

REPRODUCIBILITY AND GENERALIZABILITY
The sequencing of the human genome and the subsequent availability of inexpensive, robust methods for “omics” profiling (e.g., genome-wide association studies, gene expression microarrays, and metabolomics) have led to optimism of a new era of biomarkers that would allow for a “precision medicine” approach to critical care. Unfortunately, this promise has yielded few tangible results, as the general biomedical reproducibility crisis (1–3) is particularly troublesome in critical care (4–8) and in omics biomarker studies (9–11). There are two broad problems that lead to seemingly similar studies of biomarkers in critical care producing different results. One problem is traditional nonreproducibility due to false positive biomarker selection or nonrobust statistical models. The other, more importantly, is a lack of generalizability in moving from a narrow study population into broader applications in critical care. We present here a contextual framework for addressing these problems and for assessing new biomarker studies.
SYNDROMIC ILLNESS AND GENERALIZABLE BIOMARKERS
Many critical illnesses are defined syndromically, such as sepsis, acute kidney injury (AKI), acute respiratory distress syndrome (ARDS), and delirium. These syndromes typically have clear, though changing, clinical criteria (12–14). Still, a syndrome may arise from multiple causes; as a result, it is unclear whether all cases of the syndrome really represent the same disease. Such uncertainty raises a major problem in the field. For example, if a positive clinical trial for adults with ARDS defined by Berlin criteria has failed to reproduce in an independent population of children with ARDS also defined by Berlin criteria, was the original finding a false positive or do adults and children have a “different” version of ARDS? Our reliance on syndromic definitions and the lack of clear gold-standard diagnostics linked to pathophysiology thus makes it difficult to assess clinical trial results. In theory, if the entire clinical spectrum of a disease has a common molecular pathophysiology, then a molecular biomarker should exist that is generalizable to the disease. Thus, finding a generalizable biomarker can help to define the disease, improving both patient care and clinical trial design, and potentially moving a whole field of study forward.
There are other practical reasons to search for biomarkers that are generalizable. First, requiring context-specific biomarkers for every variant on a clinical condition (e.g., a different biomarker for different sources of sepsis, or for each different cause of kidney injury) could end up requiring dozens of tests for each critical syndrome. Tests indicated for such increasingly fragmented populations will fail to overcome barriers to market entry. In addition, those that do make it may have overly specific indications for use, leaving many patients without help. Finally, since off-label uses of tests and therapies are common, if biomarkers fail to deliver similar performance in seemingly similar conditions, patients will be harmed.
We thus argue that research should focus first on finding generalizable, disease-defining molecular biomarkers for syndromes in critical illness, or alternatively on showing that such biomarkers do not exist (evidence-of-absence studies). If no generalizable biomarker exists, then more context-specific biomarkers can direct the effort to accurately characterize clinically actionable syndromic subtypes. In other words, we need to clearly define a disease before we begin to divide it into subtypes. Both are necessary components of a precision medicine approach, but due to the high heterogeneity of critical illness, research of both types can be challenging.
HETEROGENEITY IN CRITICAL ILLNESS AND THE CHALLENGE OF CLINICAL TRIALS
Clinical trials in the critical care setting are among the hardest to carry out, for reasons of practicality, patient protection, and patient heterogeneity; this leads to smaller, mostly homogeneous cohorts that do not represent the broad spectrum of critical illness. First, as described above, similar acute syndromes (such as sepsis, AKI, and ARDS) often have multiple possible definitions and span a range of severities. Second, critically ill patients span the entire range of ages, comorbid conditions, and demographics. Third, the medical, surgical, neurologic, and pediatric pathways of critical illness have widely varying primary problems. Fourth, the practicality of conducting a trial leads to differing sampling times and stages of disease at trial enrollment. Finally, the changing treatment patterns over time (such as the change in early sepsis resuscitation with early goal-directed therapy) can lead to different outcomes for the same intervention. The logistical and budgetary constraints of trying to represent all of these sources of heterogeneity means that most single-cohort studies cannot capture the broad spectrum of critical illness, and thus may have difficulty producing generalizable results.
Still, the bedrock of continued progress toward generalizable biomarkers is continued publication of clinical trials. One way to improve trials is to focus on not just size but also heterogeneity. Single-cohort studies are more likely to yield reproducible results when they are appropriately powered, and are more likely to yield generalizable results when they are designed with broad inclusion criteria that attempt to match the full spectrum of the condition under study. Thus, a biomarker that has been tested in 500 adults with pneumonia and ARDS at admission is more reliable than one that has been tested in only 50 such patients. However, until it is tested in children, or in ARDS arising from other causes, or at other clinical timepoints, its generalizability is unknown. We thus caution against the false security of solely relying on a high sample size in evaluating the robustness of a single study.
MULTICOHORT ANALYSIS AND DATA SHARING
An efficient, inexpensive way of tackling the problem of heterogeneity is to combine studies that represent the broad spectrum of disease. At a fixed total sample size, greater reproducibility is gained when the samples are integrated from a greater number of smaller sized studies, rather than vice versa (15). Our group has worked with many collaborators in repeatedly demonstrating that leveraging biological and technical heterogeneity across multiple cohorts can identify generalizable diagnostic and prognostic biomarkers in a diverse set of diseases including organ transplant, cancer, and autoimmune and infectious diseases (16–24). These early successes of multicohort analysis are firmly rooted in the hypothesis that although a broad representation of a disease could make the discovery of a biomarker challenging; such biomarkers are more likely to be reproducible and generalizable when tested in novel circumstances (Fig. 1). On the other hand, making full use of these studies often requires making imperfect comparisons (e.g., integrating datasets that use multiple different definitions for AKI). Although no hard rule can be set, we feel it both reasonable and pragmatic to use data to their fullest extent, even if the statistical methods are simple or some assumptions are slightly violated, as long as such caveats are fully explained and discussed.
Figure 1.
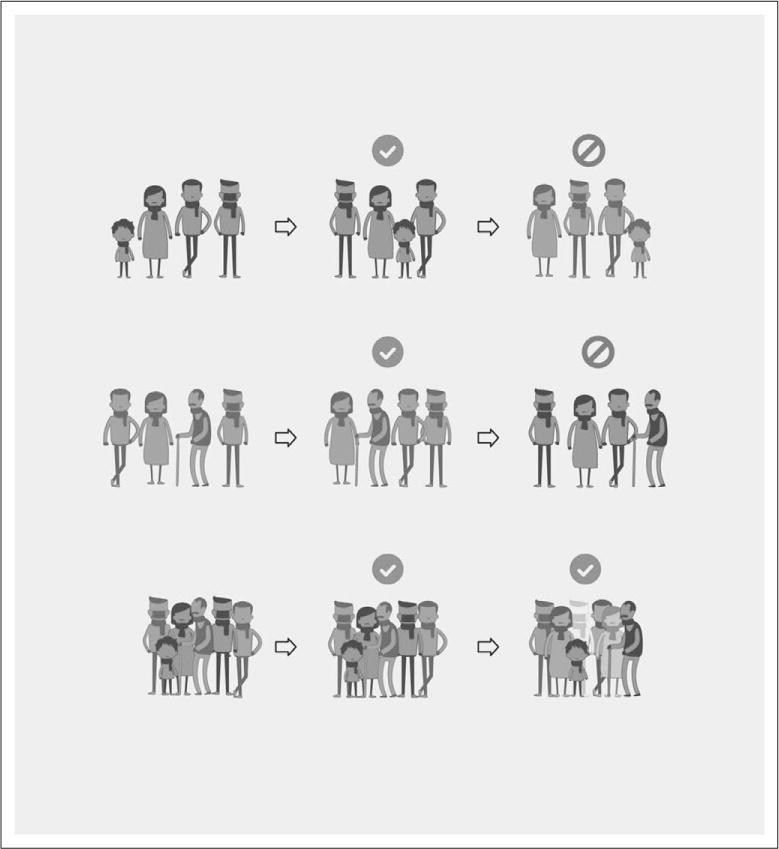
The benefit of incorporating heterogeneity. Biomarkers discovered in a homogeneous cohort are highly likely to work in external cohorts that are similar to the original cohort, but less likely to work in different settings. Biomarkers that are discovered in heterogeneous cohorts are more likely to be generalizable across a broad spectrum of patients.
However, multicohort studies are only possible when data are shared (such as is now required for most genome-wide expression studies). We thus argue for the increased appropriate sharing of molecular data from clinical trials, so that multiple cohorts can be combined in the discovery of new biomarkers (25). In many research areas, data are held privately, preventing such reuse. For instance, we searched the literature for metabolomics, clinical studies in critical care and identified 28 studies (total n = 2,322), out of which only two studies made their raw data publically available (Table 1) (26–53). Public sharing of these data would allow for meta-analysis and data-driven hypotheses generation, avoiding the need for each new cohort to “reinvent the wheel.” It is clear that studies that are performed on single cohorts can be successful at producing robust biomarkers if pitfalls are avoided; but we propose that these investigators make their data available for (and themselves take part in) efforts at later meta-analysis.
TABLE 1.
Metabolomics Studies in Critical Care
| Year | Reference | Metabolomic Method | Cases | Controls | n | Tissue | Summary Data Available | Raw Data Available |
|---|---|---|---|---|---|---|---|---|
| 2009 | Mao et al (26) | NMR | Healthy/SIRS | Multiple organ dysfunction syndrome | 78 | Serum | No | No |
| 2011 | Stringer et al (27) | NMR | Healthy | Sepsis + acute lung injury | 19 | Plasma | Partial | No |
| 2012 | Schmerler et al (28) | LC/MS/MS | SIRS | Sepsis | 143 | Plasma | No | No |
| 2013 | Antti et al (29) | GC/MS | None | Sepsis | 16 | Serum | Partial | No |
| 2013 | Blaise et al (30) | NMR | None | Trauma | 22 | Plasma | No | No |
| 2013 | Mickiewicz et al (31) | NMR | Healthy/SIRS | Sepsis | 140 | Serum | No | No |
| 2013 | Langley et al (32) | GC/MS | SIRS | Sepsis | 150 | Plasma | Yes | Deposited but not available |
| 2013 | Kamisoglu et al (33) | LC and GC/MS | Healthy | Endotoxin | 19 | Plasma | Yes | No |
| 2013 | Seymour et al (34) | UHPLC/MS/MS | Sepsis survivor | Sepsis nonsurvivor | 30 | Plasma | Partial | No |
| 2014 | Bos et al (35) | GC/MS | Ventilated non-ARDS | ARDS | 101 | Breath | No | No |
| 2014 | Dessì et al (36) | GC/MS | Healthy | Sepsis (n = 1) | 14 | Urine | No | No |
| 2014 | Evans et al (37) | LC/MS | Healthy | ARDS | 26 | Bronchoalveolar lavage fluid | Partial | No |
| 2014 | Fanos et al (38) | GC/MS and NMR | Healthy | Sepsis | 15 | Urine | No | No |
| 2014 | Mickiewicz et al (39) | NMR | SIRS | Sepsis | 59 | Serum | Partial | No |
| 2014 | Rogers et al (40) | GC/MS | SIRS | Sepsis, ± ARDS | 240 | Plasma | Yes | No |
| 2014 | Su et al (41) | LC/MS/MS | Healthy/SIRS | Sepsis | 65 | Serum | No | No |
| 2015 | Puskarich et al (42) | NMR | Sepsis | Sepsis with L-carnitine | 30 | Serum | Partial | No |
| 2015 | Garcia-Simon et al (43) | NMR | Sepsis survivor | Sepsis nonsurvivor | 64 | Urine | Partial | No |
| 2015 | Mickiewicz et al (44) | NMR | ED nonsepsis | Sepsis | 238 | Serum | No | No |
| 2015 | Su et al (45) | LC/MS | Sepsis survivor | Sepsis nonsurvivor | 35 | Serum | Yes | No |
| 2016 | Richter et al (46) | LC/MS/MS | Febrile neutropenia | Sepsis | 48 | Plasma | Partial | No |
| 2016 | Kauppi et al (47) | GC/MS | ED nonsepsis | Sepsis | 114 | Whole blood | Partial | Yes |
| 2016 | Ferrario et al (48) | LC/MS/MS | Sepsis survivor | Sepsis nonsurvivor | 20 | Plasma | Yes | Yes |
| 2016 | Neugebauer et al (49) | LC/MS/MS | SIRS | Sepsis | 406 | Serum | Yes | No |
| 2016 | Liu et al (50) | UHPLC-MS | Sepsis survivor | Sepsis nonsurvivor | 50 | Serum | Partial | No |
| 2016 | Stewart et al (51) | LC/MS/MS | Healthy neonate | Necrotizing enterocolitis/sepsis | 19 | Serum | No | No |
| 2016 | Mogensen et al (52) | LC and GC/MS | SIRS | Sepsis | 85 | Plasma | Partial | No |
| 2016 | Lau et al (53) | UHPLC-MS | Noninfected controls | Bacteremia | 76 | Plasma | Yes | No |
ARDS = acute respiratory distress syndrome, ED = emergency department, GC = gas chromatography, LC = liquid chromatography, MS = mass spectrometry, NMR = nuclear magnetic resonance, SIRS = systemic inflammatory response syndrome, UHPLC = ultra-high performance liquid chromatography.
All raw data and most summary data are unpublished, preventing their reuse.
To aid the broader community in this effort, we have made available on our website (http://khatrilab.stanford.edu/sepsis) a large number of existing studies of gene expression in sepsis along with source code for analysis (20, 21). This is one resource any researcher can use to further explore their biomarkers in broader clinical context and to test their generalizability in silico prior to embarking on a clinical trial.
BIG DATA AND BIOMARKERS
One of the biggest benefits of the data-driven omics approach to biomarker discovery is the possibility of discovering novel pathobiology in the heterogeneity of critical illness. Although hypothesis-driven studies of familiar cytokines (e.g., those resulting from activation of the nuclear factor-kB or interferon pathways) may be warranted by preclinical models, many common pathways are activated by multiple stimuli at a cellular level (54) and so are unlikely to be highly specific for a given syndrome. Similarly, clinical scores that use similar data available in an electronic health record (vitals, common laboratories, etc.) are unlikely to be highly specific for multiple conditions. An omics approach, by contrast, can sift through thousands or tens of thousands of candidate biomarkers to find the best fit for a given condition. Unfortunately, the promise of omics is also thus its major pitfall: false positives are likely when there are many more variables than samples in a study. This has contributed to some early failures in the field. It is thus worthwhile to have a general framework with which to approach biomarker development studies.
BIOMARKER STUDIES: A CONCEPTUAL FRAMEWORK
There are two excellent guidelines for how to determine the rigor of multivariable prediction models (the Transparent Reporting of a multivariable prediction model for Individual Prognosis Or Diagnosis [TRIPOD] statement [www.tripod-statement.org] [55]) and of diagnostic accuracy studies generally (the Standards for Reporting Diagnostic accuracy studies [STARD] statement [www.stard-statement.org] [56]). In addition to these reporting guidelines, we suggest the following list of questions that we use to help place a study in context:
What is the context of the reported diagnostic comparison? Are there existing comparators/gold standards for this question, and what is their diagnostic accuracy in practice? Ideally, a study will compare a new biomarker to a gold standard, possibly with a net reclassification, but this is often not possible and only reported in later validations.
How “locked down” is the reported biomarker? Are the biomarkers themselves being selected? Is a statistical model being retrained in the new cohort (i.e., if using a regression model, were the coefficients determined prior to testing)? If a cutoff is used, was it determined prospectively, and is it standard?
How generalizable is the validation cohort being studied? Is this merely a random held-out set of the original discovery cohort? Is it from the same center as the discovery cohort? Does it represent a new area of application?
Is this biomarker useful? If applied clinically, would it change practice?
Is there a link to known biology? In our opinion, this may not be necessary at first, especially if the study is searching in spaces that are not well-studied (outside the “street lamp” of common studies). Procalcitonin, for instance, had not been well-characterized as part of the immune response (and the biology remains somewhat unclear today) at its first testing as a biomarker for bacterial infection (57).
Can the biomarker be measured in a reasonable amount of time to make it useful in critical care? Although not a reason to dismiss results, many of the diagnostic applications in critical care require a rapid turnaround time. A more complex process, or one that relies on new technologies, may take longer to be clinically translated, and will be harder to replicate in validation studies. For example, neutrophilic CD64 as measured by flow cytometry is highly diagnostic for sepsis but has a turnaround time of several hours (58).
In addition, it is helpful to put a study into context in terms of biomarker development (Fig. 2). Early validation may be simply the generation of receiver-operating characteristic curves in similar cohorts to the initial discovery cohort. As evidence accumulates, however, such studies should 1) investigate the application of the biomarker in a broader variety of cohorts that represent the full spectrum of disease and 2) compare the test to known standards for easy comparison. For instance, the later-stage validation of a biomarker for the prognosis of sepsis that is not compared to either lactate or clinical severity scores (e.g., Sequential Organ Failure Assessment) is unhelpful. Similarly, a study examining the diagnostic power for a locked, commercially available biomarker is important, but not as helpful as one examining outcome after intervention.
Figure 2.
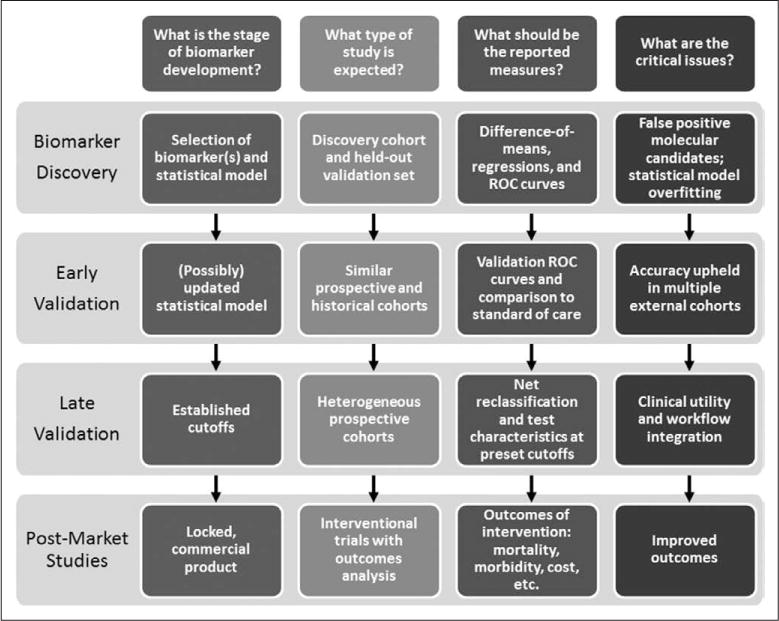
Maturity of biomarkers: a conceptual framework. ROC = receiver-operating characteristic.
MOVING FORWARD IN THE BIG DATA ERA
The promise of precision medicine is to have the right treatment for the right patient at the right time. In critical care, our immediate need is to get the basics right. For instance, we should first try to answer urgent clinical questions (such as which patients need antibiotics), and then pose new ones that may not have been previously answerable (such as whether there are molecular subtypes of sepsis). As omics and big data technologies proliferate, so too will studies utilizing them as biomarkers in critical illness (studying the genome, epigenome, transcriptome, proteome, metabolome, lipidome, microbiome, and quantified self, to name a few). In all cases, we must remember the extreme heterogeneity of critical illness, and strive for generalizable disease-defining diagnostics and robust biomarkers that can help the entire spectrum of critical care research and delivery.
Acknowledgments
We thank Dr. Thomas Weiser and Dr. Claude Piantadosi for their helpful comments in preparing this article.
Contributor Information
Timothy E. Sweeney, Stanford Institute for Immunity, Transplantation and Infection, Stanford University School of Medicine, Stanford, CA.
Purvesh Khatri, Division of Biomedical Informatics Research, Department of Medicine, Stanford University School of Medicine, Stanford, CA.
References
- 1.Ioannidis JP. Why most published research findings are false. PLoS Med. 2005;2:e124. doi: 10.1371/journal.pmed.0020124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goodman SN, Fanelli D, Ioannidis JP. What does research reproducibility mean? Sci Transl Med. 2016;8:341ps12. doi: 10.1126/scitranslmed.aaf5027. [DOI] [PubMed] [Google Scholar]
- 3.Open Science Collaboration PSYCHOLOGY. Estimating the reproducibility of psychological science. Science. 2015;349:aac4716. doi: 10.1126/science.aac4716. [DOI] [PubMed] [Google Scholar]
- 4.Opal SM, Dellinger RP, Vincent JL, et al. The next generation of sepsis clinical trial designs: What is next after the demise of recombinant human activated protein C? Crit Care Med. 2014;42:1714–1721. doi: 10.1097/CCM.0000000000000325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cohen J, Vincent JL, Adhikari NK, et al. Sepsis: A roadmap for future research. Lancet Infect Dis. 2015;15:581–614. doi: 10.1016/S1473-3099(15)70112-X. [DOI] [PubMed] [Google Scholar]
- 6.Yadav H, Thompson BT, Gajic O. Fifty years of research in ARDS. Is acute respiratory distress syndrome a preventable disease? Am J Respir Crit Care Med. 2016 Dec;:31. doi: 10.1164/rccm.201609-1767CI. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.de Caestecker M, Humphreys BD, Liu KD. Bridging translation by improving preclinical study design in AKI. J Am Soc Nephrol. 2015;26:2905–2916. doi: 10.1681/ASN.2015070832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marshall JC. Why have clinical trials in sepsis failed? Trends Mol Med. 2014;20:195–203. doi: 10.1016/j.molmed.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 9.Ioannidis JP, Ntzani EE, Trikalinos TA, et al. Replication validity of genetic association studies. Nat Genet. 2001;29:306–309. doi: 10.1038/ng749. [DOI] [PubMed] [Google Scholar]
- 10.Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput Biol. 2011;7:e1002240. doi: 10.1371/journal.pcbi.1002240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ioannidis JP. Biomarker failures. Clin Chem. 2013;59:202–204. doi: 10.1373/clinchem.2012.185801. [DOI] [PubMed] [Google Scholar]
- 12.Bone RC, Balk RA, Cerra FB, et al. Definitions for sepsis and organ failure and guidelines for the use of innovative therapies in sepsis. The ACCP/SCCM Consensus Conference Committee American College of Chest Physicians/Society of Critical Care Medicine. Chest. 1992;101:1644–1655. doi: 10.1378/chest.101.6.1644. [DOI] [PubMed] [Google Scholar]
- 13.Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med. 2003;31:1250–1256. doi: 10.1097/01.CCM.0000050454.01978.3B. [DOI] [PubMed] [Google Scholar]
- 14.Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3) JAMA. 2016;315:801–810. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sweeney TE, Haynes WA, Vallania F, et al. Methods to increase reproducibility in differential gene expression via meta-analysis. Nucleic Acids Res. 2017;45:e1. doi: 10.1093/nar/gkw797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khatri P, Roedder S, Kimura N, et al. A common rejection module (CRM) for acute rejection across multiple organs identifies novel therapeutics for organ transplantation. J Exp Med. 2013;210:2205–2221. doi: 10.1084/jem.20122709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen R, Khatri P, Mazur PK, et al. A meta-analysis of lung cancer gene expression identifies PTK7 as a survival gene in lung adenocarcinoma. Cancer Res. 2014;74:2892–2902. doi: 10.1158/0008-5472.CAN-13-2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li MD, Burns TC, Morgan AA, et al. Integrated multi-cohort transcriptional meta-analysis of neurodegenerative diseases. Acta Neuropathol Commun. 2014;2:93. doi: 10.1186/s40478-014-0093-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mazur PK, Reynoird N, Khatri P, et al. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 2014;510:283–287. doi: 10.1038/nature13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sweeney TE, Shidham A, Wong HR, et al. A comprehensive time-course-based multicohort analysis of sepsis and sterile inflammation reveals a robust diagnostic gene set. Sci Transl Med. 2015;7:287ra71. doi: 10.1126/scitranslmed.aaa5993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sweeney TE, Khatri P. Benchmarking sepsis gene expression diagnostics using public data. Crit Care Med. 2017;45:1–10. doi: 10.1097/CCM.0000000000002021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sweeney TE, Braviak L, Tato CM, et al. Genome-wide expression for diagnosis of pulmonary tuberculosis: A multicohort analysis. Lancet Respir Med. 2016;4:213–224. doi: 10.1016/S2213-2600(16)00048-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sweeney TE, Perumal TM, Henao R, et al. Mortality prediction in sepsis via gene expression analysis: A community approach. bioRxiv. 2016 Dec 19; doi: 10.1038/s41467-018-03078-2. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sweeney TE, Wong HR, Khatri P. Robust classification of bacterial and viral infections via integrated host gene expression diagnostics. Sci Transl Med. 2016;8:346ra91. doi: 10.1126/scitranslmed.aaf7165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bauchner H, Golub RM, Fontanarosa PB. Data sharing: An ethical and scientific imperative. JAMA. 2016;315:1237–1239. doi: 10.1001/jama.2016.2420. [DOI] [PubMed] [Google Scholar]
- 26.Mao H, Wang H, Wang B, et al. Systemic metabolic changes of traumatic critically ill patients revealed by an NMR-based metabonomic approach. J Proteome Res. 2009;8:5423–5430. doi: 10.1021/pr900576y. [DOI] [PubMed] [Google Scholar]
- 27.Stringer KA, Serkova NJ, Karnovsky A, et al. Metabolic consequences of sepsis-induced acute lung injury revealed by plasma ¹H-nuclear magnetic resonance quantitative metabolomics and computational analysis. Am J Physiol Lung Cell Mol Physiol. 2011;300:L4–L11. doi: 10.1152/ajplung.00231.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schmerler D, Neugebauer S, Ludewig K, et al. Targeted metabolomics for discrimination of systemic inflammatory disorders in critically ill patients. J Lipid Res. 2012;53:1369–1375. doi: 10.1194/jlr.P023309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Antti H, Fahlgren A, Näsström E, et al. Metabolic profiling for detection of Staphylococcus aureus infection and antibiotic resistance. PLoS One. 2013;8:e56971. doi: 10.1371/journal.pone.0056971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Blaise BJ, Gouel-Chéron A, Floccard B, et al. Metabolic phenotyping of traumatized patients reveals a susceptibility to sepsis. Anal Chem. 2013;85:10850–10855. doi: 10.1021/ac402235q. [DOI] [PubMed] [Google Scholar]
- 31.Mickiewicz B, Vogel HJ, Wong HR, et al. Metabolomics as a novel approach for early diagnosis of pediatric septic shock and its mortality. Am J Respir Crit Care Med. 2013;187:967–976. doi: 10.1164/rccm.201209-1726OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langley RJ, Tsalik EL, van Velkinburgh JC, et al. An integrated clinicometabolomic model improves prediction of death in sepsis. Sci Transl Med. 2013;5:195ra95. doi: 10.1126/scitranslmed.3005893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamisoglu K, Sleight KE, Calvano SE, et al. Temporal metabolic profiling of plasma during endotoxemia in humans. Shock. 2013;40:519–526. doi: 10.1097/SHK.0000000000000063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Seymour CW, Yende S, Scott MJ, et al. Metabolomics in pneumonia and sepsis: An analysis of the GenIMS cohort study. Intensive Care Med. 2013;39:1423–1434. doi: 10.1007/s00134-013-2935-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bos LD, Weda H, Wang Y, et al. Exhaled breath metabolomics as a noninvasive diagnostic tool for acute respiratory distress syndrome. Eur Respir J. 2014;44:188–197. doi: 10.1183/09031936.00005614. [DOI] [PubMed] [Google Scholar]
- 36.Dessì A, Marincola FC, Pattumelli MG, et al. Investigation of the ¹H-NMR based urine metabolomic profiles of IUGR, LGA and AGA newborns on the first day of life. J Matern Fetal Neonatal Med. 2014;27(Suppl 2):13–19. doi: 10.3109/14767058.2014.955674. [DOI] [PubMed] [Google Scholar]
- 37.Evans CR, Karnovsky A, Kovach MA, et al. Untargeted LC-MS metabolomics of bronchoalveolar lavage fluid differentiates acute respiratory distress syndrome from health. J Proteome Res. 2014;13:640–649. doi: 10.1021/pr4007624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fanos V, Caboni P, Corsello G, et al. Urinary (1)H-NMR and GC-MS metabolomics predicts early and late onset neonatal sepsis. Early Hum Dev. 2014;90(Suppl 1):S78–S83. doi: 10.1016/S0378-3782(14)70024-6. [DOI] [PubMed] [Google Scholar]
- 39.Mickiewicz B, Duggan GE, Winston BW, et al. Alberta Sepsis Network Metabolic profiling of serum samples by 1H nuclear magnetic resonance spectroscopy as a potential diagnostic approach for septic shock. Crit Care Med. 2014;42:1140–1149. doi: 10.1097/CCM.0000000000000142. [DOI] [PubMed] [Google Scholar]
- 40.Rogers AJ, McGeachie M, Baron RM, et al. Metabolomic derangements are associated with mortality in critically ill adult patients. PLoS One. 2014;9:e87538. doi: 10.1371/journal.pone.0087538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Su L, Huang Y, Zhu Y, et al. Discrimination of sepsis stage metabolic profiles with an LC/MS-MS-based metabolomics approach. BMJ Open Respir Res. 2014;1:e000056. doi: 10.1136/bmjresp-2014-000056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puskarich MA, Finkel MA, Karnovsky A, et al. Pharmacometabolomics of l-carnitine treatment response phenotypes in patients with septic shock. Ann Am Thorac Soc. 2015;12:46–56. doi: 10.1513/AnnalsATS.201409-415OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Garcia-Simon M, Morales JM, Modesto-Alapont V, et al. Prognosis biomarkers of severe sepsis and septic shock by 1H NMR urine metabolomics in the intensive care unit. PLoS One. 2015;10:e0140993. doi: 10.1371/journal.pone.0140993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mickiewicz B, Thompson GC, Blackwood J, et al. Alberta Sepsis Network Development of metabolic and inflammatory mediator biomarker phenotyping for early diagnosis and triage of pediatric sepsis. Crit Care. 2015;19:320. doi: 10.1186/s13054-015-1026-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Su L, Li H, Xie A, et al. Dynamic changes in amino acid concentration profiles in patients with sepsis. PLoS One. 2015;10:e0121933. doi: 10.1371/journal.pone.0121933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Richter ME, Neugebauer S, Engelmann F, et al. Biomarker candidates for the detection of an infectious etiology of febrile neutropenia. Infection. 2016;44:175–186. doi: 10.1007/s15010-015-0830-6. [DOI] [PubMed] [Google Scholar]
- 47.Kauppi AM, Edin A, Ziegler I, et al. Metabolites in blood for prediction of bacteremic sepsis in the emergency room. PLoS One. 2016;11:e0147670. doi: 10.1371/journal.pone.0147670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrario M, Cambiaghi A, Brunelli L, et al. Mortality prediction in patients with severe septic shock: A pilot study using a target metabolomics approach. Sci Rep. 2016;6:20391. doi: 10.1038/srep20391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Neugebauer S, Giamarellos-Bourboulis EJ, Pelekanou A, et al. Metabolite profiles in sepsis: Developing prognostic tools based on the type of infection. Crit Care Med. 2016;44:1649–1662. doi: 10.1097/CCM.0000000000001740. [DOI] [PubMed] [Google Scholar]
- 50.Liu Z, Yin P, Amathieu R, et al. Application of LC-MS-based metabolomics method in differentiating septic survivors from non-survivors. Anal Bioanal Chem. 2016;408:7641–7649. doi: 10.1007/s00216-016-9845-9. [DOI] [PubMed] [Google Scholar]
- 51.Stewart CJ, Nelson A, Treumann A, et al. Metabolomic and proteomic analysis of serum from preterm infants with necrotising entercolitis and late-onset sepsis. Pediatr Res. 2016;79:425–431. doi: 10.1038/pr.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mogensen KM, Lasky-Su J, Rogers AJ, et al. Metabolites associated with malnutrition in the intensive care unit are also associated with 28-day mortality. JPEN J Parenter Enteral Nutr. 2017;41:188–197. doi: 10.1177/0148607116656164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lau SK, Lee KC, Lo GC, et al. Metabolomic profiling of plasma from melioidosis patients using UHPLC-QTOF MS reveals novel biomarkers for diagnosis. Int J Mol Sci. 2016;17:307. doi: 10.3390/ijms17030307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mills KH. TLR-dependent T cell activation in autoimmunity. Nat Rev Immunol. 2011;11:807–822. doi: 10.1038/nri3095. [DOI] [PubMed] [Google Scholar]
- 55.Collins GS, Reitsma JB, Altman DG, et al. Transparent Reporting of a multivariable prediction model for Individual Prognosis or Diagnosis (TRIPOD): The TRIPOD statement. Ann Intern Med. 2015;162:55–63. doi: 10.7326/M14-0697. [DOI] [PubMed] [Google Scholar]
- 56.Bossuyt PM, Reitsma JB, Bruns DE, et al. STARD Group STARD 2015: An updated list of essential items for reporting diagnostic accuracy studies. BMJ. 2015;351:h5527. doi: 10.1136/bmj.h5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Assicot M, Gendrel D, Carsin H, et al. High serum procalcitonin concentrations in patients with sepsis and infection. Lancet. 1993;341:515–518. doi: 10.1016/0140-6736(93)90277-N. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang X, Li ZY, Zeng L, et al. Neutrophil CD64 expression as a diagnostic marker for sepsis in adult patients: A meta-analysis. Crit Care. 2015;19:245. doi: 10.1186/s13054-015-0972-z. [DOI] [PMC free article] [PubMed] [Google Scholar]