

Abstract
Background
Disrupting the costimulatory CD40-CD40L dyad reduces atherosclerosis, but can result in immune suppression. The authors recently identified small molecule inhibitors that block the interaction between CD40 and tumor necrosis factor receptor-associated factor (TRAF) 6 (TRAF-STOPs), while leaving CD40-TRAF2/3/5 interactions intact, thereby preserving CD40-mediated immunity.
Objectives
This study evaluates the potential of TRAF-STOP treatment in atherosclerosis.
Methods
The effects of TRAF-STOPs on atherosclerosis were investigated in apolipoprotein E deficient (Apoe−/−) mice. Recombinant high-density lipoprotein (rHDL) nanoparticles were used to target TRAF-STOPs to macrophages.
Results
TRAF-STOP treatment of young Apoe−/− mice reduced atherosclerosis by reducing CD40 and integrin expression in classical monocytes, thereby hampering monocyte recruitment. When Apoe−/− mice with established atherosclerosis were treated with TRAF-STOPs, plaque progression was halted, and plaques contained an increase in collagen, developed small necrotic cores, and contained only a few immune cells. TRAF-STOP treatment did not impair “classical” immune pathways of CD40, including T-cell proliferation and costimulation, Ig isotype switching, or germinal center formation, but reduced CD40 and β2-integrin expression in inflammatory monocytes. In vitro testing and transcriptional profiling showed that TRAF-STOPs are effective in reducing macrophage migration and activation, which could be attributed to reduced phosphorylation of signaling intermediates of the canonical NF-κB pathway. To target TRAF-STOPs specifically to macrophages, TRAF-STOP 6877002 was incorporated into rHDL nanoparticles. Six weeks of rHDL-6877002 treatment attenuated the initiation of atherosclerosis in Apoe−/− mice.
Conclusions
TRAF-STOPs can overcome the current limitations of long-term CD40 inhibition in atherosclerosis and have the potential to become a future therapeutic for atherosclerosis.
Key Words: atherosclerosis, drug development, immunology, inflammation, nanotechnology
Abbreviations and Acronyms: Apoe, apolipoprotein E; BMDM, bone marrow-derived macrophage; CVD, cardiovascular disease; DC, dendritic cell; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; rHDL, recombinant high-density lipoprotein; SMI, small molecule inhibitor; TRAF, tumor necrosis factor receptor-associated factor
Central Illustration
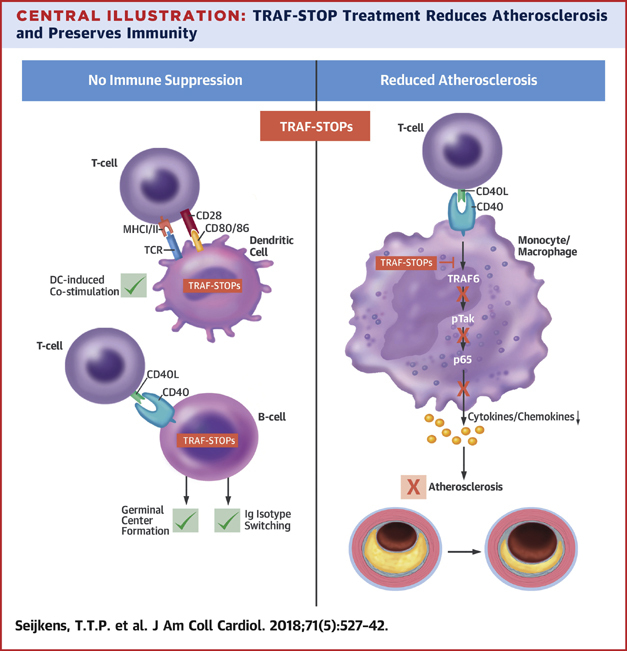
Atherosclerosis, the underlying cause of the majority of cardiovascular diseases (CVDs), is a lipid-driven inflammatory disease of the large arteries 1, 2, 3, 4. Although lipid-lowering drugs, such as 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitors and the novel proprotein convertase subtilisin/kexin 9 (PCSK9) inhibitors have helped to lower the incidence of CVD (5), a substantial part of the population still have CVD (6). Hence, additional strategies to decrease the persistent residual risk of CVD in the population are needed. In conjunction with hyperlipidemia, activation of the immune system and subsequent inflammation determines atherosclerosis progression 2, 3. Therefore, novel drug targets that block atherosclerosis-associated inflammation have great therapeutic potential. This was recently demonstrated in the CANTOS (Canakinumab Antiinflammatory Thrombosis Outcome Study) trial, in which the anti-IL1β antibody canakinumab reduced the incidence rate of the primary composite endpoint of nonfatal myocardial infarction, nonfatal stroke, or cardiovascular death (hazard ratio: 0.85; 95% confidence interval: 0.74 to 0.98; p = 0.021) in patients with a previous myocardial infarction and high residual inflammatory risk (7).
The costimulatory CD40-CD40L receptor/ligand dyad is a well-known driver of atherosclerosis and other chronic inflammatory diseases 8, 9, 10. Genetic or antibody-mediated inhibition of CD40 or CD40L strongly reduces atherosclerosis in hyperlipidemic mice 11, 12, 13, 14. However, long-term inhibition of CD40 or CD40L results in immune suppression and/or thromboembolic events, and is therefore not feasible as a therapy 10, 15, 16.
Upon activation, CD40 recruits tumor necrosis factor receptor-associated factors (TRAFs) to elicit intracellular signaling (12). The C-terminal tail of CD40 has a distal binding site that can bind TRAF2, TRAF3, and TRAF5, and a proximal binding site for TRAF6. Studies on mice with mutated CD40-TRAF binding sites revealed that CD40-TRAF6 interactions are important for atherosclerosis and restenosis 12, 17. CD40-TRAF2/3/5 interactions only play a minor role in these diseases, and are more required for CD40-associated immunity 12, 17.
We recently identified and characterized small molecule inhibitors (SMIs) that selectively block CD40-TRAF6 interactions: TRAF-STOPs (18). We found that several SMIs of the TRAF-STOP family successfully ameliorated peritonitis, sepsis, diet-induced obesity, and experimental autoimmune encephalomyelitis by reducing inflammation, without causing immune suppression 18, 19, 20, 21. Here, we further characterized the cell-type specificity and mode of action of 2 members of our TRAF-STOP family (i.e., 6677002 and 6860766) and investigated their potential for treatment of atherosclerosis.
Methods
All experiments were approved by the local animal experimentation ethics committees. C57Bl6, apolipoprotein E-deficient (Apoe−/−) (C57Bl6 background), CD40−/−, CD40-Traf wild type (Twt), CD40-T2/3/5−/−, CD40-T6−/−, Cx3cr1egfp/Apoe−/−, and OTII mice were purchased from Charles River Laboratories (Wilmington, Massachusetts), the Jackson Laboratory (Bar Harbor, Maine), or bred at a local animal facility (Maastricht University, Amsterdam Medical Center, Ludwig-Maximilians-University). Expanded methods are available in the Online Appendix.
Results
TRAF-STOP treatment hampers the initiation of atherosclerosis
To investigate the effect of TRAF-STOP treatment on the initiation of atherosclerosis, Apoe−/− mice on a normal chow diet were treated with TRAF-STOP 6877002, TRAF-STOP 6860766, or control at 10 μmol/kg/day by intraperitoneal injection for 6 weeks, starting at the age of 12 weeks, when no atherosclerotic plaques were present (Figure 1A). Treatment did not affect body weight, plasma cholesterol levels, hematologic parameters, peripheral blood leukocyte counts, or immune cell distribution in blood and lymphoid organs, and did not cause toxic effects in any of the organs analyzed (Online Figure 1). TRAF-STOP treatment reduced atherosclerotic plaque area in the aortic arch by 47% (6877002) and 67% (6860766) compared with control-treated mice (Figures 1B to 1D). Aortas from TRAF-STOP–treated mice contained relatively less fibrous cap atheromata and, correspondingly, a relative increase in early atherosclerotic plaques (intimal xanthoma and pathological intimal thickening), indicating a retarded initiation of atherosclerosis (Figures 1C and 1D). Within the plaque, the number of macrophages (Mac3+), T cells (CD3+), and neutrophils (Ly6G+) significantly decreased after TRAF-STOP treatment (Figures 1E to 1G). No changes were observed in the number of proliferating (Ki67+) or apoptotic cells (TUNEL+) in the plaque, or plaque smooth muscle cell (αSMA+) or collagen (Sirius Red+) content (Online Figure 2). Treatment with either of the 2 TRAF-STOPs thus retards early atherosclerosis development and generates atherosclerotic plaques that are low in inflammatory cells.
Figure 1.

TRAF-STOP Treatment Inhibits the Development of Atherosclerosis
(A) Twelve-week-old male Apoe−/− mice were fed a normal chow diet and were injected for 6 weeks with TRAF-STOP 6877002 (n = 13), 6860766 (n = 12) (10 μmol/kg/day in 200 μl of vehicle), or vehicle control (vehicle: phosphate-buffered saline, 0.05% Tween 80, 5% dimethylsulfoxide) (n = 15). (B) Atherosclerotic plaque area of the aortic arch had decreased after TRAF-STOP treatment. (C) Atherosclerotic plaques were classified by phenotype, intimal xanthoma (IX), pathological intimal thickening (PIT), fibrous cap atheroma (FCA), revealing less FCA after TRAF-STOP treatment. (D) Representative images (hematoxylin and eosin–stained sections) of longitudinal sections of plaques in the aortic arch (AA), including the brachiocephalic trunk (BCT), left carotid artery (LCA), and left subclavian artery (LSA) (left panel, scale bar = 2 mm), and plaques in the brachiocephalic trunk (right panel, scale bar = 100 μm) of TRAF-STOP- and control-treated Apoe−/− mice showing a decrease in plaque size after TRAF-STOP treatment. TRAF-STOP treatment decreases the amount of Mac3+ macrophages (scale bar = 70 μm) (E), CD3+ T cells (scale bar = 40 μm) (F), and Ly6G+ neutrophils (scale bar = 50 μm) (G), as shown in these representative pictures of atherosclerotic plaques of the brachiocephalic trunk. *p < 0.05. TRAF = tumor necrosis factor receptor-associated factors.
TRAF-STOP treatment prevents the progression of established atherosclerosis
Commonly, patients start receiving treatment when advanced atherosclerosis is already eminent. Therefore, we investigated the effect of TRAF-STOP treatment on established atherosclerosis in mice. Apoe−/− mice were treated with TRAF-STOP 6877002, TRAF-STOP 6860766, or control at 10 μmol/kg/day for 6 weeks, starting at the age of 22 weeks, when advanced atherosclerotic lesions were present in the aortic arch (Figure 2A). Again, treatment did not affect body weight, plasma cholesterol levels, metabolic or hematologic parameters, leukocyte counts, or immune cell composition, and did not cause abnormalities in any of the organs investigated (Online Figures 3A to 3L). Remarkably, TRAF-STOP treatment halted the progression of established atherosclerosis, as total atherosclerotic plaque area was reduced compared with control-treated mice in both the aortic arch and aortic root (Figure 2B, Online Figure 3M). After treatment with TRAF-STOP 6877002 or 6860766, atherosclerotic plaques exhibited a stable plaque phenotype. Macrophage number and macrophage proliferation (Online Figure 3N) were decreased, and plaques featured smaller necrotic cores (Figures 2C to 2E). Plaques had fewer Ly6G+ neutrophils (2.4 ± 0.6 per plaque [control] vs. 0.8 ± 0.3 per plaque [6877002; p < 0.05] vs. 1.2 ± 0.2 per plaque [6860766; p = 0.06]), fewer CD3+ T cells (4.8 ± 0.7 per plaque [control] vs. 1.6 ± 0.3 per plaque [6877002] vs. 2.3 ± 0.3 per plaque [6860766]; p < 0.05), and had an increase in collagen (Figure 2F) and αSMA+ smooth muscle cell content (2.9 ± 0.4% [control] vs. 7.2 ± 1.2% [6877002] vs. 8.0 ± 1.5% [6860766]; p < 0.05).
Figure 2.

TRAF-STOP Treatment Reduces the Progression of Established Atherosclerosis and Induces a Stable Plaque Phenotype
(A) Twenty-two-week-old male Apoe−/− mice were fed a normal chow diet and were injected for 6 weeks with TRAF-STOP 6877002 (n = 12), 6860766 (n = 11) (10 μmol/kg/day in 200 μl of vehicle), or vehicle control (phosphate-buffered saline, 0.05% Tween 80, 5% dimethylsulfoxide) (n = 11). (B) Atherosclerotic plaque area of established lesions of the aortic arch had decreased after TRAF-STOP treatment; representative pictures of plaques of the brachiocephalic trunk (scale bar = 100 μm). TRAF-STOP treatment decreases the amount of Mac3+ macrophages (scale bar = 100 μm) (C), the fraction of Ki67+ proliferating cells (scale bar = 50 μm) (D), and the lipid core content of the atherosclerotic plaque (scale bar = 100 μm) (E), and increases the percentage of SR+ plaque collagen content (scale bar = 100 μm) (F) showing that TRAF-STOP treatment not only slows down the progression of established atherosclerotic plaques, but also induces atherosclerotic plaque stabilization. HE = hematoxylin and eosin. *p < 0.05.
These results show that TRAF-STOPs 6877002 and 6860766 attenuate the progression of existing atherosclerotic plaques and induce plaque stabilization.
TRAF-STOP treatment reduces leukocyte recruitment
Although TRAF-STOP treatment did not result in changes in immune cell composition or composition of the hematopoietic stem and progenitor cell population (Online Figure 4), flow cytometry revealed that the classical, but not the non-classical monocyte population exhibited a decrease in activation markers. TRAF-STOP treatment reduced the frequency of CD40high classical monocytes, as well as the overall surface expression of CD40 (Figures 3A to 3C). Moreover, the expression of the β2-integrin subunits CD18, CD11a, and CD11b on classical monocytes was reduced, suggesting an impaired recruitment-ability (Figures 3D and 3E). Indeed, intravital microscopy of the carotid artery of 14-week-old Apoe−/− mice that were fed an atherogenic diet for 6 weeks and treated with TRAF-STOPs or control showed that 6877002 and 6860766 strongly reduced the recruitment of leukocytes to the carotid arterial wall of Apoe−/− mice (Figures 3F and 3G).
Figure 3.

TRAF-STOP Treatment Reduces Leukocyte Recruitment
(A) TRAF-STOP 6877002 reduced the frequency of CD40high classical monocytes (CD45+CD11b+Ly6G-Ly6Chigh) in the spleen, whereas nonclassical monocytes (CD45+CD11b+Ly6G-Ly6Clow) were not affected (n = 3). (B and C) TRAF-STOP 6877002 reduced the overall expression of CD40 on classical monocytes, but not on nonclassical monocytes (n = 3). (D and E) The expression of the β2-integrin subunits CD18, CD11a, and CD11b on classical monocytes was reduced by TRAF-STOP; this decrease was not observed in nonclassical monocytes (n = 3). (F) Intravital microscopy of the carotid artery in 14-week-old Cx3cr1egfp/wtApoe−/− mice fed a high-fat diet for 6 weeks (n = 5 to 8 per group). To visualize neutrophils, an antibody to Ly6G was instilled. (G) After the initial recordings, leukocytes were stained by rhodamine 6G administration. Leukocyte adhesion to the endothelium was reduced in 6877002-treated and 6860766-treated Apoe−/− mice (scale bar = 2 mm). In particular, monocyte and neutrophil adhesion was impaired in TRAF-STOP–treated mice (n = 5 to 8 per group). (H) An in vitro migration assay demonstrated that TRAF-STOP treatment of monocytes markedly reduced monocyte migration, whereas treatment of endothelial cells (ECs) with TRAF-STOP 6877002 had a minor effect. (I) HUVECs (human umbilical vein endothelial cells) were activated using the CD40-agonistic antibody G28.5. TRAF-STOP 6877002 and 6860766 reduced the expression of the cell adhesion molecules intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1 (n = 3 experiments). (J and K) Ex vivo 2-photon microscopy in aortas of Apoe−/− mice (n = 6 to 9) revealed that the en face expression of VCAM did not differ between control and TRAF-STOP 6877002-treated endothelium. VCAM-1 expression was analyzed using labeled-antibodies. *p < 0.05, **p < 0.01, ***p < 0.001. CCL2 = C-C motif chemokine ligand 2; IL = interleukin; MFI = mean fluorescence intensity; TNF = tumor necrosis factor.
In vitro monocyte migration assays using human endothelial cells and human monocytes confirmed the decrease in monocyte recruitment upon TRAF-STOP treatment (Figure 3H). When monocytes were treated with TRAF-STOP 6877002 or 6860766, recruitment was reduced by 30.5% and 49.9%, respectively (Figure 3H). However, when the endothelial cells were treated with TRAF-STOPs, monocyte recruitment was only slightly reduced by 6877002 (Figure 3H). A similar phenomenon was observed in human umbilical vein endothelial cells: TRAF-STOP treatment resulted in a small, but significant, reduction in vascular cell adhesion molecule (VCAM)-1 and intercellular adhesion molecule (ICAM)-1 expression, but did not affect other endothelial activation markers such as interleukin (IL)-6, IL-8, tumor necrosis factor (TNF)-α, and C-C motif chemokine ligand 2 (CCL2) (Figure 3I). Moreover, in ex vivo TRAF-STOP–treated aortas of Apoe−/− mice, VCAM-1 expression was not altered (Figures 3J and 3K), suggesting that TRAF-STOPs predominantly affect myeloid cells.
TRAF-STOPs reduce macrophage activation
TRAF-STOP treatment attenuates the inflammatory propensity of macrophages. The CD40-induced expression of TNF-α, IL-1β, IL-6, IL-10, IL-12, and inducible nitric oxide synthase (iNOS) was reduced in TRAF-STOP 6877002- and 6860766-treated bone marrow-derived macrophages (BMDMs) (Figure 4A). In accordance, the chemokine-chemokine receptor pairs CCL2-CCR2 and CCL5-CCR5 exhibited a similar decrease (Figure 4B), which was consistent with a reduction in CD40-induced chemotaxis of BMDMs (Figure 4C).
Figure 4.

TRAF-STOP Target Macrophage Activation
TRAF-STOP 6877002 and 6860766 reduced the CD40-induced mRNA expression of cytokines (A) and chemokines (B) in BMDMS (bone marrow-derived macrophages) (n = 3 experiments with triplicates). (C) TRAF-STOPs hampered the CD40-induced migration of BMDMs in a chemotaxis assays (n = 12). CD36 expression (n = 10) (D) and Bodipy–oxidized low-density lipoprotein uptake (n = 5) (E) in BMDMs decreased after TRAF-STOP treatment, as measured by flow cytometry. (F) Foam cell formation, indicated by Oil Red O staining, was reduced after TRAF-STOP treatment (n = 5). Antibody-mediated activation of CD40 induced CCL2 expression in CD40+/+, CD40-Twt, and CD40-T2/3/5−/− BMDMs, but not in CD40−/− and CD40-T6−/− macrophages (n = 3 experiments). (G) TRAF-STOPs inhibited the CD40-induced expression of CCL2 in CD40+/+, CD40-Twt, and CD40-T2/3/5−/− macrophages and had no additional effect in CD40−/− and CD40-T6−/− macrophages. (H) Western blot analysis of the CD40-induced signaling intermediates of the canonical and noncanonical nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways and ERK1/2 signaling in BMDMs (n = 2 to 3). Both TRAF-STOPs reduced the phosphorylation of Tak1 (I) and the NF-κB p65 subunit (J) upon antibody-mediated activation of CD40 in BMDM. TRAF-STOPs did not affect NF-κB2 p52 levels (K) and phosphorylation of ERK1/2 (L). *p < 0.05, **p < 0.01, ***p < 0.001. iNOS = inducible nitric oxide synthase; mRNA = messenger ribonucleic acid; other abbreviations as in Figure 3.
In addition to its anti-inflammatory effects, TRAF-STOPs reduced CD36 expression (Figure 4D), resulting in a decreased uptake of oxidized low-density lipoprotein (Figure 4E) and a reduction in macrophage foam cell formation (Figure 4F). Neither phagocytosis or efferocytosis nor the expression of the reverse cholesterol transporters ABCA1 or ABCG1 were affected by TRAF-STOP treatment (Online Figure 5). These data suggest that TRAF-STOP treatment retards foam cell formation by decreasing CD36-mediated lipoprotein uptake.
To confirm that TRAF-STOPs preferentially abolish CD40-TRAF6 over CD40-TRAF2/3/5 signaling, we activated BMDMs of CD40−/−, CD40+/+, CD40-Traf wild type (Twt), CD40-T2/3/5−/−, and CD40-T6−/− mice (15) with CD40-agonistic antibodies after pre-incubation with the respective TRAF-STOPs. After CD40 activation, CD40+/+, CD40-Twt, and CD40-T2/3/5−/− macrophages displayed a high expression of CCL2. Yet, CCL2 was down-regulated in CD40−/− and CD40-T6−/− macrophages, demonstrating a critical role of CD40-TRAF6 interactions in regulating CCL2 levels (Figure 4G). Upon TRAF-STOP treatment, CCL2 levels decreased in CD40+/+, CD40-Twt, and CD40-T2/3/5−/− macrophages, suggesting that TRAF-STOPs prevent the CD40-TRAF6–mediated increase in CCL2 (Figure 4G). Moreover, TRAF-STOP treatment did not further decrease CCL2 expression in CD40−/− and CD40-TRAF6−/− macrophages (Figure 4G), indicating that TRAF-STOPs abolish CD40-TRAF6, and not CD40-TRAF2/3/5, interactions.
Next, we analyzed the effects of 6877002 and 6860766 on the activation of CD40-induced signaling intermediates of the canonical and noncanonical nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways and ERK1/2 signaling in BMDMs. Both TRAF-STOPs reduced the CD40-induced phosphorylation of Tak1 and NF-κB p65 (Figures 4H to 4J), whereas the levels of NF-κB2 p52 and the phosphorylation of ERK1/2 were not affected (Figures 4H, 4K, and 4L). These results indicate that TRAF-STOPs specifically inhibit the canonical NF-κB pathway, and not the noncanonical NF-κB pathway or ERK1/2 signaling.
To unravel further transcriptional changes upon TRAF-STOP treatment, BMDMs from C57Bl6 mice were treated with either of the 2 TRAF-STOPs and activated with a CD40 agonistic antibody. As expected, transcriptional analysis (Illumina, San Diego, California) comparing TRAF-STOP–treated BMDMs with control-treated cells and subsequent Ingenuity Pathway Analysis (Qiagen, Hilden, Germany) revealed “immune reactions” as the most highly affected pathways for both TRAF-STOPs (Online Figures 6A and 6B). Moreover, other top-ranking pathways were cholesterol biosynthesis (6860766 and 6877002), and/or cell cycle progression (only 6877002) (Online Figures 6A and 6B, Online Tables 1 and 2). Validation by quantitative polymerase chain reaction confirmed that TRAF-STOPs reduce the expression of cholesterol biosynthesis genes, including acat2, hmgcr, cyp51, sqle, mvd, and dhcr24 (Online Figures 6C and 6D).
Because Ingenuity Pathway Analysis revealed that TRAF-STOP 6877002, but not 6860766, affected cell cycle–related pathways in BMDMs (Online Figures 6A and 6B, Online Tables 1 and 2), we further analyzed this phenomenon. TRAF-STOP 6877002, but not 6860766, did reduce macrophage proliferation in atherosclerotic plaques, as measured by Ki67 immunohistochemistry, and reduced the expression of Ki67 in BMDMs, suggesting reduced proliferation of these cells (Figures 2C and 2D, Online Figures 3N and 6E). However, TRAF-STOP 6877002 also increased the expression of cyclin D1, the regulator of CDK4 and CDK6, crucial for G1/S transition (Online Figure 6E). These results suggest that TRAF-STOP 6877002 may affect certain aspects of cell cycle progression, but that these effects are minor compared to the effects on inflammation.
The different effects on cell cycle parameters suggest that TRAF-STOP 6877002 and 6860766 have a slightly different mode of action (Online Figures 6A and 6B). Using several docking strategies and binding studies using mutant TRAF6 protein, we could indeed prove that 6877002 and 6860766 interact with the TRAF6 C-terminus in a different conformation (Online Figure 7, Online Table 3).
TRAF-STOP treatment does not impair classical CD40-mediated immune responses
Antibody-mediated inhibition of CD40 results in immunosuppression because it impairs CD40-mediated immune responses, including immunoglobulin isotype switching, germinal center formation, dendritic cell (DC)-mediated costimulation, and lymphocyte activation (12). We investigated whether TRAF-STOP treatment causes any of these CD40-inhibition–associated side effects.
Immunoglobulin levels were analyzed in the plasma of 28-week-old Apoe−/− mice that were treated with TRAF-STOPs for 6 weeks. No differences in IgM, IgG, and IgG subtype levels were detected (Figures 5A and 5B). Histological analysis of the spleen showed normal germinal center formation in TRAF-STOP–treated animals (Figure 5C). To address the effect of TRAF-STOPs on antigen-specific immunoglobulin responses, C57Bl6 mice were vaccinated with myelin oligodendrocyte glycoprotein peptide (MOG35-55) and immunoglobulin levels were measured. TRAF-STOP treatment did not affect IgM, IgG, IgG1, IgG2b, IgG2c, IgG3, or antigen-specific anti-MOG IgG levels (Figures 5D and 5E), indicating that TRAF-STOP treatment does not interfere with the role of CD40 signaling in Ig isotype switching.
Figure 5.

TRAF-STOPs Do Not Impair Classical CD40-Mediated Immune Responses
(A) TRAF-STOPs did not affect immunoglobulin M (IgM), IgG levels in the plasma of 28-week-old Apoe−/− mice that were treated for 6 weeks (n = 11 to 12). (B) IgG2b, IgG2c, and IgG3 ratios were not altered by TRAF-STOPs. Immunoglobulin levels were determined by enzyme-linked immunosorbent assay (ELISA) (n = 11 to 12). (C) Representative images (hematoxylin and eosin–stained sections and immunohistochemical staining of CD21 and Bcl6) of splenic germinal centers demonstrated that TRAF-STOPs did not affect germinal center formation in 28-week-old Apoe−/− mice that were treated for 6 weeks (scale bar = 100 μm) (n = 12 to 15 per group). (D and E) C57Bl6 mice were vaccinated with myelin oligodendrocyte glycoprotein peptide (MOG35-55) and antigen-specific immunoglobulin levels were determined by ELISA (n = 6 to 8 per group). TRAF-STOP treatment did not affect the levels of IgM and IgG, or the IgG1, IgG2b, IgG2c, and IgG3 ratios. Antigen-specific anti-MOG IgG levels at 16 and 28 days after vaccination were not affected by TRAF-STOP treatment (n = 8). (F) Coculture of TRAF-STOP–treated, ovalbumin peptide-loaded dendritic cells (DCs) and CFSE-labeled, OTII transgenic CD4+ T cells demonstrated that T-cell proliferation was not affected. T-cell proliferation was assessed by flow cytometry (n = 6). (G) Jurkat T cells were activated using CD3/CD28 beads. TRAF-STOP treatment did not affect T-cell cytokine expression nor expression of costimulatory molecules. (H) CA46 B-cells were activated using the CD40-agonistic antibody G28.5 and treated using TRAF-STOP 6877002 and 6860766. No effects of TRAF-STOP on B-cell activation could be detected (n = 3 experiments). (I) C57Bl6 mice (n = 8 per group) were treated with the TRAF-STOPs, and peritonitis was induced by intraperitoneal injection of Escherichia coli. Sixteen hours after inoculation the bacterial outgrowth in various tissues was analyzed. TRAF-STOP did not increase the bacterial outgrowth in the peritoneal lavage fluid, lungs, liver and blood. *p < 0.05, **p < 0.01. Abbreviations as in Figures 3 and 4.
To elucidate the effects of TRAF-STOPs on DC-mediated costimulation of T cells, ovalbumin peptide (OVA323)-loaded DCs were matured, treated with 6877002 or 6860766, and cocultured with OTII-transgenic CD4+ T cells. TRAF-STOPs did not affect antigen-specific T-cell proliferation (Figure 5F), indicating that DCs maintained their antigen presenting capabilities and were able to induce T-cell proliferation.
To test the effects of TRAF-STOPs on lymphoid cell activation, T cells (Jurkat cells) and B cells (CA46 cells) were incubated with TRAF-STOPs and, depending on the cell type, activated with a CD40 agonistic antibody (G28.5) and/or lipopolysaccharide, TNFα, or CD3/CD28 beads. This revealed that TRAF-STOPs had only minor effects on the activation status of T and B cells (Figures 5G and 5H).
Because interference with the CD40 pathway may also affect acute antibacterial immune responses, TRAF-STOP–treated C57Bl6 mice were subjected to Escherichia coli-induced peritonitis and bacterial outgrowth was analyzed in the peritoneal lavage fluid, lungs, liver, and blood. TRAF-STOP 6860766 reduced bacterial load in the lungs and peritoneal fluid, whereas 6877002 had no effect (Figure 5I), indicating that anti-bacterial responses were not affected.
Collectively, these experiments demonstrate that TRAF-STOPs do not impair the classical CD40-mediated immune responses, indicating that these inhibitors have the potential to overcome the current limitations of CD40 targeting antibodies.
Macrophage-specific targeting of TRAF-STOPs via recombinant high-density lipoprotein nanotherapy
Although the TRAF-STOPs predominantly target myeloid cells, and do not seem to impair CD40-mediated lymphocyte functions, we aimed to even improve TRAF-STOP’s in vivo specificity for macrophages by incorporating 6877002 in recombinant high-density lipoprotein (rHDL) nanoparticles (Figure 6A) 17, 18.
Figure 6.

rHDL-6877002 Nanoparticles Target Macrophages and Reduce the Initiation of Atherosclerosis
(A) A schematic representation of rHDL-6877002, which was created by combining human apoA-I, lipids (DMPC [1,2-dimyristoyl-sn-glycero-3-phosphorylcholine] and MHPC [1-myristoyl-2-hydroxy-sn-glycero-3-phosphocholine]) and TRAF-STOP 6877002. (B) Near infrared fluorescence (NIRF) imaging of DiR-labeled rHDL-6877002 distribution in mouse organs (n = 3), showing accumulation of DiR-rHDL-6877002 in the liver and spleen. (C) NIRF imaging of DiR-labeled rHDL-6877002 distribution in mouse aorta (n = 3), showing accumulation of DiR-rHDL-6877002 in the aortic root area, a region containing atherosclerosis. (D) Flow cytometry data of blood, spleen, and whole mouse aortas (n = 8) with DiO-labeled rHDL-6877002, showing rHDL accumulation in macrophages and monocytes, especially in the aorta. (E) Sections of the aortic root containing atherosclerotic plaques, showing DiO-labeled rHDL-6877002 (red) and F4/80+ macrophages (green), revealing that rHDL-6877002 is being taken up by plaque macrophages (colocalization, yellow). Plaque volume (scale bar = 200 μm) (F) and the number of plaque macrophages (scale bar = 100 μm) (G) decrease after 6 weeks of rHDL-6877002 treatment (n = 8) compared with rHDL treatment (n = 8). *p < 0.05. ApoA = apolipoprotein A; rHDL = recombinant high-density lipoprotein.
Twenty-four hours after injection of DiR-labeled rHDL-6877002 nanoparticles, we were able to show localization of rHDL-6877002 in the liver and spleen (Figure 6B), as well as in the aortic sinus region of the aorta (Figure 6C). Flow cytometric analysis of blood, spleen, and aorta showed that DiO-labeled rHDL-6877002 was predominantly present in macrophages, and to a lesser extent in monocytes and neutrophils (Figure 6D), with the highest fluorescence signal in aortic macrophages (Figure 6D). Histological analysis confirmed the presence of DiO-rHDL-6877002 in atherosclerotic plaque macrophages (Figure 6E).
Subsequently, Apoe−/− mice that were fed a normal chow diet were treated twice a week with rHDL-6877002 (10 μmol/kg) for the duration of 6 weeks, starting at the age of 12 weeks (Figure 6A). Besides a small increase in cholesterol levels and total leukocyte counts, no abnormalities in body weight, hematological parameters, and leukocyte subsets were observed (Online Figures 8A to 8F). Although pronounced uptake of rHDL-6877002 was observed in the liver and spleen, we did not find any signs of hepatotoxicity or functional hyposplenism (Online Figures 8G to 8J).
Atherosclerotic plaques were small, and only intimal xanthomas and few pathological intimal thickenings could be observed in the aortic root and aortic arch region. However, plaque volume in the aortic root had significantly decreased upon rHDL-6877002 treatment (Figure 6F), and plaques contained fewer macrophages (Figure 6G). In this initial stage, the number of CD3+ T cells, αSMC and collagen content, proliferation, and apoptosis were not affected. Compared with the administration of the bare TRAF-STOPs, this nanomedicine-based strategy allowed us to reduce the treatment frequency and cumulative dosage.
Discussion
Here, we show that blocking CD40-TRAF6 interactions by SMI (TRAF-STOP) treatment strongly reduces atherosclerosis by preventing activation of classical monocytes, leukocyte recruitment, and macrophage activation and migration in the arterial wall (Central Illustration). Importantly, TRAF-STOP treatment not only retarded the initiation of atherosclerosis, but also reduced the progression of (established) atherosclerosis and stabilized existing atherosclerotic plaques.
Central Illustration.

TRAF-STOP Treatment Reduces Atherosclerosis and Preserves Immunity
Binding of CD40L to CD40 results in the recruitment of tumor necrosis factor receptor-associated factors (TRAFs), and propagation of signaling. The C-terminal part of CD40 contains a TRAF2/3/5 binding domain and a TRAF6 binding domain that both induce nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) transcription. CD40-TRAF6 interactions are the predominant signal transduction route in macrophages, and are important in atherosclerosis, whereas CD40-TRAF2/3/5 interactions are important in CD40 driven immunity in other cell types, including dendritic cells (DCs) and B cells. Small molecule inhibitors of the CD40-TRAF6 interactions (TRAF-STOPs) inhibit CD40-induced activation of the canonical NF-κB pathway and subsequent cytokine and chemokine production. TRAF-STOPs are particularly effective in monocytes/macrophages and hamper monocyte recruitment, macrophage activation, and foam cell formation. Consequently, both de novo and established atherosclerosis are reduced upon TRAF-STOP treatment. By keeping CD40-TRAF2/3/5 interactions intact, CD40-mediated immunity is preserved, and immune suppressive side effects are prevented. MHC = major histocompatibility complex; TCR = T cell receptor.
The costimulatory receptor/ligand dyad CD40-CD40L is highly expressed in human atherosclerotic lesions, and plays a pivotal role in atherosclerosis by orchestrating the inflammatory response underlying plaque development and progression 10, 22. The therapeutic potential of CD40-CD40L targeting strategies is emphasized by the observation that genetic or antibody-mediated inhibition of CD40L in Apoe−/− mice induced a plaque phenotype that is low in inflammatory cell content and high in fibrosis, representing the clinically favorable stable phenotype 11, 12, 13, 14, 23. However, until now, no treatment options blocking CD40-CD40L in CVD were available. Clinical trials, evaluating the efficacy of anti-CD40L antibodies, were halted due to the occurrence of thromboembolic events 9, 16. Although antagonistic CD40 antibodies are available, long-term antibody-mediated inhibition of CD40 results in immune suppression and is therefore not feasible for the treatment of chronic diseases like atherosclerosis 15, 24.
Our newly developed TRAF-STOPs harbor the potential for utilization in chronic inflammatory diseases such as atherosclerosis, as classical CD40-mediated immune responses including antigen-specific T-cell proliferation and immunoglobulin isotype switching are not impaired 15, 24. We observed no thromboembolic complications in TRAF-STOP–treated mice, as these SMIs do not interfere with the CD40L-αIIbβ3 interaction and hence do not induce destabilization of arterial thrombi (16).
TRAF-STOPs have the inherent characteristic of reducing the recruitment and chemokine and cytokine release of myeloid cells, while leaving the remainder of the vascular-associated CD40-expressing cells largely unaffected. This inherent cell-type specificity reflects the CD40-TRAF interactions that occur in the different cell types. In monocytes and macrophages, the interaction of CD40 with TRAF6, but not with TRAF2 is essential to induce proinflammatory pathways (25). In monocytes/macrophages, TRAF6 acts as a critical adaptor of both the Src/ERK1/2 and IKK/NFκB signaling pathways (25). In B cells, CD40-TRAF6 interactions promote canonical NF-κB signaling via activation of transforming growth factor (TGF)-β activated kinase (TAK), and CD40-TRAF6 interactions are needed for affinity maturation and the generation of long-lived plasma cells (15). However, for normal B-cell function, CD40-TRAF2 interactions are more crucial (24). Mouse studies indeed showed that CD40-TRAF2, but not CD40-TRAF3 or -6 interactions are essential for germinal center formation, and that CD40-TRAF2 activation and TRAF3 degradation are more crucial for CD40-induced B-cell activation than CD40-TRAF6 interactions 15, 26. The divergent CD40-TRAF interactions in B cells may thus explain the limited effects of our CD40-TRAF6 SMI on B-cell activation, immunoglobulin isotype switching, and germinal center formation observed in our study. CD40 is also expressed on endothelial cells, and its activation induces expression of cell adhesion molecules and promotes leukocyte adhesion 10, 17. Upon CD40 activation, aortic endothelial cells produce CX3CL1 and TNF-α, and this process is dependent on both CD40-TRAF2 and CD40-TRAF6 interactions (27). Interestingly, TRAF3 can prevent CD40-mediated endothelial cell activation (28). Our data show that TRAF-STOP treatment of endothelial cells can induce a minor reduction in VCAM-1 and ICAM-1 expression as well as macrophage recruitment in vitro, but does not affect VCAM-1 expression ex vivo.
Although TRAF-STOPs intrinsically target macrophage function and have no effects on classical CD40 immune responses, we cannot exclude the possibility that systemic long-term administration will have undesired effects on other cell types. Therefore, we exploited the possibility of using a nanotherapeutic approach; TRAF-STOP 6877002 was packaged into rHDL, which has a high affinity for macrophages, and is not taken up by lymphoid cells 29, 30. Indeed, rHDL-6877002 specifically targets macrophages, and prevents plaque initiation, requiring less frequent drug administration and a lower cumulative dosage compared with TRAF-STOP treatment. These results show that nanotherapeutic delivery of TRAF-STOPs is promising.
Study Limitations
Here we showed that TRAF-STOPs successfully reduced both de novo and established atherosclerosis in Apoe-/- mice. Further pharmacological and toxicological studies are required before the “first in human” application of TRAF-STOPs.
Conclusions
We have identified and characterized a group of SMI of the CD40-TRAF6 interaction that successfully reduce (existing) atherosclerosis by hampering chemokine-mediated leukocyte recruitment to the arterial wall and inhibiting cytokine secretion by macrophages. These inhibitors will undoubtedly require further development and refinement before they can be applied in a clinical setting. However, our newly identified TRAF-STOPs can overcome the current limitations of long-term CD40 and CD40L inhibition in atherosclerosis and potentially in other inflammatory diseases.
Perspectives.
COMPETENCY IN MEDICAL KNOWLEDGE: Blocking CD40-CD40L interactions can reduce atherosclerosis and converts vulnerable plaques into stable plaques in laboratory models, but is not therapeutically viable because inhibition compromises long-term systemic immune responses. Two small molecule inhibitors that selectively block CD40-TRAF6 interactions (TRAF-STOPs) reduce established atherosclerosis but preserve immunity and have potential therapeutic value.
TRANSLATIONAL OUTLOOK: Clinical studies are needed to establish the safety and efficacy of targeted delivery of selective CD40-TRAF6 inhibitors to plaque macrophages through high-density lipoprotein nanoparticles.
Footnotes
This work was supported by the Netherlands CardioVascular Research Initiative: the Dutch Heart Foundation, Dutch Federation of University Medical Centres, the Netherlands Organisation for Health Research and Development, and the Royal Netherlands Academy of Sciences for the GENIUS project “Generating the best evidence-based pharmaceutical targets for atherosclerosis” (CVON2011-19). This study was also supported by the Deutsche Forschungs Gemeinschaft (SFB 1123 to Drs. Lutgens, Gerdes, Soehnlein, Megens, and Weber; and INST 409/97-1 FUGG to Drs. Weber, Megens, and Soehnlein), the Netherlands Organization for Scientific Research (NWO) (VIDI grant to Drs. Soehnlein and Mulder, and VICI grant 016.130.676 to Dr. Lutgens and Dr. Weber), the Dutch Heart Foundation (Dr. E. Dekker MD-grant to Dr. Seijkens), the EU (H2020-PHC-2015-667673, REPROGRAM to Drs. Lutgens, de Winther, Seijkens, and Atzler), National Institutes of Health (RO1 HL118440 and RO1 HL125703 to Dr. Mulder), the European Research Council (ERC consolidator grant CD40-INN 681492 to Dr. Lutgens), ERC advanced grant (to Dr. Weber), and the German Centre for Cardiovascular Research (DZHK) high-risk high-volume (HRHV) grant to Drs. Lutgens, Atzler, and Weber. Dr. Boon is an employee of Bioceros. Dr. Duivenvoorden has received a speakers honorarium from Vifor Fresenius. All other authors have reported that they have no relationships relevant to the content of this paper to disclose. Drs. van Tiel, Kusters, and Atzler contributed equally to this work and are joint second authors. Gwen Randolph, MD, served as Guest Editor for this paper.
Appendix
For an expanded Methods section, and supplemental figures and tables, please see the online version of this paper.
Appendix
References
- 1.Cybulsky M.I., Cheong C., Robbins C.S. Macrophages and dendritic cells: partners in atherogenesis. Circ Res. 2016;118:637–652. doi: 10.1161/CIRCRESAHA.115.306542. [DOI] [PubMed] [Google Scholar]
- 2.Legein B., Temmerman L., Biessen E.A., Lutgens E. Inflammation and immune system interactions in atherosclerosis. Cell Mol Life Sci. 2013;70:3847–3869. doi: 10.1007/s00018-013-1289-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libby P., Lichtman A.H., Hansson G.K. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. 2013;38:1092–1104. doi: 10.1016/j.immuni.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabas I., Bornfeldt K.E. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. 2016;118:653–667. doi: 10.1161/CIRCRESAHA.115.306256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ridker P.M., Revkin J., Amarenco P., et al. Cardiovascular efficacy and safety of bococizumab in high-risk patients. N Engl J Med. 2017;376:1527–1539. doi: 10.1056/NEJMoa1701488. [DOI] [PubMed] [Google Scholar]
- 6.Mihaylova B., Emberson J., Blackwell L., et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–590. doi: 10.1016/S0140-6736(12)60367-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ridker P.M., Everett B.M., Thuren T., et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. doi: 10.1056/NEJMoa1707914. [DOI] [PubMed] [Google Scholar]
- 8.Elgueta R., Benson M.J., de Vries V.C., Wasiuk A., Guo Y., Noelle R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol Rev. 2009;229:152–172. doi: 10.1111/j.1600-065X.2009.00782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boumpas D.T., Furie R., Manzi S., et al. A short course of BG9588 (anti-CD40 ligand antibody) improves serologic activity and decreases hematuria in patients with proliferative lupus glomerulonephritis. Arthritis Rheum. 2003;48:719–727. doi: 10.1002/art.10856. [DOI] [PubMed] [Google Scholar]
- 10.Lutgens E., Lievens D., Beckers L., Donners M., Daemen M. CD40 and its ligand in atherosclerosis. Trends Cardiovasc Med. 2007;17:118–123. doi: 10.1016/j.tcm.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Lutgens E., Gorelik L., Daemen M.J., et al. Requirement for CD154 in the progression of atherosclerosis. Nat Med. 1999;5:1313–1316. doi: 10.1038/15271. [DOI] [PubMed] [Google Scholar]
- 12.Lutgens E., Lievens D., Beckers L., et al. Deficient CD40-TRAF6 signaling in leukocytes prevents atherosclerosis by skewing the immune response toward an antiinflammatory profile. J Exp Med. 2010;207:391–404. doi: 10.1084/jem.20091293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mach F., Schonbeck U., Sukhova G.K., Atkinson E., Libby P. Reduction of atherosclerosis in mice by inhibition of CD40 signalling. Nature. 1998;394:200–203. doi: 10.1038/28204. [DOI] [PubMed] [Google Scholar]
- 14.Schonbeck U., Sukhova G.K., Shimizu K., Mach F., Libby P. Inhibition of CD40 signaling limits evolution of established atherosclerosis in mice. Proc Natl Acad Sci U S A. 2000;97:7458–7463. doi: 10.1073/pnas.97.13.7458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahonen C., Manning E., Erickson L.D., et al. The CD40-TRAF6 axis controls affinity maturation and the generation of long-lived plasma cells. Nature immunology. 2002;3:451–456. doi: 10.1038/ni792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andre P., Prasad K.S., Denis C.V., et al. CD40L stabilizes arterial thrombi by a beta3 integrin–dependent mechanism. Nat Med. 2002;8:247–252. doi: 10.1038/nm0302-247. [DOI] [PubMed] [Google Scholar]
- 17.Donners M.M., Beckers L., Lievens D., et al. The CD40-TRAF6 axis is the key regulator of the CD40/CD40L system in neointima formation and arterial remodeling. Blood. 2008;111:4596–4604. doi: 10.1182/blood-2007-05-088906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zarzycka B., Seijkens T., Nabuurs S.B., et al. Discovery of small molecule CD40-TRAF6 inhibitors. Journal of chemical information and modeling. 2015;55:294–307. doi: 10.1021/ci500631e. [DOI] [PubMed] [Google Scholar]
- 19.Chatzigeorgiou A., Seijkens T., Zarzycka B., et al. Blocking CD40-TRAF6 signaling is a therapeutic target in obesity-associated insulin resistance. Proc Natl Acad Sci U S A. 2014;111:2686–2691. doi: 10.1073/pnas.1400419111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van den Berg S.M., Seijkens T.T., Kusters P.J., et al. Blocking CD40-TRAF6 interactions by small-molecule inhibitor 6860766 ameliorates the complications of diet-induced obesity in mice. Int J Obes (Lond) 2015;39:782–790. doi: 10.1038/ijo.2014.198. [DOI] [PubMed] [Google Scholar]
- 21.Aarts S., Seijkens T.T.P., Kusters P.J.H., et al. Inhibition of CD40-TRAF6 interactions by the small molecule inhibitor 6877002 reduces neuroinflammation. J Neuroinflammation. 2017;14:105. doi: 10.1186/s12974-017-0875-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mach F., Schonbeck U., Libby P. CD40 signaling in vascular cells: a key role in atherosclerosis? Atherosclerosis. 1998;137 Suppl:S89–S95. doi: 10.1016/s0021-9150(97)00309-2. [DOI] [PubMed] [Google Scholar]
- 23.Lutgens E., Cleutjens K.B., Heeneman S., Koteliansky V.E., Burkly L.C., Daemen M.J. Both early and delayed anti-CD40L antibody treatment induces a stable plaque phenotype. Proc Natl Acad Sci U S A. 2000;97:7464–7469. doi: 10.1073/pnas.97.13.7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rickert R.C., Jellusova J., Miletic A.V. Signaling by the tumor necrosis factor receptor superfamily in B-cell biology and disease. Immunol Rev. 2011;244:115–133. doi: 10.1111/j.1600-065X.2011.01067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukundan L., Bishop G.A., Head K.Z., Zhang L., Wahl L.M., Suttles J. TNF receptor-associated factor 6 is an essential mediator of CD40-activated proinflammatory pathways in monocytes and macrophages. J Immunol. 2005;174:1081–1090. doi: 10.4049/jimmunol.174.2.1081. [DOI] [PubMed] [Google Scholar]
- 26.Lin W.W., Hildebrand J.M., Bishop G.A. A complex relationship between TRAF3 and non-canonical NF-kappaB2 activation in B lymphocytes. Front Immunol. 2013;4:477. doi: 10.3389/fimmu.2013.00477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Greene J.A., Portillo J.A., Lopez Corcino Y., Subauste C.S. CD40-TRAF signaling upregulates CX3CL1 and TNF-alpha in human aortic endothelial cells but not in retinal endothelial cells. PloS One. 2015;10 doi: 10.1371/journal.pone.0144133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Urbich C., Mallat Z., Tedgui A., Clauss M., Zeiher A.M., Dimmeler S. Upregulation of TRAF-3 by shear stress blocks CD40-mediated endothelial activation. J Clin Invest. 2001;108:1451–1458. doi: 10.1172/JCI13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Duivenvoorden R., Tang J., Cormode D.P., et al. A statin-loaded reconstituted high-density lipoprotein nanoparticle inhibits atherosclerotic plaque inflammation. Nat Commun. 2014;5:3065. doi: 10.1038/ncomms4065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang J., Baxter S., Menon A., et al. Immune cell screening of a nanoparticle library improves atherosclerosis therapy. Proc Natl Acad Sci U S A. 2016;113 doi: 10.1073/pnas.1609629113. E6731–e40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.