

Abstract
Purpose of review:
Niemann-Pick disease type C (NP-C) is a neurovisceral disorder that may be more prevalent than earlier estimates. Diagnosis of NP-C is often delayed; a key aim for clinical practice is to reduce this delay. Recently, substantial progress has been made in the field of NP-C screening and diagnosis, justifying an update to the existing recommendations for clinical practice.
Recent findings:
New biomarker profiling and genetic analysis technologies are included as first-line diagnostic tests for NP-C. Most diagnoses can now be confirmed by combination of biomarker and genetic analyses. Filipin staining may facilitate diagnosis in uncertain cases. Recommendations are provided for psychiatrists, neuro-ophthalmologists, and radiologists, and on screening within specific at-risk patient cohorts. The NP-C diagnostic algorithm has been updated and simplified.
Summary:
This publication provides expert recommendations for clinicians who may see patients presenting with the signs and symptoms of NP-C, including general practitioners, pediatricians, neurologists, and psychiatrists.
Niemann-Pick disease type C (NP-C) is a rare neurovisceral lysosomal disorder caused by autosomal recessive mutations in NPC1 or NPC2 (95% and ∼4% of patients).1,2 Analysis of 4 independent exome sequencing databases gave a conservative estimated incidence of 1/92,104 for NP-C1 and 1/2,858,998 for NP-C2, in agreement with recent clinical data. After accounting for 2 common NPC1 variants, the incidence was estimated to be potentially 1/19,000–1/36,000 for late-onset forms, suggesting that previous prevalence estimates may be too low.3 NP-C diagnosis is typically delayed by a mean of 4.1 years from first onset of neurologic symptoms,4 narrowing the therapeutic window.
The first international recommendations for clinical management of NP-C were prepared in 2009 based on a literature review and consensus among an expert panel5 with updates in 2012 after a second meeting of an NP-C Guidelines Working Group.1 Recent advances in the field of NP-C screening technologies (e.g., biomarkers) and diagnostic techniques (e.g., molecular genetics) justify an update of these recommendations. The development and availability of simple, rapid, and reliable biomarkers (oxysterols,6–8 lysosphingomyelin derivatives,9,10 and bile acids11,12) will likely increase detection of patients with NP-C. Increased availability of next-generation sequencing (NGS) (e.g., phenotype-specific gene panels13) may widen the application of genetic testing in clinical practice, particularly in at-risk patient cohorts.14
Previous recommendations focused on identifying individual patients with a high clinical suspicion of NP-C. Technologic developments may facilitate patient identification based on clinical suspicion or within larger cohorts via broad genetic or biomarker screening, which ultimately may improve patient outcomes.
Development of the updated NP-C diagnostic recommendations
The NP-C Diagnostic Recommendations Expert Panel met in May 2016 to assess the impact of recently published data and to reach consensus on the best approaches to diagnosing NP-C. The following sections of the previous recommendations were updated: differential diagnosis and initial detection, diagnosis, and the NP-C diagnostic algorithm. Three approaches to the detection and diagnosis of patients with NP-C were established: clinical assessment, biomarker testing, and genetic analysis.
Differential diagnosis and initial detection
All patients with suspicion of NP-C require rigorous and detailed clinical assessment, as described in the 2012 diagnostic recommendations.1
All undiagnosed patients with any manifestation of NP-C should be referred to regional or national centers specializing in inherited metabolic disorders.
Visceral, neurologic, ophthalmologic, and psychiatric manifestations should be explored in depth.1
Details of the signs, symptoms, and disease stages of NP-C are covered extensively elsewhere.1,2,15
Advances in psychiatric assessments
-
Psychiatric symptoms and cognitive disorders were observed in 45% and 61%, respectively, of patients with late-onset NP-C,16 and psychiatric symptoms in 86% of patients in another small cohort17; however, no specific neuropsychiatric profile was observed.
Patients with NP-C, particularly adults, can present with a range of psychiatric symptoms, most commonly cognitive impairment (decline in executive function, memory, and psychomotor speed), schizophrenia-like symptoms (frequently with atypical features), and mood disorders. These symptoms can overshadow other symptoms of NP-C.
Owing to the inherent difficulties in examining patients in psychiatric practice, and incomplete or inaccurate medical histories, populations of adult patients displaying neuropsychiatric symptoms may be enriched for NP-C.18
Advances in ocular motor assessment
Eye movements (saccades, smooth pursuit, gaze-holding function, optokinetic nystagmus, and vergence) should be examined (neurocular.com). Impaired vertical saccades, commonly referred to as vertical supranuclear gaze palsy (VSGP) yet correctly termed vertical supranuclear saccade palsy (VSSP), are of particular importance in NP-C as one of the first and most frequent signs in patients with NP-C.19
Slow vertical saccades are often masked by blinking; the eyelids must be lifted during examination.
As the disease progresses, VSGP emerges. It is typically followed by impairment in horizontal saccades and then horizontal gaze.
Video-oculography allows the recording of all types of eye movements and can be used for quantitative analysis of ocular motor function (eyeseecam.com/).
VSSP may be present before visceral, neurologic, or psychiatric manifestations occur and is sometimes the only symptom of NP-C in adults, representing a highly sensitive and specific sign.19
Advances in clinical tools
The NP-C Suspicion Index (NP-C SI)20 quantitates the diagnostic weight of NP-C signs and symptoms, individually and in combination.
The updated NP-C SI features 2 age-specific indices (≤4 and >4 years of age [npc-si.com/]).15,21
The 2-out-of-3 suspicion index can be used to provide a rapid assessment of NP-C suspicion in patients presenting with early-onset ataxia.22
At-risk clinical patient groups
The clinical manifestations of NP-C may be nonspecific (e.g., ataxia), implying that patients with NP-C may be “hidden” within larger at-risk clinical patient cohorts, termed here and elsewhere14 as clinical niches, who also present with nonspecific signs and symptoms.14 NP-C should therefore be considered as a differential diagnosis for patients in these at-risk groups.
We defined several at-risk clinical patient groups, detailed below and summarized in table 1.
Table 1.
Clinical patient groups with an increased risk of Niemann-Pick disease type C (NP-C)
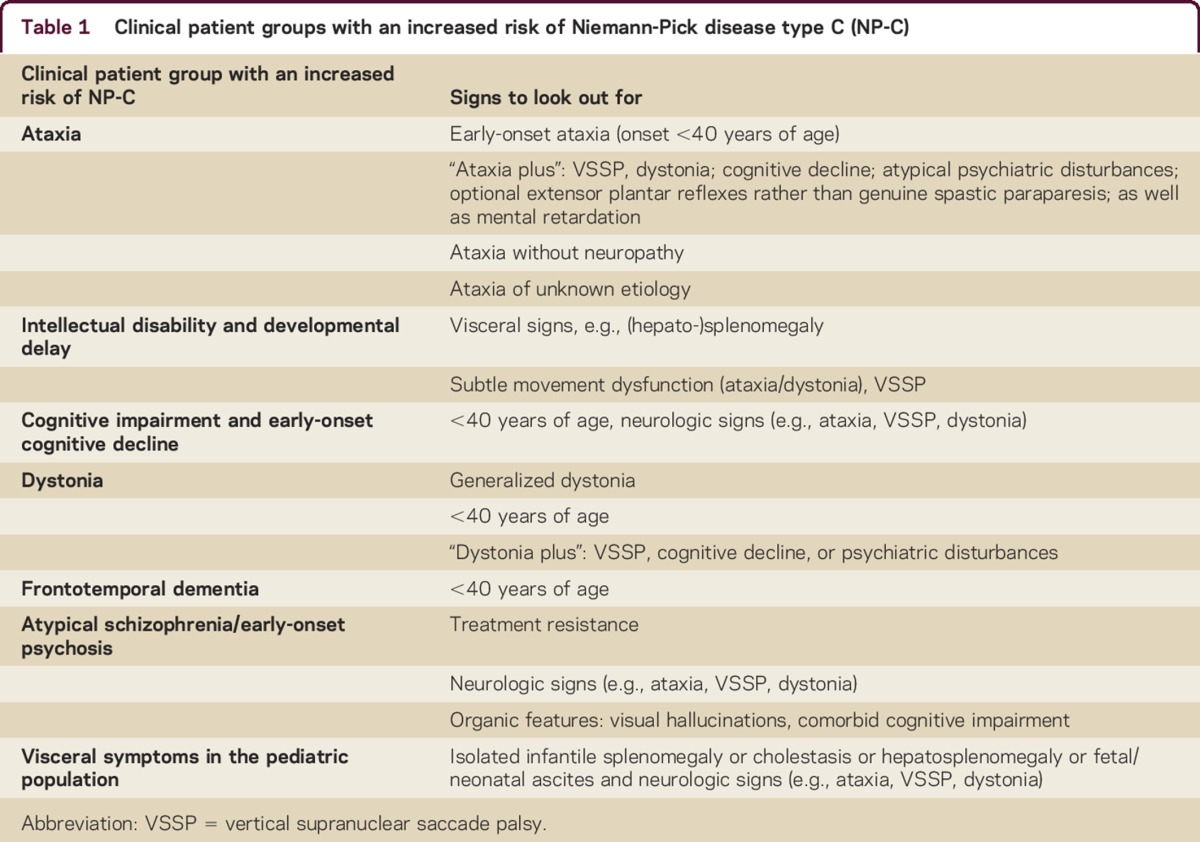
Ataxia
Intellectual disability and developmental delay
Patients with NP-C presenting with intellectual disability and developmental delay (ID/DD) typically have other manifestations (e.g., liver abnormalities in infancy, splenomegaly) or neurologic signs (e.g., ataxia or dystonia).
Detailed history and examination should exclude NP-C as a cause of ID/DD.
Further genetic and biomarker investigation are mandatory when consanguinity, family history of unexplained neurologic disease, or focal neurologic or visceral signs are present.
Cognitive impairment and early-onset cognitive decline
Dystonia
Dystonia is a relatively common presenting feature in patients with NP-C, particularly in juvenile or adult neurologic onset.4
For patients presenting with focal idiopathic dystonia, or dystonia in combination with VSSP, cognitive decline, or psychiatric disturbances (“dystonia plus”; see table 1), NP-C should be considered as a potential diagnosis.
Myoclonus, chorea, or tremor may also occur.
Frontotemporal dementia
Patients with NP-C show symptoms of progressive dementia, impaired cognitive functioning, and behavioral problems2 consistent with those observed in frontotemporal dementia.26
The degree of overlap between the symptoms of frontotemporal dementia and NP-C suggests that this patient group warrants screening for NP-C.
Atypical schizophrenia/early-onset psychosis
Schizophrenia-like psychosis is relatively common in patients with NP-C.17,27
Additional signs include early onset of mental confusion and cognitive impairment, hallucinations (visual being more important than other sensory components), catatonia, and resistance to antipsychotic treatment.27
Co-occurrence of psychosis and any other neurologic signs should raise suspicion of an organic cause, with NP-C as a differential diagnosis.
Visceral symptoms in the pediatric population
NP-C should be considered in the differential diagnosis of children with visceral symptoms such as hepatosplenomegaly, isolated splenomegaly, prolonged unexplained neonatal jaundice lasting >2 weeks with conjugated hyperbilirubinemia, and acute neonatal liver failure,1 as such symptoms can be the initial manifestation of NP-C.2
Visceral findings are common in other metabolic diseases.1
Neuroimaging tests
MRI and PET may be useful, but are not specific for NP-C.
MRI findings commonly include atrophy of the cerebellum and cortex (particularly frontal zones) in more advanced disease, but this may be absent in early stages.
Segmentation of subcortical structures may demonstrate volumetric reductions in the thalamus and hippocampus.28
PET commonly shows hypometabolism of the thalamus and cerebellum.29
Absence of abnormal MRI findings does not exclude a diagnosis of NP-C. Changes in MRI findings are highly variable, nonspecific, and of uncertain sensitivity.
Diagnosis
Biomarkers
Biomarker screening methods can now be considered a first-line step in the NP-C diagnostic process (figure).
Compared with the filipin staining test, biomarkers have several advantages, including noninvasiveness, rapidity, higher throughput, lower cost, and ease of use (regarding sample preparation, stability, and shipment). Sensitivity and specificity of these methods have been reported.30
Figure. Revised Niemann-Pick disease type C (NP-C) diagnostic algorithm for the use of biomarkers and genetic testing.

Individual steps can be omitted if not locally available. aAt-risk patient populations are defined in table 1. bNegative biomarkers may be suggestive that the diagnosis is not NP-C. cSingle-gene sequencing (exons or known mutations) or other. d+1 Variant of unknown significance optional. eCovers deep intronic sequencing and if possible gene transcription regulatory regions. fTwo different alleles. gBiomarker(s) profiling (if not initially conducted), or extended biomarkers profiling (in addition to those already conducted). Despite comprehensive investigations, it may not be possible to definitively confirm a diagnosis of NP-C in a few patients. In such cases, a thorough reappraisal of longitudinal clinical data, more in-depth genomic analyses (e.g., whole exome and whole genome sequencing), and cell biological assessments could be considered. cDNA = complementary DNA; MLPA = multiplex ligation-dependent probe amplification.
Currently available biomarkers
Oxysterols
Oxysterols (cholesterol oxidation products) are the most established, accessible, and widely used biomarkers, with the largest evidence base to support their reliability and sensitivity for NP-C.30
Cholestane-3β,5α,6β-triol (C-triol) and 7-ketocholesterol (7-KC) were shown to be elevated in plasma from patients with NP-C.6–8
C-triol is the preferred oxysterol biomarker, with superior specificity and sensitivity for NP-C compared with 7-KC.8,31 C-triol values for the upper quartile of NP-C carriers overlap with those of patients with NP-C.
Elevated C-triol and 7-KC levels occur in other diseases, mainly NP-A and NP-B, and acid lipase deficiency30–32; therefore, elevated oxysterol levels should be interpreted with caution.
Prolonged storage of samples at room temperature can lead to autoxidation of cholesterol, which may cause false-positive results.7
Other currently validated biomarkers
Newer biomarkers may one day eclipse oxysterols. Desirable qualities include increased stability, more convenient sampling methods (e.g., dried blood spot [DBS] or urine), smaller blood volumes needed (e.g., DBS with infants), more convenient handling, and characteristic NP-C profiles facilitating differential diagnosis when used in multianalyte panels.30,33
Lysosphingomyelin-509 and other lysosphingolipids
Lysosphingomyelin-509 (Lyso-SM-509) is elevated in plasma from patients with NP-C and NP-A/B compared with controls.9,33
The increase of Lyso-SM in patients with NP-C is very small compared with that of patients with NP-A/B; therefore, measurement of combinations of biomarkers, such as Lyso-SM and Lyso-SM-509, allows distinction between NP-C and sphingomyelinase deficiencies (NP-A/B).30,33 In the future, additional derivatives of Lyso-SM and multianalyte panels may be able to further differentiate between NP-A/B, NP-C, and other related diseases.
Bile acids
Specific bile acids have been found to be elevated in patients with NP-C. The analytical species of choice is 3β,5α,6β-trihydroxy-cholanoyl-glycine.11,12 The assay is applicable to DBS, plasma, and urine, and preanalytical auto-oxidation is not a concern.11,12
3β,5α,6β-trihydroxy-cholanoyl-glycine appears to be more specific for NP-C than C-triol; other than NP-C, it is only known to be elevated in NP-A/B, and it better discriminates NP-C carriers from patients.11,12
When and in whom to use biomarkers
Any/all biomarkers should be tested as early as possible in the following:
Patients presenting with splenomegaly/hepatosplenomegaly, cholestatic jaundice in neonates or young infants, or neurologic or psychiatric symptoms
Patients with a high clinical suspicion of NP-C
Patients in at-risk clinical groups
In addition:
When the initially selected biomarker does not show a profile consistent with that of NP-C, additional biomarkers should be considered.
Oxysterols may not be discriminatory in the presence of neonatal cholestasis34; bile acid biomarkers may be more suitable for diagnostics in this population.11 Although newborn screening is technically feasible and being explored, there is currently insufficient evidence to recommend implementation, and ethical considerations must first be accounted for.
Biomarkers alone provide a very high suspicion of NP-C but diagnoses must be confirmed by genetic testing.
Current place of the filipin staining test
The filipin staining test, the historical gold standard assay for NP-C diagnosis,30 is no longer favored as the initial laboratory diagnostic test.
The filipin staining test is a tool to assess the functional significance of new NPC1 or NPC2 genetic variants, and helpful for confirming a diagnosis in patients for whom genetic testing has not allowed identification of 2 pathogenic alleles30 (figure).
Molecular genetics and NP-C
It is crucial to genetically confirm a diagnosis in patients with high clinical suspicion and/or a biomarker profile consistent with NP-C.30
Almost 700 variants, 400 of which are designated pathogenic mutations, are known for NPC1 and 23 have been described for NPC2. The highly polymorphic nature of NPC1 can confound diagnostic conclusions and make interpretation of new mutations a challenge.
The estimated proportion of NP-C cases in which detection of mutations on both alleles can be reached using routine sequencing methods (exons and boundaries) is currently higher than 90%.
It should be noted that some genetic changes, for example large deletions/duplications or deep intronic changes, cannot be reliably identified by these methods.
Detection of such mutations (estimated at ∼5%) may therefore require other techniques (see below). A small percentage of mutant NPC1 alleles (below 5%) has remained unidentified in all experienced groups.35
Pathogenic variants of NPC1 and NPC2 include point mutations that are detectable by standard sequencing methods, and more complex mutations that require additional techniques, as described below.
Traditional sequencing methods
Sanger sequencing of genomic DNA (gDNA) or complementary DNA (cDNA) was traditionally recommended, in parallel with the filipin staining test, to confirm a diagnosis of NP-C.1
NGS and gene panels
NGS technologies provide accurate and sensitive methods for genetic analysis.
The use of only high-quality reads and integrated software tools is essential. To assess sensitivity, specificity, and reproducibility, all gene panels must be evaluated using standardized samples (e.g., Genome in a Bottle samples). For further information, refer to the following standards and guidelines: acmg.net/ACMG/Publications/Laboratory_Standards___Guidelines/ACMG/Publications/Laboratory_Standards___Guidelines.aspx and eurogentest.org/index.php?id=958.
Targeted gene and multigene panels including NPC1 and NPC2, with complete exons and exon–intron boundaries, can be used to screen clinical patient groups with an increased risk of NP-C.
NPC1 and NPC2 are included in several gene panels, including clinical exome, early-onset ataxia,13,22 epilepsy, dystonia, infantile cholestasis,36 specific inborn errors of metabolism (e.g., lysosomal storage diseases), organic psychosis, early-onset cognitive decline, hepatosplenomegaly, and developmental delay; new gene panels are in development.
Diagnostic yield seems to be highest among patients with early-onset ataxia, where 1%–2% of the patients have been found to have NP-C.13
Prevalence of the other phenotypes mentioned above, even in clinically distinct patient cohorts, is lower (<1%).37
Panels in which all diagnostic target regions are not covered above a threshold that ensures diagnosis must be reported, and if necessary, evaluated by another technology (e.g., Sanger sequencing) to provide full coverage.
Complementary genetic tests
In patients with high clinical suspicion and/or positive biomarker results, a negative or heterozygous result from a primary genetic test (e.g., Sanger sequencing) should always be questioned and confirmed by additional molecular studies able to detect complete or partial gene deletions/duplications and deep intronic mutations.1,30
Heterozygous complete or partial deletions/duplications of NPC1 or NPC2 require quantitative techniques to assess copy numbers.30,38,39 Array comparative genomic hybridization (CGH) can be used to detect large DNA dosage alterations. For DNA dosage alterations below the array CGH platform resolution, quantitative real-time PCR or multiplex ligation-dependent probe amplification should be used, to specifically quantify NPC1 and NPC2 copy numbers.
To detect deep intronic mutations, full-gene sequencing can be performed. Owing to the polymorphic nature of intronic regions, cDNA analysis followed by the sequencing of the specific intronic regions within the gDNA represents a highly preferred strategy.
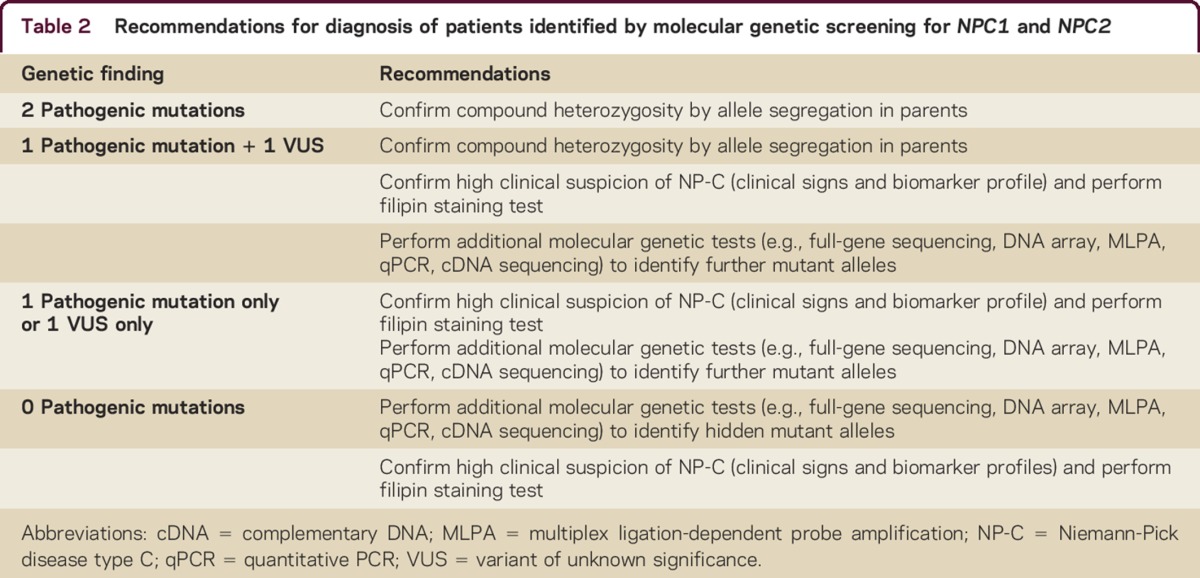
Molecular genetics and NP-C diagnosis
Table 2 shows an approach to the interpretation of molecular genetics results.
Online databases should be used to record and assess the pathogenicity of mutations (e.g., NPC-db2 database in Tübingen; see medgen.medizin.uni-tuebingen.de/NPC-db2/index.php).
Adherence to the American College of Medical Genetics and Genomics guidelines is recommended for assessment and reporting of new mutations.
Table 2.
Recommendations for diagnosis of patients identified by molecular genetic screening for NPC1 and NPC2
Mutations and genotype/phenotype associations
The 2 most frequent NPC1 mutations reported worldwide are p.I1061T (c.3182T<C in exon 21), accounting for 20% of mutant alleles in Western Europeans, and p.P1007A, also prevalent in patients of European descent.40 A number of recurrent mutations are highly prevalent in patients originating from particular countries.
Some NPC1 mutations (including p.P1007A) are associated with less pronounced cholesterol trafficking alterations (variant filipin profile).30
A few missense mutations appear to be clearly associated with a late-onset form of NP-C, even in compound heterozygosity with a very severe/null allele.
In the NPC2 gene, 23 disease-causing mutations (including a large deletion) have been described; the majority are severe, leading to a truncated protein and null alleles, but some (p.V39M and p.P120S) lead to later-onset forms.
A revised laboratory diagnostic algorithm for the use of biomarkers and genetic testing in NP-C
Clinical assessment and symptom profiling are important in guiding physicians to suspect NP-C.
The previous diagnostic algorithm has been revised to reflect our understanding of the value in screening not only individual patients with a high clinical suspicion of NP-C but also clinical patient groups with an increased risk of NP-C, the availability of diagnostic biomarkers for NP-C, and advancements in molecular genetics. An updated diagnostic algorithm reflecting current best practice is shown in the figure.
Positive genetic testing results, or positive biomarker testing results combined with molecular genetic analysis, are now deemed sufficient to diagnose NP-C in most cases.
Local factors (e.g., availability of, and access to, services) determine the choice and order of diagnostic tests.
Treatment
Miglustat (Zavesca; Actelion Pharmaceuticals Ltd., Allschwil, Switzerland) therapy for existing neurologic manifestations should be considered, as discussed in the previous recommendations.1
Disease monitoring
Neurologic function and quality of life should be monitored, as outlined in the previous recommendations.1
Future considerations
There is no single biomarker specific for NP-C. Combining biomarkers that reflect defective cholesterol trafficking/oxidative stress with lysosphingolipid biomarkers could improve screening specificity. Sensitivity has not yet been evaluated for biomarker combinations.
NP-C is a global disease, yet expert testing laboratories are not available in every country. Diagnostic workflows should include easy, robust, and inexpensive sampling, shipment, and storage technologies; DBS filtercards, shown to offer genetic and biochemical value for NP-C, should be evaluated for a more general approach.
With the advent of whole-genome sequencing, interpretation tools for variants of unknown significance require improvement by systematically establishing functional assays for missense and splicing mutations or transcriptional failure.
Incorporation of NP-C biomarker tests into routine panels (e.g., metabolic) for screening in populations with increased risk of NP-C, in particular neonates with cholestasis and hepatosplenomegaly, could facilitate earlier diagnosis.
Better understanding of the genotype–phenotype correlation in NP-C will allow a personalized approach to treatment.
Currently, miglustat is the only approved therapy for patients with NP-C. Other treatments are undergoing investigation.
AUTHOR CONTRIBUTIONS
M.C. Patterson, P. Clayton, P. Gissen, M. Anheim, P. Bauer, O. Bonnot, A. Dardis, C. Dionisi-Vici, H.-H. Klünemann, P. Latour, C.M. Lourenço, D.S. Ory, A. Parker, M. Pocovi, M. Strupp, M.T. Vanier, M. Walterfang, and T. Marquardt attended the NP-C diagnostic recommendations meeting, agreed to be accountable for all aspects of the work, assessed available literature, contributed to the drafting and revision of the recommendations publication, and approved the final version for submission.
ACKNOWLEDGMENT
The authors comprised the NP-C Diagnostic Recommendations Expert Panel. All members of the Expert Panel assessed the recently published literature and contributed to the development of the updated diagnostic recommendations. Editorial assistance was provided by Katie Bickford of Fishawack Communications GmbH, Basel, Switzerland. Editorial assistance was limited to copyediting, formatting, and administrative support, but did not contribute to development of intellectual content.
STUDY FUNDING
This work was supported by Actelion Pharmaceuticals Ltd.
DISCLOSURES
M.C. Patterson serves/has served on scientific advisory boards for Actelion, Shire, Stem Cells, Inc., Amicus, Agios, Vtesse, IntraBio, Orphazyme, and Alexion; has received funding for travel and speaker honoraria from Actelion; serves as Section Editor on Pediatric Neurology for Up-To-Date, Editor-in-Chief for Journal of Child Neurology and Child Neurology Open, and Editor of Journal of Inherited Metabolic Disease; serves as a consultant for Actelion, Agios, Novartis, Genzyme, Orphazyme, Vtesse, Alexion, and IntraBio; participates in activities for Institute of Medicine and WHO; receives research support from Actelion, Merck Serono, NIH, and National MS Society; and holds stock/stock options in IntraBio. P. Clayton serves on a scientific advisory board for and received funding for travel and speaker honoraria from Actelion; serves as Communicating Editor for Journal of Inherited Metabolic Disease; receives publishing royalties for Brain's Diseases of the Nervous System, 12th Ed. (Oxford University Press, 2009); receives research support from Actelion, NIHR, and Great Ormond Street Hospital Children's Charity. P. Gissen serves on a scientific advisory board for Synageva; has received funding for travel and/or speaker honoraria from Actelion, BioMarin, SOBI, Dipharma, and BlueBirdBio: and receives research support from Actelion, BioMarin, SOBI, Dipharma, a Wellcome Trust Senior Clinical Research Fellowship, and the European Research Council. M. Anheim serves on scientific advisory boards for Actelion and AbbVie; has received funding for travel and/or speaker honoraria from Novartis, TEVA/Lundbeck, Actelion, AbbVie, and UCB; and serves as an Associate Editor for Revue Neurologique. P. Bauer has served on a scientific advisory board/as a consultant for Centogene; has received funding for travel and speaker honoraria from Actelion and Centogene; holds stock/stock options in and serves as Chief Scientific Officer for Centogene since 2016; and receives research support from Actelion, Germany Ministry of Research (BMBF), European Commission (EC NeurOmics), and German Research Community (DFG). O. Bonnot serves on a scientific advisory board for Actelion; receives funding for travel and/or speaker honoraria from Actelion Pharma, Orphan Europe, and Shire; and receives research support from Actelion. A. Dardis has served on a scientific advisory board for Actelion; has received funding for travel from Actelion and Orphazyme; serves as an Academic Editor for PLOS ONE; and has received research support from Actelion, Orphazyme, Shire, Telethon Foundation, and the Michael J. Fox Foundation. C. Dionisi-Vici serves on scientific advisory boards for Promethera, Synageva, Sanofi Genzyme, and Nutricia; has received funding for travel and/or speaker honoraria from Actelion, Orphan Europe, and Nutricia; and receives research support from Genzyme, Actelion, Edison Pharmaceuticals, Nutricia, Fondazione Telethon Italia, Fondazione Mariani, and Associazione la Vita è un Dono. H.-H. Klünemann serves on a scientific advisory board for Actelion; has received funding for travel and/or speaker honoraria from NPC Suisse, Actelion, AstraZeneca, Lilly, and Biogen; and serves on the speakers' bureau for Actelion. P. Latour serves on a scientific advisory board for and received speaker honoraria from Actelion. C.M. Lourenco has received speaker honoraria from Actelion, Alexion, and Shire. All fees are donated to the CML Medical Foundation for Research and Diagnosis Support, helping patients without insurance to access diagnostic testing to diagnose their genetic disorder. D.S. Ory serves on scientific advisory boards for Vtesse and Ara Parseghian Medical Research Fund; serves on the Editorial Board of JCI Insight; is an author on patents re: (1) Methods of determining efficacy of Cyclodextrin Therapy and (2) Disease-specific biomarkers for Niemann-Pick C Disease; serves as a consultant for Actelion, LAM Therapeutics, Vtesse, and Calporta; receives research support from NIH (NINDS, NHLBI), Dana's Angels Research Trust, LAM Therapeutics, and Navitor; and receives license fee payments from Novus for technology re: NPC1 rabbit polyclonal antibody, and from Calporta for licensing of the NPC1 I1061T knockin mouse. A. Parker serves on scientific advisory boards for Actelion, Novartis, and GW Pharmaceuticals; and has received speaker's honoraria or consultation fees and travel expenses from Actelion. M. Pocovi has received speaker honoraria or consultation fees and travel expenses from Actelion, Shire, and Amgen; and receives research support from Instituto Aragones de Ciencias de la Salud and CIBERCV. M. Strupp serves on scientific advisory boards for Abbott, Actelion, Auris Medical, Sensorion, Heel, IntraBio, and Pierre-Fabre; has received speaker honoraria from Abbott, GlaxoSmithKline, Merck Serono, Hennig Pharma, Pierre Fabre, UCB, TEVA, Heel, Biogen Idec, Interacoustics, Otometrics, and Eisai Inc.; serves as Joint-Chief Editor of the Journal of Neurology, Editor-in-Chief of Frontiers in Neuro-otology, and Section Editor of F1000; and receives publishing royalties for Leitsymptom Schwindel (Springer, 2012) and Vertigo and dizziness: Common complaints (Springer, 2013). M.T. Vanier serves on scientific advisory boards for and has received funding for travel and/or speaker honoraria from Shire, Actelion, Vtesse, and Genzyme. M. Walterfang has served on a scientific advisory board/as a consultant for and received funding for travel from Actelion. T. Marquardt has received speaker honoraria, travel expenses, and research funding from Actelion Pharmaceuticals. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.

REFERENCES
- 1.Patterson MC, Hendriksz CJ, Walterfang M, Sedel F, Vanier MT, Wijburg F. Recommendations for the diagnosis and management of Niemann-Pick disease type C: an update. Mol Genet Metab 2012;106:330–344. [DOI] [PubMed] [Google Scholar]
- 2.Vanier MT. Niemann-Pick disease type C. Orphanet J Rare Dis 2010;5:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wassif CA, Cross JL, Iben J, et al. High incidence of unrecognized visceral/neurological late-onset Niemann-Pick disease, type C1, predicted by analysis of massively parallel sequencing data sets. Genet Med 2016;18:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Patterson MC, Mengel E, Wijburg FA, et al. Disease and patient characteristics in NP-C patients: findings from an international disease registry. Orphanet J Rare Dis 2013;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wraith JE, Baumgartner MR, Bembi B, et al. Recommendations on the diagnosis and management of Niemann-Pick disease type C. Mol Genet Metab 2009;98:152–165. [DOI] [PubMed] [Google Scholar]
- 6.Jiang X, Sidhu R, Porter FD, et al. A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J Lipid Res 2011;52:1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reunert J, Fobker M, Kannenberg F, et al. Rapid diagnosis of 83 patients with Niemann Pick type C disease and related cholesterol transport disorders by cholestantriol screening. EBioMedcine 2016;4:170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boenzi S, Deodato F, Taurisano R, et al. A new simple and rapid LC-ESI-MS/MS method for quantification of plasma oxysterols as dimethylaminobutyrate esters: its successful use for the diagnosis of Niemann-Pick type C disease. Clin Chim Acta 2014;437:93–100. [DOI] [PubMed] [Google Scholar]
- 9.Giese AK, Mascher H, Grittner U, et al. A novel, highly sensitive and specific biomarker for Niemann-Pick type C1 disease. Orphanet J Rare Dis 2015;10:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Welford RW, Garzotti M, Lourenco C, Mengel E, Marquardt T, Reunert J. Plasma lysosphingomyelin demonstrates great potential as a diagnostic biomarker for Niemann-Pick disease type C in a retrospective study. PLoS One 2014;9:e114669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang X, Sidhu R, Mydock-McGrane L, et al. Development of a bile acid-based newborn screen for Niemann-Pick disease type C. Sci Transl Med 2016;8:337ra363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mazzacuva F, Mills P, Mills K, et al. Identification of novel bile acids as biomarkers for the early diagnosis of Niemann-Pick C disease. FEBS Lett 2016;590:1651–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Synofzik M, Harmuth F, Stampfer M, Muller Vom Hagen J, Schols L, Bauer P. NPC1 is enriched in unexplained early onset ataxia: a targeted high-throughput screening. J Neurol 2015;262:2557–2563. [DOI] [PubMed] [Google Scholar]
- 14.Hendriksz CJ, Anheim M, Bauer P, et al. The hidden Niemann-Pick type C patient: clinical niches for a rare inherited metabolic disease. Curr Med Res Opin 2017;33:877–890. [DOI] [PubMed] [Google Scholar]
- 15.Pineda M, Mengel E, Jahnova H, et al. A Suspicion Index to aid screening of early-onset Niemann-Pick disease type C (NP-C). BMC Pediatr 2016;16:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sevin M, Lesca G, Baumann N, et al. The adult form of Niemann-Pick disease type C. Brain 2007;130:120–133. [DOI] [PubMed] [Google Scholar]
- 17.Maubert A, Hanon C, Sedel F. Psychiatric disorders in adult form of Niemann-Pick disease type C [in French]. Encephale 2016;42:208–213. [DOI] [PubMed] [Google Scholar]
- 18.Bauer P, Balding DJ, Klünemann HH, et al. Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: findings from the ZOOM study. Hum Mol Genet 2013;22:4349–4356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Strupp M, Kremmyda O, Adamczyk C, et al. Central ocular motor disorders, including gaze palsy and nystagmus. J Neurol 2014;261(suppl 2):S542–S558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wijburg FA, Sedel F, Pineda M, et al. Development of a suspicion index to aid diagnosis of Niemann-Pick disease type C. Neurology 2012;78:1560–1567. [DOI] [PubMed] [Google Scholar]
- 21.Hendriksz C, Pineda M, Fahey M, et al. The Niemann-Pick disease type C suspicion index: development of a new tool to aid diagnosis. J Rare Dis Diagn Ther 2015;1:11. [Google Scholar]
- 22.Synofzik M, Fleszar Z, Schols L, et al. Identifying Niemann-Pick type C in early-onset ataxia: two quick clinical screening tools. J Neurol 2016;263:1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol 2015;15:257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anheim M, Tranchant C, Koenig M. The autosomal recessive cerebellar ataxias. N Engl J Med 2012;366:636–646. [DOI] [PubMed] [Google Scholar]
- 25.Schicks J, Muller Vom Hagen J, Bauer P, et al. Niemann-Pick type C is frequent in adult ataxia with cognitive decline and vertical gaze palsy. Neurology 2013;80:1169–1170. [DOI] [PubMed] [Google Scholar]
- 26.Chow TW, Miller BL, Boone K, Mishkin F, Cummings JL. Frontotemporal dementia classification and neuropsychiatry. Neurologist 2002;8:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bonnot O. Niemann-Pick disease type C: example of an inborn error of metabolism producing psychiatric manifestations. Eur Psychiatr Rev 2011;4:84–88. [Google Scholar]
- 28.Walterfang M, Patenaude B, Abel LA, et al. Subcortical volumetric reductions in adult Niemann-Pick disease type C: a cross-sectional study. AJNR Am J Neuroradiol 2013;34:1334–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pineda M, Perez-Poyato MS, O'Callaghan M, et al. Clinical experience with miglustat therapy in pediatric patients with Niemann-Pick disease type C: a case series. Mol Genet Metab 2010;99:358–366. [DOI] [PubMed] [Google Scholar]
- 30.Vanier MT, Gissen P, Bauer P, et al. Diagnostic tests for Niemann-Pick disease type C (NP-C): a critical review. Mol Genet Metab 2016;118:224–254. [DOI] [PubMed] [Google Scholar]
- 31.Boenzi S, Deodato F, Taurisano R, Goffredo BM, Rizzo C. Evaluation of plasma cholestane-3β,5α,6β-triol and 7-ketocholesterol in inherited disorders related to cholesterol metabolism. J Lipid Res 2016;57:361–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Romanello M, Zampieri S, Bortolotti N, et al. Comprehensive evaluation of plasma 7-ketocholesterol and Cholestan-3beta,5alpha,6beta-triol in an Italian cohort of patients affected by Niemann-Pick disease due to NPC1 and SMPD1 mutations. Clin Chim Acta 2016;455:39–45. [DOI] [PubMed] [Google Scholar]
- 33.Polo G, Burlina AP, Kolamunnage TB, et al. Diagnosis of sphingolipidoses: a new simultaneous measurement of lysosphingolipids by LC-MS/MS. Clin Chem Lab Med 2016;55:403–414. [DOI] [PubMed] [Google Scholar]
- 34.Polo G, Burlina A, Furlan F, et al. High level of oxysterols in neonatal cholestasis: a pitfall in analysis of biochemical markers for Niemann-Pick type C disease. Clin Chem Lab Med 2015;54:1221–1229. [DOI] [PubMed] [Google Scholar]
- 35.Stampfer M, Theiss S, Amraoui Y, et al. Niemann-Pick disease type C clinical database: cognitive and coordination deficits are early disease indicators. Orphanet J Rare Dis 2013;8:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Herbst SM, Schirmer S, Posovszky C, et al. Taking the next step forward: diagnosing inherited infantile cholestatic disorders with next generation sequencing. Mol Cell Probe 2015;29:291–298. [DOI] [PubMed] [Google Scholar]
- 37.Yang Y, Muzny DM, Reid JG, et al. Clinical whole-exome sequencing for the diagnosis of Mendelian disorders. N Engl J Med 2013;369:1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bauer P, Knoblich R, Bauer C, et al. NPC1: complete genomic sequence, mutation analysis, and characterization of haplotypes. Hum Mutat 2002;19:30–38. [DOI] [PubMed] [Google Scholar]
- 39.Rodriguez-Pascau L, Toma C, Macias-Vidal J, et al. Characterisation of two deletions involving NPC1 and flanking genes in Niemann-Pick type C disease patients. Mol Genet Metab 2012;107:716–720. [DOI] [PubMed] [Google Scholar]
- 40.Runz H, Dolle D, Schlitter AM, Zschocke J. NPC-db, a Niemann-Pick type C disease gene variation database. Hum Mutat 2008;29:345–350. [DOI] [PubMed] [Google Scholar]