

Abstract
Dendritic cells are innate sentinels of the immune system and potent activators of naïve T cells. Mechanisms must exist to enable these cells to achieve maximal activation of T cells specific for microbial antigens, while avoiding activation of T cells specific for self-antigens. Here we discuss how a combination of signals from pattern recognition receptors and T cells co-ordinates subcellular trafficking of antigen with both major histocompatibility complex class I and class II molecules and T-cell costimulatory molecules, resulting in the preferential presentation of microbial peptides within a stimulatory context.
Keywords: antigen presentation, costimulation, cross-presentation, C-type lectin receptors, dendritic cells, MHC class I, MHC class II, pattern recognition receptors, toll-like receptors, trafficking
Essential for the induction of effective immune responses is the ability of antigen-presenting cells (APCs) to internalize antigen from the tissue microenvironment, process it into peptides and present these on surface-expressed major histocompatibility complex (MHC) molecules. This presentation of antigenic peptides on MHC molecules is central to the activation of T cells as T-cell receptors (TCRs) recognize antigens in the peptide-binding groove of MHC molecules (1).
Two major classes of T cells exist. Of these, CD4+ T cells are central controllers of the immune system. Several effector CD4+ T-cell lineages can be generated (e.g. T-helper 1, T-helper 2), each dedicated to co-ordinating responses against specific classes of microbes. CD8+ T cells, on the other hand, primarily function to kill cells infected with intracellular microbes such as viruses and certain types of bacteria. CD4+ T cells recognize peptides in MHC class II molecules (MHCII), which are mostly derived from antigens degraded in endosomal and phagosomal compartments. Peptides in MHC class I molecules (MHCI), in contrast, are generally derived from cytosolic proteins and are recognized by CD8+ T cells. MHCI are present on the surface of almost all cells, whereas MHCII are found on cells with specialized functions in antigen presentation (1).
At steady state, most MHC molecules on cells are loaded with peptides derived from self-antigens (1). Despite the existence of multiple tolerance mechanisms, T cells with specificity for such self-peptides still exist in the repertoire of mature T cells (2). As activation of such clones could lead to autoimmune disease, mechanisms must exist to prevent their activation. Indeed, the activation of naïve T cells requires not only engagement of TCRs by cognate peptide–MHC (pMHC) complexes but also delivery of additional licensing signals, which collectively provide T-cell costimulation. This constitutes a barrier against the activation of self-reactive T cells, as most cells presenting self-antigen lack expression of costimulatory molecules. Expression of costimulatory molecules, such as CD86 and CD70, is reserved for specialized APCs like dendritic cells (DCs) (3). While immature, these cells phagocytose microbial invaders in tissues and subsequently migrate to lymphoid organs to present microbial peptides to T cells. Interaction with microbes leads DCs to mature, resulting in expression of costimulatory molecules and, correspondingly, potent ability to activate naïve T cells. This process requires the activation of pattern recognition receptors (PRRs), such as toll-like receptors (TLRs), which recognize relatively invariant structures associated with microorganisms (known as pathogen-associated molecular patterns or PAMPs) (3). Activation of PRRs also stimulates DCs to produce signals instructing differentiation of T cells into effector cells. Examples are the cytokine interleukin-12 (IL-12) and ligands for Notch receptors (4). Detection of PAMPs by DCs indicates the presence of microbes and therefore justifies activation of T cells. However, even after microbial stimulation, APCs present self-peptides as well as foreign peptides on their surface. This creates two conceptual problems:
As the presentation of these self-peptides is now accompanied by the expression of costimulatory molecules, self-reactive T-cell clones could potentially be activated and cause autoimmune disease.
The presence of many irrelevant self-peptides creates a challenge for microbial peptide-specific TCRs to ‘find’ their cognate peptide.
Here, we review how microbial antigens are taken up and processed by DCs. We discuss how signals from PRRs and T cells together guide trafficking of pMHC complexes as well as accessory signals inside DCs to help solve the conceptual problems articulated above. This guidance acts at the level of antigen uptake, processing and targeting toward T cells, and results in the preferential presentation of microbial antigen in a costimulatory context.
Control of Antigen Presentation by PRRs
TLRs recognize a variety of PAMPs, including prokaryotic nucleic acids, proteins and cell wall components such as lipopolysaccharide (LPS). The efficacy of antigen processing and presentation is strongly affected by signals from TLRs. For instance, the in vivo presentation (in MHCII) of Toxoplasma gondii profilin, a TLR11 protein ligand, required TLR signaling (5). The presentation of tumor antigens in MHCI depended on secretion of the high-mobility group box 1 (HMGB1) protein by necrotic tumor cells, and its engagement of TLR4 on DCs (5). Furthermore, the presentation of peptide derived from exogenous ovalbumin protein in MHCI only occurred efficiently in the presence of LPS (6).
Another group of PRRs consists of the C-type lectin receptors (CLRs), characterized by the presence of at least one carbohydrate recognition domain (7). CLRs interact with pathogens primarily through the recognition of mannose, fucose and glucan carbohydrate structures, thereby recognizing a wide repertoire of bacteria, viruses, fungi and helminthes. Several CLRs, including Langerin, CD205, CD206, Dectin1 and DNGR1, increase phagocytosis of pathogens (reviewed in 7). In addition, these CLRs are able to deliver antigen to distinct MHCI- and MHCII-loading compartments. This property has been used to target antigens to these compartments. For instance, immunization with anti-DEC205–antigen conjugates resulted in enhanced CD4+ and CD8+ T-cell responses (8). CD206-deficient DCs could not cross-present exogenous antigen to CD8+ T cells, whereas the activation of CD4+ T cells was not affected (8). CD206 is exclusively present on monocyte-derived inflammatory DCs, which are recruited to sites of infection with gram-negative bacteria (9) and are superior at stimulating CD4+ and CD8+ T-cell responses. DNGR1, implicated in the recognition of dying cells, was also required for efficient cross-presentation of antigens from dead cells in vitro and in vivo (10).
PRRs Regulate Antigen Processing and Presentation at Multiple Levels
Control of antigen uptake
DCs have the extraordinary ability to internalize larger particulates such as microbes and apoptotic cells through phagocytosis (11). In addition, DCs internalize antigen via other processes such as macropinocytosis and different types of receptor-mediated endocytosis (12). Both phagocytosis and macropinocytosis are enhanced by TLR signaling (5,13). During internalization, the biochemical nature of the internalized cargo determines the types of PRRs engaged. While some PRRs merely facilitate macropinocytosis or phagocytosis, a subset mediates signal transduction and thereby critically impacts the fate of the cargo and the cellular responses induced to it (14,15). For example, the DC actin cytoskeleton is rapidly mobilized in response to LPS stimulation to enhance capture of soluble antigen via macropinocytosis (16). The surface TLRs, TLR2 and TLR4 accelerated the rate of phagocytosis of Salmonella and Escherichia coli, whereas the uptake of apoptotic cells, which lack the expression of PAMPs, proceeded at the constitutive rate of phagocytosis (13).
The types of receptors engaged during internalization also determine where the internalized cargo is delivered within the cell. For example, DEC205 was shown to recycle and localize to the late endosomal/lysosomal compartments, while CD206 localizes to early endosomes (8). These preferential routes of PRR internalization determine the immune response to the particular cargo delivered into DCs by these receptors. For example, DEC205-mediated delivery enhanced MHCII presentation of antigens derived from the cargo, and similarly CD206-mediated delivery enhanced the presentation of exogenously derived antigen by MHCI (8).
Control of processing and presentation in MHCI
In the classical MHCI presentation pathway, peptides generated in the cytosol are delivered into MHCI-loading compartments in the endoplasmic reticulum (ER). Antigens routed via this pathway include either normal cellular proteins or proteins made by viruses or intracellular bacteria that have infected the cells. These antigens are degraded by various peptidases and the proteasome in the cytosol. The resulting peptides are translocated into the ER by the transporter associated with antigen presentation (TAP) for loading onto nascent MHCI with the help of the peptide-loading complex (17). After release from the peptide-loading complex, pMHCI exit the ER and are routed to the plasma membrane through the Golgi apparatus via interaction with cargo receptors such as Bap31 (17). This pathway operates in all MHCI-expressing cells and has been extensively reviewed elsewhere (17,18). Here, we focus on an alternative route known as cross-presentation, a process found in dedicated APCs like DCs (19).
Cross-presentation of exogenous antigens in MHCI is considered critical for initiation of CD8+ T-cell responses against infectious agents and tumors as well as for induction of tolerance (19). How internalized antigen gains access to ER-resident MHCI has been enigmatic. Three major pathways have been proposed (Figure 1). The phagosome-to-cytosol pathway (Figure 1A) involves escape of the antigen from the phagosome into the cytosol, possibly mediated by Sec61, a translocon shown to be involved in the ER-associated degradation pathway (ERAD) (1). Once in the cytosol, the antigen could follow a fate similar to the classical MHCI pathway described earlier (1). In the highly controversial phagosome–ER fusion pathway (Figure 1B), the ER fuses with the phagosome creating a so-called ERgosome, which contains ER molecules such as TAP and MHCI (1). The ERgosome thus provides an environment optimal for cross-presentation. Antigen still escapes the ERgosome via Sec61 to be processed by the proteasome, but is then transported back into the ERgosome by TAP and loaded onto MHCI. Finally, the vacuolar pathway (Figure 1C) involves direct processing of the antigen within the phagosome by endocytic proteases such as cathepsins and subsequent loading of peptides onto MHCI (1).
Figure 1. Three proposed pathways for cross-presentation and their regulation by PRRs.
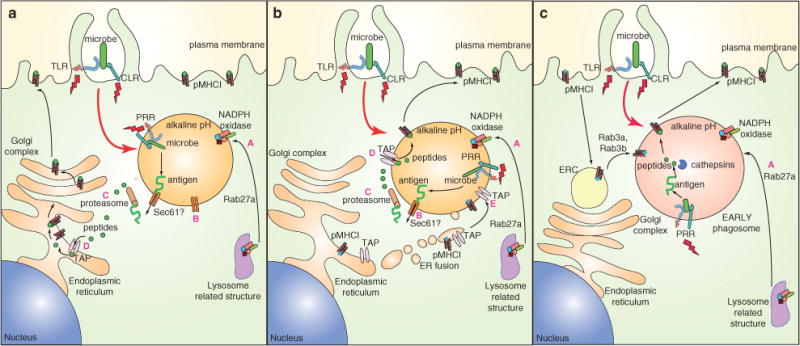
A) The phagosome-to-cytosol pathway. After phagocytosis, antigen is transported out of the phagosome, possibly through the Sec61 translocon. Once in the cytosol, antigen follows the classical MHCI route, consisting of degradation by the proteasome, TAP-mediated entry into the ER and routing to the plasma membrane via the Golgi complex. B) The ERgosome pathway. Phagosomes fuse with (components of) the ER (forming an ERgosome), which brings in TAP and pMHCI displaying self-antigen (shown in blue). Antigens are translocated to the cytosol for processing by cytosolic proteasomes, followed by re-entry into the ERgosome, preferential loading of microbial antigen (shown in green) on MHCI and migration of pMHCI complexes to the plasma membrane. C) The vacuolar pathway. Antigens can be directly processed in the early phagosome by resident cathepsins. Resultant microbial peptides (green) are loaded onto MHCI derived from the plasma membrane (presumably after exchange with self-peptides), which arrive in phagosomes via early recycling endosomes (EREs). After loading of microbial peptides, migration to the plasma membrane takes place by unknown mechanisms. PRRs (TLR and CLR) continue to be engaged (indicated by red lightning bolts) by PAMPs in phagosomes, resulting in signals that may promote these pathways via inducing Rab27a-mediated recruitment of NADPH oxidase from lysosomal structures (A) (which may help preservation of antigenic epitopes by limiting acidification), recruitment of Sec61 to facilitate translocation of antigens into the cytosol (B), enhancement of proteasomal (C) and TAP activity (D) as well as ER recruitment of TAP and MHCI to phagosomes (E).
In both the ERgosome and the vacuolar model, MHCI may be derived from the plasma membrane. Indeed, MHCI are continually endocytosed and traffic in a clathrin- and dynamin-independent pathway into early embryonic antigen-1 (EEA-1)+ and Lysosomal-associated membrane protein 1 (LAMP-1)+ endolysosomal compartments. Those MHCI can then either be degraded or traffic to endosomal recycling compartments and be rerouted to the plasma membrane. Endocytosis depends on a conserved tyrosine within the cytosolic domain of the MHCI, and to a lesser extent on a conserved serine phosphorylation site (20,21). Interestingly, the ability to cross-present was lost upon mutation of the same conserved tyrosine residue, supporting a role for recycling of MHCI in this process (21). Recycling of MHCI requires the GTPases Rab11, Rab22 as well as Arf6 (17). Rab3a and Rab3b are also involved in MHCI recycling and, importantly, their activity was also critical to maintain the ability of DCs to cross-present antigen derived from exogenous bacteria (22).
TLRs enhance cross-priming of CD8+ T cells in vitro (5,6,23). However, direct in vivo evidence implicating TLRs in enhancing the formation of pMHCI complexes is still sparse. Experiments addressing this issue are difficult to design, as several DC subsets, including conventional splenic- and lymph node-resident CD8α+ DCs as well as migratory DC populations such as lung and dermal CD103+ DCs, are able to cross-present cellular antigens at steady state (24). Recently, it was shown that an additional subset of DCs is recruited upon engagement of TLR4, TRIF and CD14 during microbial infection (9). These blood monocyte-derived ‘inflammatory’ DCs were shown to be extremely efficient at cross-presentation of soluble and particulate antigens, and we speculate that this ability is controlled by TLRs. In another study, total populations of splenic DCs in vivo were shown to retain the capacity to internalize soluble antigens by endocytosis, despite their maturation by inflammatory TLR-activating stimuli (25). These mature DCs were subsequently capable of MHCII presentation and cross-presentation of these antigens to T cells.
How TLRs regulate cross-presentation is poorly understood (Figure 1). TLR4 signaling enhanced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity in DCs (26). This enzyme provides a sustained alkaline phagosomal environment, leading to reduced proteolysis, preservation of antigen from complete degradation and, as a consequence, enhanced cross-presentation (26). DCs lacking the Rab27a GTPase experienced a 2–3-h delay in recruitment of the NADPH oxidase (NOX)-2 to phagosomes, which translated into more acidic compartments and impaired cross-presentation (26). Perhaps, TLR signaling facilitates enhanced trafficking of Rab27a at early time-points to phagosomes containing microbial signatures, thereby promoting recruitment of NOX2 to these early phagosomes and favoring antigen cross-presentation.
LPS stimulation of DCs via TLR4 and MyD88 enhanced recruitment of TAP to early endosomes, thereby allowing cross-presentation of soluble ovalbumin (6). In contrast to their MyD88-deficient counterparts, DCs lacking TRIF were still able to recruit TAP to ovalbumin-containing endosomes (6). However, these DCs failed to generate pMHCI complexes in the endocytic compartment, indicating other defects in antigen processing. Other studies have shown that LPS stimulation increases the proteasomal activity in DCs as well as the ability of TAP to actively translocate antigens from the cytosol (27). In addition, it is conceivable that upon TLR engagement, elements of the ERAD pathway such as Sec61 are recruited to phagosomes containing TLR ligands, thereby facilitating antigen escape to the cytosol.
Control of processing and presentation in MHCII
The classical route for exogenous antigens, such as those derived from phagocytosed microbial pathogens, results in presentation on MHCII for recognition by CD4+ T cells. Like MHCI, MHCII also begins its journey in the ER, where it is synthesized and associates with its chaperone invariant chain (Ii) to form a complex consisting of three αβ dimers and three Ii molecules (28). The Ii blocks the peptide-binding groove of MHCII, thus preventing premature peptide loading of the complex. The cytoplasmic domain of the invariant chain contains dileucine-based sorting signals that interact with clathrin adaptor proteins (AP)-1 or AP-2, which guide the transfer of MHCII out of the ER into the endocytic pathway either directly from the trans Golgi network or via the plasma membrane, respectively (28). RNA interference studies in human cell lines, such as HELA-CIITA, Mel JuSo and DG75, have revealed that AP-2- and not AP-1-deficient cells exhibit defective MHCII trafficking to endosomes and result in accumulation of MHCII–Ii complexes on the plasma membrane (29). These studies indicate that MHCII–Ii complexes are transported to endosomes mainly via the plasma membrane; however, whether this route is also dominant in APCs is still unclear.
Newly formed phagosomes undergo a series of fusion events with the endocytic pathway in a process termed phagosome maturation, and ultimately form phagolysosomes that intersect with the MHCII peptide-loading pathway (1). In the acidic phagolysosomal environment, the Ii is degraded by lysosomal proteases such as cathepsins (cathepsin S in particular), liberating the MHCII αβ dimers (28). The peptide-binding groove is still occupied at this point by an N-terminal fragment of the Ii chain, called the ‘class II-associated invariant chain-derived peptide’ (CLIP). CLIP is then exchanged for antigenic peptides generated from cargo proteins hydrolyzed in the same acidic compartment. Non-classical MHC molecules H2-DM and DO facilitate this exchange of peptides (30). MHCII trafficking to the plasma membrane was recently found to be regulated by the activity of actin-based motor myosin 1E (31). This motor protein is recruited by the GTPases ARL14/ARF7, which are present on phagosomes containing MHCII.
TLR signaling regulates several steps in this pathway (Figure 2). After internalization, TLRs reside on the phagosomal membrane, and their engagement by microbial cargo controls subsequent maturation of phagosomes, leading to accelerated fusion of phagosomes with lysosomal compartments (13). Furthermore, TLR signaling stimulates the degradation of Ii (32). Interestingly, signaling by TLRs promotes these processes in a phagosome-autonomous manner. Thus, within one and the same cell, phagosomes containing microbial cargo behave differently from phagosomes containing apoptotic cells resulting in efficient presentation of microbial, but not apoptotic cell-derived, peptides (32). How TLRs control degradation of Ii is not known. We speculate that TLRs may promote phagolysosomal acidification via controlling the density or the activity of NOX2 and the vacuolar proton-adenosine triphosphatase (V-ATPase). Indeed, the assembly of V-ATPase is regulated in DCs by TLR signals (33). TLRs may also stimulate the activity of various cathepsins, including the asparagine endopeptidase (AEP) cathepsins S, B, D, L and F, which contribute to degradation of the MHCII-associated Ii. Finally, TLR signals promote phosphorylation of p38 mitogen activated protein (MAP) kinases, and may enhance the activity of small GTPases like Rhos and Rabs (reviewed in 5,13,33), each of which might affect the rate of phagosome maturation.
Figure 2. Antigen processing and presentation on MHCII following phagocytosis of microbial antigen.
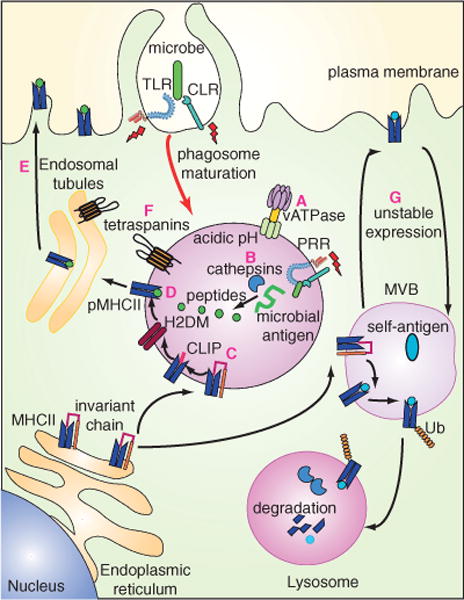
Phagosomes containing microorganisms (in green) engage TLR and CLR signals (indicated by red lightning bolts) and undergo accelerated maturation fusing with lysosomes to become phagolysosomes. PRRs may regulate assembly and function of V-ATPases to promote phagolysosomal acidification (A). This acidic compartment favors the degradation of antigens by promoting activity of resident cathepsins (B) within individual phagosomes containing PRR ligands. Perhaps as a consequence, processing of the invariant chain is favored (C), thereby allowing MHCII to acquire peptides generated from microbial antigens (D). The assembled pMHCII complexes are then routed to the plasma membrane via endolysosomal tubules (E). Tetraspanins are also selectively recruited to phagosomes containing microbial signatures and are believed to help create microdomains with superior antigen presentation capacity (F). MHCII loaded with self-peptides (in blue) in immature DCs are ubiquitinated (indicated by Ub) on their cytoplasmic tail. Although these complexes can go to the plasma membrane (G), they are rapidly internalized and routed to MVBs and end up in lysosomes, where they are degraded. As a consequence, levels of MHCII-carrying self-peptides on the cell surface are low. Upon TLR stimulation, newly synthesized MHCII loaded with microbial peptides are not ubiquitinated, allowing their stable expression on the plasma membrane.
In contrast to phagosome autonomous control, TLR stimulation favors the presentation of microbial peptides also in a more general manner by promoting the stability of pMHC complexes (Figure 2). In immature DCs, the cytoplasmic tail of MHCII is ubiquitinated (28). This modification directs these molecules into internal vesicles of multivesicular bodies (MVBs), collectively known as MHCII-enriched compartments (MIICs). Here, MHCII is targeted for degradation upon fusion with lysosomes. Ubiquitination of MHCII is reportedly preceded by degradation of Ii and loading with peptide (28), and is mediated by the membrane-associated RING-CH (MARCH) family of ubiquitin E3 ligases (28). It is believed that MHCII carrying self-peptides at the surface of immature DCs may have escaped MVB sorting. However, these escapees have a short half-life at the plasma membrane due to their ubiquitination, endocytosis and a second round of sorting to MVBs (28). Upon stimulation of DCs with LPS, the expression of MARCH1 is downregulated and ubiquitination of MHCII is reduced, allowing its stable expression at the plasma membrane (28). As a consequence, the levels of MHCII on the surface increase strongly. Interference with the biosynthetic pathway using brefeldin A or cycloheximide prevented this upregulation, suggesting that the increase in MHCII expression is due to newly synthesized MHCII (34). Indeed, it was recently shown that stably expressed surface pMHCII complexes in mature DCs derive from newly synthesized and not MVB-stored MHCII (35). In fact, the majority of self-peptide-presenting MHCII found in MVBs is not recruited to the plasma membrane even after TLR stimulation, but is destined for lysosomal degradation (35), further favoring the presentation of microbial peptides over self-peptides.
Evidence from multiple laboratories shows that pMHCII complexes are transported to the plasma membrane of DCs in endolysosomal tubules emanating from a perinuclear region (36–38). Live imaging on DCs expressing a fusion protein of MHCII and green fluorescent protein (GFP) revealed that, in contrast to immature DCs, DCs matured upon LPS stimulation are very dynamic and rapidly form such MHCII-containing tubules (37). In a separate study using LPS-primed DCs from MHCII-GFP knock-in mice, the authors further showed that these tubules were approximately up to 5 μm in length and could move at speeds up to 2 μm/second toward the plasma membrane (36). Thus, PAMP stimulation also regulates transport of MHCII to the cell surface.
Regulation of pMHC and Costimulatory Molecule Transport by T Cells
MHC molecules, loaded with self-peptides, are always present on the surface of DCs and compete with foreign antigens for binding to TCRs. T cells must have an extraordinary ability to find their specific TCR ligand, as peptides present at a density below 1 in 104 peptides are sufficient for activation of the T cell. Full activation of T cells requires engagement of several hundreds of TCRs (39), and the chance to reach this number would greatly increase by concentration of the relevant pMHC complexes at the interface between a DC and a T cell. Interactions between T cells and DCs do indeed lead to the generation of a supramolecular structure, called the immunological synapse (IS), in which many molecules involved in activation of T cells, including pMHC complexes, are clustered (39). On the T cell side, this IS includes a central domain with TCRs, costimulatory receptors (CD28, CD27) and receptors for differentiation signals, such as Notch and the interferon-γ (IFN-γ) receptor. Outside this central domain, adhesion molecules such as lymphocyte function-associated antigen 1 (LFA-1) are found. A mirror structure is formed on the surface of DCs, with MHC molecules centrally positioned together with CD86 and CD70 (ligands for CD28 and CD27) and ligands for Notch, all surrounded by intercellular adhesion molecule (ICAM)-1 and ICAM-3 ligands for LFA-1 (39,40). Although there is some controversy concerning the function of this IS, it seems likely that the clustering involved promotes the efficacy of T-cell activation.
It is clear that active processes are involved in targeting these molecules to the IS both on the T cell side as well as on the DC side (41) (Figure 3). One mechanism likely involves the directed transport of pMHC-containing tubules described above. Strikingly, T cells can actively influence this transport. Interaction of antigen-specific CD4+ T cells with DCs stimulates the reorientation of the microtubule-orienting center (MTOC) toward the T cells. From this, MHCII- and antigen-containing tubules of up to 20 μm are projected along polymerizing microtubule tracks toward the site of T cell–DC interaction (36,42–44). Induction of tubule migration to the plasma membrane by TLR signaling in the absence of T cells requires high doses of LPS. It is likely that under physiological conditions (for instance, phagocytosis of bacteria), such high concentrations are not encountered and that T cell-induced migration of tubules to the surface has a prominent role. The directed movement of MHCII is regulated at two levels. First, the ability to generate these tubular endolysosomes requires a low level of signaling from PRRs such as TLRs (43,44). Second, recruitment signals are generated at the DC surface interacting with the T cell. The antigen- specific nature of this process implies that some pioneering pMHC complexes must already be on the cell surface to initiate tubulation. The signaling pathways leading to tubule recruitment have not been fully identified, although activation of the Vav1–Rac1 axis by signaling from integrins may be involved (45). Indeed, tubulation responds to engagement of ICAM-3 on DCs by LFA-1 on T cells, and is prevented by blockade of LFA-1 (42,45). The ability of TCR signals to enhance the affinity of LFA-1 for ICAM-3 (39) may explain the antigen specificity of the process. Furthermore, TCR signaling results in surface expression of CD40L on T cells, and blockade of this molecule also inhibited the formation of tubules (42).
Figure 3. Formation of a stimulatory immunological synapse by targeted recruitment.
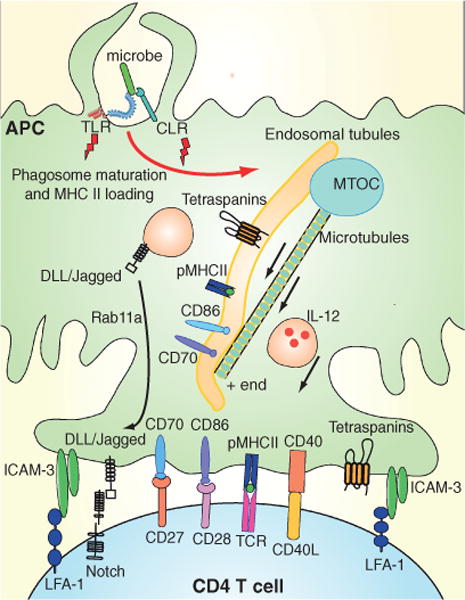
T cells actively influence the transport of pMHCII from endocytic compartments to the plasma membrane. Interaction of antigen-specific CD4+ T cells with APCs stimulates reorientation of the MTOC in the APC toward the T cells, and leads to the formation of tubules containing pMHCII complexes, tetraspanins and costimulatory molecules such as CD70 and CD86. CD40L–CD40 and LFA-1–ICAM-3 interactions promote tubule formation. Molecules instructing T-cell differentiation, including IL-12 and the Notch ligands, Delta-like ligands (DLLs) and Jagged, are also projected toward the interaction site.
Not only do tubules target pMHC to T cells but they also package these in a powerful stimulatory context. Thus, the CD63 and CD82 tetraspanins, which may help create microdomains with enhanced T-cell stimulatory capacity, are also present in these tubules (44). Furthermore, the CD86 and CD70 costimulatory molecules are found in these tubular structures (36,37,46). At least CD70 molecules are actively targeted to MIIC compartments via interaction with the Ii chaperone (47). T cells also stimulate directed secretion of the cytokine IL-12, which promotes differentiation of both CD4+ and CD8+ T cells into effector cells. This cytokine is found in the Golgi complex in a ring around the MTOC, and its production is stimulated by triggering TLR. Upon antigen-specific interaction with T cells, vesicles containing IL-12 are transported along microtubules toward the IS in a process depending on Cdc42 (48).
Although little is known about this, other molecules involved in activation or differentiation of T cells are also likely routed toward interacting T cells. For instance, ligands for Notch, which control differentiation of both CD4+ and CD8+ T cells into effector cells (4), cluster in the central area of the IS (40). Optimal activity of these ligands requires monoubiquitination-driven migration through Rab11+ recycling endosomes. Indeed, the majority of these molecules is found in such endosomes in DCs (40), and it is conceivable that this localization allows rapid targeting toward the IS, possibly together with recycling MHC molecules also found in Rab11+ vesicles (49). Finally, the targeting by Ii of CD70 to MIICs suggests that other tumor necrosis factor receptor (TNFR) family ligands, which have also been implicated in the control of T-cell activation/differentiation (50), could be subjected to similar trafficking rules.
Synthesis: How Directed Trafficking May Solve Problems Caused by Presentation of Self-peptides
In the introduction of this review, we presented two conceptual problems caused by presentation of self-peptides: on the one hand, difficulty for microbial antigen-specific T cells to find their cognate pMHC and on the other hand, a risk to activate self-reactive T cells. Specialized subcellular control of antigen processing and trafficking may help solve these problems. First, the presentation of self-peptides is kept low in immature DCs, due to constitutive targeting of pMHC (at least those containing MHCII) complexes to lysosomes. Only upon stimulation with PAMPs, coinciding with the presence of microbial cargo in phagosomes, are stable pMHC complexes formed de novo. At this point, phagosome autonomous control of MHC loading by engagement of TLR strongly favors the loading of microbial-derived antigen over self-peptides (32). Although this has not yet been tested, it would make sense that phagosome autonomous TLR signaling also controls the ability to form endolysosomal tubules, which would target a set of MHC molecules, enriched for microbial peptides, toward interacting T cells. This would diminish the competition by irrelevant pMHC complexes and promote activation of microbial antigen-specific T cells due to the inclusion of molecules providing costimulatory and differentiation signals. On the other hand, when self-reactive T cells induce tubulation, these would have a much lower chance to engage additional TCR ligands due to the underrepresentation of self-peptides targeted toward the IS and full activation would not be accomplished.
References
- 1.Vyas JM, Van der Veen AG, Ploegh HL. The known unknowns of antigen processing and presentation. Nat Rev Immunol. 2008;8:607–618. doi: 10.1038/nri2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol. 2010;11:7–13. doi: 10.1038/ni.1818. [DOI] [PubMed] [Google Scholar]
- 3.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 4.Amsen D, Antov A, Flavell RA. The different faces of Notch in T-helper-cell differentiation. Nat Rev Immunol. 2009;9:116–124. doi: 10.1038/nri2488. [DOI] [PubMed] [Google Scholar]
- 5.Blander JM. Phagocytosis and antigen presentation: a partnership initiated by Toll-like receptors. Ann Rheum Dis. 2008;67(Suppl 3):iii44–iii49. doi: 10.1136/ard.2008.097964. [DOI] [PubMed] [Google Scholar]
- 6.Burgdorf S, Scholz C, Kautz A, Tampe R, Kurts C. Spatial and mechanistic separation of cross-presentation and endogenous antigen presentation. Nat Immunol. 2008;9:558–566. doi: 10.1038/ni.1601. [DOI] [PubMed] [Google Scholar]
- 7.Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9:465–479. doi: 10.1038/nri2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burgdorf S, Kurts C. Endocytosis mechanisms and the cell biology of antigen presentation. Curr Opin Immunol. 2008;20:89–95. doi: 10.1016/j.coi.2007.12.002. [DOI] [PubMed] [Google Scholar]
- 9.Cheong C, Matos I, Choi JH, Dandamudi DB, Shrestha E, Longhi MP, Jeffrey KL, Anthony RM, Kluger C, Nchinda G, Koh H, Rodriguez A, Idoyaga J, Pack M, Velinzon K, et al. Microbial stimulation fully differentiates monocytes to DC-SIGN/CD209(+) dendritic cells for immune T cell areas. Cell. 2010;143:416–429. doi: 10.1016/j.cell.2010.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sancho D, Joffre OP, Keller AM, Rogers NC, Martinez D, Hernanz-Falcon P, Rosewell I, Reis e Sousa C. Identification of a dendritic cell receptor that couples sensing of necrosis to immunity. Nature. 2009;458:899–903. doi: 10.1038/nature07750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jutras I, Desjardins M. Phagocytosis: at the crossroads of innate and adaptive immunity. Annu Rev Cell Dev Biol. 2005;21:511–527. doi: 10.1146/annurev.cellbio.20.010403.102755. [DOI] [PubMed] [Google Scholar]
- 12.Norbury CC. Drinking a lot is good for dendritic cells. Immunol. 2006;117:443–451. doi: 10.1111/j.1365-2567.2006.02335.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 14.Mellman I, Warren G. The road taken: past and future foundations of membrane traffic. Cell. 2000;100:99–112. doi: 10.1016/s0092-8674(00)81687-6. [DOI] [PubMed] [Google Scholar]
- 15.Niedergang F, Chavrier P. Signaling and membrane dynamics during phagocytosis: many roads lead to the phagos(R)ome. Curr Opin Cell Biol. 2004;16:422–428. doi: 10.1016/j.ceb.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 16.West MA, Wallin RP, Matthews SP, Svensson HG, Zaru R, Ljunggren HG, Prescott AR, Watts C. Enhanced dendritic cell antigen capture via toll-like receptor-induced actin remodeling. Science. 2004;305:1153–1157. doi: 10.1126/science.1099153. [DOI] [PubMed] [Google Scholar]
- 17.Donaldson JG, Williams DB. Intracellular assembly and trafficking of MHC class I molecules. Traffic. 2009;10:1745–1752. doi: 10.1111/j.1600-0854.2009.00979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wearsch PA, Cresswell P. The quality control of MHC class I peptide loading. Curr Opin Cell Biol. 2008;20:624–631. doi: 10.1016/j.ceb.2008.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rock KL, Shen L. Cross-presentation: underlying mechanisms and role in immune surveillance. Immunol Rev. 2005;207:166–183. doi: 10.1111/j.0105-2896.2005.00301.x. [DOI] [PubMed] [Google Scholar]
- 20.Basha G, Lizee G, Reinicke AT, Seipp RP, Omilusik KD, Jefferies WA. MHC class I endosomal and lysosomal trafficking coincides with exogenous antigen loading in dendritic cells. PLoS ONE. 2008;3:e3247. doi: 10.1371/journal.pone.0003247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lizee G, Basha G, Tiong J, Julien JP, Tian M, Biron KE, Jefferies WA. Control of dendritic cell cross-presentation by the major histocompatibility complex class I cytoplasmic domain. Nat Immunol. 2003;4:1065–1073. doi: 10.1038/ni989. [DOI] [PubMed] [Google Scholar]
- 22.Zou L, Zhou J, Zhang J, Li J, Liu N, Chai L, Li N, Liu T, Li L, Xie Z, Liu H, Wan Y, Wu Y. The GTPase Rab3b/3c-positive recycling vesicles are involved in cross-presentation in dendritic cells. Proc Natl Acad Sci U S A. 2009;106:15801–15806. doi: 10.1073/pnas.0905684106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schulz O, Diebold SS, Chen M, Naslund TI, Nolte MA, Alexopoulou L, Azuma YT, Flavell RA, Liljestrom P, Reis e Sousa C. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–892. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 24.Segura E, Villadangos JA. Antigen presentation by dendritic cells in vivo. Curr Opin Immunol. 2009;21:105–110. doi: 10.1016/j.coi.2009.03.011. [DOI] [PubMed] [Google Scholar]
- 25.Drutman SB, Trombetta ES. Dendritic cells continue to capture and present antigens after maturation in vivo. J Immunol. 2010;185:2140–2146. doi: 10.4049/jimmunol.1000642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amigorena S, Savina A. Intracellular mechanisms of antigen cross presentation in dendritic cells. Curr Opin Immunol. 2010;22:109–117. doi: 10.1016/j.coi.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 27.Gil-Torregrosa BC, Lennon-Dumenil AM, Kessler B, Guermonprez P, Ploegh HL, Fruci D, van Endert P, Amigorena S. Control of cross-presentation during dendritic cell maturation. Eur J Immunol. 2004;34:398–407. doi: 10.1002/eji.200324508. [DOI] [PubMed] [Google Scholar]
- 28.van Niel G, Wubbolts R, Stoorvogel W. Endosomal sorting of MHC class II determines antigen presentation by dendritic cells. Curr Opin Cell Biol. 2008;20:437–444. doi: 10.1016/j.ceb.2008.05.011. [DOI] [PubMed] [Google Scholar]
- 29.Landsverk OJ, Bakke O, Gregers TF. MHC II and the endocytic pathway: regulation by invariant chain. Scand J Immunol. 2009;70:184–193. doi: 10.1111/j.1365-3083.2009.02301.x. [DOI] [PubMed] [Google Scholar]
- 30.Karlsson L. DM and DO shape the repertoire of peptide-MHC-class-II complexes. Curr Opin Immunol. 2005;17:65–70. doi: 10.1016/j.coi.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Paul P, van den Hoorn T, Jongsma ML, Bakker MJ, Hengeveld R, Janssen L, Cresswell P, Egan DA, van Ham M, Ten Brinke A, Ovaa H, Beijersbergen RL, Kuijl C, Neefjes J. A genome-wide multidimensional RNAi screen reveals pathways controlling MHC class II antigen presentation. Cell. 2011;143:416–29. doi: 10.1016/j.cell.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 32.Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature. 2006;440:808–812. doi: 10.1038/nature04596. [DOI] [PubMed] [Google Scholar]
- 33.Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–1035. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- 34.Wilson NS, El-Sukkari D, Villadangos JA. Dendritic cells constitutively present self antigens in their immature state in vivo and regulate antigen presentation by controlling the rates of MHC class II synthesis and endocytosis. Blood. 2004;103:2187–2195. doi: 10.1182/blood-2003-08-2729. [DOI] [PubMed] [Google Scholar]
- 35.Broeke TT, van Niel G, Wauben MH, Wubbolts R, Stoorvogel W. Endosomally stored MHC class II does not contribute to antigen presentation by dendritic cells at inflammatory conditions. Traffic. 2011;12:1025–36. doi: 10.1111/j.1600-0854.2011.01212.x. [DOI] [PubMed] [Google Scholar]
- 36.Boes M, Cerny J, Massol R, Op den Brouw M, Kirchhausen T, Chen J, Ploegh HL. T-cell engagement of dendritic cells rapidly rearranges MHC class II transport. Nature. 2002;418:983–988. doi: 10.1038/nature01004. [DOI] [PubMed] [Google Scholar]
- 37.Chow A, Toomre D, Garrett W, Mellman I. Dendritic cell maturation triggers retrograde MHC class II transport from lysosomes to the plasma membrane. Nature. 2002;418:988–994. doi: 10.1038/nature01006. [DOI] [PubMed] [Google Scholar]
- 38.Kleijmeer M, Ramm G, Schuurhuis D, Griffith J, Rescigno M, Ricciardi-Castagnoli P, Rudensky AY, Ossendorp F, Melief CJ, Stoorvogel W, Geuze HJ. Reorganization of multivesicular bodies regulates MHC class II antigen presentation by dendritic cells. J Cell Biol. 2001;155:53–63. doi: 10.1083/jcb.200103071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, Sacristan C, Victora GD, Zanin-Zhorov A, Dustin ML. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Luty WH, Rodeberg D, Parness J, Vyas YM. Antiparallel segregation of notch components in the immunological synapse directs reciprocal signaling in allogeneic Th:DC conjugates. J Immunol. 2007;179:819–829. doi: 10.4049/jimmunol.179.2.819. [DOI] [PubMed] [Google Scholar]
- 41.Rodriguez-Fernandez JL, Riol-Blanco L, Delgado-Martin C. What is the function of the dendritic cell side of the immunological synapse? Sci Signal. 2010;3:re2. doi: 10.1126/scisignal.3105re2. [DOI] [PubMed] [Google Scholar]
- 42.Bertho N, Cerny J, Kim YM, Fiebiger E, Ploegh H, Boes M. Requirements for T cell-polarized tubulation of class II+ compartments in dendritic cells. J Immunol. 2003;171:5689–5696. doi: 10.4049/jimmunol.171.11.5689. [DOI] [PubMed] [Google Scholar]
- 43.Boes M, Bertho N, Cerny J, Op den Brouw M, Kirchhausen T, Ploegh H. T cells induce extended class II MHC compartments in dendritic cells in a Toll-like receptor-dependent manner. J Immunol. 2003;171:4081–4088. doi: 10.4049/jimmunol.171.8.4081. [DOI] [PubMed] [Google Scholar]
- 44.Vyas JM, Kim YM, Artavanis-Tsakonas K, Love JC, Van der Veen AG, Ploegh HL. Tubulation of class II MHC compartments is microtubule dependent and involves multiple endolysosomal membrane proteins in primary dendritic cells. J Immunol. 2007;178:7199–7210. doi: 10.4049/jimmunol.178.11.7199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.de la Fuente H, Mittelbrunn M, Sanchez-Martin L, Vicente-Manzanares M, Lamana A, Pardi R, Cabanas C, Sanchez-Madrid F. Synaptic clusters of MHC class II molecules induced on DCs by adhesion molecule-mediated initial T-cell scanning. Mol Biol Cell. 2005;16:3314–3322. doi: 10.1091/mbc.E05-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keller AM, Groothuis TA, Veraar EA, Marsman M, Maillette de Buy Wenniger L, Janssen H, Neefjes J, Borst J. Costimulatory ligand CD70 is delivered to the immunological synapse by shared intracellular trafficking with MHC class II molecules. Proc Natl Acad Sci U S A. 2007;104:5989–5994. doi: 10.1073/pnas.0700946104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zwart W, Peperzak V, de Vries E, Keller AM, van der Horst G, Veraar EA, Geumann U, Janssen H, Janssen L, Naik SH, Neefjes J, Borst J. The invariant chain transports TNF family member CD70 to MHC class II compartments in dendritic cells. J Cell Sci. 2010;123:3817–3827. doi: 10.1242/jcs.068510. [DOI] [PubMed] [Google Scholar]
- 48.Pulecio J, Petrovic J, Prete F, Chiaruttini G, Lennon-Dumenil AM, Desdouets C, Gasman S, Burrone OR, Benvenuti F. Cdc42-mediated MTOC polarization in dendritic cells controls targeted delivery of cytokines at the immune synapse. J Exp Med. 2010;207:2719–2732. doi: 10.1084/jem.20100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weigert R, Yeung AC, Li J, Donaldson JG. Rab22a regulates the recycling of membrane proteins internalized independently of clathrin. Mol Biol Cell. 2004;15:3758–3770. doi: 10.1091/mbc.E04-04-0342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol. 2009;9:271–285. doi: 10.1038/nri2526. [DOI] [PMC free article] [PubMed] [Google Scholar]