

Abstract
While inflammatory phagocytosis of microbial pathogens and non-inflammatory phagocytosis of apoptotic cells have each been studied extensively, the consequences of innate immune recognition of host cells undergoing apoptosis as a direct result of infection are unclear. In this situation, the innate immune system is confronted with mixed signals, those from apoptotic cells and those from the infecting pathogen. Nuclear receptor activation has been implicated downstream of apoptotic cell recognition while Toll-like receptors are the prototypical inflammatory receptors engaged during infection. When the two signals combine, a new set of events takes place beginning with transrepression of a subset of inflammatory-response genes and ending with the induction of a T helper-17 adaptive immune response. This response is best suited for clearing the infecting pathogen and repairing the damage that occurred to the host tissue during infection.
Introduction
Maintenance of tissue homeostasis necessitates not only recognition and removal of invading microbial pathogens, but also clearance of dying cells. Cells die at different stages of life as part of embryonic development and normal tissue turnover, thus the term programmed cell death, or apoptosis, where death is expected and part of the well-being of healthy tissues [1]. The urgency of dealing with microbial pathogens is evident; rapid multiplication of pathogens, their invasion of host tissues, systemic spread, and interference with proper organ function are to be avoided at all costs. Timely death of cells in a healthy host seems harmless in comparison, but only when all steps are executed according to plan. An elegant feature of apoptosis is its tidiness: chromatin condensation, nuclear fragmentation, cell shrinkage, and plasma membrane blebbing are followed by the formation of apoptotic bodies with no leakage of cellular contents into the surrounding tissue. Despite these ordered events, apoptotic cell corpses still require prompt removal. Defects in clearance of apoptotic cells can lead to their accumulation and progression into secondary necrosis, a type of death that is inflammatory in nature with dire consequences to the host manifested by autoimmune disease such as systemic lupus erythematosus (SLE) [2•,3].
Clearance of both microbial pathogens and apoptotic cells relies on phagocytosis. A crucial difference, however, is the immunological outcome, which is determined by the type of pattern recognition receptors (PRRs) engaged during phagocytosis [4]. Microbial structures collectively known as pathogen associated molecular patterns (PAMPs) engage inflammatory PRRs such as Toll-like receptors (TLRs), leading to inflammatory phagocytosis, while apoptotic cells express ‘eat me’ signals that are recognized by a different set of non-inflammatory PRRs, leading to non-inflammatory and immunosuppressive phagocytosis. At the subcellular level, each phagosome undergoes a distinct maturation program directly dictated by the type of PRRs engaged at the outset of phagocytosis. Engagement of TLRs during phagocytosis imparts a distinct TLR based molecular signature on phagosomes carrying microorganisms different than that expected of phagosomes carrying apoptotic cells [4,5]. TLRs specifically increase the maturation kinetics of phagosomes carrying PAMPs with rapid delivery to lysosomes for degradation [4]. In addition, TLRs favor major histocompatibility complex class II (MHC II) presentation of microbial peptides derived from phagosomes carrying bacteria, while peptides derived from phagosomes carrying apoptotic cells within the same dendritic cell (DC) are not favored for presentation within the context of TLR-induced co-stimulatory molecule expression [4]. Therefore, phagocytosis can be controlled by signals from PRRs, and compartmentalized engagement of PRRs from individual phagosomes determines the fate of the cargo within those phagosomes as well as the immune responses tailored to that cargo. But what happens when phagosomes carry apoptotic cells dying from an infection, and thus contain both PAMPs and apoptotic cell components within the same space? How can the immunosuppressive clearance of apoptotic cells coexist with the necessarily inflammatory nature of infection? Can delivery of both signals trigger a new type of immune response specifically tailored to immunity against and clearance of the invading pathogen, as well as apoptotic cell clearance, all while protecting and healing host tissues? Here we consider each signal alone, and then discuss the effect of combining these signals on innate and adaptive immune outcomes.
The pro-inflammatory signal triggered by PAMPs
All TLR family members have an extracellular domain comprised of leucine rich repeats (LRRs), and a cytoplasmic domain known as the Toll/IL-1R (TIR) homology domain that is crucial for signaling [6]. TLRs are expressed on the plasma membrane and along the endocytic pathway, placing them at the proper locations for detecting microbial pathogens. Remarkably, despite conservation among LRR domains, different TLRs recognize different PAMPs. TLR5 that recognizes flagellin, TLR2 that forms heterodimers with TLR1 and TLR2 and recognizes bacterial lipoproteins and lipopeptides, and TLR4 that recognizes lipopolysaccharide (LPS) are primarily expressed on the plasma membrane. Upon binding to LPS, TLR4 is internalized into early endosomes, a process necessary for its ability to induce type I interferons. The nucleic acid sensing TLRs, TLR3, 7, 8, and 9 are confined to endolysosomal compartments, and as such are ideally suited for the detection of viruses. TLR signaling is initiated with recruitment of the sorting adaptor proteins TIRAP and TRAM and their respective signaling adaptors MyD88 and TRIF; TRIF exclusively for TLR3, MyD88 for all TLRs, while TLR4 uses both adaptors. Signaling downstream of TLRs activates the nuclear factor-κB (NF-κB) and interferon regulatory factor (IRF) families of transcription factors. TLR signaling also activates MAP kinases such as p38, JNK, and ERK1/2, which activate the AP-1 transcription factor. TLR signaling converges on the transcription of a number of inflammatory cytokines, chemokines, type I interferons and immune response genes. The impact of TLR signaling on professional phagocytes is remarkable clearly favoring inflammation and activation of the adaptive immune response. Much is known about TLR signaling pathways and their control of adaptive immunity, and we refer to a recent review on this topic [6].
The anti-inflammatory signal triggered by apoptotic cells
‘Eat me’ signals displayed by apoptotic cells include expression of new molecules as well as oxidized or modified forms of existing molecules [7]. Here we focus on the most prominent of these, phosphatidyl serine (PS) that localizes to the outer leaflet of the plasma membrane upon apoptosis [7,8]. Recognition of PS can occur via a specific ‘bridging’ molecule: as examples, milk fat globule epidermal growth factor 8 (MFG-E8) links PS to phagocyte αvβ3 integrin [7], while growth-arrest-specific 6 (GAS6) links PS to the receptor tyrosine kinase MER [7]. Three receptors that recognize PS directly have recently been identified after the first described PS receptor was found not to mediate apoptotic cell recognition [7]. PS acts as a ligand for the T-cell immunoglobulin domain and mucin domain (TIM)-4 molecule on macrophages and DC [9••], and TIM-4 helps to promote uptake of the apoptotic cell [10••]. Two other molecules, brain-specific angiogenesis inhibitor 1 (BAI1) and stabilin-2 have also been shown to mediate uptake of apoptotic cells via recognition of PS [11•,12•].
In contrast to PAMP-induced activation of TLR signaling and inflammatory immune responses, it has long been known that apoptotic cell uptake by phagocytes has immunosuppressive effects, first described as reduced inflammatory cytokine production by human monocytes in response to LPS, and upregulation of the immunoregulatory cytokine interleukin 10 (IL-10) [13•]. Apoptotic cells have been known for some time to also induce the synthesis of anti-inflammatory mediators such as TGF-β, prostaglandin E2, and platelet activating factor by macrophages [14•,15•]. Recognition of apoptotic cell ‘eat me’ signals without physical internalization seems sufficient for inducing the anti-inflammatory response from macrophages [16], suggesting that signals for synthesis of anti-inflammatory cytokines are distinct from those inducing phagocytosis, and probably diverge downstream of PRR activation at the plasma membrane.
Recent studies have shed light on the pathways leading to the anti-inflammatory response induced by apoptotic cells. Because phagocytosis of apoptotic cells involves recognition of oxidized fatty acids [7,17] and increased levels of cholesterol in phagocytes [18,19] a role was postulated for the peroxisome-proliferator-activated receptors (PPARs) and liver X receptors (LXRs), which function as sensors of modified fatty acids and sterols [20,21]. Indeed, phagocytosis of apoptotic cells was reported to induce activation and expression of PPAR-γ [22,23], PPAR-δ [24••], and LXR in macrophages [25••]. Macrophage-specific deletion of PPAR-γ led to down-regulation of CD36, AXL receptor tyrosine kinase, transglutaminase-2 (TG2), and pentraxin-3 (PTX3) genes involved in phagocytosis of apoptotic cells [23]. PPAR-δ deficient macrophages showed decreased expression of bridge molecules such as the complement component 1qb, GAS6, and Thrombsopondin, and a resultant decrease in phagocytosis of apoptotic cells [24••]. Similarly, macrophages deficient for both LXRα and LXRβ showed impaired phagocytosis of apoptotic cells, while LXR activation increased expression of MER [25••]. Global or macrophage-specific deletion of PPAR-δ or deficiency in LXRα/β led to defects in apoptotic cell clearance in vivo and consequent increases in serum levels of autoantibodies with progressive lupus-like autoimmune disease. Importantly, apoptotic cell uptake by PPAR-δ or LXRα/β deficient macrophages was unable to suppress LPS-induced IL-12, TNF-α, or IL-1β transcription and synthesis, and failed to induce TGF-β and IL-10 synthesis demonstrating that PPAR-δ and LXR contribute to the anti-inflammatory response induced by apoptotic cells. Which PRR and ‘eat me signal’ interaction could be responsible for the PPAR-δ and LXR mediated transcriptional response? Of the PS-specific PRRs, and if we assume these are the ones that primarily mediate TGF-β synthesis in response to apoptotic cells, TIM-4 lacks signaling capability [26•]. Stabilin-2 activation or the agonist anti-stabilin-2 antibody induced TGF-β, suggesting that this receptor may be involved not only in apoptotic cell clearance, but also in the activation of signaling pathways leading to TGF-β production [26•]. The ability of BAI1 to induce TGF-β was not studied.
Consequences of integrating signals from PAMPs and apoptotic cells
The link between apoptotic cell recognition and activation of the nuclear receptors PPAR-γ, PPAR-δ, and LXR has important implications on understanding the mechanistic basis for how recognition of apoptotic cells modulates inflammatory responses. What happens when apoptotic cell-induced nuclear receptor activation accompanies PAMP-induced TLR activation as might occur when apoptosis results from an infection? Our prediction is that the well known apoptotic cell-mediated suppression of TLR-induced inflammatory-response genes occurs via nuclear receptor-mediated transrepression, a process where ligand bound nuclear receptors antagonize signal-dependent activation of target genes by other transcription factors, including NF-κB [27]. Many TLR target genes in macrophages are occupied by the nuclear receptor corepressor (NCoR) or the silencing mediator of retinoic acid and thyroid hormone receptors (SMRT), in complex with histone deacetylase 3 (HDAC3) and the transducin-β like proteins TBL1 and TBLR1 [27]. NCoR is recruited by c-Jun to inflammatory-response genes that contain AP-1 and κB sites in close proximity (Figure 1). TLR4 signaling induces NF-κB p65/IKKε-dependent phosphorylation of c-Jun and initiates corepressor clearance and transcriptional activation [28••]. The clearance mechanism involves activation of TBL1 and TBLR1, which are in fact F-box/WD-40 adaptor proteins used by RING finger-containing E3 ligases to mediate substrate recognition. TBL1/TBLR1 recruit ubiquitin-conjugating enzymes, including a ubiquitin E2 ligase, UbcH5, and the 19S regulatory proteasome resulting in ubiquitination of NCoR/SMRT complexes and their subsequent removal from gene promoters [28••] (Figure 1).
Figure 1.
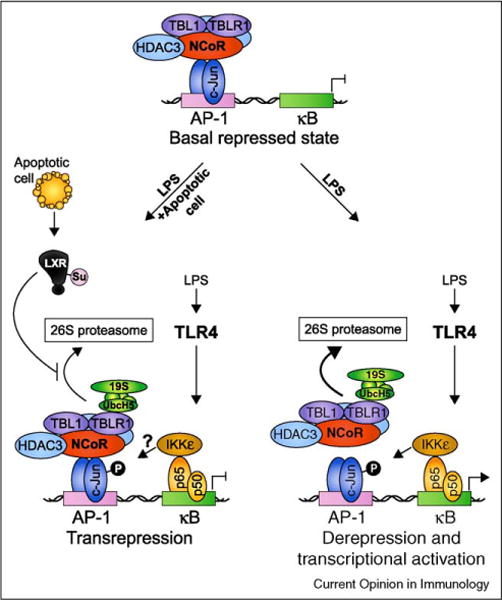
Ligand-dependent transrepression as a possible mechanism for apoptotic cell-mediated suppression of TLR-induced inflammatory gene expression. In the repressed ‘off’ state, LPS-induced genes such as the well characterized iNOS gene are actively suppressed by a multisubunit complex containing NCoR, HDAC3, TBL1, and TBLR1. When TLR4 signaling is initiated, IκB is degraded and NF-κB p50-p65 subunits enter the nucleus and bind to κB elements in the promoter. Phosphorylation of p65 on S536 creates a docking site for IκB Kinase ε (IKKε) allowing it to phosphorylate adjacent c-Jun/NCoR complexes and initiate corepressor clearance through TBLR1-mediated recruitment of UbcH5 and the 19S proteasome, ubiquitination, and subsequent degradation by the 26S proteasome. Binding of NF-κB allows recruitment of coactivators resulting in derepression and transcriptional activation. In this way, coupled AP-1/κB elements have been suggested to serve as ‘integrated circuits’ for switching promoters from a repressed to an activated state in response to an inflammatory trigger [28••]. When both TLR ligands and nuclear receptor ligands are present (depicted here as ‘LPS + apoptotic cell’ where the ligands for LXR are derived from apoptotic cells), ligand binding to LXR results in its SUMOylation by the E2 ligase Ubc9 and the E3 ligase HDAC4 and subsequent ability to interfere with NCoR corepressor clearance [27]. The result is ligand-dependent transrepression of inflammatory-response gene transcription.
When nuclear receptors are activated concurrently with TLRs, a different scenario emerges. In cells treated with PPAR-γ agonists and LPS, ligand binding mediates covalent conjugation of SUMO1 (small ubiquitin-like modifier) to PPAR-γ allowing it to bind to NCoR and interfere with clearance of the corepressor complex by blocking its degradation [27]. This maintains the promoter in the repressed state and results in suppression of inflammation (Figure 1). Similar to PPAR-γ, conjugation of SUMO2 and SUMO3 to LXR upon ligand binding also interferes with clearance of NCoR containing corepressor complexes from the promoters of distinct but overlapping inflammatory-response genes [27]. When ligand binds to PPAR-δ, on the contrary, the transcriptional repressor BCL-6 is released, which then represses inflammatory-response genes [27]. All studies described above involved the use of synthetic ligands for PPAR-γ and LXR such as rosiglitazone and GW3965, respectively, and it seems likely in light of the new studies [22,23,24••,25••], that nuclear receptor ligands derived from apoptotic cells may trigger similar mechanisms of ligand-dependent transrepression of inflammatory-response gene transcription. However, this possibility will have to await further investigation. Whether compartmentalized TLR and nuclear receptor signaling from one phagosome as in the case of phagocytosed apoptotic cells carrying TLR ligands, is equivalent to these signals being received by phagocytes in trans, that is, uninfected apoptotic cell in one phagosome and TLR signal at the plasma membrane, is an important discrimination to make as it may have different transcriptional and immunological outcomes. Alternative mechanisms of apoptotic cell-mediated suppression of inflammatory responses are also likely, as illustrated by an earlier finding that a zinc-finger transcription factor GC-binding protein binds and suppresses the promoter of the gene encoding the IL-12 p35 subunit [29].
Although we were cognizant of the reports that uptake of apoptotic cells could suppress LPS-induced production of IL-12 and TNF-α, when we considered the consequences of phagocytosis of apoptotic cells within the context of infection-induced TLR signals, we predicted that particular anti-inflammatory and pro-inflammatory cytokines could simultaneously be produced at some level by DC. This hypothesis was supported by studies showing that TLR-responsive genes showed different sensitivities to repression by different nuclear receptors: while some inflammatory genes were sensitive to repression, others were resistant demonstrating gene-specific repression of TLR-induced transcriptional responses [30••]. To test our hypothesis, we used either apoptotic neutrophils isolated following peritoneal injection of Escherichia coli, or apoptotic B cells carrying the TLR4 ligand LPS (LPS-blasts) [31••]. We found that phagocytosis of these TLR ligand-carrying apoptotic cells led to the synthesis of the inflammatory cytokines IL-6 and IL-23 at levels similar to those induced by soluble LPS alone [31••]. Remarkably, the levels of IL-12 were consistently lower when DC phagocytosed apoptotic E. coli-carrying neutrophils or LPS-blasts compared to treatment with LPS alone. This was consistent with the reported immunosuppressive effects of apoptotic cells on IL-12 production and particularly transcription of the p35 subunit of IL-12, which unlike the p40 subunit is not shared with IL-23 [29]. Furthermore, high levels of biologically active TGF-β were made specifically in response to uninfected or TLR ligand-carrying apoptotic cells, but not to soluble LPS. Thus, while TH1 promoting cytokines like IL-12 were suppressed by the uptake of apoptotic cells, the synthesis of other inflammatory cytokines like IL-23 and IL-6 were less affected.
We were interested in understanding how the cytokine profile induced in response to the phagocytosis of TLR ligand-carrying apoptotic cells could impact differentiation of naïve CD4+ T cells. Curiously, the combination of TGF-β, IL-6, and IL-23 had been reported to trigger the development of TH17 cells in vitro [32,33]. TH17 are a recently described subset of CD4+ T cells that produce the cytokine IL-17. They differentiate as a distinct lineage from TH1 or TH2 subsets, and play a crucial role in T-cell-mediated adaptive immunity [34]. TH17 cells are generally thought to be pro-inflammatory, especially through the production of IL-17A and IL-17F [34]. They have been reported to play an important role in host defense against infections with extracellular bacteria and fungi by facilitating recruitment of neutrophils and macrophages to infected tissues, promoting abscess formation, and inducing expression of anti-microbial peptides [35]. They have also been shown to participate in the development of autoimmunity and have thus received a lot of attention [36]. However, until recently, a single physiological stimulus that induced simultaneous synthesis of IL-6 and TGF-β from innate immune cells and subsequent differentiation of TH17 cells was not yet known. We thus hypothesized that DC recognition of apoptotic cells in the presence of infection might be the innate immune stimulus that induces differentiation of the TH17 lineage from naïve CD4+ T cells. We were able to definitively show that TLR ligation during phagocytosis of apoptotic cells promoted in vitro differentiation of naïve CD4+ T cells into TH17 cells. Similarly, phagocytosis of uninfected apoptotic cells in the presence of IL-6, which would be produced during infection, also promoted TH17 cell differentiation. Conversely, phagocytosis of apoptotic cells in the absence of TLR ligands or by DC deficient in both TRIF and Myd88 induced TGF-β alone, causing DC to direct naïve CD4+ T cells toward the reciprocal T regulatory (Treg) lineage [31••]. In other words, the presence or absence of TLR ligands with apoptotic cells dictated whether DC that phagocytosed those cells instructed generation of TH17 or Treg cells (Figure 2).
Figure 2.
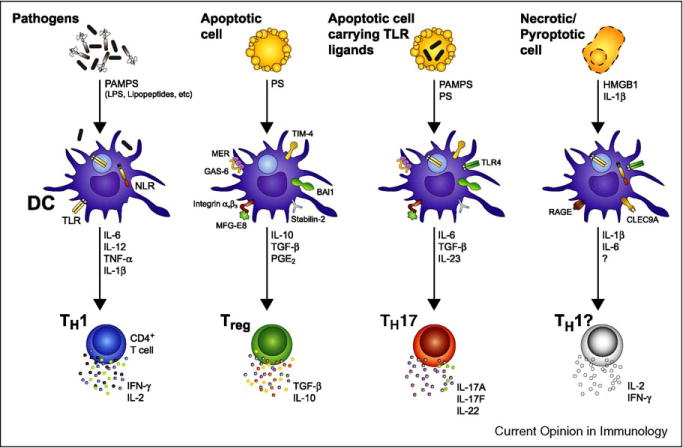
While phagocytosis of TLR ligand-carrying apoptotic cells instructs TH17 differentiation, innate recognition of other forms of cell death may have different immune consequences. DC facing an infection sense a multitude of PAMPs expressed by microbes via a variety of PRRs such as TLR and NLRs. DC undergo a maturation program leading to the expression of T-cell co-stimulatory molecules and the secretion of inflammatory cytokines including IL-6, IL-12, IL-1β, and TNF-α. IL-12 production is the main driver of CD4+ T-cell differentiation into TH1 [6]. By contrast, when DC phagocytose an apoptotic cell, they induce a non-inflammatory response associated with the differentiation of Treg cells. Cells that succumb to apoptosis express ‘eat-me’ signals including exposure of PS at the outer leaflet of the plasma membrane. PS is subsequently recognized by DC through several receptors such as TIM-4, stabilin-2, BAI1, MER, and the αvβ3 integrin. While TIM-4, stabilin-2, and BAI1 recognize PS directly, MER or the αvβ3 integrin require interaction with bridging molecules such as GAS6 or MFG-E8, respectively, to recognize PS and initiate phagocytosis. DC can then prime CD4+ T cells to differentiate into regulatory T cells by secreting anti-inflammatory cytokines such as TGF-β and IL-10. In the particular case of an apoptotic cell carrying TLR ligands, for example a neutrophil undergoing apoptosis following phagocytosis of bacteria, the combination of pro-inflammatory signals from activated TLRs with signals driven by the apoptotic cell cargo induces a unique cocktail of cytokines including IL-6, TGF-β, and IL-23, and triggers TH17 differentiation. Other types of cell death such as necrosis and pyroptosis also exist and these are thought to be immunogenic because of the release of cellular contents and inflammatory cytokines such as IL-1β that can activate DC [49]. While HMGB1 released by necrotic cells is known to result in DC maturation when recognized by RAGE, the ligand for CLEC9A, a C-type lectin receptor implicated in recognition of necrotic cells, remains to be discovered [49]. The consequence of phagocytosis of necrotic or pyroptotic cells on CD4+ T-cell differentiation is not known. HMGB1, high-mobility group box 1; RAGE, receptor for advanced glycation end-products; CLEC9A, C-type lectin domain family 9, member A.
Using Citrobacter rodentium as a model of infection that has been shown to induce TH17 immunity [37,38•,39], we also found that Citrobacter induction of host cell apoptosis was necessary to induce this response [31••]. C. rodentium is equipped with a locus for enterocyte effacement (LEE), which encodes a Type III secretion system that mediates firm adhesion of the bacteria to the host cell and injection of various bacterial effectors into the host cell cytosol to alter cellular functions and survival. One of these effectors, EPEC secreted protein F (EspF), translocates to the mitochondrion to initiate mitochondrial outer membrane permeabilization (MOMP) and subsequent apoptosis [31••]. Indeed, infection with a mutant strain of C. rodentium lacking EspF and unable to induce apoptosis, was not accompanied by the characteristic TH17 response within the intestinal lamina propria illustrating the crucial role of apoptosis for this response [31••]. Many other pathogens have been shown to elicit IL-17 responses, including not only C. rodentium, but also Klebsiella pneumoniae, Streprococcus pneumoniae, Borrelia species, Pseudomonas aeruginosa, Helicobacter pylori [35], and Staphylococcus aureus [39], and it will be interesting to investigate the relationship between the ability of these pathogens to induce apoptosis and to trigger IL-17 associated responses.
Thus, phagocytosis of apoptotic cells contributes signals that, in combination with TLR engagement, induce tailored immunity to bacterial infection through the development of TH17 cells. Extracellular bacteria that are particularly adept at inducing host cell apoptosis, via secreted toxins or Type III secretion system encoded effectors, are probably best at triggering a TH17 response. Neutrophils that are recruited to the site of infection to clear the bacteria can subsequently undergo apoptosis, and as such serve as apoptotic cells that carry TLR ligands. Phagocytosis of such ‘bacteria-loaded’ apoptotic neutrophils could then trigger a TH17 response.
It is likely that TH17 immunity is the best tailored response against bacterial infections that cause significant apoptosis and tissue injury given that TH17 cells have also been associated with tissue repair through their production of the cytokine IL-22 [40,41]. The protective roles of IL-22 in infections [38•,42•] as well as acute [43•] and chronic inflammatory conditions [44•,45•] are associated with its functions in maintaining the integrity of epithelial barriers [46]. In addition to its anti-microbial and tissue protective properties, IL-22 can also promote inflammatory pathology in murine models of asthma, psoriasis, and inflammatory bowel disease [46]. TH17 cells also produce IL-10, which limits the inflammatory response that might otherwise cause tissue damage [47,48].
Conclusions
There are many causes and consequences of inflammation [50•]. The inflammatory trigger induced by the simultaneous recognition of TLR ligands and apoptotic cells comprises elements of both infection and tissue injury (Figure 3). This combination of triggers results in TH17 immunity. The physiological purpose of TH17 immunity is not only to defend the host against infection, but also to induce tissue repair. The pathological consequence of the response could be development of autoimmune disease. The association of TH17 cells with autoimmune diseases may reflect defects in powerful regulatory mechanisms that normally focus the effector functions of these cells on defending and repairing rather than attacking host tissue. Elucidating the nature of these mechanisms will be an important area of investigation in the future.
Figure 3.
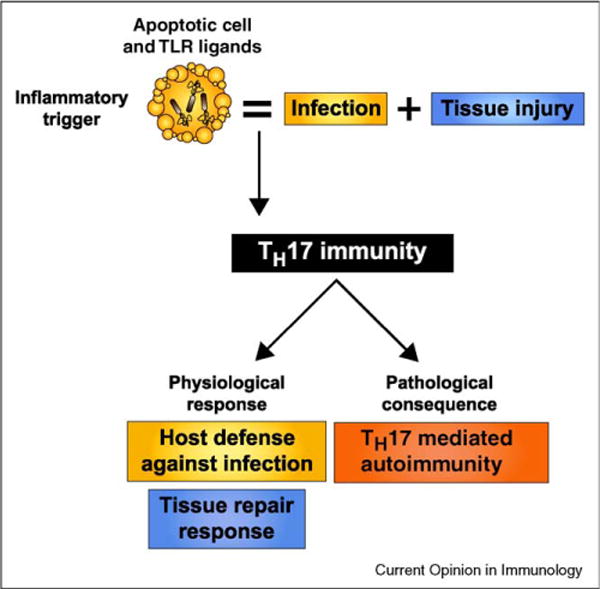
Proposed physiological and pathological outcomes of innate recognition of apoptotic cells dying from infection. When cells undergo apoptosis as a result of infection, the inflammatory trigger presented to the innate immune system is that of both infection and tissue injury. While PAMPs trigger inflammatory PRRs such as TLRs, apoptotic ‘eat-me’ signals trigger anti-inflammatory signaling pathways. Given that apoptosis frequently occurs during infection, the ability of the immune system to mount an effective response against pathogens despite the presence of immunosuppressive dying cells has been a long-standing paradox. A TH17 adaptive immune response reconciles the tolerance induced by apoptotic cell clearance with the necessarily inflammatory nature of infections. Through the release of inflammatory and reparative cytokines, TH17 cells can initiate not only defense against the pathogen, but also repair of injured tissues. Because one of the signals that trigger TH17 immunity consists of dying host cells, the pathological consequences of innate recognition of apoptotic cells during infection could be the development of autoimmunity.
Acknowledgments
We thank Blander lab members Leif Sander, Corinna Brereton, and Priyanka Nair for critical reading of the manuscript. JMB is supported by NIH grants AI073899 and AI080959A, the American Cancer Society, the Hirschl and Weill-Caulier Charitable Trust Fund, and the Kinship Foundation Searle scholar award.
References and recommended reading
Papers of particular interest, published within the period of review, have been highlighted as:
• of special interest
•• of outstanding interest
- 1.Vaux DL, Korsmeyer SJ. Cell death in development. Cell. 1999;96:245–254. doi: 10.1016/s0092-8674(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 2•.Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, Voll RE, Winkler T, Kuhn A, Kalden J, et al. Clearance deficiency and systemic lupus erythematosus (SLE) J Autoimmun. 2007;28:114–121. doi: 10.1016/j.jaut.2007.02.005. This paper showed that patients with SLE had apoptotic cell material in dendritic cells located in lymph node germinal centers, and autoantibodies that had gained autoreactivity in germinal center reactions. This supported the hypothesis that impaired clearance of apoptotic cells in patients with SLE could lead to secondary necrosis and buildup of self-antigens in tissues, contributing to autoimmunity in this disease. [DOI] [PubMed] [Google Scholar]
- 3.Janko C, Schorn C, Grossmayer GE, Frey B, Herrmann M, Gaipl US, Munoz LE. Inflammatory clearance of apoptotic remnants in systemic lupus erythematosus (SLE) Autoimmun Rev. 2008;8:9–12. doi: 10.1016/j.autrev.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 4.Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–1035. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- 5.Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol. 2008;9:781–795. doi: 10.1038/nrm2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Manicassamy S, Pulendran B. Modulation of adaptive immunity with Toll-like receptors. Semin Immunol. 2009;21:185–193. doi: 10.1016/j.smim.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ravichandran KS, Lorenz U. Engulfment of apoptotic cells: signals for a good meal. Nat Rev Immunol. 2007;7:964–974. doi: 10.1038/nri2214. [DOI] [PubMed] [Google Scholar]
- 8.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–1556. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9••.Miyanishi M, Tada K, Koike M, Uchiyama Y, Kitamura T, Nagata S. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–439. doi: 10.1038/nature06307. [10••] [DOI] [PubMed] [Google Scholar]
- 10••.Kobayashi N, Karisola P, Pena-Cruz V, Dorfman DM, Jinushi M, Umetsu SE, Butte MJ, Nagumo H, Chernova I, Zhu B, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–940. doi: 10.1016/j.immuni.2007.11.011. Together with reference [9••], this paper identified TIM-4 as well as TIM-1 as phagocytic transmembrane receptors for the ‘eat me’ signal PS on apoptotic cells. While TIM-1 is expressed by activated T cells and kidney epithelial cells, TIM-4 is expressed by macrophages and DC and is crucial for apoptotic cell clearance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11•.Park D, Tosello-Trampont AC, Elliott MR, Lu M, Haney LB, Ma Z, Klibanov AL, Mandell JW, Ravichandran KS. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–434. doi: 10.1038/nature06329. This paper links recognition of PS on apoptotic cells by BAI1 with recruitment of a Rac-GEF complex, ELMO/Dock180/Rac, showing a mechanism mediating uptake of apoptotic cells by phagocytes. [DOI] [PubMed] [Google Scholar]
- 12•.Park SY, Jung MY, Kim HJ, Lee SJ, Kim SY, Lee BH, Kwon TH, Park RW, Kim IS. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15:192–201. doi: 10.1038/sj.cdd.4402242. This paper describes a novel receptor for apoptotic cell PS, stabilin-2, showing that it plays a key role in clearance of apoptotic cells by macrophages. [DOI] [PubMed] [Google Scholar]
- 13•.Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. This study was the first demonstration that phagocytosis of apoptotic cells during phagocytes activation inhibits inflammatory cytokine secretion. [DOI] [PubMed] [Google Scholar]
- 14•.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. This paper demonstrates that phagocytosis of apoptotic neutrophils inhibits the production of interleukins such as IL-1β, IL-8, and TNF-α while inducing the secretion of anti-inflammatory TGF-β1 and PGE2. In addition, the authors demonstrate that the anti-inflammatory cytokines produced are also involved in inhibition of pro-inflammatory cytokine production. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15•.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. Using models of inflammation in the peritoneal cavity and lung, this study provides evidence that PS-mediated apoptotic cell phagocytosis by macrophages within the context of inflamed tissue, induces TGF-β1 secretion and promotes the resolution of inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cvetanovic M, Ucker DS. Innate immune discrimination of apoptotic cells: repression of proinflammatory macrophage transcription is coupled directly to specific recognition. J Immunol. 2004;172:880–889. doi: 10.4049/jimmunol.172.2.880. [DOI] [PubMed] [Google Scholar]
- 17.Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell. 2004;14:277–287. doi: 10.1016/s1097-2765(04)00237-0. [DOI] [PubMed] [Google Scholar]
- 18.Gerbod-Giannone MC, Li Y, Holleboom A, Han S, Hsu LC, Tabas I, Tall AR. TNFalpha induces ABCA1 through NF-kappaB in macrophages and in phagocytes ingesting apoptotic cells. Proc Natl Acad Sci USA. 2006;103:3112–3117. doi: 10.1073/pnas.0510345103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kiss RS, Elliott MR, Ma Z, Marcel YL, Ravichandran KS. Apoptotic cells induce a phosphatidylserine-dependent homeostatic response from phagocytes. Curr Biol. 2006;16:2252–2258. doi: 10.1016/j.cub.2006.09.043. [DOI] [PubMed] [Google Scholar]
- 20.Castrillo A, Tontonoz P. Nuclear receptors in macrophage biology: at the crossroads of lipid metabolism and inflammation. Annu Rev Cell Dev Biol. 2004;20:455–480. doi: 10.1146/annurev.cellbio.20.012103.134432. [DOI] [PubMed] [Google Scholar]
- 21.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–1870. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 22.Johann AM, von Knethen A, Lindemann D, Brune B. Recognition of apoptotic cells by macrophages activates the peroxisome proliferator-activated receptor-gamma and attenuates the oxidative burst. Cell Death Differ. 2006;13:1533–1540. doi: 10.1038/sj.cdd.4401832. [DOI] [PubMed] [Google Scholar]
- 23.Majai G, Sarang Z, Csomos K, Zahuczky G, Fesus L. PPARgamma-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol. 2007;37:1343–1354. doi: 10.1002/eji.200636398. [DOI] [PubMed] [Google Scholar]
- 24••.Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, Goh YP, Eagle AR, Dunn SE, Awakuni JU, et al. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med. 2009 doi: 10.1038/nm.2048. This paper shows that the increase in oxidized fatty acids following engulfment of apoptotic cells by macrophages is sensed by PPAR-δ which in turn orchestrates the transcriptional response to apoptotic cells. Notably, PPAR-δ activation during phagocytosis of dying cells increased expression of anti-inflammatory cytokines and conversely inhibited expression of pro-inflammatory cytokines. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25••.A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, Deniz J, Ramirez C, Diaz M, Gallardo G, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity. 2009;31:245–258. doi: 10.1016/j.immuni.2009.06.018. Together with reference [24••], these papers test the hypothesis that sensors of modified fatty acids coordinate the transcriptional response by macrophages to the phagocytosis of dying cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26•.Park D, Hochreiter-Hufford A, Ravichandran KS. The phosphatidylserine receptor TIM-4 does not mediate direct signaling. Curr Biol. 2009;19:346–351. doi: 10.1016/j.cub.2009.01.042. This study proposes that unlike BAI1 and stabilin-2, TIM-4 mediated apoptotic cell phagocytosis does not engage direct signaling. Interestingly, the cytoplasmic and transmembrane regions of TIM-4 were dispensable for apoptotic cell engulfment. [DOI] [PubMed] [Google Scholar]
- 27.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 28••.Huang W, Ghisletti S, Perissi V, Rosenfeld MG, Glass CK. Transcriptional integration of TLR2 and TLR4 signaling at the NCoR derepression checkpoint. Mol Cell. 2009;35:48–57. doi: 10.1016/j.molcel.2009.05.023. This paper illustrates differential mechanisms used by TLR4 and TLR2 to mediate NCoR/SMRT clearance from inflammatory-response genes. The authors further show that the previously recognized resistance of TLR2, but not TLR4 gene expression to LXR repression is related to activation of Ca2+/calmodulin-dependent protein kinase CaMKII. These findingsimply that the ability of LXR ligands to inhibit repression is probably context dependent. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Elkon KB, Ma X. Transcriptional suppression of interleukin-12 gene expression following phagocytosis of apoptotic cells. Immunity. 2004;21:643–653. doi: 10.1016/j.immuni.2004.09.009. [DOI] [PubMed] [Google Scholar]
- 30••.Ogawa S, Lozach J, Benner C, Pascual G, Tangirala RK, Westin S, Hoffmann A, Subramaniam S, David M, Rosenfeld MG, et al. Molecular determinants of crosstalk between nuclear receptors and toll-like receptors. Cell. 2005;122:707–721. doi: 10.1016/j.cell.2005.06.029. In this paper, the authors used a combination of gene expression profiling and molecular analysis to demonstrate that TLR-responsive genes were differentially transrepressed by nuclear receptors. This study further shows that PPAR-γ and LXR repress overlapping transcriptional targets and can function in combination with the glucocorticoid receptor to repress a variety of LPS-responsive genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31••.Torchinsky MB, Garaude J, Martin AP, Blander JM. Innate immune recognition of infected apoptotic cells directs T(H)17 cell differentiation. Nature. 2009;458:78–82. doi: 10.1038/nature07781. This paper shows that innate immune recognition of apoptotic cells within the context of infection drives TH17 differentiation while recognition of apoptotic cells drives differentiation of Treg cells. A new role for apoptotic cells as the driving force for induction of TH17 immune responses at mucosal surfaces is demonstrated. The study also points to bacterial infections that induce apoptosis of host cells as one physiological stimulus for differentiation of TH17 cells, suggesting that this helper cell has a specific role in host defense against infections with certain extracellular bacteria. [DOI] [PubMed] [Google Scholar]
- 32.Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. doi: 10.1038/nature04754. [DOI] [PubMed] [Google Scholar]
- 33.Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity. 2006;24:179–189. doi: 10.1016/j.immuni.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 34.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 35.Curtis MM, Way SS. Interleukin-17 in host defence against bacterial, mycobacterial and fungal pathogens. Immunology. 2009;126:177–185. doi: 10.1111/j.1365-2567.2008.03017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Awasthi A, Kuchroo VK. Th17 cells: from precursors to players in inflammation and infection. Int Immunol. 2009;21:489–498. doi: 10.1093/intimm/dxp021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol. 2005;6:1123–1132. doi: 10.1038/ni1254. [DOI] [PubMed] [Google Scholar]
- 38•.Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med. 2008;14:282–289. doi: 10.1038/nm1720. [42•] [DOI] [PubMed] [Google Scholar]
- 39.Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, et al. Differential roles of interleukin-17A and-17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity. 2009;30:109–119. doi: 10.1016/j.immuni.2008.11.009. [DOI] [PubMed] [Google Scholar]
- 40.Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA. Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med. 2006;203:2271–2279. doi: 10.1084/jem.20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dong C. TH17 cells in development: an updated view of their molecular identity and genetic programming. Nat Rev Immunol. 2008;8:337–348. doi: 10.1038/nri2295. [DOI] [PubMed] [Google Scholar]
- 42•.Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med. 2008;14:275–281. doi: 10.1038/nm1710. Along with reference [38•], this paper demonstrates the importance of IL-22 in host defense against mucosal infections in the lung and gastrointestinal tract. IL-17 and IL-22 are both crucial for effective control of pathogens, but IL-22 also increases epithelial resistance to injury in the lung, and is responsible for the induction of the Reg family of antimicrobial proteins in the gut, indicating a crucial role for IL-22 in tissue protection and repair during inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43•.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Karow M, Flavell RA. Interleukin-22 but not interleukin-17 provides protection to hepatocytes during acute liver inflammation. Immunity. 2007;27:647–659. doi: 10.1016/j.immuni.2007.07.023. [45•] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [45•] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45•.Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity. 2008;29:947–957. doi: 10.1016/j.immuni.2008.11.003. Together with references [43•] and [44•], these papers collectively showed that IL-22 derived from TH17 cells played a protective role in preventing tissue injury in mouse models of both acute and chronic inflammatory disease. Hepatocytes from mice deficient in IL-22 were highly sensitive to destruction by acute hepatitis, and IL-22-expressing TH17 cells provided protection in IL-22-deficient mice in this model. In a chronic model of inflammation, ulcerative colitis, IL-22 gene delivery to intestinal cells led to dramatic amelioration of local inflammation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol. 2007;8:1390–1397. doi: 10.1038/ni1539. [DOI] [PubMed] [Google Scholar]
- 48.Stumhofer JS, Silver JS, Laurence A, Porrett PM, Harris TH, Turka LA, Ernst M, Saris CJ, O’Shea JJ, Hunter CA. Interleukins 27 and 6 induce STAT3-mediated T cell production of interleukin 10. Nat Immunol. 2007;8:1363–1371. doi: 10.1038/ni1537. [DOI] [PubMed] [Google Scholar]
- 49.Green DR, Ferguson T, Zitvogel L, Kroemer G. Immunogenic and tolerogenic cell death. Nat Rev Immunol. 2009;9:353–363. doi: 10.1038/nri2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50•.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. This insightful article provides an excellent overview of the inflammatory responses triggered by infection, tissue injury, and tissue stress. [DOI] [PubMed] [Google Scholar]