

Abstract
Systemic sclerosis with pulmonary arterial hypertension (SSc-PAH) is a debilitating and frequently lethal disease of unknown cause lacking effective treatment options. Lymphocyte anomalies and autoantibodies observed in systemic sclerosis have suggested an autoimmune character. We study the clonal structure of the B cell repertoire in SSc-PAH using immunoglobulin heavy chain (IGH) sequencing before and after B cell depletion. We found SSc-PAH to be associated with anomalies in B cell development, namely, altered VDJ rearrangement frequencies (reduced IGHV2-5 segment usage) and an increased somatic mutation–fixation probability in expanded B cell lineages. SSc-PAH was also characterized by anomalies in B cell homeostasis, namely, an expanded immunoglobulin D–positive (IgD+) proportion with reduced mutation loads and an expanded proportion of highly antibody-secreting cells. Disease signatures pertaining to IGHV2-5 segment usage, IgD proportions, and mutation loads were temporarily reversed after B cell depletion. Analyzing the time course of B cell depletion, we find that the kinetics of naïve replenishment are predictable from baseline measurements alone, that release of plasma cells into the periphery can precede naïve replenishment, and that modes of B cell receptor diversity are highly elastic. Our findings reveal humoral immune signatures of SSc-PAH and uncover determinism in the effects of B cell depletion on the antibody repertoire.
INTRODUCTION
Systemic sclerosis (SSc), or scleroderma, is a rare chronic autoimmune disease that leads to tissue fibrosis and vasculopathy (1). Indications of a dysregulated immune system include reduced lymphocyte counts (2), T cell anomalies (3), and B cell anomalies, such as the presence of autoantibodies (4), increased CD19 expression (5), increased naïve B cell proportions, and diminished memory B cell proportions (5). Both preclinical and clinical studies have implicated B cell abnormalities in the pathogenesis of SSc (6, 7). A complication with particularly poor prognosis is pulmonary arterial hypertension (PAH) (8), which can arise from SSc-associated interstitial lung disease (ILD) but also directly from SSc without notable ILD; the latter case, which we will refer to as SSc-PAH, is the object of the present study. Current treatment typically focuses on pulmonary vasodilation and is not curative (9). Immunotherapy with the B cell–depleting agent rituximab, a monoclonal antibody against CD20 (10, 11), has shown promise as a potential treatment for SSc-associated skin fibrosis and/or ILD (12–17). There is early anecdotal evidence that rituximab is effective in certain refractory cases of PAH, including SSc-PAH: For example, B cell depletion with rituximab is effective in a model of T helper 2 cell–mediated PAH (18), and an SSc patient with mild ILD and very substantial PAH who was refractory to standard PAH therapy was successfully treated with rituximab twice (19). The B cell basis of SSc has previously been studied (20) with respect to subset sizes (5), cytokine levels (21), response regulators (5, 22) and autoantibody specificities (23, 24) but not at the level of sequence-based B cell phylogeny. Reconstitution of the immunoglobulin heavy chain (IGH) repertoire after rituximab administration has been studied before (25), but the sample size was small (two participants) and the sequencing depth was low (on the order of 680 sequences).
Here, we elucidate signatures of SSc-PAH and the dynamics of B cell replenishment after depletion using massively parallel sequencing of IGH transcripts found in peripheral blood mononuclear cells (PBMCs) (“antibody repertoire sequencing”), which provides a wealth of information on the processes of VDJ recombination, clonal expansion, somatic hypermutation, and isotype switching that shape the B cell repertoire (26, 27). We longitudinally studied 11 participants from an ongoing randomized, double-blind, placebo-controlled phase 2 multi-center trial of rituximab for the treatment of SSc-PAH, sequencing 85 samples and obtaining >4 million IGH sequences. Infusions of 1000 mg of rituximab or a placebo were given at weeks 0 and 2; blood was sampled at baseline (preinfusion), at weeks 2 and 4 relative to the first infusion, and roughly in 12-week increments after that (Fig. 1A). All SSc-PAH participants were females between the ages of 48 and 73, inclusive; healthy baseline controls were 15 females between the ages of 50 and 75, inclusive.
Fig. 1. Study design and comparison of study groups across time.
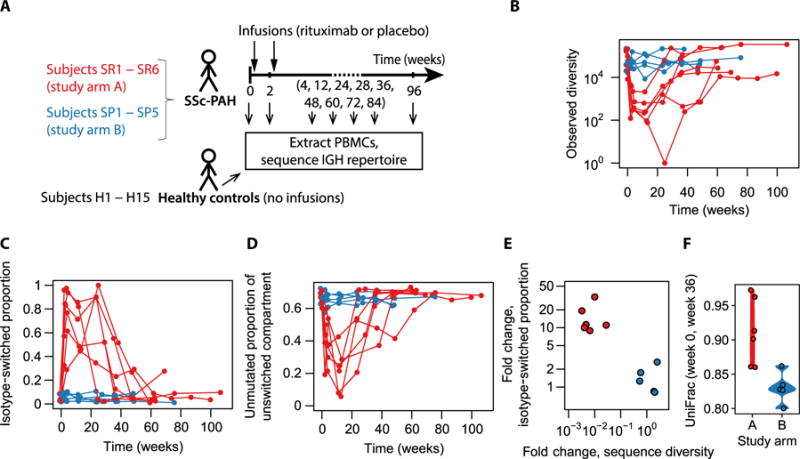
(A) Study design. Whether infusions were of rituximab or placebo depended on the study arm. (Note that not all the indicated time points were available for all participants.) (B) Number of distinct IGH sequences observed as a function of time, colored by study arm. (C) Fraction of observed sequences that had the IgA or IgG isotype as a function of time, colored by study arm. (D) Fraction of IgD or IgM sequences that displayed zero somatic mutations as a function of time, colored by study arm. (E) Summary of the signatures identified in (B) and (C): fold change in isotype-switched proportion from baseline to depletion versus fold change in sequence diversity from baseline to depletion. Here, “depletion” refers to the week 4 and week 12 visits; diversities and isotype-switched proportions were averaged across these two time points. Numeric data values are also provided in table S1. (F) Phylogenetic distance between each participant’s baseline repertoire and her repertoire at the week 36 visit. Week 36 was chosen as the common second time point because all but two study arm A recipients had recovered more than 10,000 sequences by then and times beyond week 36 had not been sampled for all participants. Numeric data values are also provided in table S2.
RESULTS
Identification of a study group undergoing B cell depletion
Because the clinical trial providing study samples is ongoing, it is blinded to whether study arm A or B corresponded to the rituximab group as opposed to placebo (Fig. 1A). Here, we show that the characteristics of group A are consistent with CD20+ B cell depletion. Group A shows a transient reduction in overall B cell receptor (BCR) diversity (Fig. 1B), and B cells that remain during the depletion period preferentially tend to have undergone isotype switching (Fig. 1C) or acquired one or more somatic mutations (Fig. 1D), consistent with the hypothesis that they are CD20− B cells in the terminal stages of differentiation [plasmablasts and plasma cells (28)]. B cell depletion was observed to be transient, ended by repopulation with new naïve cells (Fig. 1, B to D). Summarizing the above depletion signatures as fold changes between baseline and early postinfusion time points, it is evident that participants cluster into unaffected and affected groups (Fig. 1E). To compare the initial B cell repertoire with the reconstituted repertoire after depletion, we applied the phylogenetic distance metric UniFrac (29), whose use on immune repertoire data was recently demonstrated (30). As expected, we found that group A displayed greater preinfusion-to-postinfusion distances than group B (Fig. 1F), consistent with repertoire reconstitution by a fresh set of naïve B cells not clonally related to the baseline population. These observations establish group A as a cohort that can be used to study B cell depletion.
B cell signatures of SSc-PAH
The hypervariable complementarity-determining region 3 (CDR3) of antibodies is a key determinant of antigen specificity (31). Antibody repertoire analysis has been used to search for convergent CDR3 sequences that characterize the immune response to certain infectious agents, notably dengue virus (32). To investigate which antibodies may be responsible for autoimmunity in SSc, we tested all CDR3 amino acid sequences identified in this study, together with their one-mismatch derivatives, for enrichment in the SSc-PAH cohort versus the healthy cohort (Fisher’s exact tests with Benjamini-Hochberg correction). No sequences displayed a statistically significant association with SSc-PAH (fig. S1, A and B). This negative result may be due to limited statistical power or undersampling of the repertoire, but also to biological idiosyncrasy: The relevant autoantigen(s) may differ from participant to participant, and antibodies binding any given autoantigen may be encoded by distinct amino acid sequences.
We may nevertheless gain insight into the disease by studying the global structure of the antibody repertoire rather than individual antibody sequences. SSc-PAH participants tended to display slightly lower overall BCR diversity (fig. S1C), a higher proportion of immunoglobulin D (IgD) diversity, and lower mean somatic mutation loads in IgD than healthy participants (Fig. 2A), consistent with previous reports of reduced lymphocyte counts (2) and expanded naïve B cell proportions (5). This finding constitutes evidence for altered B cell homeostatic mechanisms.
Fig. 2. Signatures of SSc-PAH compared with healthy Ig repertoires at baseline.
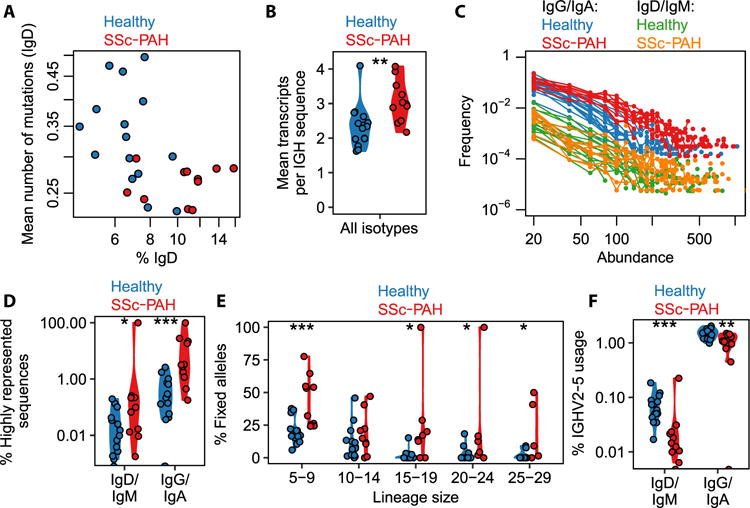
(A) IgD proportion of the IGH sequence repertoire and average mutation load in IgD. (B) Average number of transcripts recorded per distinct IGH sequence (i.e., sum total of all transcripts across all observed IGH sequences divided by the number of observed IGH sequences). Each point corresponds to one study participant. Wilcoxon–Mann-Whitney test, P = 0.0025; n = 26. (Asterisks in the figure correspond to significance codes: *0.01 ≤ P < 0.05; **0.001 ≤ P < 0.01; ***0.0001 ≤ P < 0.001.) (C) Distribution of transcript abundances, separated by isotype compartment. Each curve corresponds to one study participant. Here, “abundance” is the number of transcript copies associated with a sequence, and “frequency” refers to the number of distinct sequences that display a given abundance value. (D) Percentage of IGH sequences that had a relative transcript abundance of at least 0.05% of the total repertoire, separated by isotype compartment. Points straddling the lower edge of the panel correspond to the value 0, which cannot be rendered on the logarithmic scale. Wilcoxon–Mann-Whitney tests: P = 0.026, n = 26 for IgD/IgM; P = 0.00061, n = 26 for IgG/IgA. [Note: When the single high value in SSc-PAH IgD/IgM is removed (participant SR3), the P value for the IgD/IgM comparison changes from 0.026 to 0.049 (<0.05); there are two points at 0 for healthy IgM/IgD]. (E) Proportion of somatic mutations that were considered to have become fixed in their B cell lineage, that is, present in at least 80% of sequences in that lineage. The proportion of fixed mutations was calculated separately for each lineage and then averaged across all lineages in the relevant size bin. Here, lineage size refers to the number of distinct IGH sequences in the lineage. Wilcoxon–Mann-Whitney tests, for lineage size bins from left to right: P = 0.00040, n = 25; P = 0.27, n = 24; P = 0.015, n = 20; P = 0.022, n = 19; P = 0.023, n = 15. (Note: The two highest points for the “15–19” bin, the highest point for the “20–24” bin, and the two highest points for the “25–29” bin correspond to five distinct SSc-PAH participants, namely, SP5, SP4, SP2, SR4, and SR2.) (F) Percentage of lineages that used the IGHV2-5 segment for each isotype compartment. Points straddling the lower edge of the panel correspond to the value 0, which cannot be rendered on the logarithmic scale. Wilcoxon–Mann-Whitney tests: P = 0.00051, n = 26 for IgD/IgM; P = 0.0045, n = 26 for IgG/IgA. [Note: When the single high point for SSc-PAH IgD/IgM is removed (participant SR3), the P value for the IgD/IgM comparison changes from 5.1 × 10−4 to 7.3 × 10−6. When the single point at 0 in SSc-PAH IgG/IgA is removed (participant SP2), the P value for the IgG/IgA comparison changes from 4.4 × 10−3 to 9.6 × 10−3.]
It has also been reported that memory B cells are reduced in number but activated in scleroderma (5). Here, we are able to obtain further insight into the anomalies of the antigen-experienced compartment. We found that the average expression of distinct BCRs was higher in SSc-PAH (Fig. 2B), that this increase in average expression was due to an accumulation of BCRs at the highest observed expression levels (Fig. 2C), and that the effect was mostly driven by the isotype-switched (i.e., antigen-experienced) compartment (Fig. 2D). The effect persisted when abundances were defined in a relative rather than absolute fashion, that is, as a proportion of the total repertoire abundances, eliminating any undesired potential systematic variations in sequencing depth (Fig. 2D). Note that amplification biases were corrected for using unique molecular identifiers (UIDs) (see Materials and Methods). IGH sequences observed at high abundances are likely to correspond to plasmablasts, so the excess diversity of high-abundance BCRs suggests that B cells differentiate more frequently into plasmablasts in SSc-PAH participants than in healthy ones.
To further elucidate the importance of selection processes acting on proliferating B cell lineages, we measured what fraction of somatic mutations had become fixed in each lineage, defining a fixed mutation as one that was present in at least 80% of the lineage’s sequences. (A lineage was defined as a set of sequences with the same V segment, same J segment, same CDR3 length, and a 90% CDR3 sequence similarity in single-linkage clustering.) To eliminate artifacts due to differences in lineage sizes, we separately carried out the analyses for different ranges of lineage sizes. Germline alleles distinct from the reference could be misinterpreted as fixed somatic mutations, but no convincing evidence for the presence of novel germline alleles was found using TIgGER (33) on the present data set. We found that the SSc-PAH cohort, on average, tended to have a larger fraction of fixed mutations than the healthy cohort across lineage sizes of up to 30 sequences (larger lineages were not consistently found in participants) (Fig. 2E). This finding suggests that SSc-PAH lineages have, on average, undergone more selective sweeps, that is, have been subject to sustained affinity maturation in response to antigen. One can speculate that the antigens in question are self-antigens, perhaps triggered by molecular mimicry (34), and are not necessarily the same for each patient.
To assess whether VDJ rearrangement probabilities or postrearrangement tolerance checkpoints (35) may be affected in SSc-PAH, we tested for differential expression of V segments, which form the most diverse set of germline segments compared with D and J. To focus on original VDJ recombination events while removing the effects of clonal expansion and transcript abundance, we defined V segment counts as a number of lineages (rather than a number of sequences or a number of transcripts). We found that IGHV2-5 was used at a lower level in SSc-PAH participants (fig. S1D and Fig. 2F). Note that IGHV2-5 usage was higher for the isotype-switched than for the unswitched compartment (Fig. 2F), but differential expression between disease and healthy cohorts was observed separately in both compartments, so the effect cannot be solely a consequence of the SSc-PAH–related expansion of the unswitched compartment (Fig. 2A). Differential expression was particularly pronounced in the unswitched compartment (Fig. 2F).
Having identified these B cell signatures of SSc-PAH, we can ask how they are affected in participants undergoing B cell depletion (study arm A). In study arm A, the SSc-PAH–related excess of IgD diversity and deficit in mean IgD mutation load were transiently reversed (Fig. 3, A and B), as was the deficit in IGHV2-5 usage (Fig. 3C). By contrast, B cell depletion did not have an evident effect on the fraction of fixed mutations (Fig. 3D).
Fig. 3. Time courses of SSc-PAH signatures.
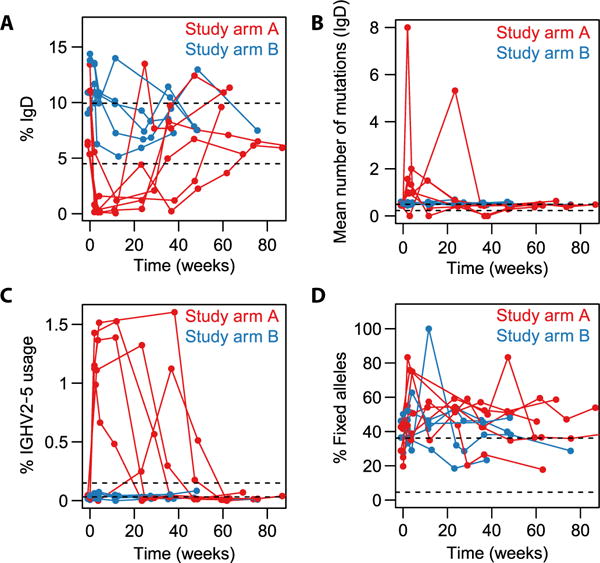
Dashed black lines indicate the range of values observed in healthy participants at baseline. Note that the data displayed for study arms A and B in the present figure (longitudinal experiment) were acquired in a separate batch from the data on healthy participants (cross-sectional experiment; already described in Fig. 2) and so may not be directly comparable to the healthy data; we nevertheless include the dashed lines as a guide. (A) IgD proportion. (B) IgD mutation loads. (C) Percentage of lineages using the IGHV2-5 segment (all isotypes combined). (D) Proportion of somatic mutations observed in a lineage that were considered “fixed,” that is, present in at least 80% of the lineage’s sequences. The proportion of fixed mutations was calculated separately for each lineage containing between 5 and 29 distinct sequences and then averaged across all those lineages.
Kinetics of repertoire reconstitution
Because we have inferred that the peripheral naïve repertoire is almost entirely wiped out in study group A (Fig. 1, B to E), we can use that group’s longitudinal repertoire data to deduce various rates governing homeostasis of the humoral immune system. Using the number of IgM sequences with zero somatic mutations as a proxy for the naïve diversity of the repertoire, we can express the rate of change of naïve diversity as
| (1) |
where Mx denotes the number of IgM sequences with x mutations, a is the generation rate of naïve sequences entering the periphery from the bone marrow, b is the mutation probability per unit time, c1 is the class switch probability per unit time, and c2 is the probability of apoptosis per unit time. Sequences that exit the M0 diversity by acquiring a mutation will increase the M1 diversity
| (2) |
Cells can leave the IgM compartment by undergoing either apoptosis (rate c2) or isotype switching (predominantly to IgA or IgG; rate c1). In the latter case, they increase the isotype-switched diversity
| (3) |
where Sx denotes the number of IgA and IgG sequences with x mutations. In Eqs. 1 and 2, we have made the simplifying assumption that the “exit” rates b, c1, and c2 applicable to the M0 diversity are the same for the M1 diversity; similarly, in Eq. 3, we have assumed that the rates b and c2 governing exits from the S0 diversity have the same values as in Eqs. 1 and 2. For a first approximation, it seems reasonable to assume that the fraction of a compartment that undergoes apoptosis, mutation, or class switching is roughly independent of the identity of the compartment, particularly if we restrict ourselves to lowly mutated compartments that likely have a smaller proportion of long-lived memory or plasma cells than more highly mutated compartments.
Given that the assessment of diversity using repertoire sequencing is a reliable measurement (fig. S2, A and B), we can determine a and d ≡ (b + c1 + c2) from the B cell replenishment time course using Eq. 1, starting at the onset of replenishment (after the more or less extended near-total depletion period). Fitting the linear model for all groups simultaneously as described in fig. S2C [after correcting observed sequence numbers for unseen sequences using the Chao1 estimator (36)] suggested that coefficient d varied little between participants, whereas coefficient a varied significantly, so we fitted the data again using a reduced model with a universal coefficient d for all participants and coefficients a that were allowed to vary from participant to participant (fits shown in Fig. 4A). As expected for a steady-state diversity controlled by these generation and exit rates, the diversity to which the M0 compartment returns after depletion is commensurate with its initial diversity (fig. S2D).
Fig. 4. Quantitative modeling of repertoire replenishment rates.
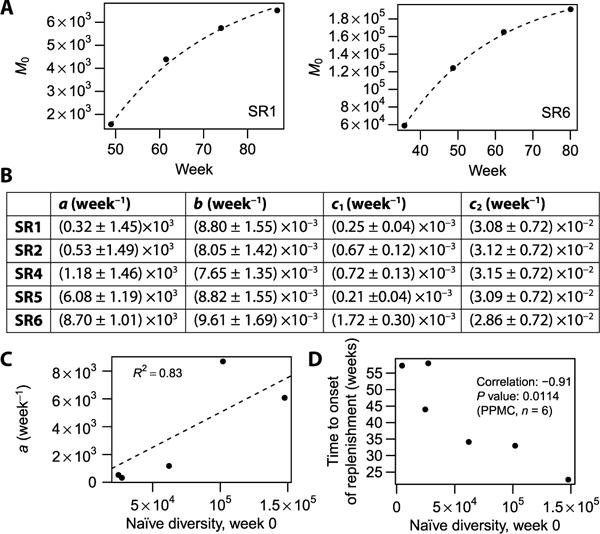
(A) Fit for the naïve diversity M0 (number of nonmutated IgM sequences, Chao1 estimate) as a function of time, after the onset of replenishment. Only the two participants (SR1 and SR6) with the largest numbers of corresponding time points are shown. Numeric data values are also provided in table S3. (B) Fit parameters describing sequence generation (a), mutation (b), class switch (c1), and apoptosis (c2) rates. SR1 to SR6 are labels for individual participants. Participant SR3 is omitted because an insufficient number of time points after the onset of depletion were available. (C) Baseline naïve diversity versus naïve generation rate. R2 value is for the linear fit shown by the dashed line performed with intercept set to 0. Numeric data values are also provided in table S4. (D) Baseline naïve diversity versus time to onset of replenishment (defined as the interval between second visit and the earliest visit exhibiting more than 5000 distinct IGH sequences). Numeric data values are also provided in table S5. PPMC, Pearson product-moment correlation.
In steady state, that is, in the baseline homeostatic regime, the time derivatives in Eqs. 2 and 3 are zero, so we can deduce
| (4) |
and
| (5) |
followed by c2 = d − b − c1 (Fig. 4B). Note that the steady-state form of Eq. 2 can be generalized to higher mutation loads
| (6) |
meaning that the distribution of mutation loads in the IgM compartment should be an exponential decay, which is roughly obeyed in our data (fig. S2E; note that the isotype-switched compartment displayed in fig. S2F follows a contrasting distribution peaked at a nonzero mutation load). Because the data show a hint that there may be a slight excess of highly mutated IgM sequences compared with the exponential law, which may be attributable to long-lived IgM memory cells and plasmablasts that do not follow our simple system of equations above, we decided to determine b on the basis of only M0 and M1 using Eq. 4 rather than fitting the entire distribution Mx using Eq. 6. The resulting sequence generation, mutation, class switch, and apoptosis rates (Fig. 4B) show that apoptosis due to the absence of activation by any cognate antigen is the most important factor limiting the naïve diversity; once a cell gets activated by a cognate antigen, a mutation event is more likely to occur first than a class switching event (c2 > b > c1).
If one wanted to predict the time course of naïve B cell replenishment after the first depleted time point, one would need three pieces: the time to onset of replenishment, the generation rate a, and the exit rate d. Now, first, the exit rate d appeared to be near-“universal” between participants. Second, in steady state (from Eq. 1 evaluated with ), the rate a is expected to be proportional to the baseline naïve diversity, a fact that was borne out by our data (Fig. 4C). Third, the time to onset of replenishment was found to be inversely correlated with baseline diversity (Fig. 4D). These observations suggest the intriguing possibility of predicting depletion and replenishment time courses from baseline measurements alone.
Similarities and perturbations of the reconstituted repertoire compared with baseline
We sought to assess which features of the BCR repertoire are elastic or plastic under B cell depletion. We found that there is a stereotypy to the repertoire that is recovered after depletion: Isotype usages, VDJ combination usages, and mutation load histograms show a marked similarity to their baseline profiles that is only temporarily lost after depletion (Fig. 5A). The mutation load profiles show that high mutation loads do not necessitate a lifetime of pathogen-induced affinity maturation but are recovered within months—so any subsequent improved immune responses are presumably not generated by B cell lineages acquiring more and more mutations ad infinitum but rather by adding breadth to the phylogenetic tree. Frequencies of VDJ segment combinations (defined here as the number of distinct IGH sequences with a given combination) exhibited a similarity between participants at baseline that was lost in depletion but recovered at the monitoring end point (fig. S3A). The set of amino acid changes in the repertoire also showed a greater degree of interparticipant similarity when repertoires were full (baseline or end point) rather than depleted (fig. S3B). This observation suggests intrinsic biases of the somatic hypermutation machinery that are conserved between participants (37), contrasted with participant-by-participant idiosyncrasy of the mutations exhibited by the (presumably highly pathogen-specific) plasmablasts that remain during depletion. Although VDJ usages, isotype usages, and mutation histograms were highly elastic, demonstrating that mechanisms of diversity generation are intact after B cell depletion, only a fraction of amino acid changes present at baseline were observed to return by the end point (Fig. 5A, “specific mutations”).
Fig. 5. Assessment of repertoire elasticity and compartment replenishment after depletion.
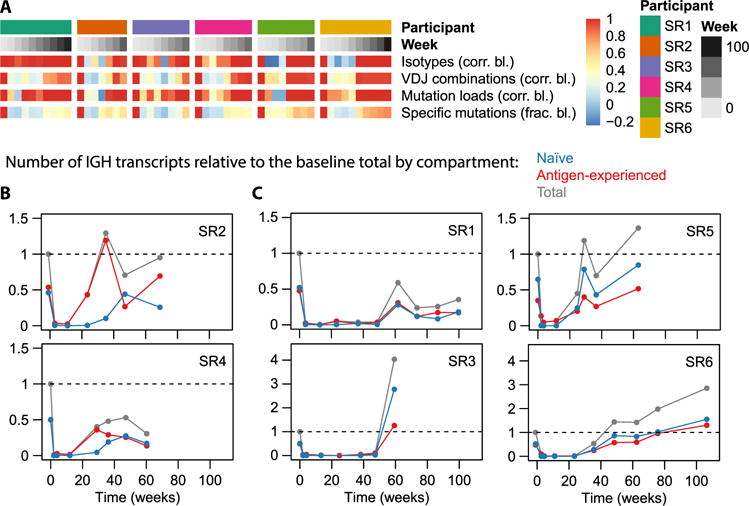
(A) Similarity of various repertoire characteristics to their baseline state on a per-participant basis. The top two rows identify participants and time points; the other rows display data as a heat map for each of the indicated variables, compared with baseline using either a correlation coefficient or a fraction as indicated. Naturally, for each participant, the baseline value is 1; values then tend to drop during repertoire depletion and gradually increase again during replenishment. For isotype usage profiles (resolved by subisotype) and VDJ usage profiles, the value shown at each time point is the correlation to the baseline profile. A repertoire’s “mutation load profile” was defined as the number of nucleotide sequences having each mutation load from 0 to 10 (nucleotide) mutations; we display the correlation of such mutation load histograms to the baseline histogram. “Specific mutations” refers to the set of distinct V-segment amino acid mutations observed in a repertoire (each defined by the identity of the V segment, the position being mutated, and the identity of the observed residue). We displayed the fraction of baseline mutations that were observed at each postbaseline time point to indicate to what extent specific mutations observed at baseline return after B cell depletion. (B and C) Number of transcripts observed as a function of time, separated by compartment (naïve or antigen-experienced), relative to the total number of transcripts (from either compartment) observed at baseline. Note that in (B), the antigen-experienced fraction increases well before the naïve fraction, whereas in (C), it does not. Sequences were considered naïve if they were IgD or IgM and nonmutated and considered antigen-experienced if they had at least one mutation or were class-switched.
Next, we disentangled the B cell populations that replenish the repertoire. First, we contrast antigen-experienced cells with naïve cells (Fig. 5, B and C), focusing on their contributions to the total transcript abundances, which should be a proxy for rough protein abundances. We observe two distinct phenotypes: In certain participants (SR2 and SR4), the first postdepletion repertoire consists almost entirely of antigen-experienced transcripts (Fig. 5B), whereas in most participants (SR1, SR3, SR5, and SR6), the naïve and antigen-experienced compartments recover simultaneously (Fig. 5C). The latter phenotype is what one might expect in a model in which new naïve B cells enter the periphery from the bone marrow, soon encounter antigen, and get activated. The former phenotype suggests a scenario in which an early surge in plasmablasts or plasma cells palliates B cell depletion until replenishment of naïve cells begins. Because plasmablasts live only a few days before dying or terminally differentiating into long-lived plasma cells in the bone marrow (38), this phenotype must be explained by long-lived plasma cells being released from the bone marrow to populate the blood. The early increase in antigen-experienced transcripts in participants SR2 and SR4 corresponded to increases in the IgA1, IgA2, and IgG2 isotypes beyond baseline levels (Fig. 6A and table S6); in participant SR2, there was also a marked increase in IGH sequences with high somatic mutation loads (in the range from six to nine mutations in the sequenced region; Fig. 6B).
Fig. 6. Comparison of repertoire distributions to baseline.
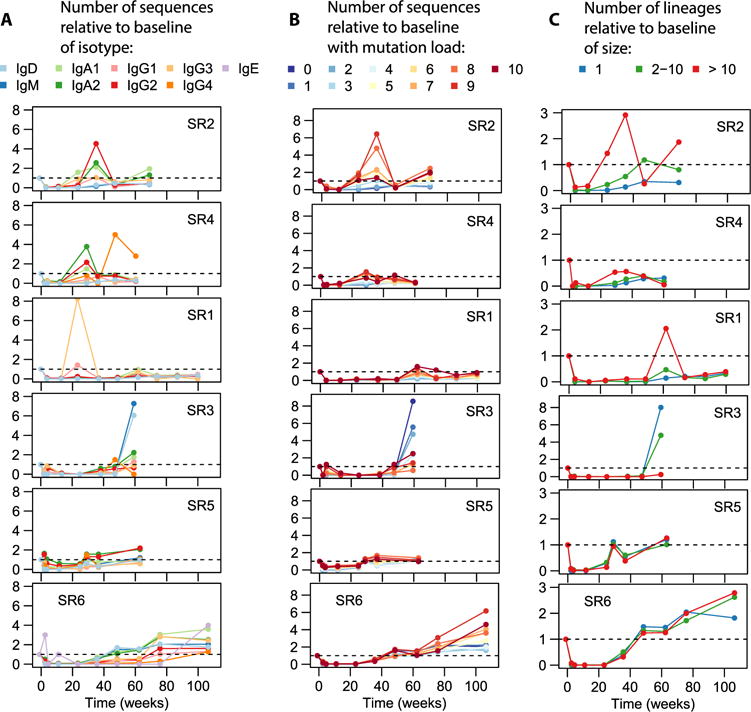
(A) Frequency of sequences with each subisotype relative to baseline. (B) Frequency of sequences with various mutation loads relative to baseline. (C) Frequency of lineages within various size bins relative to baseline. Here, lineage size refers to the number of distinct sequences in a lineage.
To characterize the role of B cell clonal proliferation in the replenishment of the repertoire, we assigned lineages to three bins depending on their degree of expansion (Fig. 6C). One can theorize two extreme modes of replenishment: In one scenario, the repertoire would get filled with a large diversity of clonally unrelated B cells, none of which proliferate much; in the other scenario, the repertoire would be seeded by a single B cell clone, which proliferates to fill the entire repertoire. In reality, we observe an intermediate regime: All three lineage size bins contribute to replenishment (Fig. 6C). Patterns of clonal expansion are observed to be idiosyncratic to individuals: Certain participants display an increased importance of highly expanded lineages by the last time point (e.g., SR2 and SR6; also transiently the case for SR1), suggesting proliferation with somatic hypermutation (compare Fig. 6B); another participant (SR3) displayed a markedly nonexpanded, naïve phenotype at the end of depletion, with mostly IgD or IgM antibodies showing few mutations (Fig. 6, A to C); in yet another group of participants, the importance of the three lineage size bins relative to each other at the last time point was comparable with baseline (SR1 and SR5 in Fig. 6C).
DISCUSSION
We have characterized the role of B cells in SSc-PAH at the level of the BCR. We identified an anomaly in the process of VDJ recombination, namely, underuse of the IGHV2-5 segment (particularly in the isotype-unswitched compartment; Fig. 2F) and noted a bimodal effect at the extremes of peripheral B cell development: SSc-PAH participants displayed elevated proportions of both naïve BCRs (Fig. 2A) and highly secreted antigen-experienced BCRs (Fig. 2, B to D). The presence of any infection at baseline would have made participants ineligible to participate in the present study, such that any excess proportion of plasmablasts cannot be the result of current infection. Instead, the excess may suggest a sustained immune response against self-antigens, a hypothesis that is further corroborated by the observation of an increased proportion of near-fixed somatic mutations likely to arise from clonal selection (Fig. 2E). Certain of these disease signatures are transiently reversed by CD20+ B cell depletion, namely, the IGHV2-5 gene usage (Fig. 3C) and the naïve proportion (Fig. 3, A and B).
We do not suggest that our findings can be used to define clinical subsets of SSc-PAH patients, and we do not analyze clinical outcomes here. Because the therapeutic trial, which is the source of samples here, is double-blinded and ongoing, it is too early to know whether the therapy stabilized or reversed the disease. Rather, our focus is understanding the basic science of the immune repertoire of SSc patients and the immune repertoire response to the B cell ablating therapy (rituximab). In this respect, although we were technically blinded to the treatment and control groups through the use of dummy identifiers, it is evident which group received rituximab from the marked and consistent changes in the repertoire.
The observation of B cell depletion in one group of study participants (Fig. 1) allowed us to measure rates governing BCR diversity generation and dissipation in humans in vivo (Fig. 4, A and B), demonstrating the high frequency of apoptosis of naïve cells compared with activation as well as the precedence of somatic mutation over class switch recombination in activation. These measurements, combined with trends relating replenishment rates and depletion durations to baseline information (Fig. 4, C and D), constitute a proof of principle for the prediction of immunologic depletion and replenishment time courses from baseline information alone. We compared the reconstituted repertoire with the baseline repertoire and noted a marked resiliency of summary properties, such as VDJ frequencies, mutation histograms, and isotype frequencies (Fig. 5A), indicating that the diversity generation mechanisms of VDJ recombination, somatic hypermutation, and class switch recombination are intact after depletion and can regenerate a normal B cell repertoire in a time span of months.
We also dissected the mechanism of B cell replenishment after depletion. We found that release of antigen-experienced cells into the periphery—likely plasma cells released from the bone marrow—does (Fig. 5B) or does not (Fig. 5C) precede the onset of naïve repopulation depending on the individual. Antibody repertoire sequencing exposed the lineage structure of the recovering B cell ensemble, revealing participant-to-participant heterogeneity in the pattern of clonal expansion after depletion (Fig. 6C).
MATERIALS AND METHODS
Study design
The present study was an exploratory study aimed at understanding how the B cell repertoire is affected in SSc-PAH and how it responds to B cell depletion. Protocols were approved by the Institutional Review Board at Stanford University and at all Autoimmunity Center of Excellence sites that provided samples for this study; participants gave informed consent.
Sequencing and analysis
IGH repertoires were obtained using previously published protocols (39, 40) with small modifications, via the following steps: reverse transcription (RT) with IGH constant domain–specific primers containing random barcodes (table S7) on 250 ng of total RNA extracted from PBMCs; second-strand synthesis with IGH variable domain–specific primers containing random barcodes (table S8); AMPure purification (Beckman Coulter); polymerase chain reaction (PCR) amplification incorporating Illumina adapters and sample multiplexing indices; 150–base pair paired-end NextSeq sequencing (Illumina); and analysis using pRESTO (41), IMGT/HighV-QUEST (42), Change-O (43), and R scripts deposited at https://github.com/cdebourcy/scleroderma. During analysis, the random barcodes contained in the primers were used as UIDs, allowing RNA transcript counting and amplification bias correction: We collapsed reads that had identical UIDs so as to count each RNA molecule of the starting material only once, regardless of the degree of PCR amplification the molecule underwent. We also used the barcodes to correct single-nucleotide errors occurring during amplification or sequencing by calling a consensus among reads with identical UIDs. Numbers of raw sequencing read pairs for each sample are displayed in table S9, alongside numbers of sequences and UIDs after processing. For more technical descriptions, see Supplementary Materials and Methods.
Statistical tests
All tests were two-sided (Wilcoxon–Mann-Whitney tests and Pearson product-moment correlation tests). Significance codes in figures are as follows: *0.01 ≤ P < 0.05; **0.001 ≤ P < 0.01; ***0.0001 ≤ P < 0.001.
Supplementary Material
Fig. S1. Comparison of healthy and SSc-PAH cohorts at baseline.
Fig. S2. Calculation of rates governing repertoire homeostasis.
Fig. S3. Assessment of repertoire elasticity under depletion.
Table S1. Data values displayed in Fig. 1E.
Table S2. Data values displayed in Fig. 1F.
Table S3. Data values displayed in Fig. 4A.
Table S4. Data values displayed in Fig. 4C.
Table S5. Data values displayed in Fig. 4D.
Table S6. Repertoire isotype percentages for each participant and time point (longitudinal experiment).
Table S7. IGH constant-region primer pool.
Table S8. IGH variable-region primer pool.
Table S9. Read counts before and after processing.
Table S10. Data values displayed in fig. S2A.
Table S11. Data values displayed in fig. S2B.
Table S12. Data values displayed in fig. S2D.
Acknowledgments
The present paper resulted from an Autoimmunity Centers of Excellence (ACE) study. ACE is a research consortium supported by the National Institute of Allergy and Infectious Disease (NIH). We thank the ACE ASC01 study group for providing the SSc-PAH samples; S. Mackey for coordinating the clinical studies that provided the healthy participants; S. Swope and M. Ugur for conducting study visits and collecting samples from healthy participants; X. He for providing access to samples from healthy participants; H. Maecker, J. Bierre, and B. Varasteh (Stanford Human Immune Monitoring Core) for sample banking; N. Neff and G. Mantalas (Stanford Stem Cell Genome Center) for assistance with sequencing; and L. Penland, D. Croote, F. Horns, and J. Glanville for discussions.
Funding: The research on SSc-PAH participants was supported by NIH grants U19 AI046374 (to M. Holers of the Denver ACE) and P01 HL014985, R01 HL122887 (to M.R.N.). The research on healthy participants was supported by NIH grant U19 AI057229 (to M.M.D.). The clinical project on healthy participants was supported by National Center for Research Resources (NIH) Clinical and Translational Science Award UL1 RR025744. The ClinicalTrials.gov numbers from the studies from which samples were used are NCT01086540 (SSc-PAH cohort) and NCT02133781, NCT03020498, NCT03022396, and NCT03022422 (healthy cohort). C.F.A.d.B. was supported by an International Fulbright Science and Technology Award and a Melvin and Joan Lane Stanford Graduate Fellowship.
Footnotes
SUPPLEMENTARY MATERIALS
immunology.sciencemag.org/cgi/content/full/2/15/eaan8289/DC1
Materials and Methods
References (44–46)
Author contributions: C.F.A.d.B., M.R.N., and S.R.Q. designed the study; M.R.N. led the ACE protocol ASC01; C.L.D. coordinated healthy participant recruitment and sample collection; C.F.A.d.B. prepared the sequencing libraries; C.F.A.d.B., M.M.D., M.R.N., and S.R.Q. analyzed the data; C.F.A.d.B. performed the statistical analyses; C.F.A.d.B., M.R.N., and S.R.Q. wrote the manuscript.
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: The sequence data sets reported in this paper have been deposited in the National Center for Biotechnology Information BioProject with accession no. PRJNA396251.
REFERENCES AND NOTES
- 1.Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360:1989–2003. doi: 10.1056/NEJMra0806188. [DOI] [PubMed] [Google Scholar]
- 2.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: A perspective. Eur Respir J. 2005;26:1110–1118. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 3.Ingegnoli F, Trabattoni D, Saresella M, Fantini F, Clerici M. Distinct immune profiles characterize patients with diffuse or limited systemic sclerosis. Clin Immunol. 2003;108:21–28. doi: 10.1016/s1521-6616(03)00062-7. [DOI] [PubMed] [Google Scholar]
- 4.Kayser C, Fritzler MJ. Autoantibodies in systemic sclerosis: Unanswered questions. Front Immunol. 2015;6:167. doi: 10.3389/fimmu.2015.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sato S, Fujimoto M, Hasegawa M, Takehara K. Altered blood B lymphocyte homeostasis in systemic sclerosis: Expanded naive B cells and diminished but activated memory B cells. Arthritis Rheum. 2004;50:1918–1927. doi: 10.1002/art.20274. [DOI] [PubMed] [Google Scholar]
- 6.Soto L, Ferrier A, Aravena O, Fonseca E, Berendsen J, Biere A, Bueno D, Ramos V, Aguillón JC, Catalán D. Systemic sclerosis patients present alterations in the expression of molecules involved in B-cell regulation. Front Immunol. 2015;6:496. doi: 10.3389/fimmu.2015.00496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshizaki A. B lymphocytes in systemic sclerosis: Abnormalities and therapeutic targets. J Dermatol. 2016;43:39–45. doi: 10.1111/1346-8138.13184. [DOI] [PubMed] [Google Scholar]
- 8.Mukerjee D, St George B, Coleiro B, Knight C, Denton CP, Davar J, Black CM, Coghlan JG. Prevalence and outcome in systemic sclerosis associated pulmonary arterial hypertension: Application of a registry approach. Ann Rheum Dis. 2003;62:1088–1093. doi: 10.1136/ard.62.11.1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Humbert M, Sitbon O, Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med. 2004;351:1425–1436. doi: 10.1056/NEJMra040291. [DOI] [PubMed] [Google Scholar]
- 10.Grillo-López AJ. Rituximab (Rituxan®/MabThera®): The first decade (1993–2003) Expert Rev Anticancer Ther. 2003;3:767–779. doi: 10.1586/14737140.3.6.767. [DOI] [PubMed] [Google Scholar]
- 11.Browning JL. B cells move to centre stage: Novel opportunities for autoimmune disease treatment. Nat Rev Drug Discov. 2006;5:564–576. doi: 10.1038/nrd2085. [DOI] [PubMed] [Google Scholar]
- 12.Fraticelli P, De Vita S, Franzolini N, Svegliati S, Scott CA, Tonnini C, Spadoni T, Gabrielli B, Pomponio G, Moroncini G, Gabrielli A. Reduced type I collagen gene expression by skin fibroblasts of patients with systemic sclerosis after one treatment course with rituximab. Clin Exp Rheumatol. 2015;33:S160–S167. [PubMed] [Google Scholar]
- 13.Daoussis D, Liossis SNC, Tsamandas AC, Kalogeropoulou C, Kazantzi A, Sirinian C, Karampetsou M, Yiannopoulos G, Andonopoulos AP. Experience with rituximab in scleroderma: Results from a 1-year, proof-of-principle study. Rheumatology (Oxford) 2010;49:271–280. doi: 10.1093/rheumatology/kep093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Daoussis D, Melissaropoulos K, Sakellaropoulos G, Antonopoulos I, Markatseli TE, Simopoulou T, Georgiou P, Andonopoulos AP, Drosos AA, Sakkas L, Liossis SNC. A multicenter, open-label, comparative study of B-cell depletion therapy with Rituximab for systemic sclerosis-associated interstitial lung disease. Semin Arthritis Rheum. 2017;46:625–631. doi: 10.1016/j.semarthrit.2016.10.003. [DOI] [PubMed] [Google Scholar]
- 15.Jordan S, Distler JHW, Maurer B, Huscher D, van Laar JM, Allanore Y, Distler O. Effects and safety of rituximab in systemic sclerosis: An analysis from the European Scleroderma Trial and Research (EUSTAR) group. Ann Rheum Dis. 2015;74:1188–1194. doi: 10.1136/annrheumdis-2013-204522. [DOI] [PubMed] [Google Scholar]
- 16.Bosello SL, De Luca G, Rucco M, Berardi G, Falcione M, Danza FM, Pirronti T, Ferraccioli G. Long-term efficacy of B cell depletion therapy on lung and skin involvement in diffuse systemic sclerosis. Semin Arthritis Rheum. 2015;44:428–436. doi: 10.1016/j.semarthrit.2014.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Lepri G, Avouac J, Airò P, Anguita Santos F, Bellando-Randone S, Blagojevic J, Garcia Hernàndez F, Gonzalez Nieto JA, Guiducci S, Jordan S, Limaye V, Maurer B, Selva-O’Callaghan A, Riccieri V, Distler O, Matucci-Cerinic M, Allanore Y. Effects of rituximab in connective tissue disorders related interstitial lung disease. Clin Exp Rheumatol. 2016;34(suppl 100):181–185. [PubMed] [Google Scholar]
- 18.Mizuno S, Farkas L, Al Husseini A, Farkas D, Gomez-Arroyo J, Kraskauskas D, Nicolls MR, Cool CD, Bogaard HJ, Voelkel NF. Severe pulmonary arterial hypertension induced by SU5416 and ovalbumin immunization. Am J Respir Cell Mol Biol. 2012;47:679–687. doi: 10.1165/rcmb.2012-0077OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McGonagle D, Tan AL, Madden J, Rawstron AC, Rehman A, Emery P, Thomas S. Successful treatment of resistant scleroderma-associated interstitial lung disease with rituximab. Rheumatology. 2008;47:552–553. doi: 10.1093/rheumatology/kem357. [DOI] [PubMed] [Google Scholar]
- 20.Kraaij MD, van Laar JM. The role of B cells in systemic sclerosis. Biologics. 2008;2:389–395. [PMC free article] [PubMed] [Google Scholar]
- 21.Sato S, Hasegawa M, Takehara K. Serum levels of interleukin-6 and interleukin-10 correlate with total skin thickness score in patients with systemic sclerosis. J Dermatol Sci. 2001;27:140–146. doi: 10.1016/s0923-1811(01)00128-1. [DOI] [PubMed] [Google Scholar]
- 22.Sato S, Hasegawa M, Fujimoto M, Tedder TF, Takehara K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J Immunol. 2000;165:6635–6643. doi: 10.4049/jimmunol.165.11.6635. [DOI] [PubMed] [Google Scholar]
- 23.Xu GJ, Shah AA, Li MZ, Xu Q, Rosen A, Casciola-Rosen L, Elledge SJ. Systematic autoantigen analysis identifies a distinct subtype of scleroderma with coincident cancer. Proc Natl Acad Sci USA. 2016;113:E7526–E7534. doi: 10.1073/pnas.1615990113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum. 2005;35:35–42. doi: 10.1016/j.semarthrit.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 25.Rouzière AS, Kneitz C, Palanichamy A, Dörner T, Tony HP. Regeneration of the immunoglobulin heavy-chain repertoire after transient B-cell depletion with an anti-CD20 antibody. Arthritis Res Ther. 2005;7:R714. doi: 10.1186/ar1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang N, He J, Weinstein JA, Penland L, Sasaki S, He XS, Dekker CL, Zheng NY, Huang M, Sullivan M, Wilson PC, Greenberg HB, Davis MM, Fisher DS, Quake SR. Lineage structure of the human antibody repertoire in response to influenza vaccination. Sci Transl Med. 2013;5:171ra19. doi: 10.1126/scitranslmed.3004794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Georgiou G, Ippolito GC, Beausang J, Busse CE, Wardemann H, Quake SR. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat Biotechnol. 2014;32:158–168. doi: 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Edwards JCW, Cambridge G. B-cell targeting in rheumatoid arthritis and other autoimmune diseases. Nat Rev Immunol. 2006;6:394–403. doi: 10.1038/nri1838. [DOI] [PubMed] [Google Scholar]
- 29.Lozupone C, Knight R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl Environ Microbiol. 2005;71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Bourcy CFA, Angel CJL, Vollmers C, Dekker CL, Davis MM, Quake SR. Phylogenetic analysis of the human antibody repertoire reveals quantitative signatures of immune senescence and aging. Proc Natl Acad Sci USA. 2017;114:1105–1110. doi: 10.1073/pnas.1617959114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xu JL, Davis MM. Diversity in the CDR3 region of VH is sufficient for most antibody specificities. Immunity. 2000;13:37–45. doi: 10.1016/s1074-7613(00)00006-6. [DOI] [PubMed] [Google Scholar]
- 32.Parameswaran P, Liu Y, Roskin KM, Jackson KKL, Dixit VP, Lee JY, Artiles KL, Zompi S, Vargas MJ, Simen BB, Hanczaruk B, McGowan KR, Tariq MA, Pourmand N, Koller D, Balmaseda A, Boyd SD, Harris E, Fire AZ. Convergent antibody signatures in human dengue. Cell Host Microbe. 2013;13:691–700. doi: 10.1016/j.chom.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gadala-Maria D, Yaari G, Uduman M, Kleinstein SH. Automated analysis of high-throughput B-cell sequencing data reveals a high frequency of novel immunoglobulin V gene segment alleles. Proc Natl Acad Sci USA. 2015;112:E862–E870. doi: 10.1073/pnas.1417683112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Smyk DS, Koutsoumpas AL, Mytilinaiou MG, Rigopoulou EI, Sakkas LI, Bogdanos DP. Helicobacter pylori and autoimmune disease: Cause or bystander. World J Gastroenterol. 2014;20:613–629. doi: 10.3748/wjg.v20.i3.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melchers F. Checkpoints that control B cell development. J Clin Invest. 2015;125:2203–2210. doi: 10.1172/JCI78083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chao A. Nonparametric estimation of the number of classes in a population. Scand J Stat. 1984;11:265–270. [Google Scholar]
- 37.Yaari G, Vander Heiden JA, Uduman M, Gadala-Maria D, Gupta N, Stern JNH, O’Connor KC, Hafler DA, Laserson U, Vigneault F, Kleinstein SH. Models of somatic hypermutation targeting and substitution based on synonymous mutations from high-throughput immunoglobulin sequencing data. Front Immunol. 2013;4:358. doi: 10.3389/fimmu.2013.00358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Murphy K, Weaver C. Janeway’s Immunobiology. Garland Science; 2016. [Google Scholar]
- 39.Vollmers C, Sit RV, Weinstein JA, Dekker CL, Quake SR. Genetic measurement of memory B-cell recall using antibody repertoire sequencing. Proc Natl Acad Sci USA. 2013;110:13463–13468. doi: 10.1073/pnas.1312146110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horns F, Vollmers C, Croote D, Mackey SF, Swan GE, Dekker CL, Davis MM, Quake SR. Lineage tracing of human B cells reveals the in vivo landscape of human antibody class switching. eLife. 2016;5:e16578. doi: 10.7554/eLife.16578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vander Heiden JA, Yaari G, Uduman M, Stern JNH, O’Connor KC, Hafler DA, Vigneault F, Kleinstein SH. pRESTO: A toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics. 2014;30:1930–1932. doi: 10.1093/bioinformatics/btu138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alamyar E, Giudicelli V, Li S, Duroux P, Lefranc MP. IMGT/HighV-QUEST: The IMGT web portal for immunoglobulin (IG) or antibody and T cell receptor (TR) analysis from NGS high throughput and deep sequencing. Immunome Res. 2012;8:26. [Google Scholar]
- 43.Gupta NT, Vander Heiden JA, Uduman M, Gadala-Maria D, Yaari G, Kleinstein SH. Change-O: A toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics. 2015;31:3356–3358. doi: 10.1093/bioinformatics/btv359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van der Loo MPJ. The stringdist package for approximate string matching. R J. 2014;6:111–122. [Google Scholar]
- 45.Köster J, Rahmann S. Snakemake—A scalable bioinformatics workflow engine. Bioinformatics. 2012;28:2520–2522. doi: 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 46.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Comparison of healthy and SSc-PAH cohorts at baseline.
Fig. S2. Calculation of rates governing repertoire homeostasis.
Fig. S3. Assessment of repertoire elasticity under depletion.
Table S1. Data values displayed in Fig. 1E.
Table S2. Data values displayed in Fig. 1F.
Table S3. Data values displayed in Fig. 4A.
Table S4. Data values displayed in Fig. 4C.
Table S5. Data values displayed in Fig. 4D.
Table S6. Repertoire isotype percentages for each participant and time point (longitudinal experiment).
Table S7. IGH constant-region primer pool.
Table S8. IGH variable-region primer pool.
Table S9. Read counts before and after processing.
Table S10. Data values displayed in fig. S2A.
Table S11. Data values displayed in fig. S2B.
Table S12. Data values displayed in fig. S2D.