

SUMMARY
Although several interleukin-17 (IL-17) family members and their receptors have been recently appreciated as important regulators in inflammatory diseases, the function of other IL-17 cytokines and IL-17 receptor-like molecules is unclear. Here we show that an IL-17 cytokine family member, IL-17C, was induced in a Th17 cell-dependent autoimmune disease and was required for its pathogenesis. IL-17C bound to IL-17RE, a member of IL-17 receptor family whose full-length isoform was selectively expressed in Th17 cells and signaled via an IL-17RARE receptor complex and the downstream adaptor Act1. IL-17C-IL-17RE induced the expression of a nuclear IkappaB family member, IκBζ, in Th17 cells to potentiate the Th17 cell response. Thus, our work has identified a cytokine-receptor pair with important function in regulating proinflammatory responses. This pathway may be targeted to treat autoimmune diseases.
INTRODUCTION
Interleukin-17 (IL-17) family cytokines have recently emerged as important players in inflammatory responses. IL-17A and IL-17F, which are most highly related among IL-17 family cytokines, are expressed by a distinct T cell subset, Th17 cells (Dong, 2008). The cytokines TGF-β and IL-6 or IL-21 have been shown to be critical in generation of Th17 cells (Bettelli et al., 2006; Korn et al., 2007; Mangan et al., 2006; Nurieva et al., 2007; Veldhoen et al., 2006), leading to the expression of lineage-specific transcription factors, RORγt (Ivanov et al., 2006) and RORα (Yang et al., 2008c). RORγt and RORα are retinoic-acid-receptor-related orphan receptors that are induced by activation of the STAT3 pathway by IL-6 (Yang et al., 2007). Other transcription factors are also reported to promote Th17 cell differentiation. Most recently, IκBζ, a nuclear IkappaB family member, was identified to be induced by STAT3 and promote Th17 cell differentiation by cooperating with RORγt and RORα (Okamoto et al., 2010). Th17 cells also express IL-1R and IL-23R, rendering the cells to selectively respond to IL-1 (Chung et al., 2009) and IL-23 (Langrish et al., 2005) for their differentiation and cytokine expression.
Th17 cells, via production of IL-17A and IL-17F, promote the development of autoimmune diseases while protecting the host against bacterial and fungal infections (Kolls and Lindén, 2004). IL-17A and IL-17F signal through the IL-17 receptor A (IL-17RA)-IL-17RC complex (Hu et al., 2010; Toy et al., 2006). Although IL-17RA is expressed ubiquitously, IL-17RC expression is dominant in nonhematopoietic cells (Kuestner et al., 2007). IL-17A and IL-17F target epithelial cells or fibroblasts to produce arrays of cytokines and chemokines including IL-6 and CXCL1 (Chang and Dong, 2009; Gaffen, 2008). A distant member of IL-17 family cytokine, IL-25 (IL-17E), is involved in allergic diseases and host defense against parasitic helminthes (Chang and Dong, 2009; Kolls and Lindén, 2004; Pappu et al., 2008). Recently, it was shown that IL-25 utilized IL-17RA and IL-17RB as a receptor complex (Angkasekwinai et al., 2010; Rickel et al., 2008). IL-17RB is expressed in Th2 (Angkasekwinai et al., 2007; Wang et al., 2007) and Th9 (Angkasekwinai et al., 2010) cells as well as a subset of NKT cells (Terashima et al., 2008). IL-25 induces the production of IL-4, IL-5, IL-13, and IL-9 in these target cell types. IL-17RA, IL-17RC, and IL-17RB recruit Act1 (Chang et al., 2006; Claudio et al., 2009; Qian et al., 2007), an intracellular adaptor molecule sharing homology with the cytoplasmic domain of the IL-17R family (Novatchkova et al., 2003), to mediate downstream signaling.
Although IL-17 family members IL-17A, IL-17F, and IL-25 (as well as IL-17 receptor family members IL-17RA, IL-17RC, and IL-17RB) have been relatively well studied in immune responses and inflammatory diseases, the function of other IL-17 family cytokines and IL-17R-like molecules is unclear. IL-17C was identified along with IL-17B by a homology search of IL-17-like cytokines and reported to stimulate THP, a monocytic cell line, to produce TNF-α and IL-1β (Yamaguchi et al., 2007). Adenoviral delivery of IL-17C to lung triggers neutrophil recruitment (Hurst et al., 2002). IL-17C-overexpressing CD4+ T cells exacerbate collagen-induced arthritis in recipient mice (Yamaguchi et al., 2007). These results suggest that IL-17C may have an important proinflammatory function.
IL-17RE belongs to the IL-17R family and was originally identified based on searches for IL-17R homologous sequences (Li et al., 2006). The closest related molecule of IL-17RE is IL-17RC. IL-17RE shares 18% of amino acid sequence identity with IL-17RC and the two genes encoding these receptors are located adjacent to each other on human chromosome 3 and mouse chromosome 6. The Il17re gene encodes a protein carrying a single transmembrane domain with various short isoforms ranging from 34 to 70 kD. However, little is known on the specific ligands and signaling functions of these proteins. It was previously reported that IL-17RE has mitogenic activity in a ligand-independent manner (Li et al., 2006). A recent genome-wide association study revealed that IL17REL, a close homolog of IL17RE, is a risk locus for ulcerative colitis (Franke et al., 2010).
In this study, we have studied the function of IL-17C and identified IL-17RE as its receptor in regulation of Th17 cell differentiation and autoimmune diseases. IL-17C-IL-17RE signaling may be a promising target in the treatment of chronic autoimmune diseases.
RESULTS
IL-17C Is Necessary for Pathogenesis of EAE
As a first step to understand the function of IL-17C, we generated Il17c−/− mice by gene targeting. A LacZ-coding sequence was inserted into exon 2 to disrupt the expression of Il17c (see Figures S1A and S1B available online). These mice developed normally and did not exhibit any pronounced immunological deficiency. Normal development of lymphocytes was observed: CD4+ and CD8+ T cells in spleen and draining lymph nodes are comparable between WT (wild-type) and Il17c−/− mice (Figure S1C). Also, the number of regulatory T cells in Il17c−/− mice was similar to that in WT mice (Figure S1C). When splenocytes were restimulated with PMA plus ionomycin, the amount of IL-17A and IFN-γ production was comparable between Il17c−/− and WT CD4+ and CD8+ T cells (Figure S1D).
To explore the role of IL-17C in inflammatory diseases, we analyzed IL-17C expression in different tissues and found that its expression was increased by approximately 15-fold in the central nervous system (CNS) of mice induced with experimental autoimmune encephalomyelitis (EAE) compared to the normal CNS. This result suggests a role of this cytokine in autoimmune diseases. To examine whether IL-17C is involved in the development of autoimmune diseases, EAE was therefore induced in Il17c−/− mice along with appropriate WT controls. Il17c−/− mice exhibited a decrease in EAE severity, with substantial differences observed in peak clinical scores of all mice as well as peak score of those exhibiting clinical symptoms, though the kinetics of EAE onset did not appear to have been affected between the two groups (Figure 1A; Table 1). Additionally, only 50% of Il17c−/− mice exhibited EAE symptoms compared to 100% of the WT mice (Figure 1A).
Figure 1. IL-17C Is Necessary for Full Development of EAE.
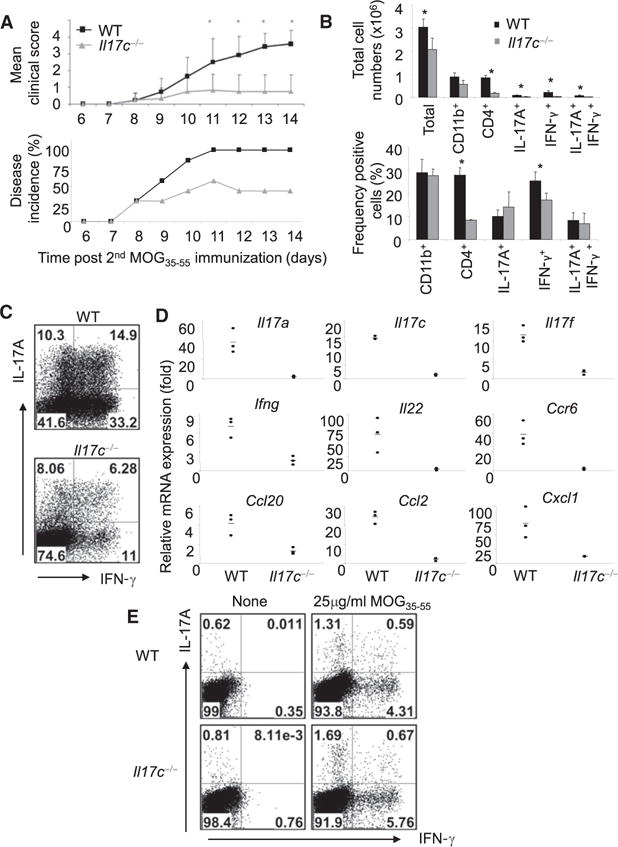
(A) EAE was induced in WT and Il17c−/− animals. Bottom graph y axis indicates ratios of animals developing EAE. Data are representative of four independent experiments.*p < 0.05 (Student’s t test).
(B) Total infiltrating CNS cell counts and percentages of infiltrating CD4+, CD11b+, IL-17A+, and IFN-γ+ cells.
(C) Representative CNS intracellular cytokine staining of WT and Il17c−/− mice after 5 hr PMA plus ionomycin restimulation.
(D) mRNA was analyzed in the CNS tissues of three individual mice per group on day 14 after EAE initiation by real-time RT-PCR. Gene expression data were normalized to actin (Actb).
(E) Splenic intracellular cytokine staining. Splenocytes derived from each group were restimulated with or without MOG35-55 peptide and intracellular cytokine staining was conducted and analyzed in CD4+ gate.
See also Figure S1.
Table 1.
MOG35-55 Induced EAE in Il17c−/− Mice
| Group | n | Day of Onset (mean ± SD) |
Peak Clinical Score (mean ± SD) |
Peak Clinical Score of Symptomatic Mice (mean ± SD) |
|---|---|---|---|---|
| WT | 16 | 8.44 ± 2.28 | 3.50 ± 1.211 | 3.50 ± 1.211 |
| Il17c−/− | 15 | 9.15 ± 3.05 | 1.833 ± 1.305* | 2.292 ± 1.010** |
EAE was induced in WT (C57BL/6 and C57BL/6 × 129 mixed) and Il17c−/− (C57BL/6 and C57BL/6 × 129 mixed backgrounds) by MOG35-55 peptide immunization. Student’s t tests were performed to compare peak clinical score of all mice in each experiment and also of only those that showed clinical symptoms of EAE.
p = 0.0009;
p = 0.0095. Data are a compilation of four independent experiments.
To analyze whether the protection by Il17c−/− mice against EAE development and severity was related to CD4+ T effector cell function, we next analyzed cell infiltration and cytokine expression in total CNS tissue in EAE mice. In the CNS, the numbers of CD4+ T cells were higher in WT mice than in Il17c−/− mice, whereas the numbers of CD11b+ macrophages were comparable between the two groups (Figure 1B). Moreover, WT CD4+ T cells in CNS contained higher numbers of IL-17A+, IFN-γ+, and IL-17A+IFN-γ + producers compared to CD4+ T cells derived from Il17c−/− animals (Figures 1B). The percentage of IFN-γ+ cells was higher in WT compared with the Il17c−/− CD4+ T cell population whereas the percentages of IL-17A+ or IL-17A+IFN-γ+ cells were not different (Figures 1B and 1C). We then directly examined cytokines and chemokines expressed at the sites of inflammation. IL-17C deficiency resulted in significant decreases in the Th17 cell-related mediators Il17a, Il17f, Il22, Ccr6, and Ccl20 (p < 0.05) (Figures 1D). Interestingly, we also observed a noticeable decrease in Ifng mRNA expression in the CNS of Il17c−/− mice. In addition to the CNS, splenocytes from EAE mice were restimulated with MOG35-55 peptide to analyze cytokine expression. WT and Il17c−/− CD4+ T cells were found to proliferate similarly (data not shown) and the cytokine response to MOG35-55 peptide was similar between WT and Il17c−/− mice by intracellular cytokine staining (Figure 1E). Thus, IL-17C appears to be necessary for the pathogenesis of EAE disease and for CNS inflammation in this model.
IL-17C Binds to IL-17RE
To identify the mechanism whereby IL-17C mediates proinflammatory function in EAE disease, we first searched for the receptor of IL-17C among IL-17R family members and the target cells for IL-17C. 293T cells were transfected with various IL-17R family members, and an IL-17C-hIg fusion protein we generated was used to detect the binding. Compared to cells transfected with an empty vector, substantial increases in binding of IL-17C-hIg was observed in 293T cells transfected with IL-17RE (Figure 2A). We did not detect any measurable binding of IL-17C-hIg to IL-17RA or other members of the IL-17 receptor family by the same system (Figure 2B). Expression of IL-17 receptor molecules was confirmed in this system via binding of positive control IL-17 cytokine fusion proteins and through real-time RT-PCR (Figure S2).
Figure 2. IL-17C Binds to IL-17RE In Vitro.
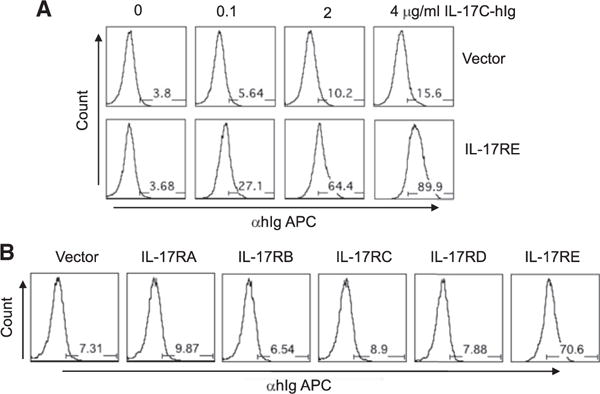
(A) Binding of IL-17C-Ig to IL-17RE. 293T cells were transfected with empty or IL-17RE expression vector (pcDNA3) and subsequently stained with IL-17C-hIg, followed by anti-hIg APC.
(B) IL-17C-hIg binds to IL-17RE but not to other IL-17R family. 293T cells were transfected with empty or IL-17R family expression vector (pcDNA3). Staining with IL-17C-hIg was performed same as (A).
See also Figure S2.
IL-17RE Is Highly Expressed in Th17 Cells
To understand how IL-17C-IL-17RE regulates pathogenesis of autoimmune disease, we further examined the expression of Il17re mRNA by RT-PCR. Based on the previous literature (Li et al., 2006), IL-17RE is predicted to have multiple isoforms. Il17re mRNA was detected in various cell types by RT-PCR but they mostly expressed an isoform lacking the transmembrane domain (data not shown). Interestingly, in our previous microarray analysis of different T cell subsets (Nurieva et al., 2008), we found Il17re mRNA to be upregulated in Th17 cells (data not shown). To further confirm this result, we isolated naive or natural regulatory T cells and prepared in vitro differentiated Th1, Th2, Th17, and inducible regulatory T cells (Figure S3). Among these subsets of CD4+ T cells, Th17 cells expressed the highest amount of full-length Il17re mRNA (Figure 3A). Because the development of Th17 cells requires transcription factors STAT3, RORγ, and RORα, we then asked whether expression of Il17re is dependent on these factors. Naive CD4+ T cells from Stat3−/−, Rorc−/−, and Rorasg/sg (Yang et al., 2007, 2008c) cultured in Th17 cell conditions expressed greatly reduced amounts of Il17re mRNA compared to their WT controls, indicating that they are necessary for Il17re expression (Figure 3B). Il17re was detected not only in in vitro cultured Th17 cells but also in in vivo derived Th17 cells. When IL-17F reporter mice, IL-17FRFP (Yang et al., 2008b), were induced with EAE, IL-17FRFP+CD4+ T cells isolated from spleen and CNS tissue expressed higher amounts of Il17re compared to that from IL-17FRFP− CD4+ T cells (data not shown). To identify what cytokine directs the expression of Il17re, we activated sorted naive CD4+ T cells in the presence of different cytokines. IL-6 and TGF-β synergistically regulate the expression of Il17re mRNA and IL-1 and IL-23 further enhanced this expression (Figure 3C). Expression of a full-length Il17re mRNA was also confirmed by RT-PCR in T cells treated with TGF-β and IL-6 (Figure 3D).
Figure 3. IL-17RE Is Highly Expressed in Th17 Cells.
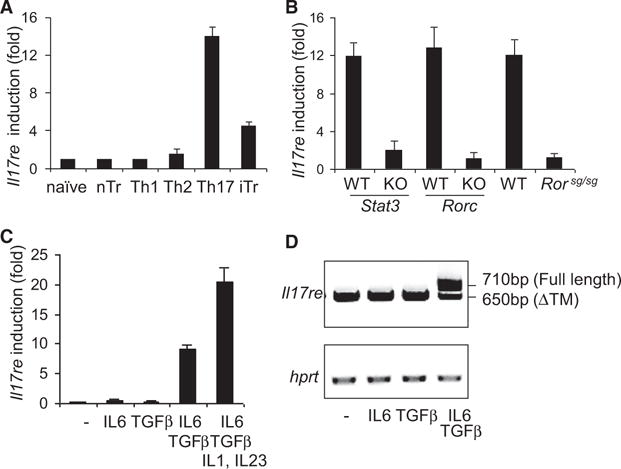
(A) Real-time RT-PCR of Il17re mRNA expression in sorted naive, nTreg, and activated CD4+ T cells differentiated under Th1, Th2, Th17, and iTreg cell conditions. Fold induction was calculated with naive cells as control and normalizing to β-actin (Actb) expression. The data are expressed as the mean ± SD of triplicate samples.
(B) Real-time RT-PCR analysis of Il17re expression in CD4+ T cells from WT, Stat3−/−, Rorc−/−, and Rorasg/sg mice. Naive CD4+ T cells from each strain were differentiated under Th17 cell conditions for 4 days.
(C) Real-time RT-PCR analysis of Il17re mRNA expression in T cells activated with the indicated cytokines for 4 days.
(D) RT-PCR analysis of Il17re in T cells activated with the indicated cytokines. The PCR products are amplified with forward primer before the transmembrane domain and reverse primer at the end of the coding sequence.
Data shown are representative of at least three independent experiments. See also Figure S3.
IL-17C-IL-17RE Regulates Th17 Cell Differentiation
Because full-length Il17re expression is selectively induced in Th17 cells, we asked whether it has any effect on the differentiation of Th17 cells. Th17 cells transduced with a retroviral vector encoding full-length IL-17RE but did not show enhanced IL-17A production compared to the control vector-infected cells (Figure 4A), suggesting that the ligand for this receptor may be absent in the culture. We therefore treated cells infected with retroviruses with different IL-17 cytokine family members and observed an enhanced production of IL-17A by intracellular cytokine staining when, and only when, the cells were cultured with IL-17C (Figure 4B). Moreover, we investigated whether IL-17C plays any direct role in Th1 cells. IL-17C did not regulate expression of IFN-γ in Th1 cells with or without IL-17RE overexpression (data not shown). Interestingly, Th17 cells cultured with IL-17C exhibited greater expression of Il17re mRNA compared to those without, suggesting that IL-17C may regulate IL-17RE expression (Figure S4A).
Figure 4. IL-17C-IL-17RE Regulates Th17 Cell Differentiation.
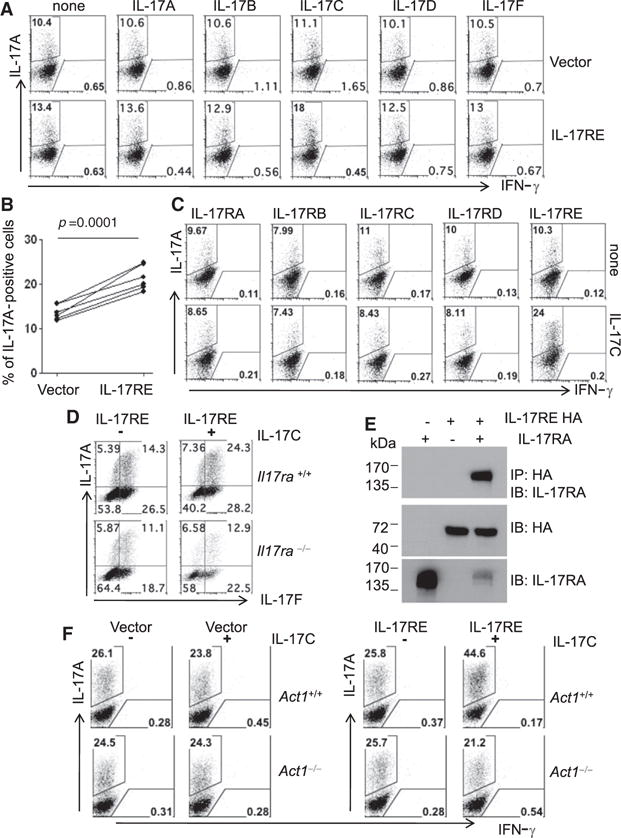
(A) Intracellular cytokine staining of IL-17A and IFN-γ. After naive CD4+ T cells were retrovirally transduced with control viral vector or IL-17RE, different IL-17 family cytokines were added during Th17 cell differentiation. The cells were restimulated with PMA and ionomycin for cytokine staining.
(B) Statistical analysis of IL-17A-positive cells.
(C) IL-17C induced IL-17A expression in Th17 cells over-expressing IL-17RE but not other IL-17R family members. CD4+ T cells were retrovirally transduced with each IL-17R-IRES-GFP and were cultured with IL-17C in Th17 cell polarizing condition. Cells were restimulated with PMA and ionomycin and GFP-positive cells were gated to show expression of IL-17A and IFN-γ.
(D) Intracellular cytokine staining of WT and Il17ra−/− T cells retrovirally transduced with IL-17RE and cultured with IL-17C for 2 days, followed by 5 hr of PMA and ionomycin. Data shown are gated GFP+ cells. Data shown are representative of at least three experiments.
(E) Coimmunoprecipitation of IL-17RA with HA-tagged IL-17RE. 293T cells were transfected with IL-17RA or HA-tagged IL-17RE or both and immunoprecipitated with anti-HA.
(F) Intracellular cytokine staining of WT and Act1−/− T cells retrovirally transduced with IL-17RE or control viral vector (RV-GFP) and followed by 5 hr of PMA and ionomycin. Data shown are gated GFP+ cells. Data shown are representative of at least three experiments.
See also Figure S4.
Our above results suggest that IL-17RE is a receptor for IL-17C but did not answer whether there could be another receptor although binding of IL-17C was restricted to IL-17RE in vitro. We therefore overexpressed various members of the IL-17 receptor family (IL-17RA through IL-17RD) in developing Th17 cells by retroviral infection and examined the effect of IL-17C in the induction of IL-17A. IL-17C enhanced IL-17A production in Th17 cells retrovirally transduced only with IL-17RE, not with other IL-17 receptor family members (Figure 4C). This observation further supports the principle that IL-17C signals via IL-17RE in Th17 cells.
In support of the above conclusion, we investigated whether IL-17C-hIg fusion protein associates with Th17 cells in vivo. Mononuclear cells from CNS of EAE mice were separated by percoll and stimulated with PMA and ionomycin. 50%–70% of IL-17A-positive cells from the CNS were CD4+ T cells and this population stained positively with IL-17C-hIg fusion protein (Figure S4B). We also found that non-CD4+ T cells, which produce IL-17A in the CNS, are positively stained with IL-17C-hIg fusion protein although the staining intensity is weaker than that of CD4+ T cells. γδ T cells are known to produce IL-17A in the CNS during EAE development (Sutton et al., 2009). When we analyzed Il17re expression from γδ T cells, we found that γδ T cells express higher amounts of Il17re than naive T cells. Furthermore, sorted IL-17FRFP+ γδ T cells express higher amounts of Il17re in comparison to IL-17FRFP− γδ T cells, indicating that IL-17A- or IL-17F-producing γδ cells also express IL-17RE. However, when we treated isolated γδ T cells with IL-17C, we did not observe any difference in cytokine production by intracellular cytokine staining, although IL-23 was able to induce IL-17A expression from γδ T cells (data not shown).
Taken together, these results indicate that IL-17C signals via IL-17RE in Th17 cells.
IL-17RE and IL-17RA Form a Signaling Complex for IL-17C and Employ Act1 Adaptor Protein
Because IL-17RA forms a complex with IL-17RC for IL-17A and IL-17F signaling (Toy et al., 2006) and with IL-17RB for IL-25 signaling (Rickel et al., 2008), we asked whether IL-17RA is required for the activity of IL-17C and IL-17RE. When naive T cells from WT and Il17ra−/− mice were transduced with an IL-17RE retroviral vector and differentiated into Th17 cells, the effect of IL-17C in inducing IL-17A was severely impaired in Il17ra−/− cells (Figure 4F). This observation prompted us to examine the physical interaction between IL-17RA and IL-17RE. When IL-17RA- and hemagglutinin (HA)-tagged IL-17RE were expressed simultaneously in 293T cells, IL-17RA coimmunoprecipitated with IL-17RE, suggesting the association of IL-17RA and IL-17RE as a signaling complex for IL-17C (Figure 4E).
IL-17RA recruits a membrane proximal adaptor protein, Act1, to mediate IL-17A, IL-17F, and IL-25 signaling (Chang et al., 2006; Claudio et al., 2009; Qian et al., 2007). In addition, IL-17RE also bears the SEFIR (similar expression to fibroblast growth factor genes and IL-17Rs) motif (Novatchkova et al., 2003), which may interact with Act1 via a homotypic interaction. Therefore, we tested whether Act1 is required for IL-17C-IL-17RE signaling with an Act1−/− mouse. Upon the addition of IL-17C in Th17 cell-polarizing conditions, IL-17RE-overexpressing Th17 cells express higher amounts of IL-17A than cells with vector alone. This effect, however, is abrogated in Act1−/− Th17 cells (Figure 4F). Act1−/− CD4+ T cells undergo normal Th17 cell development without IL-17C, suggesting no intrinsic defect in these cells (Figure 4F). Therefore, we concluded that Act1 is necessary for IL-17C-IL-17RE signaling.
IL-17C-IL-17RE Regulates IκBζ Expression
To further understand the mechanism whereby IL-17C and IL-17RE enhance Th17 cell differentiation, we further examined cytokine production and gene expression profiles of IL-17RE-transduced T cells. After T cells were retrovirally transduced with IL-17RE or empty expression vector and cultured with TGF-β and IL-6 in the presence or absence of IL-17C for 2 days, we sorted out infected cells and stimulated them with anti-CD3 for assessment of cytokine production by ELISA and gene expression profiles by real-time RT-PCR. T cells transduced with IL-17RE and cultured in the presence of IL-17C expressed higher amounts of not only IL-17A but also IL-17F and IL-22 (Figures 5A and 5B). However, the expression of Rora, Rorc, and Il23r remained similar in IL-17RE-overexpressing T cells with or without IL-17C treatment (Figure 5B).
Figure 5. IL-17C-IL-17RE Regulates IκBζ Expression.
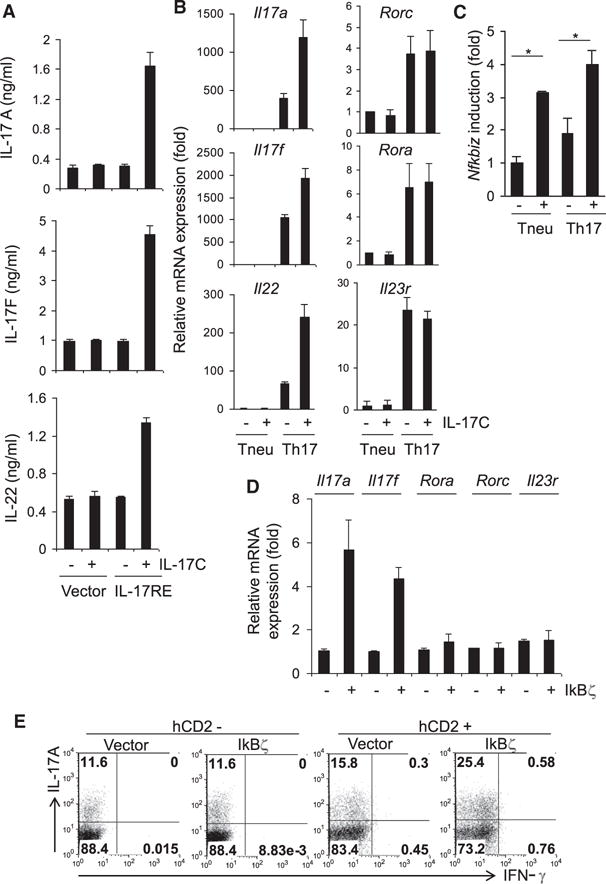
(A) ELISA of cytokine production in CD4+ T cells retrovirally transduced with IL-17RE or control viral vector and treated with IL-17C for 2 days in Th17 cell condition. Sorted GFP+ cells were activated with anti-CD3 for 24 hr.
(B) Real-time RT-PCR analysis of cytokine and transcription factor expression in CD4+ T cells retrovirally transduced and cultured either in neutral or Th17 cell conditions. The cells were activated with anti-CD3 for 4 hr for cytokine mRNA detection.
(C) Real-time RT-PCR analysis of Nfkbiz prepared as above. *p < 0.05 (Student’s t test).
(D) Real-time RT-PCR analysis of cytokine and transcription factor in CD4+ T cells retrovirally transduced with hCD2- IκBζ or control viral vector (PMIG- hCD2). Sorted hCD2+ cells were activated with anti-CD3 for 4 hr.
(E) Intracellular IL-17A and IFN-γ staining in CD4+ T cells retrovirally transduced with IκBζ or control viral vector and cultured in Th17 cell condition.
Data shown are representative of at least three independent experiments.
See also Figure S5.
Because IL-17C seems to employ IL-17RA for its signaling, we tested whether the transcription factors that are induced by IL-17RA-dependent IL-17A or IL-17F stimulation, C/EBPβ, C/EBPδ, and IκBζ (encoded by the Nfkbiz gene) (Shen et al., 2005; Yang et al., 2008a), could be induced by IL-17C. Although the expression of Cebpb and Cebpd remained unchanged (Figure S5), Nfkbiz expression was increased in cells treated with IL-17C, even when T cells were activated under neutral conditions and Th17 cell differentiation did not occur (Figure 5C), suggesting that IκBζ is a direct target of IL-17RE signaling.
Similar to a recent report (Okamoto et al., 2010), when we transduced T cells with a retroviral vector expressing IκBζ and cultured these cells in Th17 cell conditions, we found that IκBζ -overexpressing Th17 cells produced enhanced expression of IL-17A compared to controls (Figure 5E). Also, they expressed higher amounts of mRNA for Il17a and Il17f while expressing similar amounts of Rora, Rorc, and Il23r compared to control cells (Figure 5D). Therefore, IκBζ may be a downstream transcription factor in Th17 cells induced by IL-17C and IL-17RE. Because IκBζ has been recently shown to be important in Th17 cell differentiation and autoimmune disease, our study identifies a physiological regulator of its expression.
Enhanced IL-17RE Signaling In Vivo Leads to Greater Susceptibility to EAE
To confirm our in vitro data and further demonstrate the function of IL-17RE in T cells in vivo, we generated transgenic IL-17RE mice by using the CD4 minigene. Insertion of a full-length cDNA into this transgenic cassette that contains the CD4 promoter, enhancer, and first intron without the silencer results in transgene expression in all T cells (Sawada et al., 1994, Angkasekwinai et al., 2010). Of four founder lines, three were transmitted to progeny. We compared the expression of Il17re mRNA in naive CD4+ T cells from these transgenic mice to that in their control littermates (Figure 6A). Lines 1 and 4, expressing enhanced amounts of Il17re mRNA, were selected for further characterization. The CD4-IL-17RE transgenic mice showed normal populations of T and B cells in lymphoid organs (data not shown). When naive CD4+ T cells were isolated and cultured in Th17 cell polarizing condition in the presence of IL-17C, CD4+ T cells from IL-17RE transgenic mice produced enhanced amounts of IL-17A (Figure 6B).
Figure 6. CD4-IL-17RE Transgenic Mice Develop Exacerbated EAE Symptoms and Enhanced IκBζ Expression in CNS.
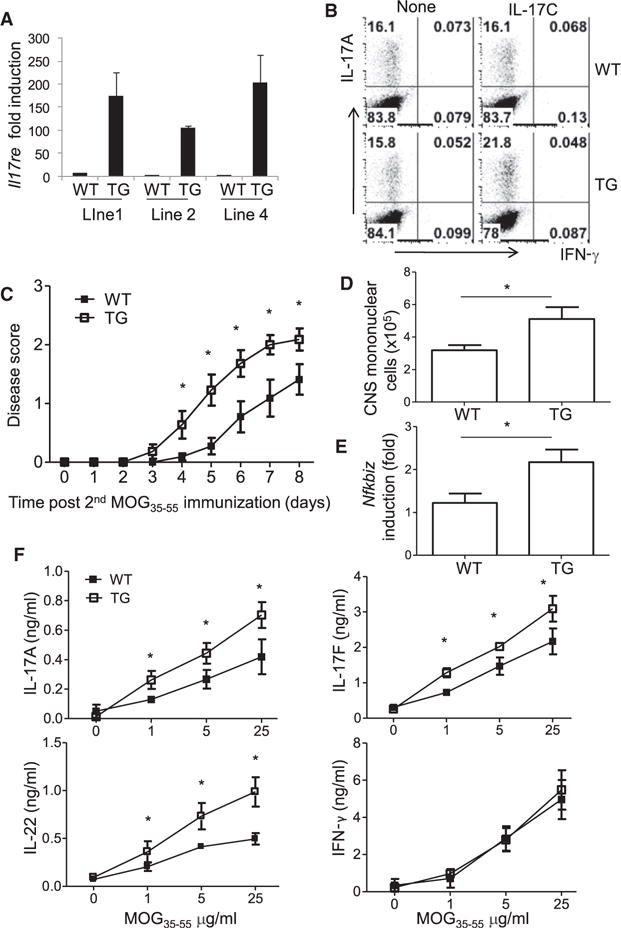
(A) Real-time RT-PCR analysis of Il17re mRNA expression in naive T cells isolated from WT or different lines of transgenic mice.
(B) Frequency of IL-17A- and IFN-γ-producing cells in naive T cells isolated from WT or CD4-IL-17RE (TG) mice and activated with anti-CD3, anti-CD28, TGF-β, and IL-6 in the presence or absence of IL-17C for 4 days, followed by 5 hr of PMA and ionomycin stimulation in the presence of protein transport inhibitor (BD GolgiStop).
(C) EAE was induced in WT and TG animals. Data represent the mean ± SD of n = 11 (WT) and n = 12 (TG) from three independent experiments.
(D) Total number of mononuclear cells recovered from CNS.
(E) Real-time RT-PCR analysis of Nfkbiz mRNA expression. Data represent the mean ± SD from two independent experiments. *p < 0.05 (Student’s t test).
(F) ELISA of cytokine production from splenocytes expanded with MOG35-55 peptide for 3 days. All comparisons presented were statistically significant (p < 0.05).
We next analyzed CD4-IL-17RE transgenic mice with an EAE model. CD4-IL-17RE transgenic or control littermate mice were immunized with MOG35-55 to induce EAE. When we reduced the concentration of complete Freund’s adjuvant (CFA) from 5 to 3 mg/ml during the second MOG immunization so that WT mice did not develop robust EAE, CD4-IL-17RE transgenic mice exhibited a moderate increase in EAE symptoms (Figure 6C) associated with an increase in total numbers of infiltrated mononuclear cells in CNS (Figure 6D). At the peak of disease, infiltrated leukocytes in CNS were isolated and subjected to RT-PCR analysis. Whereas other Th17 cell transcription factors remain similar between two groups of animals (data not shown), we observed about 2-fold induction of Nfkbiz in CNS-infiltrating cells in CD4-IL-17RE transgenic mice compared to littermate mice (Figure 6E). In these EAE mice, MOG35-55-specific Th17 cytokine expression was moderately increased in transgenic mice although proliferation and IL-2 and IFN-γ production remained similar between two groups (Figure 6F). Therefore, enhanced signaling of IL-17RE in CD4+ T cells leads to greater EAE disease as well as enhanced Th17 cell response and increased expression of IκBζ.
IL-17C-IL-17RE Enhanced Cytokine Expression by Effector Th17 Cells
Because IL-17RE was induced after the differentiation of Th17 cells and IL-17C expression was detected in inflamed tissues such as CNS in EAE, we investigated whether IL-17C has any role to promote Th17 cytokine production in effector Th17 cells. To this end, naive CD4+ T cells from IL-17FRFP mice (Yang et al., 2008b) were differentiated with TGF-β and IL-6. At day 5, RFP-positive cells were sorted (Figure 7A) and cultured in the presence of increasing amounts of anti-CD3 for 3 days. Increased production of IL-17A was observed when IL-17C was added into the culture where anti-CD3 concentration was between 0.06 and 0.25 μg/ml (Figure 7B), suggesting that IL-17C synergizes with TCR signaling to potentiate IL-17A production. In a different setting, polarized Th17 cells were cultured in the presence of irradiated splenocytes. Similarly, we observed enhanced IL-17A and IL-17F production from the effector Th17 cells in an IL-17C concentration-dependent manner (Figure 7C). Therefore, IL-17C enhances Th17 cytokine production in effector Th17 cells. Interestingly, we found that expression of Il17c is much more robustly induced in inflamed CNS lesions compared to the T cell priming site spleen from EAE mice (Figure 7D). Therefore, it is likely that the effector Th17 cells migrate to CNS and upon the encounter with IL-17C in local tissues, Th17 cells secrete enhanced amounts of effector cytokines.
Figure 7. IL-17C-IL-17RE Enhanced Polarized Th17 Cell Response.
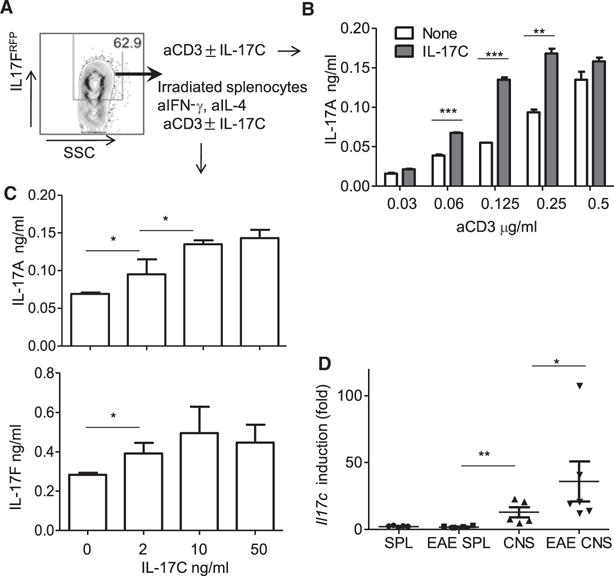
(A) Representative flow cytometry plot of Th17 cells from IL-17FRFP mice polarized with TGF-β and IL-6 at day 5.
(B) ELISA of IL-17A production in polarized Th17 cells with or without IL-17C in the presence of increasing concentration of αCD3.
(C) ELISA of cytokine production in polarized Th17 cells cultured with irradiated splenocytes in the presence of increasing concentration of IL-17C.
(D) Real-time RT-PCR analysis of Il17c in CNS and spleen from normal and EAE mice.
All comparisons presented were statistically significant (***p < 0.001, **p < 0.01, *p < 0.05).
DISCUSSION
In the current study, we have found that a cytokine-cytokine receptor pair, IL-17C and IL-17RE, regulates inflammatory disease and promotes Th17 cell differentiation (via signaling components IL-17RA and Act1) and induction of IκBζ.
Since the discovery of the cytokine IL-17A and its receptor, IL-17RA, many molecules that bear homology to IL-17A and IL-17RA have been reported. Now, the IL-17 cytokine family is composed of six members (IL-17A, IL-17B, IL-17C, IL-17D, IL-17F, and IL-25). The IL-17R family has also been extended to five members (IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE). The IL-17 cytokine family is evolutionally conserved. The S. purpuratus genome sequence contains 30 IL-17-like genes as the most diverse gene family among cytokines and growth factors (Hibino et al., 2006). Therefore, elucidating the biological roles and regulation of IL-17 and IL-17R family members are critical to understand inflammatory responses in host defense against infections and in autoimmunity. The least-understood members of IL-17 cytokine family are IL-17B, IL-17C, and IL-17D. Although some in vitro analyses indicate that they may promote inflammation, genetic approaches are needed to provide a better answer to the biological function of molecules.
Il17c−/− mice developed normally but were found to be resistant to EAE, indicating its role as a proinflammatory cytokine. To understand which cells are the targets of IL-17C and how IL-17C functions in vivo to promote inflammation, we searched for its receptor and identified IL-17RE as its receptor. IL-17RE is induced in a subset of differentiated CD4+ T cells, Th17 cells. mRNA of a shorter isoform lacking the transmembrane domain of IL-17RE was also detected in other types of CD4+ T cells as well as other, non-CD4+ cell types. It is unknown at this stage whether the shorter isoform is secreted because of a lack of trans-membrane domain or whether it has any biological function.
We show that IL-17C promotes the development of Th17 cells. Intriguingly, IL-17RE partners with IL-17RA to be an effective signaling receptor for IL-17C. IL-17RA interacts with IL-17RC to provide signaling for IL-17A and IL-17F to promote tissue inflammation, mostly by nonhematopoietic cells such as fibroblasts and epithelial cells. Whereas IL-17RA is expressed universally, the role of IL-17RA in hematopoietic cells is less understood. We recently reported that IL-17RA interacts with IL-17RB in CD4+ T cells and that the IL-17RA-IL-17RB signaling pathway is essential in the induction of Th2 and Th9 cells in response to IL-25 (Angkasekwinai et al., 2010). Our current study shows that another IL-17 receptor family, IL-17RE, can interact with IL-17RA and that this pair is functioning in Th17 cells. Additionally, we report here that Act1, a membrane-proximal adaptor molecule, is required for IL-17C signaling. Activation of the IL-17RA-RE and Act1 complex by IL-17C leads to induction of IκBζ. IκBζ was recently reported to be a positive regulator of Th17 cells and is indispensable for the development of EAE. IκBζ expression can be induced upon the activation of IL-17RA in fibroblasts or epithelial cells in response to IL-17A and IL-17F. Although the signaling mechanisms of IL-17C-IL-17RE are yet to be determined in detail, overlap of the IL-17C signaling pathway with other IL-17 cytokine family members may exist because they share a common receptor, IL-17RA. IL-25, on the other hand, utilizes IL-17RA-IL-17RB and Act1 but does not induce IκBζ in differentiated Th2 cells (data not shown).
Although we showed that IL-17C is necessary for EAE, we did not find any effect on Th17 cell generation in Il17c−/− mice. IL-17C does not directly regulate Th1 cell differentiation or function, as shown by the fact that IL-17RE is not upregulated during Th1 cell differentiation and IL-17C did not have any effect on Th1 cell differentiation, even when IL-17RE was overexpressed in these cells. In contrast, IL-17RE is upregulated in Th17 cells after their differentiation. These data suggest a model whereby IL-17C-IL-17RE regulates effector Th17 cell function in the inflamed tissues. This model is supported by our following results: (1) IL-17C is upregulated in CNS but not in spleen during EAE; (2) IL-17RE is not expressed by naive T cells but upregulated in effector Th17 cells; (3) effector Th17 but not naive T cells respond to IL-17C by upregulating IL-17A and IL-17F production; and (4) Th17 cytokine and downstream chemokine production was inhibited in CNS of EAE mice. All these results support a direct regulation of IL-17C on effector Th17 cells. However, it is possible that there are additional cell types regulated by IL-17C in EAE. Additional experiments via selective ablation of IL-17RE will conclusively resolve this issue.
The expression, regulation, and role of IL-17C and IL-17RE in the immune system deserve further exploration. Nonetheless, our study has provided insight into the physiological role of IL-17C in autoimmune disease, owing, at least in part, to its ability in promote Th17 cell development by interacting with its receptor, IL-17RE.
EXPERIMENTAL PROCEDURES
Induction of EAE
C57BL/6 mice were purchased from Jackson Laboratories. Mice were housed in the specific-pathogen-free (SPF) animal facility at M.D. Anderson Cancer Center, and the animal experiments were performed at the age of 6–10 weeks with protocols approved by the Institutional Animal Care and Use Committee. EAE disease was induced with MOG35-55/CFA as previously described (Reynolds et al., 2010). Clinical scores were as follows: 0, no disease; 1, tail paralysis; 2, wobbly gait; 3, hind limb paralysis; 4, forelimb paralysis; 5, moribund or dead. 0.5 gradations were assigned to animals exhibiting symptoms falling between two of the above listed scores.
CNS mRNA Analysis
A section of brain and spinal cord was taken from the same location of each mouse after perfusion on day 14 after EAE induction. Tissues were homogenized and mRNA was isolated with Trizol reagent (Invitrogen). cDNA was synthesized as described above. Real-time RT-PCR was performed with duplicate samples via previously described primers with β-actin as a reference gene (Reynolds et al., 2010).
Retroviral Transduction and Th Cell Differentiation
pGFP-RV-IL-17RE (GenBank accession number NM_145826) was generated. The plasmid pGFP-RV contains an internal ribosomal entry site (IRES)-regulated green fluorescent protein gene (IRES-GFP). Naive CD4+CD25− CD44loCD62Lhi T cells from C57BL/6 mice, Il17ra−/− (provided by Amgen), Act1−/− mice were sorted by flow cytometry and activated with late-bound anti-CD3 (0.5 μg/ml; 2C11; BioXcell) plus soluble anti-CD28 (0.5 μg/ml; 37.51, BioXcell) in the presence of anti-IFN-γ (XMG 1.2 [BE0055], BioXcell) and 10 μg/ml anti-IL-4 (11B11 [BE0045]; BioXcell). 36 hr after activation, cells were spin-infected with retrovirus expressing IL-17RE or control empty vector. After 4 hr of infection, medium was replaced with and without IL-17C either in neutral condition with anti-IL-4 and anti-IFN-γ or Th17 cell condition medium (10 ng/ml IL-6 [216-16; Peprotech], 2 ng/ml TGF-β). Also, different IL-17 family cytokines (IL-17A, IL-17B, IL-17D, IL-17F [R&D]) were added to culture after retroviral infection. For IκBζ overexpression, Nfkbiz (GenBank accession number NM_030612) was cloned into retroviral vector, hCD2-pMIGR2. Two days after infection, GFP+ or hCD2+ cells were sorted by flow cytometry and restimulated with plate-bound anti-CD3 (1 μg/ml) for 4 hr, and cells were then collected for RNA extraction. For cytokine measurement by ELISA, culture supernatants were collected at 24 hr. For intracellular cytokine analysis, cells were restimulated with 500 ng/ml of ionomycin and 50 ng/ml of PMA in the presence of Golgi Stop (BD Phar-Mingen) for 5 hr. Cells were then permeabilized with Cytofix/Cytoperm Kit (BD PharMingen) and analyzed for the expression of IL-17A or IFN-γ (BD PharMingen). Th cell differentiation to Th1, Th2, and iTreg cells was described previously (Yang et al., 2008c).
Expression of IL-17C-Ig Protein and Binding to IL-17RE
IL-17C-hIg protein was generated by PCR amplification of a sequence coding aa 15–194 (GenBank accession number NM_145834), then cloned into the DES-Ig vector. IL-17C-hIg expression vector was stably transfected into Drosophila S2 cells, and the secreted IL-17C-hIg fusion protein was purified with a protein A column. For detection of IL-17RE binding, 293 cells were transfected with full length of IL-17RE or empty vector (pcDNA). IL-17C-hIg fusion protein or control human IgG1 was added to the transfected 293 cells, followed by anti-human Ig conjugated with APC. Before staining, cells were preblocked with human IgG1 (Sigma-Aldrich).
Immunoprecipitation
293T cells expressing HA-IL-17RE and/or IL-17RA were washed once with ice-cold PBS and lysed in lysis buffer. Lysates were used for immunoblot or immunoprecipitation. For immunoprecipitation, cleared cell lysates were incubated with HA antibody for 120 min, followed by four washes in lysis buffer. Immunoprecipitates were denatured with 2 × SDS sample buffer before SDS-PAGE. Protein transfer was followed by overnight incubation with anti-HA or anti-IL-17RA (R&D). After incubation with HRP-conjugated secondary antibody, the signal was detected with ECL reagent (Promega).
RT-PCR Analysis
Total RNA extracted by Trizol reagent (Invitrogen) was used to generate cDNA via oligo(dT), random hexamers, and MMLV reverse transcriptase (Invitrogen). For quantitation of cytokine, cDNA samples were amplified in IQ SYRB Green Supermix (Bio-Rad Laboratories). The data were normalized to actin as a reference gene. The primer pairs for real-time RT-PCR analysis of Il17a, IL17f, IL-22, Rora, Rorc, Il23r, and Nfkbiz (Shen et al., 2005; Yang et al., 2008c) were previously described. Primer pair for detection of Il17re is (forward) 5′-CAGTCCCAGTGACGCTAGAC-3′ and (reverse) 5′-ACCCACTA GAGCGGTGAGAG-3′. Primer pair for RT-PCR analysis to detect full-length Il17re and Il17re lacking transmembrane domain is (forward) 5′-ATGTCC ATTTTGCCTGGAAG-3′ and (reverse) 5′-CTACAGACAGCTGAAACCACA-3′. Primer pair for detection of Il17c is (forward) 5′-GCTCCTCCACACCTGC TAAC-3′ and (reverse) 5′-CTGTGGGTAGCGGTTCTCAT-3′.
Generation of CD4-IL-17RE Transgenic Mice
A cDNA encoding full-length mouse IL-17RE was cloned into SalI site of the CD4 minigene plasmid, which contains CD4 promoter/enhancer and the first intron without the silencer element (a gift from D. Littman). The IL-17RE transgene construct was digested with NotI and microinjected into C57BL/6 mice at the Transgenic Mouse Facility at MD Anderson Cancer Center. Subsequent screening of CD4-IL-17RE transgenic mice was carried out by genomic PCR with the following primers: forward, 5′-GCCTTGGCCTCCTAGCTACT-3′; reverse, 5′-CTTCCTCTGGAAGGATGCTG-3′.
Generation of Il17c Gene-Targeted Mice
Il17c−/− mice were generated by introducing a LacZ reporter cassette into the Il17c locus in the 129/TC1 embryonic stem cell line. The targeting vector contained puromycin as a positive selection marker and the diphtheria toxin A gene as a negative selection marker. Targeted ES clones were selected and injected into C57BL/6 blastocysts to generate chimeras. High-percentage chimeras were bred with female CMV-Cre transgenic mice to obtain Il17c+/− mice. Heterozygous mice were then bred to obtain homozygous knockout mice and WT littermates for experiments. The genotyping primers for Il17c−/− mice were as follows: forward, 5′-GCCTATTTGCCCACCTACAA-3′; reverse 1, 5′-AGTGTCAGCTTCCAGCACCT-3′; reverse 2, 5′-AAATTCAGAC GGCAAACGAC-3′. The forward primer and the reverse 1 primer amplify a 410 bp WT band, whereas the forward and reverse 2 primers (located in the LacZ region) give a 650 bp knockout band.
Act1 Gene-Targeted Mice
A new Act1−/− mouse generated by gene trap mutagenesis was obtained from Texas Institute for Genomic Medicine. Mouse genomic sequence surrounding the gene trap insertion site identified in the C57BL/6 gene trap ES cell clone IST11484G7 was Traf3ip2. The genotyping primers for Act1−/− mice were as follows: forward 1, 5′-TGCTCACCTGGAATATGCTG-3′; reverse 1, 5′-TTGTTTGGTGGGGGTACAGT-3′; reverse 2, 5′-CCAATAAACCCTCTTGCAGTTGC-3′. The forward 1 and reverse 1 primers, surrounding LTR region of gene trap vector, amplify a 485 bp WT band, whereas the forward 1 and reverse 2 primers, flanking the deleted exons, give a 300 bp knockout band.
Supplementary Material
Acknowledgments
We thank MD Anderson Genetic Engineering Mouse Facility for their assistance in generation of Il17c gene-targeted animals, K. Murphy for RV-GFP vector, D. Littman for the CD4 minigene construct, and the C.D. lab members for their help. The work is supported by research grants from NIH (to C.D.). J.M.R. receives an NCI training grant and C.D. receives a Research Trust Fellowship, is Olga and Harry Wiess Distinguished University Chair in Cancer Research of the University of Texas MD Anderson Cancer Center, and is a Leukemia and Lymphoma Society Scholar.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures and can be found with this article online at doi:10.1016/j.immuni.2011.09.010.
References
- Angkasekwinai P, Park H, Wang YH, Wang YH, Chang SH, Corry DB, Liu YJ, Zhu Z, Dong C. Interleukin 25 promotes the initiation of proallergic type 2 responses. J Exp Med. 2007;204:1509–1517. doi: 10.1084/jem.20061675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angkasekwinai P, Chang SH, Thapa M, Watarai H, Dong C. Regulation of IL-9 expression by IL-25 signaling. Nat Immunol. 2010;11:250–256. doi: 10.1038/ni.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- Chang SH, Dong C. IL-17F: regulation, signaling and function in inflammation. Cytokine. 2009;46:7–11. doi: 10.1016/j.cyto.2008.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–587. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudio E, Sønder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, et al. The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J Immunol. 2009;182:1617–1630. doi: 10.4049/jimmunol.182.3.1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C. Regulation and pro-inflammatory function of interleukin-17 family cytokines. Immunol Rev. 2008;226:80–86. doi: 10.1111/j.1600-065X.2008.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke A, Balschun T, Sina C, Ellinghaus D, Häsler R, Mayr G, Albrecht M, Wittig M, Buchert E, Nikolaus S, et al. IBSEN study group Genome-wide association study for ulcerative colitis identifies risk loci at 7q22 and 22q13 (IL17REL) Nat Genet. 2010;42:292–294. doi: 10.1038/ng.553. [DOI] [PubMed] [Google Scholar]
- Gaffen SL. An overview of IL-17 function and signaling. Cytokine. 2008;43:402–407. doi: 10.1016/j.cyto.2008.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino T, Loza-Coll M, Messier C, Majeske AJ, Cohen AH, Terwilliger DP, Buckley KM, Brockton V, Nair SV, Berney K, et al. The immune gene repertoire encoded in the purple sea urchin genome. Dev Biol. 2006;300:349–365. doi: 10.1016/j.ydbio.2006.08.065. [DOI] [PubMed] [Google Scholar]
- Hu Y, Ota N, Peng I, Refino CJ, Danilenko DM, Caplazi P, Ouyang W. IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol. 2010;184:4307–4316. doi: 10.4049/jimmunol.0903614. [DOI] [PubMed] [Google Scholar]
- Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, Menon S, Seymour B, Jackson C, Kung TT, et al. New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol. 2002;169:443–453. doi: 10.4049/jimmunol.169.1.443. [DOI] [PubMed] [Google Scholar]
- Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell. 2006;126:1121–1133. doi: 10.1016/j.cell.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity. 2004;21:467–476. doi: 10.1016/j.immuni.2004.08.018. [DOI] [PubMed] [Google Scholar]
- Korn T, Bettelli E, Gao W, Awasthi A, Jäger A, Strom TB, Oukka M, Kuchroo VK. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 2007;448:484–487. doi: 10.1038/nature05970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K, Harder B, Okada S, Ostrander CD, Kreindler JL, et al. Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J Immunol. 2007;179:5462–5473. doi: 10.4049/jimmunol.179.8.5462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med. 2005;201:233–240. doi: 10.1084/jem.20041257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li TS, Li XN, Chang ZJ, Fu XY, Liu L. Identification and functional characterization of a novel interleukin 17 receptor: a possible mitogenic activation through ras/mitogen-activated protein kinase signaling pathway. Cell Signal. 2006;18:1287–1298. doi: 10.1016/j.cellsig.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- Novatchkova M, Leibbrandt A, Werzowa J, Neubüser A, Eisenhaber F. The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci. 2003;28:226–229. doi: 10.1016/S0968-0004(03)00067-7. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature. 2007;448:480–483. doi: 10.1038/nature05969. [DOI] [PubMed] [Google Scholar]
- Nurieva RI, Chung Y, Hwang D, Yang XO, Kang HS, Ma L, Wang YH, Watowich SS, Jetten AM, Tian Q, Dong C. Generation of T follicular helper cells is mediated by interleukin-21 but independent of T helper 1, 2, or 17 cell lineages. Immunity. 2008;29:138–149. doi: 10.1016/j.immuni.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto K, Iwai Y, Oh-Hora M, Yamamoto M, Morio T, Aoki K, Ohya K, Jetten AM, Akira S, Muta T, Takayanagi H. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature. 2010;464:1381–1385. doi: 10.1038/nature08922. [DOI] [PubMed] [Google Scholar]
- Pappu BP, Angkasekwinai P, Dong C. Regulatory mechanisms of helper T cell differentiation: new lessons learned from interleukin 17 family cytokines. Pharmacol Ther. 2008;117:374–384. doi: 10.1016/j.pharmthera.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- Reynolds JM, Pappu BP, Peng J, Martinez GJ, Zhang Y, Chung Y, Ma L, Yang XO, Nurieva RI, Tian Q, Dong C. Toll-like receptor 2 signaling in CD4(+) T lymphocytes promotes T helper 17 responses and regulates the pathogenesis of autoimmune disease. Immunity. 2010;32:692–702. doi: 10.1016/j.immuni.2010.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, Anders PM, Tocker JE, Comeau MR, Budelsky AL. Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol. 2008;181:4299–4310. doi: 10.4049/jimmunol.181.6.4299. [DOI] [PubMed] [Google Scholar]
- Sawada S, Scarborough JD, Killeen N, Littman DR. A lineage-specific transcriptional silencer regulates CD4 gene expression during T lymphocyte development. Cell. 1994;77:917–929. doi: 10.1016/0092-8674(94)90140-6. [DOI] [PubMed] [Google Scholar]
- Shen F, Ruddy MJ, Plamondon P, Gaffen SL. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J Leukoc Biol. 2005;77:388–399. doi: 10.1189/jlb.0904490. [DOI] [PubMed] [Google Scholar]
- Sutton CE, Lalor SJ, Sweeney CM, Brereton CF, Lavelle EC, Mills KH. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity. 2009;31:331–341. doi: 10.1016/j.immuni.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Terashima A, Watarai H, Inoue S, Sekine E, Nakagawa R, Hase K, Iwamura C, Nakajima H, Nakayama T, Taniguchi M. A novel subset of mouse NKT cells bearing the IL-17 receptor B responds to IL-25 and contributes to airway hyperreactivity. J Exp Med. 2008;205:2727–2733. doi: 10.1084/jem.20080698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J. Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol. 2006;177:36–39. doi: 10.4049/jimmunol.177.1.36. [DOI] [PubMed] [Google Scholar]
- Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Wang YH, Angkasekwinai P, Lu N, Voo KS, Arima K, Hanabuchi S, Hippe A, Corrigan CJ, Dong C, Homey B, et al. IL-25 augments type 2 immune responses by enhancing the expansion and functions of TSLP-DC-activated Th2 memory cells. J Exp Med. 2007;204:1837–1847. doi: 10.1084/jem.20070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi Y, Fujio K, Shoda H, Okamoto A, Tsuno NH, Takahashi K, Yamamoto K. IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol. 2007;179:7128–7136. doi: 10.4049/jimmunol.179.10.7128. [DOI] [PubMed] [Google Scholar]
- Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem. 2007;282:9358–9363. doi: 10.1074/jbc.C600321200. [DOI] [PubMed] [Google Scholar]
- Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C. Regulation of inflammatory responses by IL-17F. J Exp Med. 2008a;205:1063–1075. doi: 10.1084/jem.20071978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity. 2008b;29:44–56. doi: 10.1016/j.immuni.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 2008c;28:29–39. doi: 10.1016/j.immuni.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.