

Abstract
Background:
To prospectively evaluate the progression of cognitive-behavioral function in amyotrophic lateral sclerosis (ALS) and examine the association of cognitive-behavioral deficits with disease progression, patient quality of life (QOL), and caregiver burden.
Methods:
We evaluated cognitive-behavioral function using the Amyotrophic Lateral Sclerosis Cognitive Behavioral Screen at enrollment and after 7 months in a cohort of patients with ALS. Paired t tests were used to evaluate the change in the 2 assessments. Linear regression and Kruskal-Wallis tests were applied to investigate how initial cognitive or behavioral status related to outcomes.
Results:
The mean test-retest interval was 6.8 months (SD 1.6). Cognitive status of the study population (n = 49) overall did not change over the study period (p = 0.06) despite progression of motor weakness (p < 0.001), though small subsets of the sample demonstrate cognitive change. Patients initially classified as behaviorally normal showed increased behavioral problems over time (t = −2.8, p = 0.009). Decline in cognitive (β = −1.3, p = 0.03) and behavioral (β = −0.76, p = 0.002) status predicted increasing caregiver burden. Behavioral abnormalities predicted decline in forced vital capacity and ALS Functional Rating Scale–Revised score (p = 0.008, 0.012) in the study population and patient QOL in the most severely affected group (t = 4.3, p = 0.003).
Conclusions:
Cognitive-behavioral change is a key aspect of disease heterogeneity in ALS. Executive function in ALS overall remains stable over 7 months as detected by an administered screening tool. However, patients may develop caregiver-reported behavioral symptoms in that time period. Screening for caregiver-reported symptoms has a particular utility in predicting future clinical decline, increased caregiver burden, and worsening patient QOL.
In recent years, there has been greater appreciation for the cognitive and behavioral manifestations of diseases traditionally associated with motor weakness. Amyotrophic lateral sclerosis (ALS), Parkinson disease, and multiple sclerosis are now considered multisystem disorders with important neuropsychological symptoms.1 For ALS, there is evidence for a clinicopathologic continuum with frontotemporal dementia (FTD).2–4 Up to half of patients with ALS exhibit deficits in executive function.5
Investigations of the progression of cognitive-behavioral deficits in patients with ALS have been limited to small numbers of patients who undergo a full neuropsychological battery.6,7 There have not been rigorous evaluations of the progression of cognitive-behavioral changes as detected by a neuropsychological screening tool, which may be more practical for routine use. Though cognitive-behavioral deficits are a harbinger of clinical decline8 and screening is currently recommended in patients with ALS,9 a better understanding of the progression and effects of cognitive-behavioral changes is needed to inform recommendations for optimal screening intervals, the timing of medical interventions, and the design of supportive interventions.
The aims of this study are twofold. We prospectively evaluate the progression of cognitive and behavioral function in a clinically diverse population of patients with ALS using a validated neuropsychological screening tool and we examine the association of cognitive-behavioral deficits with disease progression, patient quality of life (QOL), and caregiver burden.
METHODS
Study design and participants
This prospective cohort study involved 2 assessments over the course of 7 months. All patients who attended an appointment between July 2014 and January 2015 at the University of California, San Francisco (UCSF) ALS Center or 2 affiliated satellite clinics were screened for eligibility. A diagnosis of definite, probable, or possible ALS by the El Escorial criteria10 and English language fluency were required for study enrollment. Patients were excluded if they carried a premorbid diagnosis of non-FTD dementia or a comorbid neurologic diagnosis, received a new ALS diagnosis on the day of enrollment, or lacked a caregiver who had known the patient prior to symptom onset. At the enrollment appointment, patients underwent a cognitive screening assessment and completed a survey with measures of QOL, depression, and pseudobulbar affect. In addition, caregivers completed a behavioral assessment of the patient and a survey regarding their level of burden. All measures were repeated 7 months later.
Data collection tools
Cognitive function was evaluated using the Amyotrophic Lateral Sclerosis Cognitive-Behavioral Screen (ALS CBS), a screening tool designed for the ALS population that minimizes unintended measurement effects due to motor weakness (appendix e-1 at Neurology.org/cp).11 The total score is calculated from 4 subtests in initiation and retrieval, concentration, attention, and tracking/monitoring. Using prespecified normative cutoffs, patients were divided into 3 categories based on their total score (maximum 20): normal, ALS-cognitive impairment (ALS-Ci), or possible FTD-level impairment.11 Patients also completed a verbal fluency test (C-words), which was used as an internal validation of study measures.12 Patients completing the follow-up assessment were given the option to take the validated telephone version of the ALS CBS.13
Patient behavior was assessed with the caregiver survey component of the ALS CBS, which queries caregivers about signs of apathy, disinhibition, difficulty problem-solving, and language difficulties in the patient. Using prespecified normative cutoffs, patients were divided into 3 categories based on their total score (maximum 45): normal, behavioral impairment (ALS-Bi), or possible FTD-level impairment.11
The ALS Functional Rating Scale–Revised (ALSFRS-R) was used to quantify disease severity.14 Pseudobulbar affect (Center for Neurologic Study Lability Scale) and depressive symptoms (Geriatric Depression Scale [GDS]) were both measured as potential confounders of cognition.15,16 The McGill Quality of Life Single-Item Scale (MQOL-SIS),17 a single self-report item that correlates with more extensive QOL measures in the ALS population,18,19 was used to rate patient QOL. Caregiver strain, isolation, disappointment, emotional involvement, and environment were evaluated by administering the 22-item Caregiver Burden Scale.20
Statistical analyses
Descriptive statistics were calculated for all variables using means and SDs for continuous variables, medians and interquartile ranges for continuous variables with skewed distributions, and counts and percentages for categorical data. Pearson correlation coefficients and analysis of variance tests were used to internally validate study measurements across sites and evaluators. Paired t tests were used to evaluate changes between the first and second assessment for selected variables of interest. Linear regression was used to investigate whether change in outcomes of interest varied by initial cognitive or behavioral category. To investigate predictors of cognitive and behavioral decline, multiple regression was used with a prespecified set of covariates considered to be potential confounders of cognitive-behavioral status (educational level, age, sex, ALSFRS-R score, anatomic region of onset, forced vital capacity [FVC], and GDS score). Kruskal-Wallis tests were used to compare variation of clinical variables across cognitive and behavioral groups. All statistical analyses were conducted using Stata/SE 13.1 (StataCorp, College Park, TX).
Standard protocol approvals, registration, and patient consents
This study was approved by the UCSF Committee on Human Research. Informed consent was obtained from all participating patients and caregivers.
RESULTS
Study population
Baseline characteristics of the study population have previously been reported.21 Of 141 consecutive eligible patients, 86 (61%) participated. No differences in mean age (t = −0.6, p = 0.55) or FVC (t = −0.7, p = 0.50) were noted between eligible participants and nonparticipants. Of the 86 patients initially enrolled, one was subsequently excluded due to a revised diagnosis, 19 died by the end of the study period, and 1 underwent mechanical ventilation. Five patients were lost to follow-up and 11 declined the follow-up assessment. The follow-up rate was 57% (49/86) in the overall sample and 74% (49/66) in the cohort that survived the study period. Of the 49 patients who repeated the cognitive testing, 6 had caregivers who could not be located or declined to complete the follow-up behavioral questionnaire.
The mean follow-up period was 6.8 months (SD 1.6). Ten of 49 patients opted to use the telephone version of the ALS CBS for their second assessment. Using a Wilcoxon-Mann-Whitney test, no difference was found in ALS CBS cognitive subscore between patients who completed in-person and telephone follow-up (z = −0.2, p = 0.9). Additional characteristics of the study population are displayed in table 1.
Table 1.
Baseline characteristics of the analysis cohort (n = 49)

Change in cognition over time
Figure 1 shows cognitive decline over time. The median change in ALS CBS cognitive subscore was 0 in the overall study sample over time (t = 1.9, p = 0.06). When stratified by baseline cognitive category, only the ALS-Ci group had a significant change in cognitive function over time. This group showed a median improvement in ALS CBS cognitive subscore of 1.0 point (t = 3.0, p = 0.007). Compared to the ALS-Ci group, patients who were initially categorized as normal declined by a median of 1.0 point on the ALS CBS cognitive subscore (β = 1.6, p = 0.004). Fifteen percent (5/34) of patients initially categorized as normal were subsequently classified as cognitively impaired (ALS-Ci). Two out of the 49 patients exhibited a decline in ALS CBS cognitive subscore greater than 1 SD from the mean. No patients exhibited a decline in cognitive subscore greater than 2 SDs from the mean.
Figure 1. Change in Amyotrophic Lateral Sclerosis Cognitive Behavioral Screen (ALS CBS) cognitive subscore over time.

(A) Normal. (B) Cognitive impairment. (C) Frontotemporal dementia (FTD). Change in ALS CBS cognitive subscore from initial to follow-up assessment stratified by cognitive category at baseline. Changes for individual participants are shown with gray lines and the average change for the group is shown as a red line. Dotted lines indicate normative cutoffs between cognitive categories (ALS CBS cognitive score ≥17 is normal, 11–16 is cognitive impairment, and ≤10 is FTD range). *Change is significant (p < 0.05).
There were no identifiable patient characteristics (including FVC, age, education, depressive symptoms, disease stage, disease duration, or region of onset) that predicted cognitive change.
Change in behavior over time
Figure 2 shows the progression of behavioral deficits over time. In the overall study population, the ALS CBS behavioral subscore showed a median decline of 1.0 point over time (t = −1.6, p = 0.1). Patients initially classified as behaviorally normal worsened in behavioral status by 2.0 points over time (t = −2.8, p = 0.009). There was no significant change in the behavioral status of patients in the ALS-Bi or FTD groups, which showed a more variable course. When regressing the total change in CBS behavioral score over initial behavioral category, the difference in behavioral change was not different between groups (p = 0.3, 0.1). Of the 31 patients initially categorized as behaviorally normal, 5 (16%) scored in the behaviorally impaired range at follow-up. Behavioral subscores for 10 patients (20%) declined more than 1 SD during the study period. Seven out of these 10 patients were initially classified as behaviorally normal.
Figure 2. Change in Amyotrophic Lateral Sclerosis Cognitive Behavioral Screen (ALS CBS) behavioral subscore over time.

(A) Normal. (B) Behavioral impairment. (C) Frontotemporal dementia (FTD). Change in ALS CBS behavioral subscore from initial to follow-up assessment stratified by behavioral category at baseline. Changes for individual participants are shown with gray lines and the average change for the group is shown as a red line. Dotted lines indicate normative cutoffs between cognitive categories (CBS behavioral score ≥37 is normal, 33–36 is behavioral impairment, and ≤32 is FTD range). *Change is significant (p < 0.05).
When considering predictors of change in behavior, longer disease duration was associated with an improvement in CBS behavioral score (β = 0.03, p = 0.046). There was no association between age, educational level, region of onset, FVC, depressive symptoms, or disease stage and change in behavioral function.
Change in disease stage over time
Disease stage as measured by ALSFRS-R (t = −5.8, p < 0.001) and FVC (t = −3.8, p = 0.001) declined over the course of the study period.
Using the Kruskal-Wallis test, there was no difference found in disease severity over time as measured by change of ALSFRS-R or FVC (p = 0.6, 0.3) when comparing patients initially classified as cognitively normal compared to those with any level of impairment at baseline. Change in FVC (p = 0.008) and ALSFRS-R score (p = 0.012) differed between initial behavioral groups. For patients with behavioral impairment (an initial ALS CBS behavioral subscore less than 37), the average decrease in FVC was 22.2% compared to a decrease of 6.8% in the behaviorally normal group.
Change in QOL over time
Figure 3 shows the change in QOL over time stratified by baseline cognitive and behavioral subgroups. Patient QOL did not change over time in the overall study population (t = −0.85, p = 0.4) or in any subcategory of baseline cognitive impairment. When considering behavioral subcategories, QOL declined in patients who initially met criteria for FTD-level behavioral deficits (t = −4.3, p = 0.003). Change in QOL was associated with initial ALS CBS behavioral subscore in univariate analysis (β = 0.09, p = 0.02), which remained robust after controlling for education, age, sex, region of onset, ALSFRS-R, FVC, and depressive symptoms (β = 0.12, p = 0.009). Worsening apathy (β = 2.0, p = 0.001) and irritability (β = 1.3, p = 0.001) correlated with decreased patient QOL at the second assessment.
Figure 3. Change in patient quality of life (QOL) over time.

(A) Change in QOL by cognitive category. (B) Change in QOL by behavior category. Change in patient QOL as measured by the McGill Quality of Life Single-Item Score over time stratified by initial cognitive and behavioral category among 46 patients with amyotrophic lateral sclerosis. FTD = frontotemporal dementia.
Change in caregiver burden over time
There was an overall increase in caregiver burden as measured by the Caregiver Burden Scale (t = 0.01, p = 0.01). When considering baseline subcategories, the burden of caregivers of patients initially categorized as cognitively normal (t = 2.9, p = 0.012) or behaviorally normal (t = 2.6, p = 0.019) increased over time. For patients with moderate or severe cognitive or behavioral impairment at baseline, caregiver burden did not significantly change. Worsening in behavioral symptoms was associated with increase in caregiver burden over time in univariate analysis (β = −0.76, p = 0.002).
Initial ALS CBS cognitive subscore was not associated with change in caregiver burden in univariate or multivariate analyses (p = 0.2, 0.3). Change in ALS CBS cognitive subscore over time was associated with change in caregiver burden in univariate analysis (β = −1.3, p = 0.03).
See tables 2 and 3 for more detail about the mean change in ALSFRS-R, MQOL-SIS, and caregiver burden over time.
Table 2.
Comparisons of mean change in ALSFRS-R, MQOL-SIS, and caregiver burden over time stratified by initial Amyotrophic Lateral Sclerosis Cognitive-Behavioral Screen cognitive subscore category among 49 patients with ALS
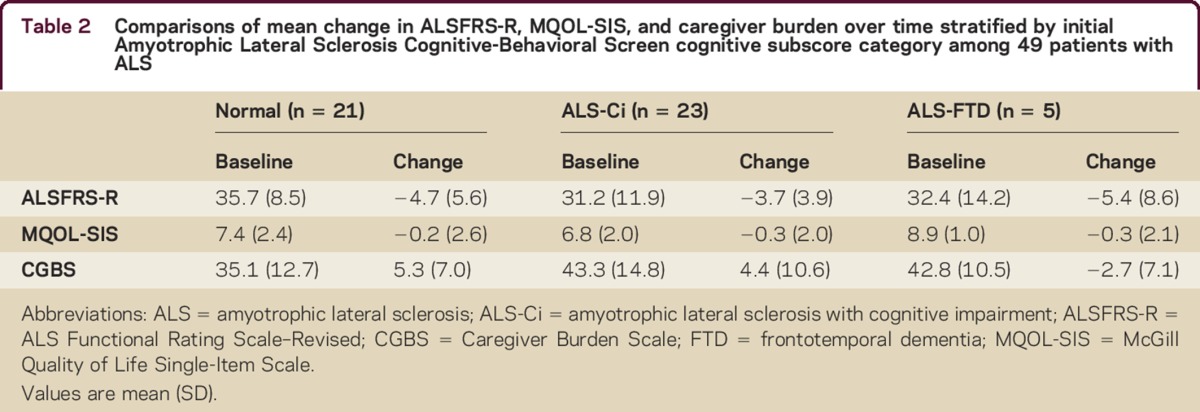
Table 3.
Comparisons of mean change in ALSFRS-R, MQOL-SIS, and caregiver burden over time stratified by initial Amyotrophic Lateral Sclerosis Cognitive-Behavioral Screen behavioral subscore category among 43 patients

DISCUSSION
We prospectively evaluated the use of a neuropsychological screening tool in a typical ALS cohort and found that cognition as detected by the ALS CBS remained stable in the setting of major declines in motor weakness over the study period. Behavioral status had a more variable trajectory depending on the initial level of abnormality. Patients initially classified as behaviorally normal significantly declined between assessments. Patients with moderate or severe behavioral impairment at baseline showed slight improvement overall, but with noticeable individual variability.
Other studies investigating cognitive change over time in patients with ALS have yielded inconsistent results. Most studies did not find a change in cognition in patients with ALS over similar time periods, with the exception of deterioration in word generation7,22,23 or a visuoconstructive task.6 However, several of these studies were performed in restricted samples, did not use neuropsychological assessments specifically validated for this patient population or behavioral assessments, or did not measure patient- or caregiver-reported outcomes of particular interest in this terminal disease.
This study extends our current understanding by applying a practical cognitive screening tool to a clinically diverse study population and by incorporating a measure of behavioral deficits that can be prominent in this patient population. Though routine cognitive-behavioral screening in this population is recommended,9 the ideal screening interval is unknown. Based on our results, a time interval of 7 months does not capture an appreciable decline in cognitive status for most patients. This lack of change (and improvement noted in the cognitively impaired group) could be attributable to a learning effect, regression to the mean, or lack of notable progression of frontotemporal degeneration during that interval. Though 15% of patients who were initially normal met criteria for cognitive impairment at the second assessment, only 2 patients changed more than 1 SD from the mean and no patients changed more than 2 SDs from the mean. It is reasonable to consider a baseline cognitive screen and repeat screening based on clinically apparent changes. Patients initially categorized as behaviorally normal may benefit from routine rescreening in 7 months since significant change in behavioral abnormalities may develop in that time period and behavioral function is highly predictive of several outcomes of interest. Identifying at-risk patients is particularly valuable as they may be appropriate candidates for interventions that have shown promise in preliminary studies.24–26
In this study, a significant relationship was found between initial behavioral symptoms and change in FVC and ALSFRS-R score over the study period. Frontotemporal involvement is a negative prognostic indicator, either as a marker of disease subtype27 or a cause of treatment noncompliance.8 This study highlights the need for clinician awareness of executive dysfunction as a harbinger of clinical decline and the utility of caregiver report as a quick method to ascertain clinically meaningful executive dysfunction.
This study adds to previous literature by evaluating the effect of cognitive-behavioral changes on the psychosocial well-being of patients and caregivers. Previous studies suggest overall patient QOL remains stable over time and can be related to individual patient characteristics, ability to communicate, and social support.28–31 This study evaluates the change of patient QOL in ALS in the context of both objectively measured cognitive changes and caregiver-reported behavioral abnormalities. In our study, cognitive change was not associated with change of patient QOL in the overall study population. However, patients with the most severe behavioral deficits had a significant QOL decline over the course of the study period and change in QOL was significantly predicted by initial behavioral symptoms (particularly apathy and irritability). Given the protective influence of social factors on patient QOL identified by past studies,32 behavioral symptoms and compromised social cognition may strain relationships and adversely affect patient well-being.
Behavioral symptoms were strongly associated with caregiver burden at enrollment21 and change in behavior predicted change in caregiver burden over time. An association between neuropsychiatric symptoms and caregiver burden over time has been shown in studies of patients with FTD33–35 and cross-sectional studies of patients with ALS.36,37 We extend this finding to a longitudinally assessed cohort of caregivers confronting a range of cognitive-behavioral impairment.
Interestingly, burden increased for caregivers of patients initially categorized as normal but remained stable for those taking care of patients with moderate or severe behavioral impairment. Caregivers may have a stable level of burden over time with established behavioral deficits or there may be a floor effect of these particular measurements. The needs of caregivers are understudied,38 though initial studies are investigating interventions to improve coping of those caring for patients with FTD.39
Caregiver-administered surveys are useful in identifying initial behavioral abnormalities and their early changes, but these tools may have limited utility in tracking behavior over longer periods of time. Several patients in this study showed unexpected improvement in behavioral symptoms. This may reflect an actual improvement as a result of medical interventions or increased social support. Alternatively, behavioral symptoms may fluctuate or progress from more disruptive behavioral symptoms (such as disinhibition) to less disruptive ones (such as apathy) that are not as easily captured by caregiver survey. The correlation between longer disease duration and an improvement in CBS behavioral subscore suggests that caregivers may become acclimated to changes over time or develop better behavioral management strategies. Other tools used to track frontally mediated behavioral symptoms are also limited in detecting progression over time.40 The analysis of the subset of patients who improved in this study was limited by sample size and further study is needed. Until the tracking of behavioral symptoms is optimized in ALS, it is important for clinicians to be aware of this plateau effect of most screening measures.
Regarding limitations to this study, it is important to note that the ALS CBS is a neuropsychological screen rather than a diagnostic tool. However, our aim was to evaluate the progression of cognitive-behavioral deficits over time using a practical and validated screening tool since this can inform clinical trial design and may be more easily applied in clinical settings. Study participants were drawn from an academic center and satellite clinics and therefore may not be representative of the entire ALS population. Only one patient required artificial respiration during the study period. There is the possibility of ceiling and floor effects on the caregiver survey component of the ALS CBS, but even considering this limitation we found that this measure was strongly predictive of burden for all caregivers and patient QOL in the most impaired groups. Because the identification of a caregiver was required for study enrollment, the sample may exclude patients with lesser social support, an important determinant of QOL. Finally, the MQOL-SIS is a single item assessment, though it correlates with longer measures in this patient population18,19 and we intentionally chose a tool that would measure global QOL. This measure still displayed informative associations with cognitive-behavioral status that future studies can investigate with more nuance.
Cognitive-behavioral changes are a key aspect of disease heterogeneity in ALS. Furthering our understanding of the trajectory and effects of cognitive-behavioral change is critical to inform our understanding of disease pathogenesis, optimize stratification for clinical trials, and guide the timing of medical or supportive interventions. Using this brief and feasible screening tool, we found that cognition in ALS overall remains stable over a course of 7 months, whereas patients may develop behavioral symptoms in that time period. Behavioral symptoms have particular utility in predicting clinical decline, increased caregiver burden at all time points studied, and decreased patient QOL over time for those with the highest levels of impairment.
AUTHOR CONTRIBUTIONS
M. Bock: Study concept and design, data acquisition and management, statistical analysis and interpretation, writing of manuscript and incorporation of revisions. Y-N. Duong: Data acquisition. A. Kim: Statistical analysis and interpretation, revision of manuscript for important intellectual content. I. Allen: Statistical analysis and interpretation. J. Murphy: Study design, statistical interpretation, revision of manuscript for important intellectual content. C. Lomen-Hoerth: Study concept, revision of manuscript for important intellectual content, study supervision.
ACKNOWLEDGMENT
Susan Woolley-Levine provided use of the ALS Cognitive-Behavioral screen. Betty Smoot provided feedback on project design. Jennifer Creasman assisted with data management. UCSF ALS Center multidisciplinary team provided feedback on project design. Danny Yagan provided informal guidance on statistical analysis.
Footnotes
Supplemental data at Neurology.org/cp
STUDY FUNDING
This publication was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through UCSF-CTSI Grant Number TL1 TR000144. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
DISCLOSURES
M. Bock receives research support from NIH/NCATS. Y.-N. Duong reports no disclosures. A. Kim serves on a DSMB for Neuroavi; serves as Associate Editor for New England Journal of Medicine JournalWatch: Neurology; receives research support from SanBio, Inc., Biogen, and NIH/NINDS; and has served as an expert in medico-legal cases. I. Allen reports no disclosures. J. Murphy holds a paid position with INC Research (a contract research organization) and her husband is employed by Genentech. C. Lomen-Hoerth reports no disclosures. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
REFERENCES
- 1.Murphy J, Factor-Litvak P, Goetz R, et al. Cognitive-behavioral screening reveals prevalent impairment in a large multicenter ALS cohort. Neurology 2016;86:813–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy J, Henry R, Lomen-Hoerth C. Establishing subtypes of the continuum of frontal lobe impairment in amyotrophic lateral sclerosis. Arch Neurol 2007;64:330–334. [DOI] [PubMed] [Google Scholar]
- 3.Strong MJ. The syndromes of frontotemporal dysfunction in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2008;9:323–338. [DOI] [PubMed] [Google Scholar]
- 4.Wilson CM, Grace GM, Munoz DG, He BP, Strong MJ. Cognitive impairment in sporadic ALS: a pathologic continuum underlying a multisystem disorder. Neurology 2001;57:651–657. [DOI] [PubMed] [Google Scholar]
- 5.Lomen-Hoerth C, Murphy J, Langmore S, Kramer JH, Olney RK, Miller B. Are amyotrophic lateral sclerosis patients cognitively normal? Neurology 2003;60:1094–1097. [DOI] [PubMed] [Google Scholar]
- 6.Elamin M, Bede P, Byrne S, et al. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology 2013;80:1590–1597. [DOI] [PubMed] [Google Scholar]
- 7.Gordon PH, Goetz RR, Rabkin JG, et al. A prospective cohort study of neuropsychological test performance in ALS. Amyotroph Lateral Scler 2010;11:312–320. [DOI] [PubMed] [Google Scholar]
- 8.Olney RK, Murphy J, Forshew D, et al. The effects of executive and behavioral dysfunction on the course of ALS. Neurology 2005;65:1774–1777. [DOI] [PubMed] [Google Scholar]
- 9.Miller RG, Jackson CE, Kasarskis EJ, et al. ; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter update: the care of the patient with amyotrophic lateral sclerosis: multidisciplinary care, symptom management, and cognitive/behavioral impairment (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology 2009;73:1227–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 11.Woolley SC, York MK, Moore DH, et al. Detecting frontotemporal dysfunction in ALS: utility of the ALS Cognitive Behavioral Screen (ALS-CBS). Amyotroph Lateral Scler 2010;11:303–311. [DOI] [PubMed] [Google Scholar]
- 12.Abrahams S, Leigh PN, Harvey A, Vythelingum GN, Grise D, Goldstein LH. Verbal fluency and executive dysfunction in amyotrophic lateral sclerosis (ALS). Neuropsychologia 2000;38:734–747. [DOI] [PubMed] [Google Scholar]
- 13.Christodoulou G, Gennings C, Hupf J, et al. Telephone based cognitive-behavioral screening for frontotemporal changes in patients with amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener 2016;17:482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Amyotrophic Lateral Sclerosis Functional Rating Scale. Assessment of activities of daily living in patients with amyotrophic lateral sclerosis: The ALS CNTF Treatment Study (ACTS) Phase I-II Study Group. Arch Neurol 1996;53:141–147. [PubMed] [Google Scholar]
- 15.Moore SR, Gresham LS, Bromberg MB, Kasarkis EJ, Smith RA. A self report measure of affective lability. J Neurol Neurosurgery Psychiatry 1997;63:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yesavage JA. Geriatric Depression Scale. Psychopharmacol Bull 1988;24:709–711. [PubMed] [Google Scholar]
- 17.Cohen SR, Mount BM, Strobel MG, Bui F. The McGill Quality of Life Questionnaire: a measure of quality of life appropriate for people with advanced disease: a preliminary study of validity and acceptability. Palliat Med 1995;9:207–219. [DOI] [PubMed] [Google Scholar]
- 18.Chio A, Gauthier A, Montuschi A, et al. A cross sectional study on determinants of quality of life in ALS. J Neurol Neurosurg Psychiatry 2004;75:1597–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lou JS, Moore D, Gordon PH, Miller R. Correlates of quality of life in ALS: lessons from the minocycline study. Amyotroph Lateral Scler 2010;11:116–121. [DOI] [PubMed] [Google Scholar]
- 20.Elmstahl S, Malmberg B, Annerstedt L. Caregiver's burden of patients 3 years after stroke assessed by a novel caregiver burden scale. Arch Phys Med Rehabil 1996;77:177–182. [DOI] [PubMed] [Google Scholar]
- 21.Bock M, Duong YN, Kim A, Allen I, Murphy J, Lomen-Hoerth C. Cognitive-behavioral changes in amyotrophic lateral sclerosis: screening prevalence and impact on patients and caregivers. Amyotroph Lateral Scler Frontotemporal Degener 2016;17:366–373. [DOI] [PubMed] [Google Scholar]
- 22.Abrahams S, Leigh PN, Goldstein LH. Cognitive change in ALS: a prospective study. Neurology 2005;64:1222–1226. [DOI] [PubMed] [Google Scholar]
- 23.Strong MJ, Grace GM, Orange JB, Leeper HA, Menon RS, Aere C. A prospective study of cognitive impairment in ALS. Neurology 1999;53:1665–1670. [DOI] [PubMed] [Google Scholar]
- 24.Averill AJ, Kasarskis EJ, Segerstrom SC. Expressive disclosure to improve well-being in patients with amyotrophic lateral sclerosis: a randomised, controlled trial. Psychol Health 2013;28:701–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gould RL, Coulson MC, Brown RG, Goldstein LH, Al-Chalabi A, Howard RJ. Psychotherapy and pharmacotherapy interventions to reduce distress or improve well-being in people with amyotrophic lateral sclerosis: a systematic review. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:293–302. [DOI] [PubMed] [Google Scholar]
- 26.van Groenestijn AC, Schroder CD, Visser-Meily JM, Reenen ET, Veldink JH, van den Berg LH. Cognitive behavioural therapy and quality of life in psychologically distressed patients with amyotrophic lateral sclerosis and their caregivers: results of a prematurely stopped randomized controlled trial. Amyotroph Lateral Scler Frontotemporal Degener 2015;16:309–315. [DOI] [PubMed] [Google Scholar]
- 27.Irwin DJ, McMillan CT, Brettschneider J, et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2013;84:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Caligari M, Godi M, Guglielmetti S, Franchignoni F, Nardone A. Eye tracking communication devices in amyotrophic lateral sclerosis: impact on disability and quality of life. Amyotroph Lateral Scler Frontotemporal Degener 2013;14:546–552. [DOI] [PubMed] [Google Scholar]
- 29.Gauthier A, Vignola A, Calvo A, et al. A longitudinal study on quality of life and depression in ALS patient-caregiver couples. Neurology 2007;68:923–926. [DOI] [PubMed] [Google Scholar]
- 30.Korner S, Sieniawski M, Kollewe K, et al. Speech therapy and communication device: impact on quality of life and mood in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler Frontotemporal Degener 2013;14:20–25. [DOI] [PubMed] [Google Scholar]
- 31.Robbins RA, Simmons Z, Bremer BA, Walsh SM, Fischer S. Quality of life in ALS is maintained as physical function declines. Neurology 2001;56:442–444. [DOI] [PubMed] [Google Scholar]
- 32.Goldstein LH, Atkins L, Landau S, Brown RG, Leigh PN. Longitudinal predictors of psychological distress and self-esteem in people with ALS. Neurology 2006;67:1652–1658. [DOI] [PubMed] [Google Scholar]
- 33.Hsieh S, Leyton CE, Caga J, et al. The evolution of caregiver burden in frontotemporal dementia with and without amyotrophic lateral sclerosis. J Alzheimer's Dis 2015;49:875–885. [DOI] [PubMed] [Google Scholar]
- 34.Mioshi E, Foxe D, Leslie F, et al. The impact of dementia severity on caregiver burden in frontotemporal dementia and Alzheimer disease. Alzheimer Dis Assoc Disord 2013;27:68–73. [DOI] [PubMed] [Google Scholar]
- 35.Mourik JC, Rosso SM, Niermeijer MF, Duivenvoorden HJ, Van Swieten JC, Tibben A. Frontotemporal dementia: behavioral symptoms and caregiver distress. Dement Geriatr Cogn Disord 2004;18:299–306. [DOI] [PubMed] [Google Scholar]
- 36.Lillo P, Mioshi E, Hodges JR. Caregiver burden in amyotrophic lateral sclerosis is more dependent on patients' behavioral changes than physical disability: a comparative study. BMC Neurol 2012;12:156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tremolizzo L, Pellegrini A, Susani E, et al. Behavioural but not cognitive impairment is a determinant of caregiver burden in amyotrophic lateral sclerosis. Eur Neurol 2016;75:191–194. [DOI] [PubMed] [Google Scholar]
- 38.Nunnemann S, Kurz A, Leucht S, Diehl-Schmid J. Caregivers of patients with frontotemporal lobar degeneration: a review of burden, problems, needs, and interventions. Int Psychogeriatr 2012;24:1368–1386. [DOI] [PubMed] [Google Scholar]
- 39.Mioshi E, McKinnon C, Savage S, O'Connor CM, Hodges JR. Improving burden and coping skills in frontotemporal dementia caregivers: a pilot study. Alzheimer Dis Assoc Disord 2013;27:84–86. [DOI] [PubMed] [Google Scholar]
- 40.Boutoleau-Bretonniere C, Lebouvier T, Volteau C, et al. Prospective evaluation of behavioral scales in the behavioral variant of frontotemporal dementia. Dement Geriatr Cogn Disord 2012;34:75–82. [DOI] [PubMed] [Google Scholar]