

Abstract
Cardiac metabolism is highly adaptive in response to changes in substrate availability, as occur during fasting. This metabolic flexibility is essential to the maintenance of contractile function and is under the control of a group of select transcriptional regulators, notably the nuclear receptor family of factors member PPARα. However, the diversity of physiologic and pathologic states through which the heart must sustain function suggests the possible existence of additional transcriptional regulators that play a role in matching cardiac metabolism to energetic demand. Here we show that cardiac KLF15 is required for the normal cardiac response to fasting. Specifically, we find that cardiac function is impaired upon fasting in systemic and cardiac specific Klf15-null mice. Further, cardiac specific Klf15-null mice display a fasting-dependent accumulation of long chain acylcarnitine species along with a decrease in expression of the carnitine translocase Slc25a20. Treatment with a diet high in short chain fatty acids relieves the KLF15-dependent long chain acylcarnitine accumulation and impaired cardiac function in response to fasting. Our observations establish KLF15 as a critical mediator of the cardiac adaptive response to fasting through its regulation of myocardial lipid utilization.
Introduction
The adult heart sustains contractile function regardless of fuel supply, in the face of enormous metabolic demand. Indeed, from an ATP pool of approximately 5 μmol/g wet weight, the heart uses 0.5 μmol/g wet weight per second and therefore perpetually verges on ATP depletion [1]. Although the heart demonstrates a preference for the oxidation of fatty acids, as approximately 70% of the ATP generated in the myocardium is derived from them, ~20% comes from glucose and lactate, and the remainder from diverse substrates (e.g. ketones) [2]. Furthermore, the heart is endowed with the capacity to adapt substrate utilization to match the systemic environment. During fasting, the myocardium increases its dependence on oxidative catabolism of fatty acids and inhibits glycolysis, events which occur simultaneously with a rise in expression of genes involved in fatty acid oxidation [3, 4]. The importance of this adaptive capacity to enhance lipid utilization is illustrated by the clinical features of patients with inborn errors of myocardial fatty acid oxidation, where episodic manifestations of the disease often occur following fasting, presumably as the heart relies more heavily on fatty acid oxidation to meet its energetic demands. [5]
Myocardial lipid utilization is a complex multi-step process [2]. Fatty acids enter the myocardium via passive diffusion or facilitated by carriers including fatty acid translocase (FAT)/CD36, the plasma membrane isoform of fatty acid binding protein (FABPpm), or fatty acid transport protein (FATP) 1/6, with CD36 accounting for 50–60% of fatty acid oxidation [6]. Long chain cytosolic fatty acids are esterified by fatty acyl CoA synthetase (FACS) and, if destined for oxidation, further transferred to carnitine via the mitochondrial enzyme carnitine palmitoyltransferase 1 (CPT1) and shuttled across the outer mitochondrial membrane. Translocation of acylcarnitines across the inner mitochondrial membrane is accomplished by a carnitine:acylcarnitine translocase (Slc25a20), moving the fatty acid moiety into the mitochondrial matrix, and simultaneously generating free carnitine. Carnitine palmitoyltransferase 2 (CPT2) subsequently converts long-chain acylcarnitines into acyl CoA species, which are free to enter the β-oxidation pathway.
Fasting induced alterations in myocardial lipid utilization are achieved, in part, through the activity of several key transcriptional regulators. Work performed by others over the last decade has identified roles for nuclear receptors including the peroxisome proliferator-activated receptors (PPARs), estrogen-related receptors (ERRs), and nuclear respiratory factors 1 and 2 (NRF-1 and -2) in the regulation of genes, among others, involved in cardiac fatty acid oxidation [7]. Recently, we have provided evidence that a zinc-finger DNA-binding transcription factor, Kruppel-like factor 15 (KLF15), is also a critical regulator of myocardial substrate metabolism, especially lipid utilization, and cooperates with PPARα to coordinately regulate gene expression [8, 9]. Further, KLF15 is required for PPARα mediated induction of canonical fasting-inducible gene targets [8, 9].
Given our observations, we therefore postulated that KLF15 regulation of cardiac metabolism might be necessary for metabolic adaptation to substrate availability, as occurs under fasting regimens. Here, we demonstrate that Klf15-null hearts exhibit impaired function during fasting, coincident with an accumulation of long chain acyl-carnitines and decrease in transcript levels of Slc25a20. These phenotypes are rescued by a diet high in short chain fatty acids, which do not require transport into the mitochondria. Together, our findings suggest KLF15 is critical to the cardiac fasting response and point to a model whereby KLF15 regulation of Slc25a20 during the fasting state enables transport of long chain acylcarnitine species into mitochondria for oxidation.
Materials and methods
Animal models
Animal protocols were approved by the Institutional Animal Care and Use Committee at Case Western Reserve University. Protocols were in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Klf15-/- and cardiac-specific KLF15-cKO mice have been previously described [10, 11]. Male, age-matched controls on a C57Bl/6 genetic background were used. Mice were maintained with ad libitum access to water and standard laboratory chow (Laboratory diet P3000; 4.5% fat by weight, 14% kilocalories). Barrier facility had a 12 hour light/dark cycle and was temperature and humidity controlled. For fasting experiments, food was removed at indicated times and lasted for up to 48 hours in duration. Where indicated, mice were fed a short chain fatty acid enriched diet (SCD: Harlan Teklad TD.09849) for 10-weeks as previously described [12].
Plasma parameters and tissue lipids
Hearts and plasma were rapidly harvested from WT and KO mice fed ad libitum or fasted for two days. Lipids were extracted from ventricles by the Folch method. Cardiac fatty acid and triglyceride content was quantified by thin-layer chromatography by the NIH-MMPC / Lipid Analytic Core at Vanderbilt University. Plasma free-fatty acids and triglycerides were quantified by the Vanderbilt NIH Mouse Metabolic Phenotyping Core. Plasma glucose measurements were performed using the glucose colorimetric / fluorometric assay kit from BioVision according to the manufacturer’s instructions.
Acylcarnitine measurements
Hearts were rapidly harvested from WT and KO mice fed ad libitum or fasted for two days. Tissue homogenates (50 mg/mL) were prepared using a polytron in 50% acetonitrile and 0.3% formic acid. 45 acylcarnitine species (including short, medium, and long-chain) were analyzed by tandem mass spectrometry (MS/MS) using sample preparation methods described previously [13, 14]. The data were acquired using a Waters triple quadrupole detector equipped with AcquityTM UPLC system and controlled by MassLynx 4.1 software platform (Waters, Milford, MA).
Echocardiographic analysis
Transthoracic echocardiography was performed in anesthetized mice using the Vevo 770 High Resolution Imaging System (Visual Sonics) and the RMV-707B 30-MHz probe as previously described [10]. Cardiac function was assessed at the mid papillary muscle level in M-mode short axis images.
RNA extraction and QPCR
Heart tissue samples were disrupted in PureZOLTM (Biorad) in a Tissue-lyzer (Qiagen) using stainless steel beads (30 Hz for a total of 4 min). AurumTM (Biorad) RNA isolation kit was used to isolate total RNA according to manufacturer’s directions. For qPCR analysis, total RNA was loaded onto columns and DNase treated before being transcribed to complementary DNA using iScriptTM (Biorad) according to manufacturer’s directions. TaqMan method was used for qPCR analysis (using the Roche Universal Probe Library System) using an ABI Step One Plus Real-Time PCR System. ΔΔCt method (with normalization genes indicated in figure legend)was used to assess relative expression. Primer sequence for Slc25a20: Forward 5’-ATCCGCGGCTTCTACAAAG-3’, Reverse 5’-TACATCCCACTGGCAGGAAC-3’. Primer sequence for Cpt1b: Forward 5’-TGCCTTTACATCGTCTCCAA-3’, Reverse 5’-GGCTCCAGGGTTCAGAAAGT-3’. Primer sequence for Cpt2: Forward 5’-CCAAAGAAGCAGCGATGG-3’, Reverse 5’-TAGAGCTCAGGCAGGGTGA-3’. Primer sequence for Fabp3: Forward 5’-CTTTGTCGGTACCTGGAAGC-3’, Reverse 5’-TGGTCATGCTAGCCACCTG-3’. Primer sequence for Glut4: Forward 5’-TCGTCATTGGCATTCTGGT-3’, Reverse 5’-AGCAGTGGCCACAGGGTA-3’. Primer sequence for Glut1: Forward 5’-ATGGATCCCAGCAGCAAG-3’, Reverse 5’-CCAGTGTTATAGCCGAACTGC-3’. Primer sequence for Ppib: Forward 5’-TTCTTCATAACCACAGTCAAGACC-3’, Reverse 5’-ACCTCCGTACCACATCCAT-3’.
Western blot analysis
Protein was extracted and homogenized from heart tissue samples in RIPA buffer (Sigma-Aldrich, R0278) containing proteinase/phosphatase inhibitor cocktail (Roche). Samples in SDS sample buffer were run on SDS-PAGE and immunoblotted with anti-CACT (Sigma, 1:1000) and anti-α-tubulin (Cell Signaling, 1:2000). Secondary HRP-conjugated antibodies were from Cell Signaling and chemiluminescent detection reagent was from Thermo Scientific. Bio-Rad Quantity One software was used for quantitation. Two-tailed Student's t-test for unpaired data was used
Data analysis
All error bars depict SEM, and data is expressed as means. In experiments comparing the means of two normally distributed groups, two-tailed Student's t-test for unpaired data was used. In experiments comparing the means of normally distributed groups with multiple treatments, one-way analysis of variance (ANOVA) with the Tukey post hoc test was used. Statistical significance defined as p < 0.05.
Results
Cardiac KLF15 is required for the heart’s functional adaptation in response to fasting
Cardiac KLF15 is highly responsive to diverse physiologic conditions requiring enhanced fatty acid utilization / oxidation, including fasting, postnatal cardiac maturation, and the circadian sleep/wake transition [11, 15, 16]. To gain initial insights into the functional role of KLF15 in controlling lipid metabolism, we reasoned that its absence under fasting conditions might render the heart susceptible to dysfunction. As such, we first utilized systemic Klf15-null animals and examined cardiac function under fed and fasted conditions. As shown in Fig 1A & 1B, cardiac function was significantly reduced in the fasted state in the absence of KLF15. This decreased cardiac function occurred in the absence of overt signs of remodeling / hypertrophy (Fig 1C). However, given the numerous metabolic disturbances observed in the systemic Klf15-null line following fasting (e.g. alterations in glucose and amino acid levels), we performed a similar set of experiments in the cardiac restricted KLF15 knockout model. As shown in Fig 2A–2C, we observed a similar KLF15-dependecy on cardiac function following fasting suggesting a cardiac specific and cell autonomous role.
Fig 1. Systemic KLF15 is required for the heart’s functional adaptation in response to fasting.

(A) Left ventricular fractional shortening from echocardiography performed in wild-type (WT) vs. systemic Klf15-null (Klf15-/-) under fed vs. 48 hours fasting conditions, (n = 5), *P,0.05 vs. WT Fast. (B) Representative echocardiography image from WT vs. Klf15-/- following a 48 hour fast. (C) Tabular representation of echocardiography data in WT vs. Klf15-/- under fed vs. 48 hour fasting conditions.
Fig 2. Cardiac KLF15 is required for the heart’s functional adaptation in response to fasting.

(A) Left ventricular fractional shortening from echocardiography performed in control (MHC-Cre) vs KLF15-cKO under fed vs. 48 hours fasting conditions, (n = 5), *P<0.05 vs. MHC-Cre Fast. (B) Representative echocardiography image from MHC-Cre vs. KLF15-cKO following a 48 hour fast. (C) Tabular representation of echocardiography data in MHC-Cre vs. KLF15-cKO under fed vs. 48 hour fasting conditions.
Cardiac specific deletion of KLF15 alters lipid utilization
To further investigate the cardiac specific role of KLF15 in controlling lipid metabolism, cardiac and plasma samples were harvested from both control (MHC-Cre) and KLF15-cKO mice in the fed and fasted state. As shown in Fig 3A & 3B, the accumulation of both free fatty acids (FFA) and triglycerides (TG) following fasting is blunted in the absence of cardiac KLF15 expression. Additionally, while we observed the expected increase in circulating FFA and TG levels following fasting, we did not observe a cardiac KLF15 dependency on circulating FFA and TG following fasting (Fig 3C & 3D). Taken together, these data suggest that cardiac KLF15 controls, at least in part, lipid uptake / utilization in the context of physiologic fasting.
Fig 3. Cardiac specific deletion of KLF15 alters tissue and plasma levels of free fatty acids and triglycerides.
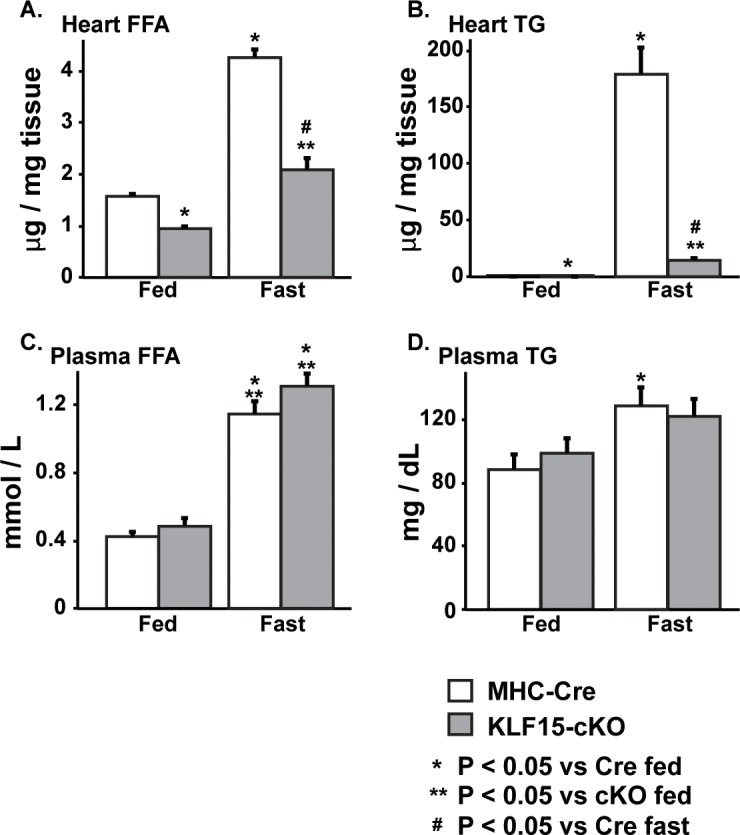
Cardiac FFA (A) and TG (B) levels in control (MHC-Cre) vs. KF15-cKO following 48 hours fasting, (n = 5), *P<0.05 vs. Cre Fed, **P<0.05 vs. CKO Fed, # P<0.05 vs. Cre Fast. Plasma FFA (C) and TG (D) levels in control (MHC-Cre) vs. KLF15-cKO following 48 hours fasting, (n = 5), *P<0.05 vs. Cre Fed, **P<0.05 vs. CKO Fed, # P<0.05 vs. Cre Fast.
To further test this hypothesis, we undertook a metabolomics approach and assessed the cardiac acylcarnitine (AC) profile in both control and KLF15-cKO under fed and fasted conditions. As described previously, AC species (ranging in chain length from 2 to 22 carbons) are formed from their acyl-CoA intermediates by carnitine acyltransferases (CACT; e.g. Slc25a20) prior to β-oxidation [17]. These ACs are membrane permeable and can accumulate in both tissue and circulation [18]. Therefore, profiling the AC pool provides a reliable index of lipid flux with even-chain species ranging in length from C6 to C22 representing incomplete utilization of fatty acids [18]. As shown in Fig 4A & 4B, we observed an expected increase in long-chain AC species in MHC-Cre hearts following fasting. Additionally, we found that loss of cardiac KLF15 expression in the fed state gave rise to augmented long-chain AC species, and thus incomplete β-oxidation, an effect that was exacerbated in the fasted state (Fig 4A & 4B). Taken together, these data suggest KLF15 is an important regulator of lipid uptake and utilization in the heart under physiologic states of stress such as fasting.
Fig 4. Cardiac specific deletion of KLF15 alters lipid profile.

Metabolomic analysis of long chain acylcarnitines in cardiac tissue from control (MHC-Cre) vs. KLF15-cKO with and without 48 hour fast, (n = 5), *P<0.05 by one-way analysis of variance (ANOVA) with the Tukey post hoc test.
Short-chain diet rescues the KLF15-dependent attenuation of cardiac function in response to fasting
Given the above results, we next sought to rescue the fasting and KLF15 dependent cardiac dysfunction. Towards this end, we fed control and KLF15-cKO mice a diet rich in short-chain fatty acids which bypass the Slc25a20 transporter and drive β-oxidation. We assessed the expression of Slc25a20, whose gene product is the rate-limiting step in converting long-chain acyl-CoAs to the cell permeable acylcarnitine species. As shown in Fig 5A, mRNA transcript levels of other transporters involved in glucose transport and fatty acid transport into mitochondria, are either unaffected or slightly increased by cardiac specific loss of KLF15 under fed or fasted conditions. In contrast, the fasting induction of Slc25a20 gene expression is dependent on KLF15 (Fig 5B). Levels of CACT, the gene product of Slc25a20 are also lower (Fig 5C and 5D). Furthermore, following 10 weeks of short-chain diet, cardiac function was assessed in both the fed and fasted states. As shown in Fig 5E–5G, 10 weeks of SCD rescued the KLF15-dependent cardiac dysfunction following fasting. Importantly, we then performed metabolomics to assess AC species, and, as shown in Fig 6, the augmentation of long-chain AC species in the fed state after loss of cardiac KLF15 expression is nearly abolished with 10 weeks of SCD and is strongly suppressed in the fasted state (Table 1). Taken together, these data suggest KLF15 is necessary in the cardiac adaptive response to fasting through a mechanism involving coordinated expression of Slc25a20, which drives appropriate AC conversion for β-oxidation (Fig 7).
Fig 5. Short-chain diet rescues the KLF15-dependent attenuation of cardiac function in response to fasting.

(A) qPCR analysis of expression of transporter genes in MHC-Cre vs. KLF15-cKO under fed vs. 48 hour fasting conditions. *P<0.05 vs. Cre Fed, **P<0.05 vs. CKO Fed, # P<0.05 vs. Cre Fast. Values normalized to Ppib. (B) Slc25a20 expression (qPCR) in MHC-Cre vs. KLF15-cKO under fed vs. 48 hour fasting conditions. *P<0.05 vs. Cre Fed, **P<0.05 vs. CKO Fed, # P<0.05 vs. Cre Fast. Values normalized to Ppib. (C) Western blot analysis of CACT levels in MHC-Cre vs KLF15-cKO under fed and 48 hour fasting conditions. α-tubulin used as loading control. (D) Quantification of data in C (n = 3 per group). Two-tailed Student's t-test for unpaired data was used. *P<0.05. (E) Left ventricular fractional shortening from echocardiography performed in control (MHC-Cre) vs. KLF15-cKO under fed vs. 48 hours fasting conditions following 10 weeks of short-chain fatty acid diet, (n = 10). (F) Representative echocardiography image from MHC-Cre vs. KLF15-cKO following 48 hours fasting and 10 weeks of short-chain fatty acid diet. (G) Tabular representation of echocardiography data in MHC-Cre vs. KLF15-cKO under fed vs. 48 hour fasting conditions following 10 weeks of short-chain fatty acid diet.
Fig 6. Short-chain diet rescues the KLF15-dependent accumulation of long chain acylcarnitines in response to fasting.

Metabolomic analysis of long chain acyl-carnitines in cardiac tissue from control (MHC-Cre) vs. KLF15-cKO with and without 48 hour fast, (n = 6) following 10 weeks of short-chain fatty acid diet, *P<0.05 by one-way analysis of variance (ANOVA) with the Tukey post hoc test.
Table 1. Change in long chain acylcarnitine profiles after loss of cardiac KLF15 expression.
| Diet | Normal chow | Normal chow | Short chain fatty acid enriched | Short chain fatty acid enriched |
|---|---|---|---|---|
| Condition | Fed | Fast | Fed | Fast |
| Genotype comparison | MHC-Cre vs KLF15-cKO | |||
| Long chain acylcarnitines | ||||
| C14 | NC | NC | NC | + |
| C14:1 | NC | NC | NC | + |
| C14:2 | NC | ++ | NC | NC |
| C14:3 | NC | NC | NC | NC |
| C14-OH/C12-DC | NC | + | NC | + |
| C14:1-OH/C12:1-DC | NC | ++ | NC | + |
| C14:2-OH/C12:2-DC | NC | + | NC | NC |
| C14:3-OH/C12:3-DC | NC | NC | NC | NC |
| C16 | NC | + | NC | ++ |
| C16:1 | NC | NC | NC | + |
| C16:2 | NC | ++ | NC | + |
| C16:3 | NC | ++ | NC | NC |
| C16-OH/C14-DC | NC | NC | NC | + |
| C16:1-OH/C14:1-DC | NC | + | NC | + |
| C16:2-OH/C14:2-DC | NC | + | NC | NC |
| C16:3-OH/C14:3-DC | + | + | NC | NC |
| C18 | ++ | NC | NC | + |
| C18:1 | NC | + | NC | + |
| C18:2 | NC | + | NC | ++ |
| C18:3 | NC | + | NC | NC |
| C18:1-OH/C16:1-DC | NC | + | NC | + |
| C18:2-OH/C16:2-DC | NC | ++ | NC | + |
| C18:3-OH/C16:3-DC | NC | ++ | NC | NC |
| C20 | NC | ++ | NC | NC |
| C20:1 | ++ | ++ | NC | NC |
| C20:2 | ++ | NC | NC | NC |
| C20:3 | ++ | ++ | NC | ++ |
| C20:4 | NC | ++ | NC | NC |
| C20-OH/C18-DC/C22:6 | NC | NC | NC | NC |
| C20:1-OH/C18:1-DC | NC | NC | NC | NC |
| C20:2-OH/C18:2-DC | NC | NC | NC | NC |
| C20:3-OH/C18:3-DC | NC | ++ | NC | NC |
| C22 | NC | ++ | NC | NC |
| C22:1 | NC | NC | NC | NC |
| C22:2 | + | NC | NC | NC |
| C22:3 | ++ | NC | NC | NC |
| C22:4 | ++ | NC | NC | NC |
| C22:5 | NC | NC | NC | NC |
Changes in all measured long chain acylcarnitine species after loss of KLF15 expression in fed and fasted conditions, with and without SCD diet. No statistically significant difference between MHC-Cre and KLF15-cKO is represented by (NC), a less than 2 fold increase is represented by (+) and a more than 2 fold increase is represented by (++).
Fig 7. Schematic representing KLF15 as a regulator of the cardiac adaptive response to fasting.

Discussion
Cardiac metabolic plasticity during fasting, namely the augmentation of lipid utilization, is crucial for its function [2, 19]. Transcriptional regulators coordinate this augmentation, and we establish here that KLF15 is critically required for cardiac lipid flux and function during fasting. In systemic and cardiac specific Klf15-null mice, we observe decreased fractional shortening under fasting conditions. Additionally, metabolomics analysis reveals an accumulation of long chain acylcarnitine species in hearts of cardiac specific Klf15-null mice during fasting, suggesting an inability of these hearts to adequately manage lipid flux during a nutrient deprived state. This theory is bolstered by our finding that a diet rich in short chain fatty acids, which cross easily into the mitochondria and therefore can act as an alternative energy source, can rescue both the long chain acyl-carnitine abnormalities as well as cardiac fractional shortening. Finally, we find by qPCR analysis that transcript levels of a key transporter of acyl-carnitines, Slc25a20, are lowered in cardiac specific Klf15-null mice hearts. Together with prior work, these results implicate cardiac KLF15 in the fasting response through its regulation of lipid flux and illustrate the significance of transcriptional control of cardiac metabolism in governing cardiac function.
While coordinated alterations in cardiac metabolism are critical to the normal fasting response, metabolic derangements have also been shown to contribute to various disease states. In diabetic hearts, glucose oxidation is inhibited and there is decreased activation of PI3K-Akt signaling in response to insulin, while fatty acid oxidation is enhanced through increased circulating levels of free fatty acids as well as the aberrant action of PPARα, changes contributing to the development of a diabetes associated cardiomyopathy [20, 21]. Interestingly, the overexpression of PPARα in cardiomyocytes alone is sufficient to phenocopy many of the features of this cardiomyopathy, while PPARα deficiency is protective against a streptozotocin-induced model of diabetic cardiomyopathy [22, 23]. Given our prior description of a KLF15-PPARα molecular module regulating fatty acid oxidation, KLF15 may also play a role in diabetes-induced cardiac dysfunction [9]. Additionally, alterations in substrate usage and their attendant consequences have also been noted in the failing heart, namely a relative, although not absolute, increase in glucose oxidation and decrease in fatty acid oxidation, particularly in late stages of disease [24–28]. Notably, although fatty acid oxidation is reduced, lipid uptake into the cytosol is in fact enhanced, resulting in an accumulation of triglycerides in the cardiomyocyte as well as shunting of lipids into maladaptive non-oxidative pathways, broadly termed lipotoxicity [29, 30]. Previous work from our group has shown that mRNA transcripts of cardiac KLF15 are lowered in rodent and human samples of heart failure, and that the mouse with global loss of KLF15 is more susceptible to heart failure following pressure overload, suggesting a role for KLF15 regulation of fatty acid oxidation in the context of heart failure [8, 16, 31, 32].
The selective accumulation of unoxidized long chain acylcarnitines in the cardiac KLF15 knockout mouse, its exacerbation upon fasting, and its rescue with a diet of short chain fatty acids is reminiscent of the clinical phenotype of patients affected by genetic defects in long chain fatty acid oxidation pathways. These patients, who can harbor mutations in genes of fatty acid transport across the mitochondrial membrane or in specific enzymes of the β-oxidation pathway, exhibit metabolic decompensation during fasting which is alleviated with a diet enriched in medium chain fatty acids and depleted in long chain fatty acids [33–37]. The cardiac sequelae of an inborn error of fatty acid oxidation have been theoretically attributed to toxicity of the long chain acylcarnitine, either through the generation of reactive oxygen species, or through incorporation in the sarcolemma leading to generation of arrhythmias due to its amphiphilic nature; these mechanisms may also be operative in the KLF15 deficient mouse [38]. Indeed, in the diabetic cardiomyopathy model mouse overexpressing PPARα in the heart, Finck et al. observe an accumulation of triacylglycerides containing long chain fatty acids which is rescued upon treatment with a medium chain fatty acid diet [23]. Since, in our model, the relief of long chain acylcarnitine burden occurs simultaneously with the return of normal cardiac function, it seems likely that this accumulation of long chain acylcarnitines is also key to the deterioration in cardiac function of KLF15 knockout mice. Additionally, it remains unclear what mechanisms mediate the decrease in long chain acylcarnitine accumulation in the presence of an abundant alternative lipid energy source such as short chain fatty acids. However, as diets rich in short to medium chain fatty acids have been shown to be useful in some diseases of inborn defects of fatty acid oxidation, it will be valuable to consider their therapeutic value in other diseases in which fatty acid oxidation pathways are dysregulated, including diabetes and advanced heart failure.
Acknowledgments
This work was supported, in whole or in part, by National Institutes of Health Grants F32HL110538 and K01DK104922 (to D.A.P.), R01HL112486, R01HL119195, and R01HL123098 (to M.J.K.), F30AG054237 (to P.N.H), along with generous gifts by Tom F. Peterson (TFP Lab).
Data Availability
All relevant data are within the paper.
Funding Statement
This work was supported, in whole or in part, by National Institutes of Health Grants F32HL110538 and K01DK104922 (to D.A.P.), R01HL112486, R01HL119195, and R01HL123098 (to M.J.K.), F30AG054237 (to P.N.H), along with generous gifts by Tom F. Peterson (TFP Lab). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, et al. Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with (31)P-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol. 2002;40(7):1267–74. Epub 2002/10/18. . [DOI] [PubMed] [Google Scholar]
- 2.Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90(1):207–58. Epub 2010/01/21. doi: 10.1152/physrev.00015.2009 . [DOI] [PubMed] [Google Scholar]
- 3.Denton RM, Randle PJ. Concentrations of glycerides and phospholipids in rat heart and gastrocnemius muscles. Effects of alloxan-diabetes and perfusion. The Biochemical journal. 1967;104(2):416–22. Epub 1967/08/01. ; PubMed Central PMCID: PMCPMC1270602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Suzuki J, Shen WJ, Nelson BD, Selwood SP, Murphy GM Jr., Kanehara H, et al. Cardiac gene expression profile and lipid accumulation in response to starvation. American journal of physiology Endocrinology and metabolism. 2002;283(1):E94–e102. Epub 2002/06/18. doi: 10.1152/ajpendo.00017.2002 . [DOI] [PubMed] [Google Scholar]
- 5.Kelly DP, Strauss AW. Inherited cardiomyopathies. The New England journal of medicine. 1994;330(13):913–9. Epub 1994/03/31. doi: 10.1056/NEJM199403313301308 . [DOI] [PubMed] [Google Scholar]
- 6.Kuang M, Febbraio M, Wagg C, Lopaschuk GD, Dyck JR. Fatty acid translocase/CD36 deficiency does not energetically or functionally compromise hearts before or after ischemia. Circulation. 2004;109(12):1550–7. Epub 2004/03/17. doi: 10.1161/01.CIR.0000121730.41801.12 . [DOI] [PubMed] [Google Scholar]
- 7.Leone TC, Kelly DP. Transcriptional control of cardiac fuel metabolism and mitochondrial function. Cold Spring Harbor symposia on quantitative biology. 2011;76:175–82. Epub 2011/11/19. doi: 10.1101/sqb.2011.76.011965 ; PubMed Central PMCID: PMCPMC3340448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prosdocimo DA, Anand P, Liao X, Zhu H, Shelkay S, Artero-Calderon P, et al. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. The Journal of biological chemistry. 2014;289(9):5914–24. Epub 2014/01/11. doi: 10.1074/jbc.M113.531384 ; PubMed Central PMCID: PMC3937660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prosdocimo DA, John JE, Zhang L, Efraim ES, Zhang R, Liao X, et al. KLF15 and PPARalpha Cooperate to Regulate Cardiomyocyte Lipid Gene Expression and Oxidation. PPAR research. 2015;2015:201625 Epub 2015/03/31. doi: 10.1155/2015/201625 ; PubMed Central PMCID: PMCPMC4357137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Haldar SM, Lu Y, Jeyaraj D, Kawanami D, Cui Y, Eapen SJ, et al. Klf15 deficiency is a molecular link between heart failure and aortic aneurysm formation. Science translational medicine. 2010;2(26):26ra Epub 2010/04/09. doi: 10.1126/scitranslmed.3000502 ; PubMed Central PMCID: PMCPMC3003709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang L, Prosdocimo DA, Bai X, Fu C, Zhang R, Campbell F, et al. KLF15 Establishes the Landscape of Diurnal Expression in the Heart. Cell reports. 2015;13(11):2368–75. Epub 2015/12/22. doi: 10.1016/j.celrep.2015.11.038 . [DOI] [PubMed] [Google Scholar]
- 12.York B, Reineke EL, Sagen JV, Nikolai BC, Zhou S, Louet JF, et al. Ablation of steroid receptor coactivator-3 resembles the human CACT metabolic myopathy. Cell Metab. 2012;15(5):752–63. Epub 2012/05/09. doi: 10.1016/j.cmet.2012.03.020 ; PubMed Central PMCID: PMCPmc3349072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrara CT, Wang P, Neto EC, Stevens RD, Bain JR, Wenner BR, et al. Genetic networks of liver metabolism revealed by integration of metabolic and transcriptional profiling. PLoS genetics. 2008;4(3):e1000034 Epub 2008/03/29. doi: 10.1371/journal.pgen.1000034 ; PubMed Central PMCID: PMCPMC2265422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.An J, Muoio DM, Shiota M, Fujimoto Y, Cline GW, Shulman GI, et al. Hepatic expression of malonyl-CoA decarboxylase reverses muscle, liver and whole-animal insulin resistance. Nat Med. 2004;10(3):268–74. Epub 2004/02/11. doi: 10.1038/nm995 . [DOI] [PubMed] [Google Scholar]
- 15.Prosdocimo DA, Anand P, Liao X, Zhu H, Shelkay S, Artero-Calderon P, et al. Kruppel-like factor 15 is a critical regulator of cardiac lipid metabolism. The Journal of biological chemistry. 2014;289(9):5914–24. Epub 2014/01/11. doi: 10.1074/jbc.M113.531384 ; PubMed Central PMCID: PMCPMC3937660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fisch S, Gray S, Heymans S, Haldar SM, Wang B, Pfister O, et al. Kruppel-like factor 15 is a regulator of cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2007;104(17):7074–9. doi: 10.1073/pnas.0701981104 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramsay RR. The carnitine acyltransferases: modulators of acyl-CoA-dependent reactions. Biochemical Society transactions. 2000;28(2):182–6. Epub 2000/05/18. . [DOI] [PubMed] [Google Scholar]
- 18.Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56. Epub 2008/01/08. doi: 10.1016/j.cmet.2007.10.013 . [DOI] [PubMed] [Google Scholar]
- 19.Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell metabolism. 2012;15(6):805–12. Epub 2012/06/12. doi: 10.1016/j.cmet.2012.04.006 ; PubMed Central PMCID: PMC3387529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miki T, Yuda S, Kouzu H, Miura T. Diabetic cardiomyopathy: pathophysiology and clinical features. Heart failure reviews. 2013;18(2):149–66. Epub 2012/03/29. doi: 10.1007/s10741-012-9313-3 ; PubMed Central PMCID: PMCPMC3593009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Abel ED, O'Shea KM, Ramasamy R. Insulin resistance: metabolic mechanisms and consequences in the heart. Arteriosclerosis, thrombosis, and vascular biology. 2012;32(9):2068–76. Epub 2012/08/17. doi: 10.1161/ATVBAHA.111.241984 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, et al. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109(1):121–30. Epub 2002/01/10. doi: 10.1172/JCI14080 ; PubMed Central PMCID: PMC150824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, et al. A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(3):1226–31. Epub 2003/01/29. doi: 10.1073/pnas.0336724100 ; PubMed Central PMCID: PMC298755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ingwall JS. Energy metabolism in heart failure and remodelling. Cardiovasc Res. 2009;81(3):412–9. Epub 2008/11/07. doi: 10.1093/cvr/cvn301 ; PubMed Central PMCID: PMCPMC2639129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neubauer S. The failing heart—an engine out of fuel. N Engl J Med. 2007;356(11):1140–51. Epub 2007/03/16. doi: 10.1056/NEJMra063052 . [DOI] [PubMed] [Google Scholar]
- 26.Rosca MG, Hoppel CL. Mitochondria in heart failure. Cardiovasc Res. 2010;88(1):40–50. Epub 2010/07/30. doi: 10.1093/cvr/cvq240 ; PubMed Central PMCID: PMCPMC3025720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abel ED, Doenst T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res. 2011;90(2):234–42. Epub 2011/01/25. doi: 10.1093/cvr/cvr015 ; PubMed Central PMCID: PMCPMC3115280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nickel A, Loffler J, Maack C. Myocardial energetics in heart failure. Basic research in cardiology. 2013;108(4):358 Epub 2013/06/07. doi: 10.1007/s00395-013-0358-9 . [DOI] [PubMed] [Google Scholar]
- 29.Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta. 2010;1801(3):311–9. Epub 2009/10/13. doi: 10.1016/j.bbalip.2009.09.023 ; PubMed Central PMCID: PMCPMC2823976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, et al. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2004;18(14):1692–700. Epub 2004/11/04. doi: 10.1096/fj.04-2263com . [DOI] [PubMed] [Google Scholar]
- 31.Haldar SM, Ibrahim OA, Jain MK. Kruppel-like Factors (KLFs) in muscle biology. J Mol Cell Cardiol. 2007;43(1):1–10. doi: 10.1016/j.yjmcc.2007.04.005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prosdocimo DA, Sabeh MK, Jain MK. Kruppel-like factors in muscle health and disease. Trends in cardiovascular medicine. 2014. Epub 2014/12/23. doi: 10.1016/j.tcm.2014.11.006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hale DE, Bennett MJ. Fatty acid oxidation disorders: a new class of metabolic diseases. The Journal of pediatrics. 1992;121(1):1–11. Epub 1992/07/01. . [DOI] [PubMed] [Google Scholar]
- 34.Touma EH, Rashed MS, Vianey-Saban C, Sakr A, Divry P, Gregersen N, et al. A severe genotype with favourable outcome in very long chain acyl-CoA dehydrogenase deficiency. Archives of disease in childhood. 2001;84(1):58–60. Epub 2000/12/22. doi: 10.1136/adc.84.1.58 ; PubMed Central PMCID: PMCPMC1718627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brown-Harrison MC, Nada MA, Sprecher H, Vianey-Saban C, Farquhar J Jr., Gilladoga AC, et al. Very long chain acyl-CoA dehydrogenase deficiency: successful treatment of acute cardiomyopathy. Biochemical and molecular medicine. 1996;58(1):59–65. Epub 1996/06/01. . [DOI] [PubMed] [Google Scholar]
- 36.Cox GF, Souri M, Aoyama T, Rockenmacher S, Varvogli L, Rohr F, et al. Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long-chain acyl-coenzyme A dehydrogenase deficiency. The Journal of pediatrics. 1998;133(2):247–53. Epub 1998/08/26. . [DOI] [PubMed] [Google Scholar]
- 37.Gillingham MB, Scott B, Elliott D, Harding CO. Metabolic control during exercise with and without medium-chain triglycerides (MCT) in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Molecular genetics and metabolism. 2006;89(1–2):58–63. Epub 2006/08/01. doi: 10.1016/j.ymgme.2006.06.004 ; PubMed Central PMCID: PMCPMC2706834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bonnet D, Martin D, Pascale De L, Villain E, Jouvet P, Rabier D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999;100(22):2248–53. Epub 1999/12/01. . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data are within the paper.