per deciliter (2.6 mmol per liter) or greater.
STATISTICAL ANALYSIS
We used linear mixed models to test for associations of levels of total cholesterol, LDL cholesterol, log10 HDL cholesterol, and log10 triglycerides with genotype (for aggregated ANGPTL3 loss-of-function variants and the individual variants p.Ser17Ter, p.Asn121fs, p.Asn147fs, and c.495+6T→C) in the DiscovEHR study. Odds ratios for coronary artery disease were estimated in each study with the use of logistic regression or Firth’s penalized likelihood method of logistic regression,32 as described in the Supplementary Appendix. We then performed an inverse-variance–weighted fixed-effects meta-analysis and a Mantel–Haenszel fixed-effects meta-analysis of associations with coronary artery disease.
For the study in mice, the significance of the differences between groups was determined with the use of the nonparametric Mann–Whitney U test for independent samples. For the clinical trial, an analysis of covariance (ANCOVA) model was used to compare the treatment effect in the evinacumab groups and the pooled placebo group; in the case of triglycerides, this was a rank-based ANCOVA. We used the following software programs for the statistical analyses of genetic data: GCTA software, version 1.2.433; PLINK software, version 2.0 (www.cog-genomics.org/plink/2.0/); R software, versions 3.2.1 and 3.3.1 (R Project for Statistical Computing); SAS software, version 9.2; and SPSS software, version 22. All reported P values are two-sided, and an alpha level of 0.05 was considered to indicate statistical significance.
RESULTS
LOSS-OF-FUNCTION VARIANTS IN ANGPTL3
In 58,335 adult DiscovEHR study participants of European ancestry, we identified and confirmed (through Sanger sequencing) 13 distinct loss-of-function variants in ANGPTL3, including 4 different premature stop variants, 6 different open reading frame–shifting insertion–deletion variants, and 3 splice variants (Table S3 in the Supplementary Appendix). The demographic and clinical characteristics of the study population according to variant status are shown in Table S4 in the Supplementary Appendix.
The most frequently observed ANGPTL3 loss-of-function variants were p.Ser17Ter (in 21 persons), which has been previously described to segregate with combined hypolipidemia in family studies14,16; p.Asn121fs (in 91 persons), which has been previously observed in a population sample and associated with reduced LDL cholesterol levels16; and a novel rare frame-shifting insertion–deletion variant at position 147 (p.Asn147fs, in 52 persons). A splice region variant, c.495+6T→C, was identified in 57 persons and was found to be associated with a decreased concentration of ANGPTL3 protein, similar to the other loss-of-function variants; thus, it was included in all loss-of-function analyses. A total of 246 persons were heterozygous for loss-of-function variants, corresponding to a total carrier frequency of 1 in 237 DiscovEHR study participants of European ancestry. As expected, given the allele frequencies, we did not find homozygotes or compound heterozygotes.
The median concentration of ANGPTL3 protein among 125 carriers of loss-of-function variants was 50 ng per milliliter (interquartile range, 38 to 62) and among 53 matched noncarriers was 99 ng per milliliter (interquartile range, 80 to 127) (P = 9.4×10−16 by Mann–Whitney U test). Protein abundance according to allele is shown in Figure 1.
Figure 1. Loss-of-Function Variants in ANGPTL3 and Lipid Levels in Humans.
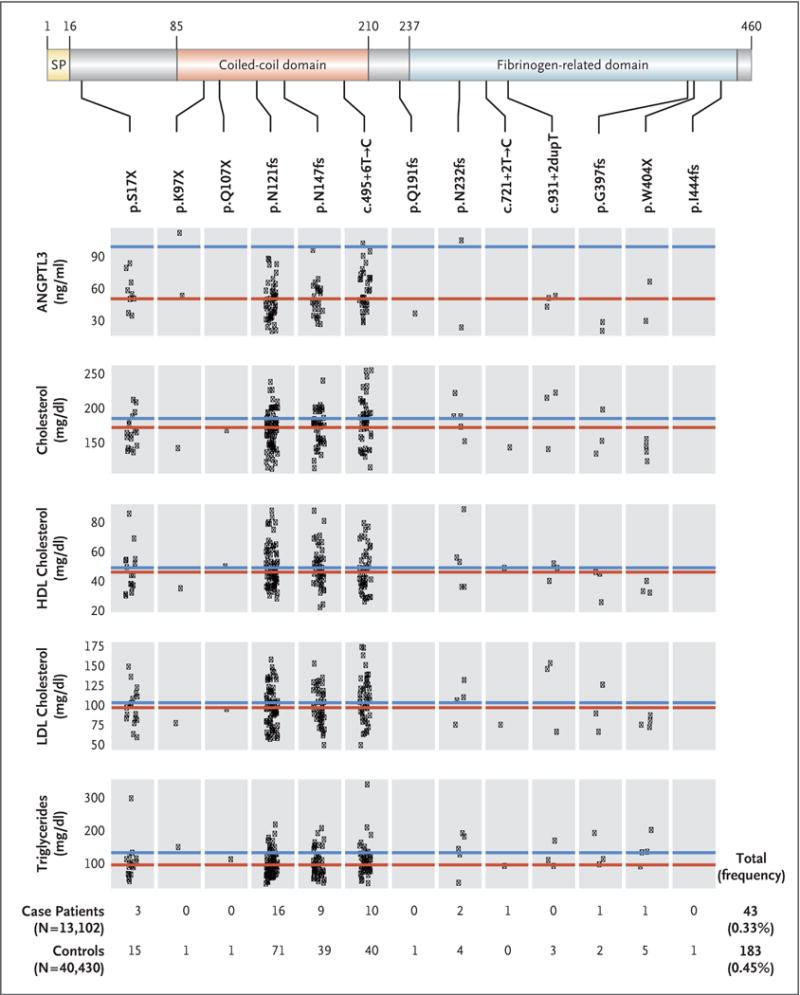
The top of the diagram shows the organization of the ANGPTL3 protein, which is 460 amino acids long. Functional sections of the protein are marked in varying colors and demarcated with lines labeled to show the numbering of the amino acid sequence for each section. Beneath the protein diagram, the individual protein-sequence variants are listed in order of their position in the protein. Beneath the protein-sequence variants are plots of ANGPTL3 concentration and lipid levels. The red lines indicate the median value for each level among all carriers of loss-of-function variants. Each point represents a trait value for a single carrier of the loss-of-function variant specified above each box. The blue line indicates the median value for each trait for all sequenced persons not carrying a loss-of-function variant in ANGPTL3. To convert the values for cholesterol to millimoles per liter, multiply by 0.02586. To convert the values for triglycerides to millimoles per liter, multiply by 0.01129. HDL denotes high-density lipoprotein, LDL low-density lipoprotein, and SP signal peptide.
LOSS-OF-FUNCTION VARIANTS IN ANGPTL3, LIPID LEVELS, AND CORONARY ARTERY DISEASE
We performed tests of association between loss-of-function variants (both individually and aggregated over ANGPTL3) and median serum lipid measures in up to 45,226 persons in the DiscovEHR study who had documented lipid levels in the electronic health record (Table 1 and Fig. 1, and Tables S5 and S6 in the Supplementary Appendix). Overall, carriers of loss-of-function variants had 27% lower triglyceride levels than noncarriers (P = 2.5×10−21), 9% lower LDL cholesterol levels (P = 2.8×10−5), and 4% lower HDL cholesterol levels (P = 0.02), after adjustment for covariates.
Table 1.
Associations between ANGPTL3 Predicted Loss-of-Function Variants and Lipid Levels in DiscovEHR Study Participants.*
| Trait | Noncarriers | Carriers of ANGPTL3 Loss-of-Function Variants | P Value† | ||
|---|---|---|---|---|---|
| No. of Participants | Median Level (IQR) mg/dl |
No. of Participants | Median Level (IQR) mg/dl |
||
| Triglycerides | 45,015 | 130 (94–179) | 191 | 94 (75–125) | 2.5×10−21 |
|
| |||||
| HDL cholesterol | 45,036 | 49 (40–59) | 190 | 46 (38–56) | 0.02 |
|
| |||||
| LDL cholesterol | 44,629 | 121 (100–146) | 190 | 112 (90–136) | 2.8×10−5 |
|
| |||||
| Total cholesterol | 44,877 | 204 (179–232) | 191 | 179 (160–203) | 1.7×10−17 |
Values for low-density lipoprotein (LDL) cholesterol and total cholesterol were adjusted for the use of lipid-lowering medication by dividing by 0.7 and 0.8, respectively. To convert the values for cholesterol to millimoles per liter, multiply by 0.02586. To convert the values for triglycerides to millimoles per liter, multiply by 0.01129. HDL denotes high-density lipoprotein, and IQR interquartile range.
P values were calculated by means of a mixed linear model association analysis of lipid residuals after adjustment for age, age squared, sex, and the first five principal components of ancestry. A genetic-relatedness matrix was included as a random-effects covariate. Values for triglycerides and HDL cholesterol were log10-transformed before residual calculation.
We then tested for association between ANGPTL3 loss-of-function variants and coronary artery disease. Overall, 43 of the 13,102 persons with coronary artery disease (cumulative carrier frequency, 0.33%) and 183 of the 40,430 controls (cumulative carrier frequency, 0.45%) carried a loss-of-function variant. After adjustment for age, sex, and the principal components of ancestry, the presence of an ANGPTL3 loss-of-function variant was associated with a 41% lower odds of coronary artery disease (adjusted odds ratio for any loss-of-function variant, 0.59; 95% confidence interval [CI], 0.41 to 0.85; P = 0.004); these variants were also associated with numerically, but not significantly, lower odds of myocardial infarction (carrier frequencies, 0.36% among persons with myocardial infarction and 0.45% among controls; odds ratio, 0.66; 95% CI, 0.39 to 1.06; P = 0.09). Individually, each of the four most abundant loss-of-function variants (p.Ser17Ter, p.Asn121fs, p.Asn147fs, and c.495+6T→C) was less frequent among case patients with coronary artery disease than among controls (Table S7 in the Supplementary Appendix). In a subgroup of the sequenced cohort in which only 1 person in every first-degree and second-degree relationship was retained (9261 case patients and 29,778 controls), ANGPTL3 loss-of-function variants were associated with lower odds of coronary artery disease (carrier frequencies, 0.31% among persons with coronary artery disease and 0.44% among controls; odds ratio, 0.61; 95% CI, 0.39 to 0.94; P = 0.03).
We sought additional evidence of an association between ANGPTL3 loss-of-function variants and coronary artery disease in 23,317 case patients with coronary artery disease and 107,166 controls from four independent population studies (Copenhagen General Population Study, Duke, Penn, and TAICHI) (Table S2 in the Supplementary Appendix). In these studies, we observed a numerically, but not significantly, lower odds of coronary artery disease among carriers of these variants (odds ratio, 0.63; 95% CI, 0.39 to 1.02; P = 0.06). An inverse-variance–weighted fixed-effects meta-analysis of the association of ANGPTL3 loss-of-function variants with coronary artery disease in the DiscovEHR study and these other population studies yielded an overall odds ratio of 0.61 (95% CI, 0.45 to 0.81; P<0.001) (Fig. 2). A Mantel–Haenszel fixed-effects meta-analysis, a method that is robust to low counts, yielded similar results (odds ratio, 0.69; 95% CI, 0.53 to 0.89; P = 0.006) without evidence of significant heterogeneity (Q = 3.94, I2 = 0%, P = 0.40 for heterogeneity).
Figure 2. Association of ANGPTL3 Loss-of-Function Variants and Coronary Artery Disease.
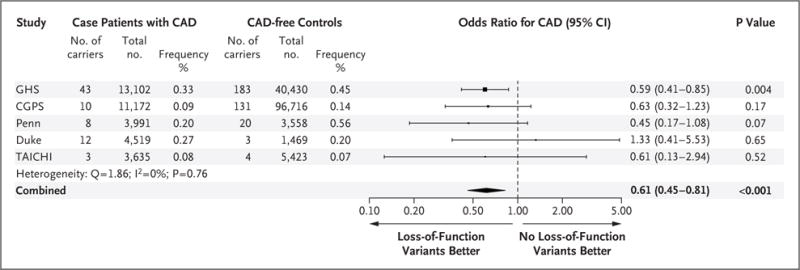
The association between the ANGPTL3 loss-of-function variants and coronary artery disease (CAD) was tested in each study with the use of logistic regression or Firth’s penalized likelihood logistic regression. Combined effects were determined by inverse-variance– weighted fixed-effects meta-analysis. For each study, the square indicates the odds ratio and the line indicates the 95% confidence interval. The square size is proportional to the precision of the estimate. For the combined estimate, the center of the diamond indicates the odds ratio for the meta-analysis, with the left and right corners of the diamond indicating the boundaries of the 95% confidence interval. ANGPTL3 loss-of-function variants were ascertained by means of targeted genotyping in the Copenhagen General Population Studies (CGPS) and by means of exome sequencing in the other studies. Duke denotes the Duke Catheterization Genetics cohort, GHS Geisinger Health System (DiscovEHR study), Penn the University of Pennsylvania Medicine BioBank, and TAICHI the Taiwan Metabochip consortium.
INHIBITION OF ANGPTL3 IN A MOUSE MODEL OF ATHEROSCLEROSIS
We evaluated the effect of an anti-ANGPTL3 monoclonal antibody candidate therapeutic30 on atherosclerosis in APOE*3Leiden.CETP mice. Evinacumab administration was associated with a significantly lower average total cholesterol level than the control antibody (difference, −52%; P<0.001) and a significantly lower triglyceride level (difference, −84%; P<0.001) by a reduction of the apoB-containing atherogenic lipoproteins (Fig. 3A through 3D). Evinacumab was associated with a significantly greater decrease in atherosclerotic lesion size than the control antibody (difference, −39%; P<0.001) (Fig. 3E and 3F). Treatment with evinacumab was associated with a significantly greater decrease in necrotic content in severe type IV and V lesions than the control antibody (difference, −45%; P = 0.001), but there were no significant differences in macrophage content, collagen content, or smooth muscle cell area (Fig. 3G and 3H).
Figure 3. (facing page). Effect of Inhibition of Angptl3 with a Monoclonal Antibody in APOE*3Leiden.CETP Mice.
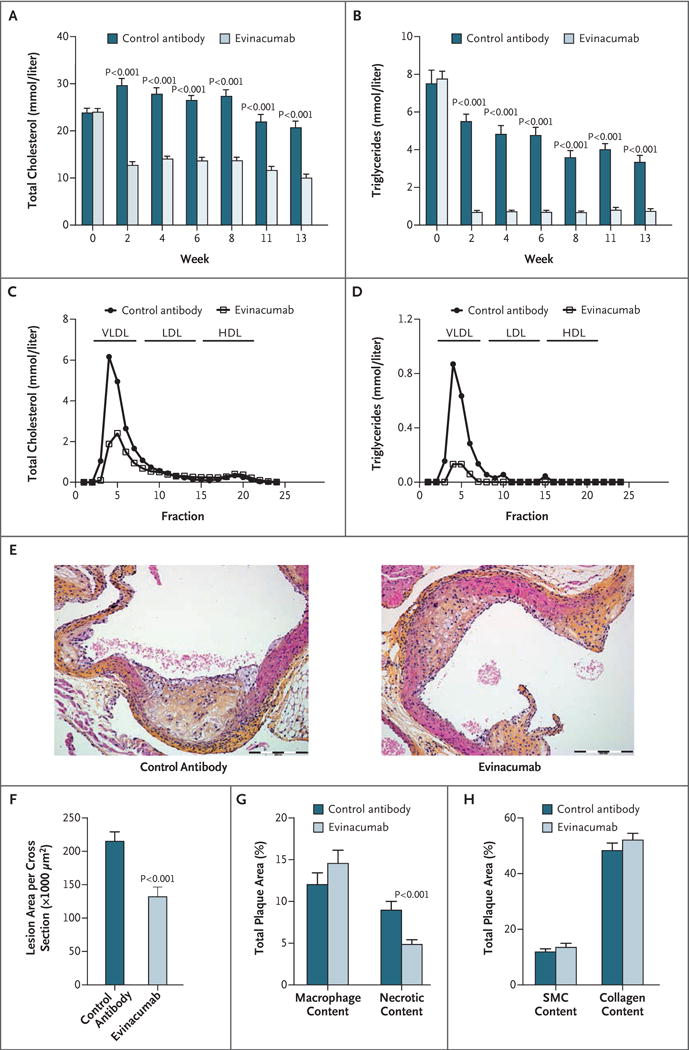
Evinacumab reduced plasma levels of total cholesterol (Panel A) and triglycerides (Panel B) in APOE*3Leiden.CETP mice throughout the 13 weeks of the study. Plasma lipoproteins were separated on fast protein liquid chromatography at the end of the study and assessed for total cholesterol (Panel C) and triglycerides (Panel D). Representative images of atherosclerotic lesions (hematoxylin–phloxine–saffron stain) in a cross section of the aortic root area that was used for area quantification are shown (Panel E); the total atherosclerotic lesion area per cross section was evaluated at the end of the study (Panel F). Macrophage and necrotic content (indicators of lesion inflammation) (Panel G) and smooth muscle cells (SMC) and collagen content (indicators of lesion stability) (Panel H) were determined in the severe (type IV and V) lesions and normalized to lesion size. All values in Panels A, B, and F through H are means ±SE. For Panels A through D, 14 mice were given the control antibody and 22 were given evinacumab. The analysis conducted in Panels E through H involved 14 mice given the control antibody and 15 given evinacumab. VLDL denotes very-low-density lipoprotein.
ANGPTL3 INHIBITION WITH EVINACUMAB IN HEALTHY HUMAN VOLUNTEERS
We evaluated the effects of evinacumab on lipid levels in healthy human volunteers with mildly to moderately elevated levels of triglycerides (150 to 450 mg per deciliter) or LDL cholesterol (≥100 mg per deciliter). A total of 83 participants at three U.S. sites entered a single-ascending-dose trial involving randomization to evinacumab (administered subcutaneously or intravenously) or placebo in a 3:1 ratio (see the Methods section and Fig. S1 in the Supplementary Appendix). The baseline characteristics of the participants are presented in Tables S8 and S9 in the Supplementary Appendix.
The most frequent adverse event among the evinacumab-treated patients was headache (seven patients; 11%). Transient, single elevations of the alanine aminotransferase level to more than 3 times the upper limit of the normal range were observed in two evinacumab-treated patients (3%), both of whom had an alanine aminotransferase level above the upper limit of the normal range at baseline or screening. There were no discontinuations due to adverse safety events (Tables S10 and S11 in the Supplementary Appendix).
The time course of triglyceride, LDL cholesterol, and HDL cholesterol responses to escalating doses of evinacumab or placebo are shown in Figure 4, and in Figures S2 and S3 in the Supplementary Appendix. The magnitude and duration of lipid reductions (placebo-corrected) were dose-proportional. The maximal changes in lipid levels found among patients who received a dose of 20 mg per kilogram intravenously were as follows: triglycerides, −76.0% (day 4); directly measured LDL cholesterol, −23.2% (day 15); and HDL cholesterol, −18.4% (day 15). A full summary of treatment effects is presented in Tables S12 through S17 in the Supplementary Appendix.
Figure 4. Effects of Inhibition of ANGPTL3 with a Monoclonal Antibody on Triglyceride Levels in Human Volunteers.
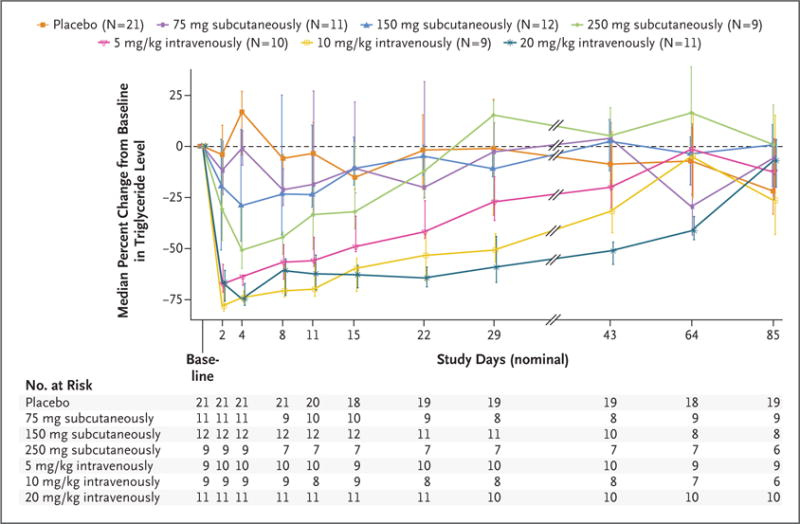
The median percent change from baseline in the triglyceride level (measured in milligrams per deciliter) is shown for the placebo group and for the groups receiving evinacumab at various doses. Individual data points are median values, with I bars showing interquartile ranges.
DISCUSSION
We performed whole-exome sequencing in 58,335 adult participants of European ancestry in the DiscovEHR study, which links genetic data to electronic health records in a large integrated health system, along with targeted genotyping of ANGPTL3 loss-of-function variants and whole-exome sequencing in four other population cohorts comprising 130,483 persons. Using these methods, we identified more than 400 persons who harbor heterozygous loss-of-function variants in ANGPTL3. Persons heterozygous for ANGPTL3 loss-of-function variants had approximately 50% lower ANGPTL3 levels than noncarriers and 39% lower odds of coronary artery disease. These results mirror the reduction in plasma lipid levels and atherosclerosis progression that we observed in dyslipidemic mice treated with evinacumab, a human monoclonal antibody inhibitor of ANGPTL3. The pattern and magnitude of reduction in atherosclerotic lesion area that was observed in this mouse model treated with evinacumab was similar to that reported previously in the same mouse model treated with atorvastatin.34 Our results provide evidence that the combined hypolipidemia profile associated with genetic or therapeutic antagonism of ANGPTL3, which includes a reduction in levels of HDL cholesterol in addition to LDL cholesterol and triglycerides, is antiatherogenic.
ANGPTL3 inhibition affects lipid levels in several ways. Triglyceride reduction is mediated at least in part through disinhibition of LPL and subsequent hydrolysis of triglyceride-rich lipoproteins. HDL cholesterol reduction is mediated by disinhibition of endothelial lipase.13,30 The reduction in LDL cholesterol levels with ANGPTL3 antagonism occurs through a mechanism that has yet to be elucidated. However, studies in Ldlr-deficient mice indicate that ANGPTL3 modulates LDL cholesterol in a manner that is independent of the LDL receptor and other known mechanisms responsible for clearance of plasma LDL cholesterol.35 These findings suggest that therapeutic antagonism of ANGPTL3 should be efficacious in lowering LDL cholesterol levels in persons with a complete deficiency in LDL receptor–mediated LDL cholesterol uptake, such as persons affected by homozygous familial hypercholesterolemia. Additional human genetics support for targeting this pathway is provided by evidence of similar reductions in coronary artery disease risk that are associated with inactivating variants in ANGPTL4,5 a related inhibitor of LPL, and genetic variants in LPL that increase LPL activity.6,7 Evidence to date does not suggest that genetic perturbation of LIPG, the gene encoding endothelial lipase, is associated with coronary artery disease, despite robust associations with HDL cholesterol levels.36
We also report here clinical pharmacology results from a trial involving therapeutic antagonism of ANGPTL3 in human volunteers treated with evinacumab. We found dose-dependent reductions in levels of triglycerides and LDL cholesterol that appear to recapitulate the lipid phenotype of patients with ANGPTL3 loss-of-function variants. In addition to the ANGPTL3 loss-of-function heterozygotes in this study, more than 200 heterozygous carriers and more than 20 homozygotes of ANGPTL3 loss-of-function variants have been reported by others with no apparent negative effects on cardiometabolic traits and diseases evaluated in these participants.14,37,38 A recent study has shown an association between ANGPTL3 deficiency and protection from coronary artery disease,38 a finding consistent with those of our study. In aggregate, these data support further investigation of inhibition of ANGPTL3 as a therapeutic target for lipid modification and possible reduction in the risk of atherosclerotic cardiovascular disease in humans.
Our study has limitations. Our human genetics studies largely involved persons of European ancestry; it is not known whether our observations will generalize to persons of other ancestries. We did not directly evaluate coronary atherosclerosis progression; instead, we evaluated aortic atherosclerosis progression in a mouse model. The sample size of our clinical trial was limited, and the trial included healthy human volunteers, not those selected for severe dyslipidemias or established cardiovascular disease. Finally, the associations with lipid levels and coronary artery disease in our human genetics studies were observed in heterozygotes who had a reduction in ANGTPL3 levels of approximately 50%, which had probably been present since birth. Whether such observations accurately reflect the phenotypic effects of a more complete blockade of ANGPTL3 later in life, with the use of a monoclonal antibody, is not clear.
In conclusion, we observed that loss-of-function variants in ANGPTL3 were associated with a reduced risk of coronary artery disease, mirroring the antiatherogenic effects of inhibition of Angptl3 by monoclonal antibody in mice. In human volunteers, monoclonal antibody inhibition of ANGPTL3 was associated with dose-dependent reductions in LDL cholesterol and triglyceride levels.
Supplementary Material
Acknowledgments
The DiscovEHR study was partially funded by Regeneron Pharmaceuticals. The evinacumab clinical trial was funded by Regeneron Pharmaceuticals. The Penn Medicine BioBank was funded by the Perelman School of Medicine and a gift from the Smilow family. Dr. Shah (Duke Catheterization Genetics biorepository) was supported by a grant (R01-HL127009-01A1) from the National Institutes of Health. The TAICHI study was supported by grants from the National Health Research Institutes, Taiwan (PH-099-PP-03, PH-100-PP-03, and PH-101-PP-03); the National Science Council, Taiwan (NSC 101-2314-B-075A-006-MY3, MOST 104-2314-B-075A-006-MY3, MOST 104-2314-B-075A-007, and MOST 105-2314-B-075A-003); and the Taichung Veterans General Hospital, Taiwan (TCVGH-1020101C, TCVGH-1020102D, TCVGH-1023102B, TCVGH-1023107D, TCVGH-1030101C, TCVGH-1030105D, TCVGH-1033503C, TCVGH-1033102B, TCVGH-1033108D, TCVGH-1040101C, TCVGH-1040102D, TCVGH-1043504C, and TCVGH-1043104B); it was also supported in part by the National Center for Advancing Translational Sciences (CTSI grant UL1TR001881).
We thank the DiscovEHR study and clinical trial participants for their collaboration; Gary Swergold for important contributions to the clinical trial of evinacumab; the participants in the Penn Medicine BioBank, as well as JoEllen Weaver, David Birtwell, Heather Williams, and Stephanie DerOhannessian; and the staff and participants of the Copenhagen General Population Study, the Copenhagen City Heart Study, and the Copenhagen Ischemic Heart Disease Study.
APPENDIX
The authors’ full names and academic degrees are as follows: Frederick E. Dewey, M.D., Viktoria Gusarova, Ph.D., Richard L. Dunbar, M.D., Colm O’Dushlaine, Ph.D., Claudia Schurmann, Ph.D., Omri Gottesman, M.D., Shane McCarthy, Ph.D., Cristopher V. Van Hout, Ph.D., Shannon Bruse, Ph.D., Hayes M. Dansky, M.D., Joseph B. Leader, B.A., Michael F. Murray, M.D., Marylyn D. Ritchie, Ph.D., H. Lester Kirchner, Ph.D., Lukas Habegger, Ph.D., Alex Lopez, M.S., John Penn, M.S., An Zhao, Ph.D., Weiping Shao, Ph.D., Neil Stahl, Ph.D., Andrew J. Murphy, Ph.D., Sara Hamon, Ph.D., Aurelie Bouzelmat, M.S., Rick Zhang, Ph.D., Brad Shumel, M.D., Robert Pordy, M.D., Daniel Gipe, M.D., Gary A. Herman, M.D., Wayne H.H. Sheu, M.D., Ph.D., I-Te Lee, M.D., Ph.D., Kae-Woei Liang, M.D., Xiuqing Guo, Ph.D., Jerome I. Rotter, M.D., Yii-Der I. Chen, Ph.D., William E. Kraus, M.D., Svati H. Shah, M.D., M.H.S., Scott Damrauer, M.D., Aeron Small, B.A., Daniel J. Rader, M.D., Anders Berg Wulff, M.D., Ph.D., Børge G. Nordestgaard, M.D., D.M.Sc., Anne Tybjærg-Hansen, M.D., D.M.Sc., Anita M. van den Hoek, Ph.D., Hans M.G. Princen, Ph.D., David H. Ledbetter, Ph.D., David J. Carey, Ph.D., John D. Overton, Ph.D., Jeffrey G. Reid, Ph.D., William J. Sasiela, Ph.D., Poulabi Banerjee, Ph.D., Alan R. Shuldiner, M.D., Ingrid B. Borecki, Ph.D., Tanya M. Teslovich, Ph.D., George D. Yancopoulos, M.D., Ph.D., Scott J. Mellis, M.D., Ph.D., Jesper Gromada, Ph.D., D.M.Sc., and Aris Baras, M.D.
The authors’ affiliations are as follows: Regeneron Genetics Center (F.E.D., C.O., C.S., O.G., S.M., C.V.V.H., S.B., L.H., A.L., J.P., N.S., A.J.M., J.D.O., J.G.R., A.R.S., I.B.B., T.M.T., G.D.Y., S.J.M., A. Baras) and Regeneron Pharmaceuticals (V.G., H.M.D., A.Z., W.S., N.S., A.J.M., S.H., A. Bouzelmat, R.Z., B.S., R.P., D.G., G.A.H., W.J.S., P.B., G.D.Y., S.J.M., J.G.) Tarrytown, NY; the Department of Medicine, Division of Translational Medicine and Human Genetics (R.L.D.), and Departments of Surgery (S.D.) and Genetics and Medicine (A.S., D.J.R.), Perelman School of Medicine, University of Pennsylvania, Philadelphia, and Geisinger Health System, Danville (J.B.L., M.F.M., M.D.R., H.L.K., D.H.L., D.J.C.) — both in Pennsylvania; the Division of Endocrinology and Metabolism, Department of Internal Medicine (W.H.H.S., I.-T.L.) and Cardiovascular Center (K.-W.L.), Taichung Veterans General Hospital, Institute of Medical Technology, National Chung-Hsing University (W.H.H.S.), School of Medicine, Chung Shan Medical University (I.-T.L.), and the Department of Medicine, China Medical University (K.-W.L.), Taichung, and School of Medicine, National Yang-Ming University (W.H.H.S., I.-T.L., K.-W.L.), and School of Medicine, National Defense Medical Center (W.H.H.S.), Taipei — all in Taiwan; Institute for Translational Genomics and Population Sciences, Los Angeles Biomedical Research Institute and Department of Pediatrics, Harbor– UCLA Medical Center, Torrance, CA (X.G., J.I.R., Y.-D.I.C.); the Division of Cardiology, Department of Medicine, Molecular Physiology Institute, School of Medicine, Duke University, Durham, NC (W.E.K., S.H.S.); the Department of Clinical Biochemistry, Rigshospitalet (A.B.W., B.G.N., A.T.-H.), the Copenhagen General Population Study (B.G.N., A.T.-H.) and Department of Clinical Biochemistry (B.G.N.), Herlev and Gentofte Hospital, and the Copenhagen City Heart Study, Frederiksberg Hospital, Copenhagen University Hospital, and Faculty of Health and Medical Sciences, University of Copenhagen (B.G.N., A.T.-H.) — all in Copenhagen; and TNO Metabolic Health Research, Gaubius Laboratory, Leiden, the Netherlands (A.M.H., H.M.G.P.).
Footnotes
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
- 1.Romeo S, Pennacchio LA, Fu Y, et al. Population-based resequencing of ANGPTL4 uncovers variations that reduce triglycerides and increase HDL. Nat Genet. 2007;39:513–6. doi: 10.1038/ng1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Romeo S, Yin W, Kozlitina J, et al. Rare loss-of-function mutations in ANGPTL family members contribute to plasma triglyceride levels in humans. J Clin Invest. 2009;119:70–9. doi: 10.1172/JCI37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Folsom AR, Peacock JM, Demerath E, Boerwinkle E. Variation in ANGPTL4 and risk of coronary heart disease: the Atherosclerosis Risk in Communities Study. Metabolism. 2008;57:1591–6. doi: 10.1016/j.metabol.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang H, Eckel RH. Lipoprotein lipase: from gene to obesity. Am J Physiol Endocrinol Metab. 2009;297:E271–E288. doi: 10.1152/ajpendo.90920.2008. [DOI] [PubMed] [Google Scholar]
- 5.Dewey FE, Gusarova V, O’Dushlaine C, et al. Inactivating variants in ANGPTL4 and risk of coronary artery disease. N Engl J Med. 2016;374:1123–33. doi: 10.1056/NEJMoa1510926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jensen MK, Rimm EB, Rader D, et al. S447X variant of the lipoprotein lipase gene, lipids, and risk of coronary heart disease in 3 prospective cohort studies. Am Heart J. 2009;157:384–90. doi: 10.1016/j.ahj.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sagoo GS, Tatt I, Salanti G, et al. Seven lipoprotein lipase gene polymorphisms, lipid fractions, and coronary disease: a HuGE association review and meta-analysis. Am J Epidemiol. 2008;168:1233–46. doi: 10.1093/aje/kwn235. [DOI] [PubMed] [Google Scholar]
- 8.Khera AV, Won HH, Peloso GM, et al. Association of rare and common variation in the lipoprotein lipase gene with coronary artery disease. JAMA. 2017;317:937–46. doi: 10.1001/jama.2017.0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kersten S. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta. 2014;1841:919–33. doi: 10.1016/j.bbalip.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, McNutt MC, Banfi S, et al. Hepatic ANGPTL3 regulates adipose tissue energy homeostasis. Proc Natl Acad Sci U S A. 2015;112:11630–5. doi: 10.1073/pnas.1515374112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tikka A, Jauhiainen M. The role of ANGPTL3 in controlling lipoprotein metabolism. Endocrine. 2016;52:187–93. doi: 10.1007/s12020-015-0838-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu J, Afroza H, Rader DJ, Jin W. Angiopoietin-like protein 3 inhibits lipoprotein lipase activity through enhancing its cleavage by proprotein convertases. J Biol Chem. 2010;285:27561–70. doi: 10.1074/jbc.M110.144279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimamura M, Matsuda M, Yasumo H, et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol. 2007;27:366–72. doi: 10.1161/01.ATV.0000252827.51626.89. [DOI] [PubMed] [Google Scholar]
- 14.Minicocci I, Montali A, Robciuc MR, et al. Mutations in the ANGPTL3 gene and familial combined hypolipidemia: a clinical and biochemical characterization. J Clin Endocrinol Metab. 2012;97:E1266–E1275. doi: 10.1210/jc.2012-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pisciotta L, Favari E, Magnolo L, et al. Characterization of three kindreds with familial combined hypolipidemia caused by loss-of-function mutations of ANGPTL3. Circ Cardiovasc Genet. 2012;5:42–50. doi: 10.1161/CIRCGENETICS.111.960674. [DOI] [PubMed] [Google Scholar]
- 16.Musunuru K, Pirruccello JP, Do R, et al. Exome sequencing, ANGPTL3 mutations, and familial combined hypolipidemia. N Engl J Med. 2010;363:2220–7. doi: 10.1056/NEJMoa1002926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Noto D, Cefalù AB, Valenti V, et al. Prevalence of ANGPTL3 and APOB gene mutations in subjects with combined hypolipidemia. Arterioscler Thromb Vasc Biol. 2012;32:805–9. doi: 10.1161/ATVBAHA.111.238766. [DOI] [PubMed] [Google Scholar]
- 18.Martín-Campos JM, Roig R, Mayoral C, et al. Identification of a novel mutation in the ANGPTL3 gene in two families diagnosed of familial hypobetalipoproteinemia without APOB mutation. Clin Chim Acta. 2012;413:552–5. doi: 10.1016/j.cca.2011.11.020. [DOI] [PubMed] [Google Scholar]
- 19.Helgadottir A, Gretarsdottir S, Thorleifsson G, et al. Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat Genet. 2016;48:634–9. doi: 10.1038/ng.3561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ando Y, Shimizugawa T, Takeshita S, et al. A decreased expression of angiopoietin-like 3 is protective against atherosclerosis in apoE-deficient mice. J Lipid Res. 2003;44:1216–23. doi: 10.1194/jlr.M300031-JLR200. [DOI] [PubMed] [Google Scholar]
- 21.Carey DJ, Fetterolf SN, Davis FD, et al. The Geisinger MyCode community health initiative: an electronic health record-linked biobank for precision medicine research. Genet Med. 2016;18:906–13. doi: 10.1038/gim.2015.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jørgensen AB, Frikke-Schmidt R, West AS, Grande P, Nordestgaard BG, Tybjærg-Hansen A. Genetically elevated non-fasting triglycerides and calculated remnant cholesterol as causal risk factors for myocardial infarction. Eur Heart J. 2013;34:1826–33. doi: 10.1093/eurheartj/ehs431. [DOI] [PubMed] [Google Scholar]
- 23.Varbo A, Benn M, Tybjærg-Hansen A, Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG. Remnant cholesterol as a causal risk factor for ischemic heart disease. J Am Coll Cardiol. 2013;61:427–36. doi: 10.1016/j.jacc.2012.08.1026. [DOI] [PubMed] [Google Scholar]
- 24.Kraus WE, Granger CB, Sketch MH, Jr, et al. A guide for a cardiovascular genomics biorepository: the CATHGEN experience. J Cardiovasc Transl Res. 2015;8:449–57. doi: 10.1007/s12265-015-9648-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kuo JZ, Sheu WH, Assimes TL, et al. Trans-ethnic fine mapping identifies a novel independent locus at the 3′ end of CDKAL1 and novel variants of several susceptibility loci for type 2 diabetes in a Han Chinese population. Diabetologia. 2013;56:2619–28. doi: 10.1007/s00125-013-3047-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Assimes TL, Lee IT, Juang JM, et al. Genetics of coronary artery disease in Taiwan: a CardioMetaboChip Study by the Taichi Consortium. PLoS One. 2016;11(3):e0138014. doi: 10.1371/journal.pone.0138014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Vlijmen BJ, van den Maagdenberg AM, Gijbels MJ, et al. Diet-induced hyperlipoproteinemia and atherosclerosis in apolipoprotein E3-Leiden transgenic mice. J Clin Invest. 1994;93:1403–10. doi: 10.1172/JCI117117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Macdonald LE, Karow M, Stevens S, et al. Precise and in situ genetic humanization of 6 Mb of mouse immunoglobulin genes. Proc Natl Acad Sci U S A. 2014;111:5147–52. doi: 10.1073/pnas.1323896111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Murphy AJ, Macdonald LE, Stevens S, et al. Mice with megabase humanization of their immunoglobulin genes generate antibodies as efficiently as normal mice. Proc Natl Acad Sci U S A. 2014;111:5153–8. doi: 10.1073/pnas.1324022111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gusarova V, Alexa CA, Wang Y, et al. ANGPTL3 blockade with a human monoclonal antibody reduces plasma lipids in dyslipidemic mice and monkeys. J Lipid Res. 2015;56:1308–17. doi: 10.1194/jlr.M054890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Vlijmen BJ, van ’t Hof HB, Mol MJ, et al. Modulation of very low density lipoprotein production and clearance contributes to age- and gender-dependent hyperlipoproteinemia in apolipoprotein E3-Leiden transgenic mice. J Clin Invest. 1996;97:1184–92. doi: 10.1172/JCI118532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinze G, Schemper M. A solution to the problem of separation in logistic regression. Stat Med. 2002;21:2409–19. doi: 10.1002/sim.1047. [DOI] [PubMed] [Google Scholar]
- 33.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82. doi: 10.1016/j.ajhg.2010.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kühnast S, van der Hoorn JW, Pieter-man EJ, et al. Alirocumab inhibits atherosclerosis, improves the plaque morphology, and enhances the effects of a statin. J Lipid Res. 2014;55:2103–12. doi: 10.1194/jlr.M051326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Gusarova V, Banfi S, Gromada J, Cohen JC, Hobbs HH. Inactivation of ANGPTL3 reduces hepatic VLDL-triglyceride secretion. J Lipid Res. 2015;56:1296–307. doi: 10.1194/jlr.M054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Voight BF, Peloso GM, Orho-Melander M, et al. Plasma HDL cholesterol and risk of myocardial infarction: a mendelian randomisation study. Lancet. 2012;380:572–80. doi: 10.1016/S0140-6736(12)60312-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Minicocci I, Santini S, Cantisani V, et al. Clinical characteristics and plasma lipids in subjects with familial combined hypolipidemia: a pooled analysis. J Lipid Res. 2013;54:3481–90. doi: 10.1194/jlr.P039875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stitziel NO, Khera AV, Wang X, et al. ANGPTL3 deficiency and protection against coronary artery disease. J Am Coll Cardiol. 2017;69:2054–63. doi: 10.1016/j.jacc.2017.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.